

Abstract
The expression of hepatic transporters, including organic anion transporting polypeptides (OATPs) and multi-drug resistance-associated proteins (MRPs), is altered in nonalcoholic steatohepatitis (NASH), however, functional data in humans are lacking. In this study, 99mTc-mebrofenin (MEB) was used to evaluate OATP1B1/1B3 and MRP2 function in NASH patients.
Healthy subjects (n=14) and NASH patients (n=7) were administered MEB (~2.5mCi). A population pharmacokinetic model was developed to describe systemic and hepatic MEB disposition. Study subjects were genotyped for SLCO1B1 variants. NASH increased systemic and hepatic exposure (median±2SE, healthy vs. NASH) of MEB [AUC0-300,blood: 1780±242 vs. 2440±775μCi*min/L, p=0.006; AUC0-180,liver: 277±36.9 vs. 433±40.3kcounts*min/sec, p <0.0001] due to decreased biliary clearance (0.035±0.008 vs. 0.017±0.002L/min, p=0.0005) and decreased Vcentral (11.1±0.57 vs. 6.32±1.02L, p<0.0001). MEB hepatic CLuptake was reduced in NASH and also in healthy subjects with SLCO1B1 *15/*15 and *1A/*15 genotypes. The pharmacokinetics of drugs that are OATP1B1/1B3 and MRP2 substrates may be substantially altered in NASH.
Keywords: pharmacokinetics, imaging, organic anion transporters, transporters, liver
Introduction
The prevalence of non-alcoholic fatty liver disease (NAFLD) is growing at an alarming rate. Recent literature reports estimate that 20-30% of adults worldwide have NAFLD.1,2 Some patients with NAFLD will develop nonalcoholic steatohepatitis (NASH), an advanced form of NAFLD characterized by hepatic steatosis, hepatocyte ballooning, and lobular inflammation.3,4 NASH is a significant health concern that can progress to advanced fibrosis, cirrhosis, hepatocellular carcinoma, and liver failure, and is predicted to become the leading cause of liver transplantation.3,5–8 Common comorbid conditions in this patient population include obesity, diabetes, hyperlipidemia, hypertriglyceridemia, metabolic syndrome, and hypertension.2 Many medications used to treat these conditions (e.g., statins, angiotensin II receptor antagonists) are substrates of hepatic transporters. Therefore, potential NASH-mediated changes in hepatic transporter function could alter the pharmacokinetics and efficacy of these drugs in this patient population.
The importance of hepatic transporters in determining the systemic and hepatic exposure to drugs is well established. Changes in transporter function due to drug-drug interactions, genetic variants, or diseases can significantly alter the pharmacokinetics, and in some cases, the pharmacodynamics of drugs.9–12 Pre-clinical models of NASH, and liver tissue from patients with NASH, have shown altered expression of important hepatic uptake and efflux transporters. For example, recent data suggest that organic anion transporting polypeptide (OATP)1B1 is upregulated while OATP1B3 is downregulated in NASH.13 Since there is substantial overlap in substrate selectivity between these two uptake transporters, the functional impact of NASH-mediated alterations in hepatic uptake transporters remains to be established in humans. A clinical study evaluating the pharmacokinetics of morphine and metabolites in patients with NASH demonstrated increased serum morphine glucuronide concentrations, consistent with increased MRP3-mediated basolateral efflux in NASH.14 However, the expression/function of MRP2, a biliary efflux transporter with similar substrate specificity as MRP3, in patients with NASH is unclear. Previous studies have shown increased MRP2 expression, but this canalicular transporter appears to be mislocalized in liver tissue from patients with NASH.13,15 Preclinical models of NASH have shown reduced Mrp2 function and an increased risk of methotrexate-induced hepatic and renal toxicity in NASH.16 Additionally, both OATP1B1 and OATP1B3, and the efflux transporter MRP2, exhibited decreased levels of the glycosylated protein in liver tissue from patients with NASH compared to control tissues, which may explain the mislocalization of some transporters.17–19 The impact of impaired function of efflux transporters can be difficult to study in the clinic because only small changes may occur in systemic concentrations even though there may be extensive changes in hepatic exposure.20 Hepatic exposure can be assessed using imaging modalities and well-characterized transporter probes [i.e., gamma scintigraphy and 99mTechnetium–mebrofenin (MEB)]
MEB is a nuclear imaging agent used to diagnose or differentiate hepatobiliary abnormalities. MEB is highly extracted by the liver and undergoes negligible metabolism; ~98% of the dose is excreted into bile with minimal urinary excretion (~1-2% in 24hr).21 MEB is primarily taken up by OATP1B1 and OATP1B3, and is rapidly excreted by the canalicular transporter MRP2 into bile.22–24 Inhibition of OATPs by co-administered drugs can increase MEB systemic exposure.25 Furthermore, hepatic exposure to MEB and other iminodiacetic acid analogs is prolonged due to genetic impairment of MRP2/Mrp2 in patients with Dubin-Johnson syndrome and in TR− rats.26–29 MRP3 also has been shown to transport MEB, but MRP3-mediated basolateral clearance of MEB is substantially lower than MRP2-mediated biliary clearance.23,25
The aim of this study was to evaluate the systemic and hepatic disposition of MEB in patients with NASH utilizing population pharmacokinetic (PopPK) modeling to elucidate NASH-mediated changes in hepatic transporter function (i.e., OATPs, MRP2).
