

Abstract
The cellular response to hypoxia is orchestrated by HIF-1, a heterodimeric transcription factor composed of an α and a β subunit that enables cell survival under low oxygen conditions by altering the transcription of over 300 genes. There is significant evidence that inhibition of HIF-1 would be beneficial for cancer therapy. We recently reported a cyclic hexapeptide that inhibits the HIF-1α/HIF-1β protein–protein interaction in vitro and prevents HIF-1-mediated hypoxia-response signaling in cells. This cyclic peptide was identified from a library of 3.2 × 106 members generated using SICLOPPS split-intein mediated protein splicing. With a view to demonstrating the potential for encoding the production of a therapeutic agent in response to a disease marker, we have engineered human cells with an additional chromosomal control circuit that conditionally encodes the production of our cyclic peptide HIF-1 inhibitor. We demonstrate the conditional production of our HIF-1 inhibitor in response to hypoxia, and its inhibitory effect on HIF-1 dimerization and downstream hypoxia-response signaling. These engineered cells are used to illustrate the synthetic lethality of inhibiting HIF-1 dimerization and glycolysis in hypoxic cells. Our approach not only eliminates the need for the chemical synthesis and targeted delivery of our HIF-1 inhibitor to cells, it also demonstrates the wider possibility that the production machinery of other bioactive compounds may be incorporated onto the chromosome of human cells. This work demonstrates the potential of sentinel circuits that produce molecular modulators of cellular pathways in response to environmental or cellular disease stimuli.
Keywords: HIF-1, hypoxia, protein−protein interaction, cellular reprogramming, cyclic peptide, SICLOPPS
Introduction
Transcription factors are master regulators of cellular fate and function that orchestrate a coordinated response to a variety physiological stimuli. Exogenous modulation of transcription factor activity therefore holds much therapeutic potential, and is a critical tool for deciphering complex cellular networks. The absolute requirement of assembly for function means that protein–protein interaction (PPI) inhibition is the optimal strategy for intervention, but transcription factors are considered to be one of the most chemically intractable targets in drug discovery.1 More generally, the challenge of identifying PPI inhibitors means that the majority of tools employed for studying these complexes are nucleic acid-based (e.g., siRNA, or CRISPR) and function to eliminate the targeted protein from the cell. Despite their widespread use, these methods have several drawbacks for studying PPIs; removal of a protein from a system eliminates all of its known and unknown interactions and functions. Thus, an observed phenotype may not necessarily be attributed to a given PPI. In cases where validated PPI inhibitors are available, the need for chemical synthesis and intracellular delivery of such compounds places limitations on their adaptation and use. We sought an alternative approach by introducing the components necessary to synthesize a PPI inhibitor onto the genome of a human cell line. Under this scenario, the intracellular production of a non-native molecule is selectively induced by one or more disease specific signals via expression of the machinery needed for its production. This approach would have the advantage that it eliminates the need for chemical synthesis and intracellular delivery of the therapeutic agent. To demonstrate the viability of the proposed approach, we turned to our recently reported cyclic peptide inhibitor of hypoxia inducible factor 1 (HIF-1) heterodimerization.2
HIF-1 is a heterodimeric transcription factor that drives the cellular response to hypoxia,3,4 by altering the transcription of over 300 genes,5 enabling cell survival and growth in a low oxygen microenvironment. HIF-1 is composed of an oxygen-regulated α-subunit (HIF-1α) and a constitutively expressed β-subunit (HIF-1β). HIF-1α is marked for degradation by prolyl hydroxylases that use oxygen as a substrate.6,7 Reduced oxygen levels lead to the stabilization and nuclear translocation of HIF-1α, where it binds HIF-1β to form the active HIF-1 transcription factor. HIF-1α mounts an immediate response to reductions of intracellular oxygen,8 while two closely related isoforms, HIF-2α (also known as EPAS1) and HIF-3α, are thought to regulate the response to prolonged hypoxia.9 The intricate interplay between HIF-α isoforms in cancer is complex and yet to be fully deciphered, but the role of HIF-1 activity in angiogenesis, tumor growth and metastasis is well established.10,11 Tumours grow rapidly, outstripping the capacity of the local vasculature, which results in a hypoxic microenvironment; HIF-1α is overexpressed in many cancers,12 and oncogene activation and loss of tumor suppressor function is shown to be associated with HIF-1.13
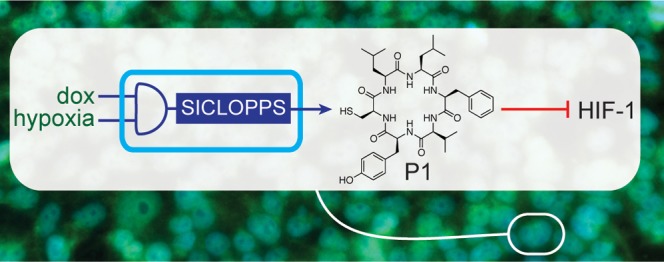
We recently reported an inhibitor of the HIF-1α/HIF-1β PPI;2 this molecule (cyclo-CLLFVY, named P1) was identified from a genetically encoded library of 3.2 million cyclic hexapeptides generated using split-intein circular ligation of peptides and proteins (SICLOPPS).14,15 P1 selectively binds to the PASB domain of HIF-1α with a Kd of 124 nM, disrupts the HIF-1α/HIF-1β PPI in vitro and in cells, and inhibits HIF-1 signaling in hypoxic cells.2 P1 is isoform-specific and does not bind to, or affect the function of the closely related HIF-2 isoform in vitro or in cells.2 SICLOPPS generates cyclic peptides viaSynechocystis sp PCC6803 (Ssp) DnaE split inteins;16 the SICLOPPS protein is composed of rearranged N-terminal and C-terminal split inteins flanking a peptide extein sequence in the form of IC-extein-IN. The N- and C-terminal split inteins combine to form an active intein that splices to cyclize the extein (Figure S1A). By altering the sequence of the SICLOPPS extein at the DNA level, a variety of cyclic peptides and proteins, including randomized cyclic peptide libraries, may be produced by this approach.17
Given our goal of incorporating the machinery required for the generation of a molecular PPI modulator into cells, and the significance of HIF-1 in tumor survival and growth, we set out probe the possibility of engineering the conditional production of P1 (via the corresponding SICLOPPS inteins) onto the chromosome of human HEK-293 cells, and to assess the effect of genetically encoded P1 on HIF-1-mediated hypoxia response in these cells.
