

Abstract
A net redox-neutral method for the decarboxylative alkylation of heteroarenes using photoredox catalysis is reported. Additionally, this method features the use of simple, commercially available carboxylic acid derivatives as alkylating agents, enabling the facile alkylation of a variety of biologically-relevant heterocyclic scaffolds under mild conditions.
Graphical Abstract
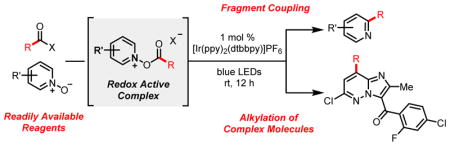
The direct C–H functionalization of N-heteroarenes provides expedient access to an array of heterocyclic motifs prevalent throughout many natural products, advanced materials, and pharmaceuticals.1 In the context of drug discovery, C–H (perfluoro)alkylation strategies can facilitate both the diversification of structural libraries, as well as the late-stage modification of clinical candidates.2 To this end, our group has reported a radical trifluoromethylation strategy that enables the direct trifluoromethylation of a range of vinyl, aryl, and heteroaryl substrates using photoredox catalysis.3 The employment of pyridine N-oxide as a redox auxiliary provides a novel mode of activating carboxylic acid derivatives, such as trifluoroacetic anhydride, by inducing reductive decarboxylation to generate the trifluoromethyl radical. This reaction paradigm of coupling electron-deficient radicals with electron-rich heterocyles (Figure 1A) has been further elaborated upon in accessing the chlorodifluoromethyl radical for the synthesis and diversification of difluoromethyl-heteroarenes, as detailed in the preceding manuscript. A separate, orthogonal mode of reactivity achievable by altering the electronics of this system involves the direct coupling of electron-deficient heterocyclic N-oxides with electron-rich alkyl radicals. This fragment coupling approach invokes the use of a heterocyclic N-oxide reagent as both a transient redox auxiliary, as well as a coupling partner for electron-rich alkyl radicals. Specifically, the decarboxylation of electron-rich carboxylic acid derivatives through reduction of electron-poor heterocyclic N-oxides generates an electronically matched pair of reactants, which can combine and provide coupled products in a selective fashion. We envisioned the immediate applicability of this reaction paradigm toward the design of a facile Minisci alkylation reaction. The Minisci reaction (Figure 1B), which involves the direct C–H alkylation of heteroarenes by a carbon-centered radical, has garnered much attention and has undergone significant development in recent years.4–6 Minisci’s original protocol for the decarboxylative alkylation of heterocycles relied on the use of stoichiometric silver salts, persulfate oxidants, and elevated temperatures.4a Since this initial publication, methods for achieving the Minisci alkylation have evolved to incorporate a diverse range of alkylating agents, including trifluoroborate salts,5a,b sulfinates,5c,d and alcohols.6a In particular, the application of photoredox catalysis for Minisci alkylations has led to significant improvements in the sustainability of this transformation, with the development of conditions that require lower reagent loadings and non-oxidative routes for alkyl radical generation.6
Figure 1.

(A) Reaction design principles (B) Oxidative alkyl radical generation (C) Reductive alkyl radical generation
While a multitude of protocols have been developed for the oxidative generation of alkyl/fluoroalkyl radicals for Minisci-type transformations, methods that promote the reductive radical alkylation of N-heteroarenes are comparatively less established (Figure 1C).6b,g In the context of photoredox catalysis, the formation of alkyl radicals through a reductive pathway would enable a net redox-neutral catalytic cycle, thereby eliminating the need for a stoichiometric terminal oxidant. Notably, in 2014, DiRocco and co-workers reported the photocatalytic alkylation of N-heterocycles through the reductive generation of alkyl radicals using perester reagents.6b Recently, the groups of Sherwood and Proctor have independently demonstrated the usage of N-(acyloxy)phthalimides for the reductive decarboxylation of alkyl carboxylic acids in Minisci-type transformations.6g,h In comparison with previously reported Minisci alkylation methods, the decarboxylative alkylation strategy disclosed herein precludes the use of strongly acidic conditions and a sacrificial redox auxiliary. In using a fragment coupling approach, waste production can be inevitably decreased, as the mutual substrate activation through acylation avoids the use of stoichiometric additives for inherent reactivity. Reduced heterocyclic N-oxides can additionally act as endogenous bases, and reactive radical intermediates can be controlled based upon the electronics of reagents in the system.