RESULTS
Study Participants
Twenty-one study participants (14 healthy subjects and 7 patients with biopsy-confirmed NASH) were enrolled in this study, and all completed the study. The two cohorts had similar age and sex distributions, albumin, and creatinine, however the NASH cohort had a higher body weight and body mass index (BMI) (Table 1). Based on biopsy results, patients with NASH had a NAFLD activity score (NAS) of 4 or 5, and fibrosis (F1-F3) (Table 1).
Table 1.
Baseline demographic and clinical characteristics of study subjects.
| Characteristics | Healthy (n=14) | NASH (n=7) |
|---|---|---|
| Male : Female | 8:6 | 4:3 |
| Hispanic : Non-Hispanic | 0:14 | 1:6 |
| Asian : Black : White | 1:3:10 | 1:0:6 |
| Age (yrs) mean (min, max) | 39 (21,64) | 37 (21,61) |
| Weight (kg) | 72 (12) | 102 (17) |
| BMI (kg/m2) | 24 (2) | 33 (5) |
| Albumin (mg/dl) | 4.2 (0.2) | 4.5 (0.5) |
| Creatinine (mg/dl) | 0.86 (0.17) | 0.83 (0.15) |
| ALP (U/L) | 56 (18) | 68 (20) |
| ALT (U/L) | 29 (10) | 110 (60) |
| AST (U/L) | 25 (8) | 73 (34) |
| Bilirubin, total (mg/dL) | 0.7 (0.2) | 1.0 (0.4) |
| NASa | 4 (4-5) | |
| Fibrosisa | 2 (1-3) | |
| NAS+Fibrosisa | 6 (5-8) |
Data are presented as mean (standard deviation) except where otherwise noted.
Data presented as median (range). ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; NAS, NAFLD activity score.
MEB Pharmacokinetics
Blood concentrations and liver count data obtained from gamma scintigraphy after administration of a ~2.5mCi intravenous dose of MEB are shown in Fig. 1. The amount of MEB recovered in urine at 180min was <1% of the administered dose for both groups, and protein binding of MEB was unchanged in NASH patients compared to healthy subjects (Table 2). Pharmacokinetic parameters derived from NCA of the blood and liver data are summarized in Table 2. The Cmax and AUC0-300,blood were increased by 2.1- and 1.4-fold respectively in the NASH cohort. The terminal elimination half-life of MEB in blood increased from 190 min to 365 min in the NASH cohort.
Fig. 1. 99mTc-mebrofenin blood concentration vs. time curves and liver scintigraphy vs. time curves in healthy subjects (blue) and patients with NASH (red).
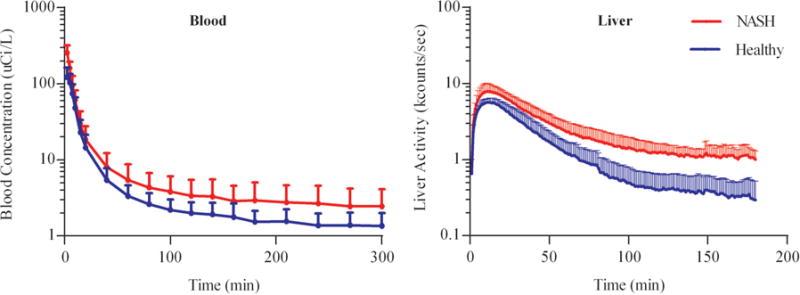
Data represent mean±SD.