Results
Expression and Processing of SICLOPPS Constructs in HEK-293 Cells
We began by constructing a cell line capable of conditional P1 production and assessing the ability of the SICLOPPS construct to correctly function in human cells. To achieve inducible expression of the SICLOPPS construct encoding our HIF-1 inhibitor, we used a cassette containing a CMV promoter, followed by two copies of the tetracycline operator (tetO), enabling regulation of transcription with doxycycline (dox), followed by the gene for SICLOPPS (Figure 1A). Engineered Nostoc punctiforme DnaE (Npu) inteins that splice significantly faster than the Ssp inteins typically used in SICLOPPS18−20 were utilized for the production of P1, with CLLFVY as the extein to be cyclized. We used flippase-flippase recognition target (Flp-FRT) recombination21 to stably integrate this cassette onto the chromosome of human HEK-293 cells (T-REx-293) to give T-REx-P1 cells. We first sought to demonstrate the production of functional P1 from the chromosome of human cells. Intein production was probed by immunoblotting with an antibody against the chitin-binding domain (CBD) present on the C-terminus of the N-terminal intein; we only observed the CBD band in the integrated cells, and only when cultured with dox (Figure S2A). The change in transcription of the chromosomal SICLOPPS construct in response to dox was quantified by RT-qPCR as ∼37-fold in both normoxia and hypoxia (Figure 1B), which was also reflected at the protein level (Figure 1C). A time course measuring intein protein production over 24 h in hypoxic cells illustrated the steady buildup of SICLOPPS inteins (Figure 1D). The splicing efficiency of the SICLOPPS protein encoding P1 was measured by immunoblot analysis for the CBD. We only observed a single band at ∼18 kDa corresponding to the spliced N-intein, suggesting full splicing of the SICLOPPS protein (Figure S2B). We next sought to directly detect the presence of P1 in T-REx-P1 cells. A synthetic sample of P1 was prepared as a standard, and a peak with identical retention time as this sample was observed in the lysate of T-REx-P1 cells treated with dox (Figure 1E). Mass spectrometric analysis of this fraction from the cell lysate revealed peaks corresponding to the mass of P1 (Figure 1F) and the same as observed for the synthetic standard (Figure S3). This data demonstrates that the Npu SICLOPPS inteins incorporated into the chromosome of human HEK-293 cells are selectively produced in the presence of dox, splicing post translation to give detectable levels of P1 in the engineered human cell line.
Figure 1.

Production of a functional cyclic peptide HIF-1 inhibitor from the chromosome of human HEK-293 cells. (A) The chromosomally integrated cassette enables the conditional expression of the SICLOPPS gene, which encodes the Npu inteins with CLLFVY as the extein. Splicing of these inteins gives P1. (B) RT-qPCR of intein expression in T-REx-P1 cells incubated in normoxia or hypoxia (24 h). (C) Immunoblot of T-REx-293 and T-REx-P1 cells treated as in B. (D) Immunoblot of T-REx-HRE cells incubated with dox in hypoxia for 0–24 h. (E) P1 produced in T-REx-P1 cells has the identical HPLC retention time as the synthetic standard. (F) The mass spectrum (ESI+) of fraction shown in F from T-REx-P1 cell lysate shows the presence of P1. (G) Firefly luciferase activity in T-REx-P1 and T-REx-Scram cells transfected with TK-HRE-luciferase and incubated for under normoxia or hypoxia (16 h). Data are means (n = 3) ± SEM, **p < 0.01.
Genetically Encoded P1 Inhibits HIF-1 Activity
With the engineered cell line in hands and having demonstrated the conditional production of P1, the functionality of the genetically encoded cyclic peptide HIF-1 inhibitor was next probed. T-REx-P1 cells were transfected with a HIF-dependent luciferase reporter plasmid, where activation of HIF results in increased luciferase expression.22 As expected, there was no change in the luciferase signal with dox in normoxia, while an ∼8-fold increase in luciferase activity was observed in hypoxic T-REx-P1 cells without dox (Figure 1G). Induction of P1 production with dox in these cells resulted in a 50% decrease in luciferase activity (Figure 1G), suggesting that chromosomally encoded P1 inhibits HIF-1 function. To demonstrate that our observations are due to P1 and not the SICLOPPS inteins, we generated a negative control cell line (T-REx-Scramble) that chromosomally encoded cyclo-CFVLYL (a scrambled variant of P1) as the extein of Npu SICLOPPS inteins. Splicing and conditional production of this scrambled peptide was demonstrated by immunoblotting (Figure S4). The above luciferase assay was repeated in this cell line, and a ∼7-fold increase in luciferase activity was observed upon induction of hypoxia without dox. There was, however, no change in luciferase activity when these cells were incubated with dox in hypoxia (Figure 1G), indicating that the scrambled peptide, or the SICLOPPS inteins do not affect HIF-1 dimerization. To validate that the effect from P1 was on HIF-1 rather than on luciferase, we used a control SV40-luciferase plasmid, and did not see any significant change in luciferase signal upon induction of P1 in T-REx-P1 cells with dox (Figure S5).
The effect of chromosomally produced P1 on HIF-1 activity was further assessed via its target genes vascular endothelial growth factor (VEGF) and carbonic anhydrase IX (CAIX). Chromosomally produced P1 reduced VEGF transcription by ∼30% (Figure S6A) and CAIX transcription by ∼45% (Figure S6B), with no effect of the scrambled peptide observed on either gene (Figure S6). Together, the above data demonstrates that chromosomally encoded P1 is functional, and able to inhibit HIF-1 signaling in hypoxia as expected from our previous studies with the synthetic, tat-tagged variant of the compound.