Pivaloyl chloride and 4-phenylpyridine N-oxide were chosen as model substrates for initial pyridine alkylation evaluation. Upon screening several solvents and photocatalysts,7 we discovered that a combination of acetonitrile and 1 mol % [Ir(ppy)2(dtbbpy)]PF6 furnished the desired 2-tert-butyl-4-phenylpyridine product in 75% yield, as well as the 2,4-di-tert-butylated product in 5% yield (Scheme 1, 1). Under the optimized conditions, the scope of decarboxylative alkylations was examined with a number of alkyl carboxylic acid derivatives. Successful methylation (2) of 4-phenylpyridine N-oxide was achieved, albeit at a lower yield (20%), due to decreased radical stability and nucleophilicity.8 Difluoromethylation (3) was also achieved with the use of difluoromethylacetic anhydride as a source of the CF2H radical. Moreover, propionic acid could be used for the ethylation (4) of 4-phenylpyridine in modest yield (50%). This protocol proved amenable to the coupling of 4-phenylpyridine with a wide range of secondary and tertiary cyclic alkanes (5–13), including the cyclohexyl (9) motif, which has been demonstrated to be a bioisostere of the phenyl functionality.9 Linear alkyl chains (14) could also be successfully accessed in modest yields, along with bridged cyclic alkane motifs such as the medicinally relevant norbornene bicycle (12).10 In contrast, the benzylation (15) of 4-phenylpyridine N-oxide proceeded with significantly diminished yields. A predominant side reaction that was observed involved the formal decarbonylation of phenylacetyl chloride, yielding 73% formation of benzyl chloride. Furthermore, these conditions enabled the successful appendage of medicinally-relevant fluorinated isosteres11 onto heteroarenes, including the first example of incorporating the 1-fluorocyclopropane motif (17) onto a heterocyclic scaffold in one step from its carboxylic acid precursor. A variety of heterocyclic motifs (19–24) successfully underwent cross coupling with 4-phenylpyridine N-oxide, including the tetrahydropyranyl (16) and piperidinyl (22–24) ring systems, which have been used as H-bond donor/acceptor probes in SAR studies.12 Most notably, we have demonstrated the unique coupling of a tertiary azetidine-derived radical (20) with an unactivated heteroarene, a transformation otherwise inaccessible to traditional transition metal-mediated cross-coupling methods. Overall, a variety of alkyl substrates, differing in size and electronic properties, have been demonstrated to be successful coupling partners in this transformation. An added benefit to this methodology entails the direct in situ formation of non-commercially available acid chloride reagents from the corresponding carboxylic acid (via oxalyl chloride and catalytic DMF), followed by addition of the heterocyclic N-oxide, without the need for any additional purification or isolation steps.
Scheme 1.

Alkyl carboxylic acid derivative scope.
[a]Isolated yields of reactions run on a 0.8 mmol scale with 1.6 mL of acetonitrile (0.5 M) and 1.1 equiv of acid chloride reagent; [b] isolated as a mixture with 4-phenylpyridine [c] run using difluoroacetic anhydride (1.1 equiv); [d] endo isomer obtained; [e] see SI experimental procedures for overall mass balance details.
In the next phase of this study, we evaluated a variety of diverse and pharmaceutically relevant heterocyclic motifs (Scheme 2). The tert-butylation of mildly electron-deficient pyridine N-oxide derivatives, such as ethyl isonicotinate N-oxide (25), as well as 4-acetylpyridine N-oxide (26) proceeded in good yields (75% and 52% respectively). Additionally, our studies showed that electron-rich pyridine N-oxides, such as alkylated N-oxides and protected alcohols (27–29), could undergo tert-butylation in good to modest yields. 7-Azaindole, which can be considered as a bioisostere of the indole and purine motifs and constitutes an essential subunit of many pharmacophores,13 could also be functionalized regioselectivity (30) using this protocol. Quinoline N-oxide was successully tert-butylated in 76% yield, leading to a 4:3 mixture of regioisomers (33). Several quinoline N-oxide derivatives containing methyl, methoxy, bromo, and chloro substituents, in addition to benzoquinoline, were alkylated in modest yields (31, 34–36). The lower yields observed in the tert-butylation of lepidine N-oxide (34) can be attributed to competing deprotonation of the methyl substituent upon generation of the acylated complex, which results in displacement of pivalic acid and precludes reductive alkyl radical generation. Furthermore, difluoromethylation of 6-methoxyquinoline (37) exclusively resulted in functionalization at the 4-position.
Scheme 2.

Heterocyclic N-oxide Scope.
[a]Isolated yields of reactions run on a 0.8 mmol scale with 1.6 mL of acetonitrile (0.5 M) and 1.1 equiv of acid chloride or anhydride reagent; [b] asterisks denote additional sites of functionalization; [c] see SI experimental procedures for overall mass balance details.