Table 2.
Parameters describing 99mTc-mebrofenin disposition in blood and liver after an intravenous bolus dose.
| Parameter | Healthy | NASH | Differencea | p-value |
|---|---|---|---|---|
| Administered dose (mCi)b | 2.44 (2.15-2.86) |
2.50 (1.99-2.68) |
0.0203 [−0.131, 0.169] |
0.636 |
| Unbound fraction (fu)c | 0.0520 (0.0170-0.153) |
0.0710 (0.0220-0.122) |
0.00500 [−0.0270, 0.0410] |
0.642 |
| Urinary recoveryd (% of dose) | 0.576 (0.199-0.923) |
0.694 (0.535-1.56) |
0.256 [−0.0311, 0.569] |
0.079 |
| Cmax blood (μCi/L) | 136 (101-180) |
286 (141-318) |
135 [65.9, 176] |
<0.001* |
| AUC0-300,blood (μCi* min/L) | 1780 (837-2630) |
2440 (1660-4870) |
845 [342, 1580] |
0.006* |
| Vss (L) | 133 (83.8-491) |
198 (96.7-394) |
−4.70 [−172, 84.2] |
0.971 |
| λe,blood (1/min) | 0.00365 (0.00100-0.0124) |
0.00190 (0.00130-0.00300) |
−0.00145 [−0.00310, −0.000100] |
0.040* |
| Xmax,liver (kcounts/sec) | 6.00 (4.46-6.74) |
8.81 (5.66-10.6) |
2.61 [0.197, 4.14] |
0.025* |
| tmax,liver (min) | 13.0 (9.00-21.0) |
13.0 (10.0-21.0) |
−1.00 [−3.00, 3.00] |
0.672 |
| AUC0-180,liver (kcounts* min/sec) | 277 (146-374) |
433 (387-519) |
203 [126, 253] |
<0.0001* |
| λe,liver (1/min) | 0.0183 (0.00910-0.117) |
0.0140 (0.0127-0.0385) |
−0.00420 [−0.0431, 0.000200] |
0.054 |
Data presented as median (range).
Median difference with 95% confidence interval (by Hodges-Lehmann estimation).
Administered dose was calculated by measuring the amount of radioactivity in the syringe containing the prepared dose and subtracting the amount of radioactivity remaining in the syringe, catheter and tubing after administration.
Data were not available to calculate unbound fraction for 1 healthy subject.
Total mass excreted from 0-180 min.
CLblood was not reported because the % extrapolated AUC0-∞,blood (median±2SE) was 17±6.4% in the healthy cohort and 29±7.2% in the NASH cohort.
Maximal blood concentration (Cmax), area under the blood concentration-time curve (AUCblood), blood clearance (CLblood), area under the liver activity-time curve (AUCliver), maximal liver activity (Xmax), elimination rate constant (λe), time to maximum liver activity (tmax,liver). Mann-Whitney-Wilcoxon test, NASH vs. healthy,
p<0.05
Gamma scintigraphy of MEB over 180min revealed rapid uptake of MEB into the liver. In both cohorts the time-to-maximal activity in the liver (tmax,liver) was ~13min (Table 2). Maximal liver activity (Xmax) and the area under the MEB liver-activity time curve (AUC0-180,liver) were significantly increased by 1.5-fold and 1.6-fold in the NASH cohort, respectively (Table 2). The terminal elimination half-life of MEB in the liver (t1/2,liver) increased from 38 to 50 min in the NASH cohort.
A three-compartment model with blood, liver, and peripheral compartments (Fig. 2) best described the systemic and hepatic MEB concentration-time profiles. NASH was a statistically significant covariate on hepatic uptake clearance (CLuptake), biliary clearance (CLbile), and volume of the central compartment (Vcentral). The model fit to the individual data is shown in Fig. 3. Individual pharmacokinetic parameter estimates from the model, stratified by study cohort, are shown in Table 3. The Vcentral of MEB was significantly decreased (~50% lower) in patients with NASH. CLuptake of MEB was reduced ~1.6-fold and CLbile was reduced by more than 2-fold in patients with NASH compared to healthy subjects.