Engineering Physiological Control of Peptide Expression
We next sought to engineer an additional layer of control into the above system by limiting the dox-dependent production of P1 to hypoxic cells. The motivation for constructing this dual-control system was our long-term goal of generating an in vivo model that contains such a circuit on its chromosome. Such a system would not only allow expression of the HIF-1 inhibitors in hypoxic tissues (as an HRE-only promoter would), but also allow temporal control over initiation of P1 production via addition of dox. Thus, the effect of HIF-1 inhibition at various stages of the tumor development may be assessed. A hybrid promoter was designed and constructed; three copies of the HRE from the inducible nitric synthase promoter22 were placed upstream of two copies of TetO, resulting in a dual physiological and chemically controlled conditional promoter that would only function in hypoxia and with dox (Figure 2A). This cassette was incorporated onto the chromosome of HEK-293 cells by Flp-FRT recombination (as above) to give T-REx-HRE cells. Analysis of SICLOPPS cassette transcription by RT-qPCR showed ∼9-fold upregulated transcription in hypoxic cells that were incubated with dox (Figure 2B). Immunoblot analysis showed the presence of SICLOPPS protein only in cells cultured in hypoxia and with dox (Figure 2C), further illustrating that the dual conditional promoter was functioning as designed. The quantity of SICLOPPS protein was next compared with T-REx-P1 cells. Interestingly, we observed higher levels of inteins in the CMV-promoted cell line than the HRE-promoted cells incubated in hypoxia and with dox (Figure S7). Analysis of these bands by densitometry indicated that there was ∼7-fold more SICLOPPS intein in T-REx-P1 cells than in T-REx-HRE cells after 24 h. There are two reasons for this difference; first, the transgene expression rate from the HRE promoter is known to be lower than that from a CMV promoter,23,24 and second, the spliced product of the promoted protein (P1) is an inhibitor of HIF-1 dimerization. Therefore, as P1 builds up, it will also inhibit the transcription factor promoting its own production. To further assess the effect of this feedback loop on P1 production, a time course analysis of intein production was conducted. We observed the steady buildup of SICLOPPS mRNA (Figure 2D) and protein (Figure 2E) in hypoxic T-REx-HRE cells over 24 h, with a noticeable increase in both after 8 h in hypoxia.
Figure 2.

Conditional production of P1 in human cells. (A) The hybrid HRE/TetO promoter requires dual input signals of HIF-1 and dox in an AND process for expression of the SICLOPPS construct. Intein splicing produces P1, which inhibits HIF-1 dimerization. (B) RT-qPCR analysis of intein expression in T-REx-HRE cells incubated for 24 h in normoxia or hypoxia, with or without 1 μg/mL dox. (C) Immunoblot of T-REx-HRE cells treated as in panel B. (D) RT-qPCR analysis of SICLOPPS mRNA levels in T-REx-HRE cells incubated in hypoxia for 0–24 h and treated with 1 μg/mL dox. (E) Immunoblot for production of SICLOPPS protein over time. T-REx-HRE cells treated as in panel D. (F) PLA of T-REx-HRE cells treated with vehicle (left panel) or dox (right panel) and incubated in hypoxia for 24 h. (G and H) RT-qPCR analysis of (G) VEGF and (H) CAIX expression in T-REx-HRE cells incubated in hypoxia for 24 h with or without 1 μg/mL dox. Data are means (n = 3) ± SEM, **p < 0.01, ****p < 0.0001.
The effect of P1 on the interaction of HIF-1α and HIF-1β in hypoxic T-REx-HRE cells was next directly probed using an in situ proximity ligation assay (PLA).2,25 A PLA signal was observed in hypoxic T-REx-HRE cells incubated without dox (Figure 2F, left-hand panel), corresponding to the hypoxia-induced stabilization of HIF-1α and subsequent dimerization of HIF-1α and HIF-1β. The PLA signal was not observed in these cells when incubated with dox (Figure 2F, right-hand panel), nor in normoxic cells with or without dox (Figure S8). This data demonstrates the disruption of HIF-1 dimerization by genetically encoded P1 in cells.
The downstream effect of disrupting HIF-1 dimerization with chromosomally encoded P1 was elucidated via analysis of the transcription of HIF-1 target genes VEGF and CAIX. The expression of both genes was measured by RT-qPCR in cells incubated in hypoxia for 24 h with or without dox. Induction of P1 with dox resulted in a ∼40% reduction in VEGF mRNA (Figure 2G) and a ∼50% reduction in CAIX mRNA in hypoxic T-REx-HRE cells (Figure 2H). It should be noted that although lower amounts of the SICLOPPS protein are produced in the T-REx-HRE cells than in T-REx-P1 cells, the extent of the effect of P1 on these HIF-1 reporter genes was similar in both cell lines, indicating that P1 concentration is not a limiting factor in the observed inhibition of HIF-1 signaling.
Endogenous P1 Expression Alters Transcriptional Response to Hypoxia
We broadened our analysis of the effect of P1 on HIF-1 signaling by using a focused microarray assessing expression of 43 hypoxia-associated genes (Table S1) in T-REx-HRE cells cultured in normoxia, hypoxia, and hypoxia with dox. The data showed altered expression of these genes in T-REx-HRE cells that were incubated in hypoxia with dox (Figure 3A), illustrating the reprogramming of hypoxia response by P1 in these cells. We have previously demonstrated (in vitro and in cells) that cyclo-CLLFVY is a specific inhibitor of HIF-1 dimerization that does not affect the interaction of HIF-2α and HIF-1β.2 We therefore aimed to illustrate the potential of using genetically encoded P1 to separate the effect of HIF-1 inhibition on hypoxia signaling from that of the closely related transcription factor HIF-2. Our control experiments showed that treatment of T-REx-HRE cells with dox had no effect on HIF-2α mRNA levels (Figure S9A), and that siRNA knockdown of HIF-2α did not significantly affect the expression of HIF-1α mRNA or protein (Figure S9B and S9C) or SICLOPPS protein (Figure S9D).
Figure 3.