While a variety of pyridine and quinoline-based heterocyclic scaffolds could be accessed as coupling partners, functionalization of other five- and six-membered heterocyclic N-oxides, including benzylimidazole, quinoxaline, pyrimidine, and pyridazine N-oxide derivatives could not be achieved using this fragment coupling approach. As is evidenced by the significant recovery of N-oxide starting material, the observed lack of reactivity is suspected to be due to the diminished nucleophilicity of the N-oxide motif rather than inefficient radical addition.14 tert-Butylation of quinoxaline did proceed successfully, however, in the presence of pyridine N-oxide as a redox auxiliary (Scheme 3, 38). Furthermore, transitioning from simple heterocyclic substrates to more complicated, biologically relevant molecular scaffolds presents further challenges, as both the selective formation of the N-oxide functionality and the ability to predict the nucleophilicity of the N-oxide increase in complexity. We envisioned that the use of pyridine N-oxide as a sacrificial redox auxiliary would be an ideal platform for the alkylation of complex pharmacophore molecules. This concept came to fruition as subjection of brimonidine, a drug molecule used for the treatment of rosacea and open-angle glaucoma, with decarboxylative alkylation conditions furnished the tert-butylated derivative in 11% yield (39). Additionally, we were able to tert-butylate (44% yield) the imidazopyridazine core structure (41).15 Further diversification of the scaffold gave rise to the methylated (42) (16% yield) and difluoromethylated (43) (13% yield) analogs.
Scheme 3.

Alkylation of Heterocycles with Pyridine N-oxide.
[a]Isolated yields of reactions run on a 0.8 mmol scale with 5 equiv of pyridine N-oxide, 5.5 equiv of acid chloride or anhydride reagent and 1.6 mL of acetonitrile (0.5 M).
Quantum yield studies indicate that radical chain processes are operative in our system, as evidenced by a ϕ of 1.7.7 Crossover experiments demonstrate the viability of both intramolecular fragment coupling, as well as intermolecular radical propagation pathways, as the decarboxylative alkylation of two differentially functionalized acylated species gave rise to an approximately 1:1 distribution of self-functionalized and cross-functionalized products.7 Finally, we have demonstrated the capability to run this decarboxylative alkylation reaction on gram scale both in batch and in flow, suggesting that this methodology may translate beyond discovery scale. Using a 900 μL flow reactor, 1 gram of quinoline N-oxide was tert-butylated in an overall 71% yield (relative to 68 % yield on a 1 gram scale in batch), with a residence time of 2.25 min.7
In conclusion, we have reported an operationally simple and visible light-mediated method for the decarboxylative alkylation of heterocyclic N-oxides. Most significantly, this protocol offers a platform for the reductive generation of alkyl radicals without the reliance on stoichiometric additives, harsh reagents, and sacrificial redox auxiliaries. We envision this methodology to be of significant utility and practicality for the diversification of heterocyclic scaffolds in a multitude of medicinal applications.
Supplementary Material
Acknowledgments
The authors acknowledge the financial support of this research from the NIH NIGMS (R01-GM096129), the Camille Dreyfus Teacher-Scholar Award Program, and the University of Michigan. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship For A.C.S. (supported under Grant No DGE 1256260).
Footnotes
Notes
The authors declare no competing financial interests.
ADDITIONAL CONTENT
The Supporting Information is available free of charge on the ACS publications website. Please see SI for descriptions of batch and flow photochemical setups, additional data, procedures, and characterization data for all compounds (PDF).
References
- 1.Recent reviews and select examples: Seregin IV, Gevorgyan V. Chem Soc Rev. 2007;36:1173. doi: 10.1039/b606984n.Welsch ME, Synder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347. doi: 10.1016/j.cbpa.2010.02.018.Vitaku E, Smith DT, Njardarson JT. J Med Chem. 2014;57:10257. doi: 10.1021/jm501100b.Bering L, Antonchick AP. Org Lett. 2015;17:3134. doi: 10.1021/acs.orglett.5b01456.Jo W, Kim J, Choi S, Cho SH. Angew Chem Int Ed. 2016;55:9690. doi: 10.1002/anie.201603329.Hilton MC, Dolewski RD, McNally Andrew. J Am Chem Soc. 2016;138:13806. doi: 10.1021/jacs.6b08662.Fier PS. J Am Chem Soc. 2017;139:9499. doi: 10.1021/jacs.7b05414.