Fig. 2. Schematic of the pharmacokinetic model describing 99mTc-mebrofenin disposition.
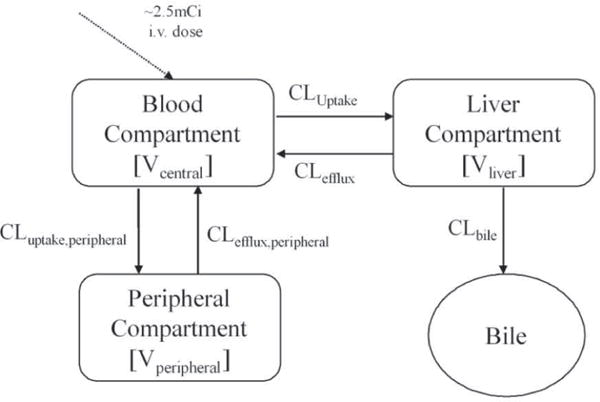
The final structural model that best described the data included a central, peripheral and a liver compartment. 99mTc-mebrofenin was excreted into bile from the liver compartment.
Fig. 3. Concentration-time profiles of observed data and model predictions.

Observed data are depicted by circles and the model predictions for each individual subject are depicted by the curves.
Table 3.
Parameter estimates obtained from the population pharmacokinetic model stratified by disease state.
| Description of PK Parameter | PK Parameter | Healthy | NASH | Difference | p-value |
|---|---|---|---|---|---|
| Uptake clearance from blood to liver | CLuptake (L/min) | 1.14 (0.732–2.27) |
0.731 (0.382–1.04) |
−0.398 [−0.679, −0.202] |
0.0008* |
| Efflux clearance from liver to blood | CLefflux (L/min) | 0.00800 (0.00481–0.0139) |
0.00579 (0.00475–0.00903) |
−0.00192 [−0.00384, 0.000137] |
0.0667 |
| Clearance from liver to bile | CLbile (L/min) | 0.0354 (0.0157–0.0728) |
0.0171 (0.0110–0.0207) |
−0.0172 [−0.0268, −0.00467] |
0.0005* |
| Uptake clearance from blood to the peripheral compartment | CLuptake,peripheral (L/min) | 0.754 (0.353–1.01) |
0.491 (0.317–0.811) |
−0.118 [−0.372, 0.0780] |
0.322 |
| Efflux clearance from the peripheral compartment to blood | CLefflux,peripheral (L/min) | 0.00517 (0.00434–0.00708) |
0.00418 (0.00311–0.00555) |
−0.000980 [−0.00175, −0.000356] |
0.004* |
| Central volume of distribution | Vcentral (L) | 11.1 (9.55–12.5) |
6.32 (5.69–9.69) |
−4.42 [−5.54, −3.24] |
<0.0001* |
| Volume of distribution in the liver | Vliver (L) | 0.958 (0.527–1.39) |
0.891 (0.648–1.43) |
−0.0424 [−0.355, 0.225] |
0.913 |
Data presented as median (range). Median difference with 95% confidence interval (by Hodges-Lehmann estimation). Mann-Whitney-Wilcoxon test, NASH vs. healthy,
p<0.05
In addition to disease state (healthy vs. NASH), the effect of genetic variants on MEB disposition also was evaluated. Genetic data were available for 12 healthy subjects and 5 NASH patients. SLCO1B1 (the gene encoding for OATP1B1) haplotypes were assigned functional classification based on clinical guidelines and literature data.30,31 In the healthy cohort, one subject had a low function genotype, three subjects had an intermediate function genotype, and the remaining subjects had normal function genotypes; all 5 NASH patients had normal function genotypes (Table S1). Hepatic uptake clearance estimates for individual subjects obtained from the pharmacokinetic model were stratified by disease state and OATP1B1 genotype (Fig. 4). The reduction in CLuptake in patients with NASH and healthy subjects with the low/intermediate function SLCO1B1 genotype is consistent with decreased function of OATP1B1.
Fig. 4. Hepatic uptake clearance (CLuptake) and SLCO1B1 genotype in healthy subjects and NASH patients.