Expression of genetically encoded P1 alters the transcriptional profile of hypoxic HEK-293 cells. (A) Heat map of relative gene expression in T-REx-HRE cells transfected incubated in normoxia, or 24 h in hypoxia with or without 1 μg/mL dox. Details of genes are given in Table S1. (B–I) RT-qPCR analysis of T-REx-HRE cells incubated in normoxia, or 24 h in hypoxia with or without HIF-2α siRNA, and with 1 μg/mL dox with or without HIF-2α siRNA. Fold change in gene expression is shown relative to normoxic expression (dotted line). Data are means (n = 3) ± SEM, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The isoform-specificity of a number of HIF-target genes was next assessed using the above approach. While there was no effect on HIF-1α mRNA from either HIF-1 or HIF-2 (Figure 3B), HIF-3α expression seemed to be primarily under the control of HIF-2 (Figure 3C). We next assessed the role of HIF isoforms on genes involved in angiogenesis (ANGPLT4 and VEGF) and erythropoiesis (EPO). Following dox treatment, expression of EPO was decreased to normoxic levels, whereas HIF-2α siRNA treatment caused no significant change, indicating EPO is a HIF-1 specific target (Figure 3D). In contrast, although inhibition of HIF-1 significantly reduced the expression of ANGPLT4 (Figure 3E) and VEGF (Figure S9E), a combination of both dox and HIF-2α siRNA treatments was required to reduce gene expression to normoxic levels. This data suggests that expression of ANGPLT4 and VEGF is transactivated by both HIF-1 and HIF-2. Together, the inhibitory effect of P1 expression on genes involved in the promotion of angiogenesis and erythropoiesis supports previous assertions for the potential of targeting HIF-1 dimerization as a therapeutic strategy to prevent tumor vascularisation.
Inhibition of HIF-1 dimerization and HIF-2α siRNA treatment also differentially impacted upstream effectors of oxygen-dependent regulation of HIF-1. Induction of P1 expression resulted in a decrease in PHD2 and PHD3 mRNA whereas HIF-2α siRNA had no significant effect (Figure 3F and 3G). These observations are in line with the theory that HIF-1 mediates the acute response to hypoxia whereas HIF-2 is the prominent driver of adaptation to prolonged periods of hypoxic conditions.26,27 Another gene primarily controlled by HIF-1 was CAIX, with no significant effect from HIF-2α siRNA alone (Figure S9F). This data was in agreement with reports of CAIX as a HIF-1 specific target.28 Interestingly, although the stress response gene DDIT4 was upregulated in hypoxia, and inhibition of HIF-1 and HIF-2 significantly reduced this induction, neither treatment was sufficient to reduce DDIT4 expression to normoxic levels (Figure 3H). DDIT4 is also induced in response to endoplasmic reticulum stress and DNA damage related to the regulation of reactive oxygen species,29,30 which may be a result of hypoxic exposure, particularly when HIF response pathways are disrupted.30 Another gene of note was NOTCH-1, whose expression was halved in hypoxic cells treated with dox, but doubled in hypoxic cells treated with HIF-2α siRNA (Figure 3I), illustrating the opposing regulatory roles of HIF-1 and HIF-2 on this gene. The above data not only demonstrates reprogramming of hypoxia response in our engineered human cell line, but also illustrates the potential utility of using genetically encoded P1, a HIF-1 specific inhibitor, to decipher the role of HIF isoforms in hypoxia signaling in a variety of cell lines.
Inhibition of HIF-1 Dimerization Confers Synthetic Lethality to Glycolysis Inhibitor
The effect on cell viability from the disruption of hypoxia-response signaling via inhibition of the HIF-1α/HIF-1β PPI by P1 was next probed. The viability of T-REx-HRE cells cultured in normoxia or hypoxia, with or without dox was assessed over 72 h using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). No difference was observed in the viability of cells cultured with dox versus those that were not (Figure 4A and Figure S10), indicating that inhibition of the HIF-1α/HIF-1β PPI in hypoxic HEK-293 cells does not affect viability. Nonetheless, previous studies have shown that knockdown of HIF-1α by siRNA sensitizes cells to the glycolysis inhibitor 2-deoxyglucose (2DG), leading to cytotoxicity.31 We questioned whether inhibition of HIF-1α/HIF-1β PPI with P1 would render HEK-293 cells susceptible to 2DG. This was initially assessed using MTT assays as above. We observed a 37% reduction in viability after 72 h in hypoxic T-REx-HRE cells incubated with dox (Figure 4B). This observation was further probed with a triplex assay that uses protease biomarkers to assess viability, cytotoxicity and apoptosis.32 We observed a 45% decrease in the ratio of viable to cytotoxic T-REx-HRE cells after 72 h (Figure 4C), in line with our observations from the MTT assay. Interestingly, no significant increase in caspase-3/7 activity was observed in these cells (Figure 4D), suggesting that the observed synthetic lethality of 2DG combined with P1 is not driven by apoptosis.33
Figure 4.

Assessing the effect of HIF-1 inhibition on T-REx-HRE cell viability. (A) Cells were incubated in hypoxia for up to 72 h with or without 1 μg/mL dox. Cell viability was assessed with an MTT assay. (B) Cells were treated as in panel A with or without the addition of 3 mg/mL 2DG to the culture media. (C) Cells were treated as in panel B and incubated in hypoxia for 72 h. The ratio of cell viability to cytotoxicity was determined using a triplex reporter assay. (D) Cells were treated as in panel C and apoptotic cell death was assessed via a caspase 3/7 activity assay. Data are means (n = 3) ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
The conditional production of P1, an inhibitor of the HIF-1α/HIF-1β PPI, has been encoded onto the chromosome of a human cell line, and demonstrated to be a viable approach for the inhibition of HIF-1 signaling. Two versions of this sentinel circuit are reported, one that initiates P1 production in response to a chemical trigger (doxycycline), and a second that requires an environmental signal (hypoxia) in addition to doxycycline. In addition, the synthetic lethality of HIF-1 dimerization inhibition (by P1) and inhibition of glycolysis (with 2DG) is demonstrated. There is a growing body of evidence that inhibition of HIF-1, or redirection of cellular pathways away from HIF-1 controlled mechanisms, may improve the anticancer effects of current chemotherapeutic agents.34−37 Using the T-REx-HRE cell line reported here, a variety of chemotherapeutic agents may be screened for synthetic lethality, or increased potency when combined with HIF-1 inhibition.38,39
This system has the potential to address several key questions about HIF-1 and its necessity for the survival and growth of tumors. Since its discovery, a large body of evidence has suggested that the HIF-1 transcription factor plays a significant and critical role in cancers, enabling survival and adaptation in the hypoxic tumor microenvironment. However, recent studies have indicated that HIF-1α also functions in the cell as a monomer.40−42 Approaches that enable disruption of the HIF-1 transcription factor, without reducing the cellular level of HIF-1α have the potential to help decipher the role of HIF-1 in cancer. The sentinel circuit reported above may, for example, be incorporated onto the genome of a variety of cancer cell lines that are used in xenograft models. P1 production may be initiated at various points during the tumor lifecycle, either globally or only within the hypoxic regions of the tumor, illustrating the significance (or not) of HIF-1 as a target for cancer therapy. Another possibility is the generation of in vivo models that contain the sentinel P1-encoding circuit on their chromosome, enabling the effect of long-term HIF-1 inhibition on tumor formation and maintenance to be studied; the dual control promoter limits P1 production to hypoxic cells and allows temporal control over P1 production, enabling the study of HIF-1 inhibition at various stages of the tumor lifecycle.