- 2.(a) Schonherr H, Cernak T. Angew Chem Int Ed. 2013;52:12256. doi: 10.1002/anie.201303207. [DOI] [PubMed] [Google Scholar]; (b) Alexandria PT, Robinson RP, Fobian YM, Blakemore DC, Jones LH, Fadeyi O. Org Biomol Chem. 2016;14:6611. doi: 10.1039/c6ob00936k. [DOI] [PubMed] [Google Scholar]; (c) Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW. Chem Soc Rev. 2016;45:546. doi: 10.1039/c5cs00628g. [DOI] [PubMed] [Google Scholar]
- 3.(a) Beatty JW, Douglas JJ, Cole KP, Stephenson CRJ. Nature Comm. 2015;6:7919. doi: 10.1038/ncomms8919. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beatty JW, Douglas JJ, Miller R, McAtee RC, Cole KP, Stephenson CRJ. Chem. 2016;1:456. doi: 10.1016/j.chempr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Minisci F, Bernardi R, Bertini F, Galli R, Perchinummo M. Tetrahedron. 1971;27:3575. [Google Scholar]; (b) Langlois BR, Laurent E, Roidot N. Tetrahedron Lett. 1991;32:7525. [Google Scholar]; (c) Punta C, Minisci F. Trends Heterocycl Chem. 2008;13:1. [Google Scholar]
- 5.Select recent examples of Minisci alkylations: Molander GA, Colombel V, Braz VA. Org Lett. 2011;13:1852. doi: 10.1021/ol2003572.Presset M, Fleury-Bregeot N, Oehlrich D, Rombouts F, Molander GA. J Org Chem. 2013;78:4615. doi: 10.1021/jo4005519.Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc Natl Acad Sci USA. 2011;108:14411. doi: 10.1073/pnas.1109059108.Fujiwara Y, Dixon JA, O’Hara F, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herle B, Sach N, Collins HR, Ishihara Y, Baran PS. Nature. 2012;492:95. doi: 10.1038/nature11680.
- 6.Select examples of photoredox catalysis-based alkylations: Jin J, MacMillan DWC. Nature. 2015;525:87. doi: 10.1038/nature14885.DiRocco DA, Dykstra K, Krska S, Vachal P, Conway DV, Tudge M. Angew Chem Int Ed. 2014;53:4802. doi: 10.1002/anie.201402023.Li GX, Molares-Rivera C, Wang Y, Fang Gao, He G, Liu P, Chen G. Chem Sci. 2016;7:6407. doi: 10.1039/c6sc02653b.McCallum T, Barriault L. Chem Sci. 2016;7:4754. doi: 10.1039/c6sc00807k.Matsui JK, Primer DN, Molander GA. Chem Sci. 2017;8:3512. doi: 10.1039/c7sc00283a.Garza-Sanchez RA, Tlahuext-Aca A, Tavakoli G, Glorius F. ACS Catal. 2017;7:4057.Sherwood TC, Li N, Yazdani AN, Murali Dhar TG. J Org Chem. 2018;83:3000–3012. doi: 10.1021/acs.joc.8b00205.Proctor RSJ, Davis HJ, Phipps RJ. Science. 2018 doi: 10.1126/science.aar6376.
- 7.See Supporting Information file for experimental details.
- 8.De Vleeschouwer F, Van Speybroeck V, Waroquier M, Geerlings P, De Proft F. Org Lett. 2007;9:2721. doi: 10.1021/ol071038k. [DOI] [PubMed] [Google Scholar]
- 9.Gunaydin H, Bartberger MD. ACS Med Chem Lett. 2016;7:341. doi: 10.1021/acsmedchemlett.6b00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stockdale TP, Williams CM. Chem Soc Rev. 2015;44:7737. doi: 10.1039/c4cs00477a. [DOI] [PubMed] [Google Scholar]
- 11.(a) Gillis EP, Eastman KJ, Hill MD, Donelly DJ, Meanwell NA. J Med Chem. 2015;58:8315. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]; (b) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (c) Ritchie TJ, Macdonald SJF. Eur J Med Chem. 2016;124:1057. doi: 10.1016/j.ejmech.2016.10.029. [DOI] [PubMed] [Google Scholar]
- 12.Murphy-Benenato KE, Olivier N, Choy A, Ross PL, Miller MD, Thresher J, Gao N, Hale MR. ACS Med Chem Lett. 2014;5:1213. doi: 10.1021/ml500210x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Popowycz F, Routier S, Joseph B, Merour JY. Tetrahedron. 2007;63:1031. [Google Scholar]; (b) Taylor AP, Robinson RP, Fobian YM, Blakemore DC, Jones LH, Fadeyi O. Org Biomol Chem. 2016;14:6611. doi: 10.1039/c6ob00936k. [DOI] [PubMed] [Google Scholar]
- 14.Baidya M, Brotzel F, Mayr H. Org Biomol Chem. 2010;8:1929. doi: 10.1039/c000965b. [DOI] [PubMed] [Google Scholar]
- 15.Douglas JJ, Cole KP, Stephenson CRJ. J Org Chem. 2014;79:11631. doi: 10.1021/jo502288q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.