The subgroups with intermediate function (IF) and low function (LF) genotypes were combined (due to the limited sample sizes) for comparison to the NASH and healthy subgroups with normal function (NF) genotypes. The null hypothesis “no distributional differences among the three populations” was tested using a Kruskal-Wallis procedure of size α=0.05 conditionally followed by paired comparisons using Dunn’s procedure. Legend: * p < 0.05, n.s. p ≥ 0.05.
Discussion
This is the first study evaluating NASH-mediated alterations in hepatic transporter function utilizing MEB, an OATP/MRP2 substrate, as a clinical probe. Systemic and hepatic exposure (AUC0-last) to MEB after administration of a ~2.5mCi dose was increased in the NASH cohort by 1.4- and 1.6-fold in blood and liver, respectively. Observed changes in MEB disposition were not due to NASH-mediated alterations in urinary excretion or protein binding. Blood and liver concentration-time profiles of MEB were analyzed using a PopPK modeling approach to elucidate the underlying mechanisms responsible for the differences in systemic and hepatic exposure between the populations. The NASH cohort exhibited increased systemic and hepatic exposure to MEB due to disease-mediated reductions in the central volume of distribution, hepatic uptake clearance, and biliary clearance of MEB.
The Cmax of MEB after intravenous administration of a ~2.5mCi dose was higher in the NASH cohort due to a reduced volume of distribution of MEB in blood. The volume of distribution is a hypothetical volume that represents the extent of drug distribution in the body rather than a physiological volume. The estimated Vcentral in healthy subjects (~11L) was similar to previously reported literature values.23 However, in the NASH cohort the Vcentral of MEB was ~50% lower than in healthy subjects, likely due to impaired hepatic uptake of MEB in patients with NASH rather than other underlying physiological changes. Changes in hepatic uptake clearance can alter the volume of distribution of medications, as discussed previously by Benet et al.; for example, inhibition of OATP1B1-mediated uptake of atorvastatin by rifampin decreased the atorvastatin volume of distribution.32
Hepatic uptake can be a major determinant of systemic pharmacokinetics for a drug. The key drug uptake transporters OATP1B1 and OATP1B3 are responsible for the hepatic uptake of numerous medications (including statins, enalapril, and valsartan) and endogenous compounds. These two transporters share 80% amino acid homology and have a high degree of substrate overlap.33 Interestingly, in liver tissue from patients with NASH, OATP1B1 expression was increased but OATP1B3 expression was decreased; however, OATP1B1 glycosylation was decreased.13,17 Additionally, a decrease in hepatic blood flow in patients with NASH34 could also contribute to a reduced hepatic clearance. In the present study, the hepatic uptake clearance of MEB, a substrate of both OATP1B1 and OATP1B3, was reduced in the NASH cohort. Medications used in patients with NASH to treat common comorbid conditions such as hyperlipidemia (rosuvastatin and pravastatin) and hypertension (enalapril, olmesartan, and valsartan) are also substrates of both OATP1B1 and OATP1B3. The hepatic uptake of these commonly used medications also may be decreased in patients with NASH, as shown with MEB.
Genetic polymorphisms can alter the function of OATP1B1 in addition to NASH-mediated changes in OATP1B1 function. The *15 haplotype is associated with reduced OATP1B1 function, and homozygous patients are characterized as low function; patients that are heterozygous for the *15 allele are classified as exhibiting intermediate OATP1B1 function.30,31 Healthy subjects with either the low or intermediate function OATP1B1 genotypes exhibited a lower hepatic uptake clearance of MEB compared to healthy subjects with the normal function genotype (Fig. 4). Surprisingly, NASH patients with a normal OATP function genotype exhibited a lower hepatic uptake clearance of MEB than the healthy subjects with impaired function genotypes (Fig. 4). Although genetic polymorphisms in OATP1B1 can reduce the uptake of MEB, the magnitude of this decrease appears to be less than the reduction in hepatic uptake of MEB due to NASH-related changes. The synergistic effect of genetic polymorphisms and NASH on transporter function was evaluated in Oatp1b2-knockout mice with diet-induced NASH; pravastatin plasma exposure was highest in Oatp1b2 knockout mice with diet-induced NASH compared to control mice or mice with either diet-induced NASH or Oatp1b2 knockout alone.13 Additional studies should be conducted to evaluate the interaction and functional impact of genetic polymorphisms and NASH-mediated alterations in hepatic transporter function.