The concept of encoding the conditional production of a non-native small molecule from the genome of engineered human cells has applications beyond the HIF-1α/HIF-1β inhibitor described here. There are a multitude of natural products used as therapeutics that may be hard-coded onto cells via their biosynthetic machinery, and produced as required. The use of sentinel circuits to induce apoptosis in cells in response to specific DNA sequences,43 or encoding an environment-coupled kill-switch in bacteria44 attests to the possibilities. Other recent examples include the introduction of new biological components into cells for therapeutic purposes.45−50 As the field of synthetic biology advances, such examples will grow in number and complexity.
Methods
Cell Culture
All cell culture reagents were purchased from Life Technologies unless otherwise stated. All cells were cultured at 37 °C in a humidified 5% CO2 atmosphere. T-REx-293 cells were maintained in DMEM containing 10% fetal bovine serum (FBS), 100 μg/mL zeocin and 15 μg/mL blasticidin and integrated T-REx cell lines were cultured in DMEM containing 10% FBS, 100 μg/mL hygromycin B and 15 μg/mL blasticidin. Unless otherwise stated, T-REx cells were dosed with 1 μg/mL doxycycline (dox) to induce expression of integrated constructs. Hypoxia treatment was achieved in a Don Whitley Scientific H35 Hypoxystation with a humidified atmosphere containing 1% O2 and 5% CO2. Transfection of plasmids was carried out using FuGENE HD (Promega) according to the manufacturer’s instructions and experiments were carried out 24 h after transfection.
Transfection and Selection of Stable Clones
Stable mammalian expression cell lines were generated by Flp recombinase-mediated integration. Flp-In T-REx-293 (T-REx-293) cells and plasmid vectors pcDNA5/FRT/TO and pOG44 (kind gift of Dr. Noel Wortham) are available commercially from Life Technologies. pOG44 and pcDNA5/FRT vectors were transfected, at a ratio of 9:1, into T-REx-293 cells with Fugene HD for the generation of stable cell lines. Polycolonal selection was carried out with 200 μg/mL hygromycin in high dilution culture. Integration was confirmed by western immunoblotting.
Quantitative PCR
Total RNA was extracted from cells using ReliaPrep RNA Cell Miniprep System (Promega) and quantified using a Nanodrop ND-1000 spectrophotometer. Complementary cDNA was synthesized in a 20 μL reaction using 1 μg of total RNA with GoScript Reverse Transcriptase (Promega) according to the manufacturer’s instructions. Quantitative real-time PCR (RT-qPCR) was performed using Universal Taqman PCR master mix (Applied Biosystems) and the TaqMan gene expression assays of interest (Applied Biosystems) on a CFX-connect 96 Real-Time PCR system (Bio-Rad). Expression assays used in this study were: 18S (Hs99999901_s1), ActB (Hs99999903_m1), VEGF (Hs00900055_m1), CAIX (Hs00154208_m1), HIF-1α (Hs00153153_m1) and EPAS1 (Hs01026149_m1). Expression values were expressed as ΔΔCT normalized to expression of 18S and β-Actin and normoxic gene expression. Hypoxia focused microarray was conducted using TaqMan Array Human Hypoxia plates (Applied Biosystems). Expression values were expressed as ΔΔCT normalized to expression of 18S and normoxic gene expression.
Western Immunoblotting
For visualization of expression of CBD tagged inteins, cells were lysed in intein extraction buffer (20 mM Tris.HCl, 1 mM TCEP, 0.5 mM NaCl, pH 7.8) on ice for 15 min. For visualization of HIF-1α protein, cells were lysed by incubation on ice with radioimmunoprecipitation assay buffer (50 mM Tris (pH 7.4), 150 μM NaCl, 1 mM EDTA, 1% v/v Triton X-100), and 1× protease inhibitor cocktail (Sigma) for 20 min.
Cell lysates were sonicated in an ice water bath then centrifuged at 10 000 rpm for 20 min at 4 °C, and the protein concentration in the supernatant quantified by Bradford assay. Proteins were separated on an SDS-PAGE gels (15% for CBD, 10% for HIF-1α), transferred to PVDF membranes (Invitrogen) and subjected to immunoblot analysis. Mouse monoclonal anti-CBD (E8034S, 1:250, New England Biolabs) anti-HIF-1α (610958, 1:250 BD Biosciences) were diluted in PBS containing 5% nonfat powdered milk and 0.1% Tween-20 and incubated with the membrane overnight at 4 °C overnight. Horseradish peroxidase conjugated antimouse antibody was used as the secondary antibody, and monoclonal anti-βactin-peroxidase antibody (A3854, 1:100 000, Sigma) served as a loading control. Bound immunocomplexes were detected using ECL prime Western blot detection reagent (RON2232, GE Healthcare) and analyzed using a ChemiDoc Imaging System (Bio-Rad) and Image Lab 4.0 software (Bio-Rad).
Detection of Peptide in Cell Lysates by HPLC and MS
T-REx-P1 cells were scraped in ice cold PBS and the cell pellet froze in N2 (l). The pellet was thawed and lysed in PMSF lysis buffer (5 mM EDTA, 2 mM EGTA, 0.4 mM PMSF in PBS) containing protease inhibitor cocktail (Sigma) by three freeze thaw cycles. TFA (10 μL) was added to precipitate proteins and the lysate centrifuged (8000 rpm, 30 min, 4 °C). The supernatant was passed through a 10 kDa cut off filter and the flow through collected and analyzed by reverse phase HPLC on a Waters HPLC system equipped with a Waters Atlantis T3, Amide capped C18 5 μm, 6 × 100 mm column. Samples were manually injected into a Waters flex inject system into the HPLC system containing a Waters 1525 binary pump. One-minute fractions were collected in the 5 min window around the elution time of the synthetic peptide and analyzed by LC–MS.
HRE Luciferase Reporter Assay
T-REx-P1, T-REx-Scramble, or T-REx-HRE cells were transiently transfected with a HIF dependent firefly luciferase reporter construct (pGL2-TK-HRE) or a HIF independent firefly luciferase reporter construct (pGL3-SV40) as a control. Transfected cells were incubated in the presence or absence of 1 μg/mL dox. After 24 h, cells were recovered and plated at 25 000 cells/well in 96 well plates and incubated for 5 h before either hypoxic or aerobic incubation for 16 h. Firefly luciferase activity was determined using Bright-Glo Reagent (Promega) according to the manufacture’s instructions. Luciferase signal was normalized using the corresponding no-transfection controls for each plate.
Duolink Proximity Ligation Assay
Duolink proximity ligation assay (PLA) was conducted using the in situ PLA Kit (O-Link Bioscience, Uppsala, Sweden) according to the manufacturer’s instructions. The antibodies used were rabbit monoclonal anti-HIF-1α (NB100–449, Novus Biologicals) and mouse monoclonal anti-HIF-1β (H00000405- B01P, Abnova). Cells were treated with 1 μg/mL dox in normoxia or hypoxia for 24 after which they were fixed with 2% formaldehyde in PBS for 10 min and permeabilized with 0.5% Triton (diluted in PBS) for 10 min. After preincubation with the Duolink Blocking Reagent for 1 h, samples were incubated overnight with the primary antibodies to HIF-1α (1:500) and HIF-1β (1:500). Duolink PLA probes and reagents were added as recommended by the manufacture’s instructions. Cells were imaged with a fluorescent microscope (Zeiss Axio Vert.A1).
HIF-2α Knockdown
T-REx-HRE cells were seeded at 20 000 cells/well on 6-well plates and incubated for 24 h such that cell density reached 50–70% confluence just prior to transfection with siRNA. Cells were transfected with siRNA using Lipofectamine RNAiMAX transfection reagent (Life Technologies) according to the manufacturer’s instructions for “forward transfection”. Briefly, cell culture media was removed from cells and replaced with serum-free OptiMEM cell culture medium (Life Technologies). siRNA and Lipofectamine were separately, diluted in a volume of OptiMEM equivalent to 10% of the final volume of cells, and incubated at RT for 5 min. The diluted oligonucleotides and transfection reagent were then combined, mixed gently, and incubated at RT for 10 min. The siRNA-Lipofectamine complexes were added dropwise to cells which were then incubated at 37 °C for 24 h. The final concentration of siRNA was 5 nM and the final amount of Lipofectamine was 0.2% v/v for all experiments. Cells were either transfected with EPAS1 (HIF-2α) siRNA (Silencer Select predesigned annealed human oligonucleotide duplex, s4700, Life Technologies), scrambled siRNA (Silencer Select negative control number 2, Life Technologies) or vehicle alone. Following transfection, cells were incubated in hypoxic or aerobic conditions for 24 h in the presence or absence of 1 μg/mL for 24 h, then harvested for total RNA extraction.
Cell Viability Assays
T-REx-HRE cells were seeded in triplicate at 5000 cells per well on 96 well plates 24 h prior to dosing with 1 μg/mL dox in 100 uL fresh DMEM, DMEM without glucose or DMEM containing 3 mg/mL 2-deoxy-glucose. MTT-based cell proliferation assays were performed on untreated cells on the day of treatment or treated cells 24, 48, or 72 h after treatment, as follows: MTT (Sigma) was prepared in sterile PBS added to cells at a final concentration of 1 mM (10% v/v). Cells were then incubated at 37 °C for 4 h until intracellular punctate purple precipitated were clearly visible under the microscope. 75 μL of the culture medium was the removed from each well and 100 μL DMSO added. The cells were incubated for 10 min in the dark, with agitation to dissolve the insoluble formazan particles. Absorbance was measured at 570 nm on a microplate reader (Tecan Infinite M200 Pro). ApoToxGlo assays (Promega) were performed on cells 48 or 72 h after treatment with 1 μg/mL dox according to the manufacturer’s instructions.
Statistical Analysis
Data analysis was performed with Prism 6 (Graphpad Software). Statistical significance was evaluated with an unpaired t-test for comparison between two means and analysis of variance followed by Bonferroni method for multiple comparisons. A value of p < 0.05 was considered to denote statistical significance.
Acknowledgments
The authors thank Cancer Research UK (A20185) and the Engineering and Physical Sciences Research Council (EP/K503150/1) for funding this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.6b00219.
Figures S1–S10; Table S1 (PDF)
The authors declare no competing financial interest.
Notes
Data supporting this study are openly available from the University of Southampton repository at http://doi.org/10.5258/SOTON/403010.