Although changes in hepatic uptake clearance can cause important differences in systemic pharmacokinetics, alterations in hepatic efflux transporters typically have the most significant impact on hepatic exposure.35 Patients with NASH exhibited a ~1.6-fold increase in hepatic exposure to MEB that was due primarily to decreased biliary excretion. Median (±2SE) CLbile was 0.017±0.002L/min in the NASH cohort compared to 0.035±0.008L/min in the healthy cohort. Biliary excretion of MEB is mediated by MRP2, an ATP-dependent canalicular transporter with broad specificity for both endogenous substrates as well as drugs and metabolites (glutathione and glucuronide conjugates). Reduced function of this canalicular transporter may alter the pharmacokinetics of medications that are MRP2 substrates including methotrexate, statins, enalapril, olmesartan, valsartan, fexofenadine, and glucuronide metabolites of mycophenolic acid.36,37 In liver tissue from patients with NASH, although MRP2 expression was increased, it appeared to be mislocalized from the canalicular membrane, likely due to reduced glycosylation15,17; unglycosylated MRP2 may not be functional.18,19 These findings are consistent with reduced MRP2 function observed in the present study. Impaired MRP2 function (due to diseases, genetic variants, or drug interactions) can have a substantial impact on the hepatic exposure of a compound as shown for the first time in this clinical study. This may have significant clinical implications in the treatment of NASH patients with drugs that are MRP2 substrates and that have a site of action (efficacy or toxicity) in the liver.
MRP3, a hepatic basolateral efflux transporter that exhibits extensive substrate overlap with MRP2, can serve as a compensatory pathway in conditions where MRP2 is downregulated.38,39 Patients with NASH have increased MRP3 expression15 and function as demonstrated by an increase in systemic concentrations of glucuronide metabolites of morphine.14 However, the two cohorts were similar in median basolateral efflux clearance of MEB (CLefflux). Although in vitro membrane vesicle studies have shown that MEB is a substrate for MRP3, the overall contribution of MRP3-mediated basolateral efflux clearance in the disposition of MEB is minimal. The ratio of biliary to basolateral efflux clearance for healthy subjects was ~4.5, consistent with our previous study, and indicates that MEB is not a sensitive clinical probe for assessing MRP3 function.23,25
To define a homogenous population and reduce the impact of confounding variables, subjects were excluded if they had evidence of cirrhosis, portal hypertension, decompensated liver disease, a history of immune-related disease, bariatric surgery, a history of drugs associated with fatty liver disease or steatohepatitis, or if they were taking any Type 2 diabetes medications other than metformin, or any medication that might interact with OATPs/MRP2. Patients with a histological confirmation of NASH with an NAS score of >3 were enrolled because changes in transporter expression are expected after the onset of inflammation.15 Additionally, intensive sampling was conducted to help reduce the number of patients required to develop the PopPK model.
In conclusion, patients with biopsy-confirmed NASH displayed increased systemic and hepatic exposure to MEB, and biliary clearance was decreased by more than 2-fold, compared to age- and sex-matched healthy subjects. Specifically, these changes are consistent with NASH-associated decreases in hepatic uptake (mediated by OATP1B1 and OATP1B3) and biliary clearance (mediated by MRP2). Given the increasing prevalence of NASH, the impact of hepatic transporter dysregulation on the systemic and hepatic exposure to drugs and their metabolites should be considered carefully when treating patients with steatohepatitis. Medications with characteristics similar to MEB (high protein binding, substrates of OATP1B1/1B3 and/or MRP2, biliary excretion with negligible renal elimination) that are likely to be prescribed to patients with NASH include statins, repaglinide, olmesartan, and fexofenadine. Clinical data regarding altered pharmacokinetics of these drugs in patients with NASH is currently lacking, and additional studies should be conducted. Importantly, the impact of decreased hepatic uptake and/or impaired biliary excretion mediated by OATP1B1 and MRP2, respectively, in patients with biopsy-confirmed NASH on compounds in drug development should be considered carefully. NASH-mediated changes in transporter function could affect the systemic concentrations, pharmacodynamic effects and/or toxicity of medications that have pharmacokinetic properties similar to MEB, especially when the liver is the site of action. Further understanding how NASH affects hepatic and extrahepatic clearance mechanisms will improve our ability to predict drug disposition, efficacy, and safety of new and existing medications in this patient population.