Supplementary Material
References
- Darnell J. E. Jr. (2002) Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2, 740–749. 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- Miranda E.; Nordgren I. K.; Male A. L.; Lawrence C. E.; Hoakwie F.; Cuda F.; Court W.; Fox K. R.; Townsend P. A.; Packham G. K.; Eccles S. A.; Tavassoli A. (2013) A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. J. Am. Chem. Soc. 135, 10418–10425. 10.1021/ja402993u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert B. L.; Bunn H. F. (1998) Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol. Cell. Biol. 18, 4089–4096. 10.1128/MCB.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. L.; Jiang B. H.; Rue E. A.; Semenza G. L. (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U. S. A. 92, 5510–5514. 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schodel J.; Oikonomopoulos S.; Ragoussis J.; Pugh C. W.; Ratcliffe P. J.; Mole D. R. (2011) High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117, e207–217. 10.1182/blood-2010-10-314427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakkola P.; Mole D. R.; Tian Y. M.; Wilson M. I.; Gielbert J.; Gaskell S. J.; Kriegsheim A.; Hebestreit H. F.; Mukherji M.; Schofield C. J.; Maxwell P. H.; Pugh C. W.; Ratcliffe P. J. (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Ivan M.; Kondo K.; Yang H.; Kim W.; Valiando J.; Ohh M.; Salic A.; Asara J. M.; Lane W. S.; Kaelin W. G. Jr. (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468. 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jewell U. R.; Kvietikova I.; Scheid A.; Bauer C.; Wenger R. H.; Gassmann M. (2001) Induction of HIF-1alpha in response to hypoxia is instantaneous. FASEB J. 15, 1312–1314. 10.1096/fj.00-0732fje. [DOI] [PubMed] [Google Scholar]
- Keith B.; Johnson R. S.; Simon M. C. (2012) HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 12, 9–22. 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A. L. (2002) Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47. 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Semenza G. L. (2003) Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732. 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Giaccia A.; Siim B. G.; Johnson R. S. (2003) HIF-1 as a target for drug development. Nat. Rev. Drug Discovery 2, 803–811. 10.1038/nrd1199. [DOI] [PubMed] [Google Scholar]
- Bardos J. I.; Ashcroft M. (2004) Hypoxia-inducible factor-1 and oncogenic signalling. BioEssays 26, 262–269. 10.1002/bies.20002. [DOI] [PubMed] [Google Scholar]
- Scott C. P.; Abel-Santos E.; Wall M.; Wahnon D. C.; Benkovic S. J. (1999) Production of cyclic peptides and proteins in vivo. Proc. Natl. Acad. Sci. U. S. A. 96, 13638–13643. 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli A.; Benkovic S. J. (2007) Split-intein mediated circular ligation used in the synthesis of cyclic peptide libraries in E. coli. Nat. Protoc. 2, 1126–1133. 10.1038/nprot.2007.152. [DOI] [PubMed] [Google Scholar]
- Wu H.; Hu Z. M.; Liu X. Q. (1998) Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. U. S. A. 95, 9226–9231. 10.1073/pnas.95.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennard K. R.; Tavassoli A. (2014) Peptides come round: using SICLOPPS libraries for early stage drug discovery. Chem. - Eur. J. 20, 10608–10614. 10.1002/chem.201403117. [DOI] [PubMed] [Google Scholar]
- Iwai H.; Zuger S.; Jin J.; Tam P. H. (2006) Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. FEBS Lett. 580, 1853–1858. 10.1016/j.febslet.2006.02.045. [DOI] [PubMed] [Google Scholar]
- Lockless S. W.; Muir T. W. (2009) Traceless protein splicing utilizing evolved split inteins. Proc. Natl. Acad. Sci. U. S. A. 106, 10999–11004. 10.1073/pnas.0902964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townend J. E.; Tavassoli A. (2016) Traceless Production of Cyclic Peptide Libraries in E. coli. ACS Chem. Biol. 11, 1624–1630. 10.1021/acschembio.6b00095. [DOI] [PubMed] [Google Scholar]
- Schlake T.; Bode J. (1994) Use of mutated FLP recognition target (FRT) sites for the exchange of expression cassettes at defined chromosomal loci. Biochemistry 33, 12746–12751. 10.1021/bi00209a003. [DOI] [PubMed] [Google Scholar]
- Rapisarda A.; Uranchimeg B.; Scudiero D. A.; Selby M.; Sausville E. A.; Shoemaker R. H.; Melillo G. (2002) Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 62, 4316–4324. [PubMed] [Google Scholar]
- Cao Y. J.; Shibata T.; Rainov N. G. (2001) Hypoxia-inducible transgene expression in differentiated human NT2N neurons - a cell culture model for gene therapy of postischemic neuronal loss. Gene Ther. 8, 1357–1362. 10.1038/sj.gt.3301536. [DOI] [PubMed] [Google Scholar]
- Post D. E.; Van Meir E. G. (2001) Generation of bidirectional hypoxia/HIF-responsive expression vectors to target gene expression to hypoxic cells. Gene Ther. 8, 1801–1807. 10.1038/sj.gt.3301605. [DOI] [PubMed] [Google Scholar]
- Soderberg O.; Gullberg M.; Jarvius M.; Ridderstrale K.; Leuchowius K. J.; Jarvius J.; Wester K.; Hydbring P.; Bahram F.; Larsson L. G.; Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000. 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Koh M. Y.; Lemos R. Jr.; Liu X.; Powis G. (2011) The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 71, 4015–4027. 10.1158/0008-5472.CAN-10-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida T.; Rossignol F.; Matthay M. A.; Mounier R.; Couette S.; Clottes E.; Clerici C. (2004) Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: implication of natural antisense HIF-1alpha. J. Biol. Chem. 279, 14871–14878. 10.1074/jbc.M400461200. [DOI] [PubMed] [Google Scholar]
- Grabmaier K.; MC A. d. W.; Verhaegh G. W.; Schalken J. A.; Oosterwijk E. (2004) Strict regulation of CAIX(G250/MN) by HIF-1alpha in clear cell renal cell carcinoma. Oncogene 23, 5624–5631. 10.1038/sj.onc.1207764. [DOI] [PubMed] [Google Scholar]
- Wang Z. Q.; Malone M. H.; Thomenius M. J.; Zhong F.; Xu F.; Distelhorst C. W. (2003) Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J. Biol. Chem. 278, 27053–27058. 10.1074/jbc.M303723200. [DOI] [PubMed] [Google Scholar]
- Ellisen L. W.; Ramsayer K. D.; Johannessen C. M.; Yang A.; Beppu H.; Minda K.; Oliner J. D.; McKeon F.; Haber D. A. (2002) REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 10, 995–1005. 10.1016/S1097-2765(02)00706-2. [DOI] [PubMed] [Google Scholar]
- Maher J. C.; Wangpaichitr M.; Savaraj N.; Kurtoglu M.; Lampidis T. J. (2007) Hypoxia-inducible factor-1 confers resistance to the glycolytic inhibitor 2-deoxy-D-glucose. Mol. Cancer Ther. 6, 732–741. 10.1158/1535-7163.MCT-06-0407. [DOI] [PubMed] [Google Scholar]
- Niles A. L.; Moravec R. A.; Eric Hesselberth P.; Scurria M. A.; Daily W. J.; Riss T. L. (2007) A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers. Anal. Biochem. 366, 197–206. 10.1016/j.ab.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Nicholson D. W.; Thornberry N. A. (1997) Caspases: killer proteases. Trends Biochem. Sci. 22, 299–306. 10.1016/S0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Kumar K.; Wigfield S.; Gee H. E.; Devlin C. M.; Singleton D.; Li J. L.; Buffa F.; Huffman M.; Sinn A. L.; Silver J.; Turley H.; Leek R.; Harris A. L.; Ivan M. (2013) Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J. Mol. Med. 91, 749–758. 10.1007/s00109-013-0996-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher J. C.; Wangpaichitr M.; Savaraj N.; Kurtoglu M.; Lampidis T. J. (2007) Hypoxia-inducible factor-1 confers resistance to the glycolytic inhibitor 2-deoxy-D-glucose. Mol. Cancer Ther. 6, 732–741. 10.1158/1535-7163.MCT-06-0407. [DOI] [PubMed] [Google Scholar]
- Burroughs S. K.; Kaluz S.; Wang D.; Wang K.; Van Meir E. G.; Wang B. (2013) Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med. Chem. 5, 553. 10.4155/fmc.13.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D.; Gilkes D. M.; Chaturvedi P.; Xiang L.; Semenza G. L. (2014) Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. U. S. A. 111, E5429–5438. 10.1073/pnas.1421438111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li J.; Shi M. X.; Cao Y.; Yuan W. S.; Pang T. X.; Li B. Z.; Sun Z.; Chen L.; Zhao R. C. H. (2006) Knockdown of hypoxia-inducible factor-1 alpha in breast carcinoma MCF-7 cells results in reduced tumor growth and increased sensitivity to methotrexate. Biochem. Biophys. Res. Commun. 342, 1341–1351. 10.1016/j.bbrc.2006.02.094. [DOI] [PubMed] [Google Scholar]
- Sun X.; Kanwar J. R.; Leung E.; Lehnert K.; Wang D.; Krissansen G. W. (2001) Gene transfer of antisense hypoxia inducible factor-1 alpha enhances the therapeutic efficacy of cancer immunotherapy. Gene Ther. 8, 638–645. 10.1038/sj.gt.3301388. [DOI] [PubMed] [Google Scholar]
- Hubbi M. E.; Kshitiz; Gilkes D. M.; Rey S.; Wong C. C.; Luo W.; Kim D. H.; Dang C. V.; Levchenko A.; Semenza G. L. (2013) A nontranscriptional role for HIF-1alpha as a direct inhibitor of DNA replication. Sci. Signaling 6, ra10. 10.1126/scisignal.2003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns R. A.; Takimoto E.; Meuchel L. W.; Elsaigh E.; Zhang A.; Heller N. M.; Semenza G. L.; Yamaji-Kegan K. (2016) Hypoxia-Inducible Factor 1alpha Is a Critical Downstream Mediator for Hypoxia-Induced Mitogenic Factor (FIZZ1/RELMalpha)-Induced Pulmonary Hypertension. Arterioscler., Thromb., Vasc. Biol. 36, 134–144. 10.1161/ATVBAHA.115.306710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang L.; Gilkes D. M.; Hu H.; Luo W.; Bullen J. W.; Liang H.; Semenza G. L. (2015) HIF-1alpha and TAZ. serve as reciprocal co-activators in human breast cancer cells. Oncotarget 6, 11768–11778. 10.18632/oncotarget.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomovic S.; Collins J. J. (2015) DNA sense-and-respond protein modules for mammalian cells. Nat. Methods 12, 1085–1090. 10.1038/nmeth.3585. [DOI] [PubMed] [Google Scholar]
- Chan C. T.; Lee J. W.; Cameron D. E.; Bashor C. J.; Collins J. J. (2016) ’Deadman’ and ’Passcode’ microbial kill switches for bacterial containment. Nat. Chem. Biol. 12, 82–86. 10.1038/nchembio.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway K. E.; Franco E.; Smolke C. D. (2013) Dynamically reshaping signaling networks to program cell fate via genetic controllers. Science 341, 1235005. 10.1126/science.1235005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng B. C.; Aubel D.; Fussenegger M. (2015) Prosthetic gene networks as an alternative to standard pharmacotherapies for metabolic disorders. Curr. Opin. Biotechnol. 35, 37–45. 10.1016/j.copbio.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Kojima R.; Aubel D.; Fussenegger M. (2015) Novel theranostic agents for next-generation personalized medicine: small molecules, nanoparticles, and engineered mammalian cells. Curr. Opin. Chem. Biol. 28, 29–38. 10.1016/j.cbpa.2015.05.021. [DOI] [PubMed] [Google Scholar]
- Morsut L.; Roybal K. T.; Xiong X.; Gordley R. M.; Coyle S. M.; Thomson M.; Lim W. A. (2016) Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164, 780–791. 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal K. T.; Rupp L. J.; Morsut L.; Walker W. J.; McNally K. A.; Park J. S.; Lim W. A. (2016) Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779. 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena P.; Charpin-El Hamri G.; Folcher M.; Zulewski H.; Fussenegger M. (2016) Synthetic gene network restoring endogenous pituitary-thyroid feedback control in experimental Graves’ disease. Proc. Natl. Acad. Sci. U. S. A. 113, 1244–1249. 10.1073/pnas.1514383113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.