METHODS
Study Participants
This single-center, observational, cohort-comparison study enrolled healthy subjects and patients with biopsy-confirmed NASH between 18 and 65 years of age. Subjects with self-reported drinking >20g/day of alcohol, or a history of gastrointestinal surgery, autoimmune disease, or other gastrointestinal/liver abnormalities were excluded. At screening, healthy subjects had clinical chemistry and coagulation panel measurements within the normal range as determined by the McLendon Clinical Laboratories at UNC Medical Center, Chapel Hill, NC. All enrolled subjects exhibited normal values for serum creatinine, total bilirubin, and negative results for HIV antigen, hepatitis B surface antigen, and hepatitis C antibody. Additionally, the enrolled healthy subjects had a homeostatic model assessment for insulin resistance (HOMA-IR) score <2.5, BMI ≤ 30kg/m2, and were not taking any medications or herbal products other than oral contraceptives or a daily multivitamin.
Patients with biopsy-confirmed, noncirrhotic NASH were recruited from the UNC-CH Hepatology Clinic and were enrolled if they met the following inclusion criteria: nonalcoholic fatty liver disease activity score (NAS) >3, BMI ≤ 45kg/m2, no treatment for Type 2 diabetes other than metformin, no milk thistle products or high-dose antioxidant treatment during the prior 30 days (e.g., Vitamin E), and absence of prior treatment with NASH-associated drugs (i.e., tamoxifen, amiodarone, methotrexate, prednisone, tetracyclines, or valproic acid). If NASH patients were taking medications that might interact with OATPs/MRP2, these medications were withheld for a washout period of 5 half-lives, if determined clinically appropriate by the study physician. Otherwise, the patient was excluded from the study.
Written informed consent was obtained from all subjects. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the UNC-CH Biomedical Institutional Review Board, and was registered on ClinicalTrials.gov (NCT02235233).
Study Design
Subjects and patients who met all inclusion/exclusion criteria were admitted to the Clinical and Translational Research Center at UNC-CH Hospitals after fasting overnight. Vital signs were assessed, and an intravenous catheter was placed in one arm delivering a normal saline drip, which was used for drawing blood samples during the study. Prior to MEB administration, a transmission acquisition was performed using a cobalt-57 flood source (Isotope Products Laboratories, Burbank, CA) positioned posteriorly, and gamma rays were detected in the cobalt window (122 KeV±15%) for 5-10min on the anterior detector in the absence and presence of the study participant to obtain a subject-specific attenuation correction factor. A ~2.5mCi intravenous bolus dose of MEB was administered via an indwelling catheter placed in a contralateral forearm or hand vein, which was removed after drug administration. Blood samples (3mL) were collected at baseline (0), 2.5, 5, 7.5, 10, 15, 20, 40, 60, 80, 100, 120, 140, 160, 180, 210, 240, 270, and 300min after administration; urine samples were collected at baseline (0) and at 180min after administration. Anterior and posterior scintigraphic images of the abdomen were acquired dynamically in the 99mTc window (140 KeV±15%) at 1-min intervals using a dual headed gamma camera up to 180 min after MEB administration. Liver activity-time curves were generated as described previously using Syngo MI applications version 6.5.9.19 (Siemens, New York, NY).25 Prior to discharge, vital signs were assessed and study participants were administered a safety questionnaire.
Blood and urine samples were analyzed for MEB radioactivity with a sodium iodide well counter and corrected for decay (99mTc t1/2 = 6.01hr). A blood sample (3mL) collected at 20min post-dose was centrifuged to obtain serum; 0.6ml of serum was filtered through a CentrifreeT ultrafiltration membrane system (Millipore Ireland Ltd, Tullagreen, Cork, Ireland) according to the manufacturer’s instructions to obtain ~10% of the initial volume as ultrafiltrate to quantify the unbound fraction of MEB in serum as described previously.23 Genomic DNA was extracted using the Maxwell 16 LEV Blood DNA Kit on a Maxwell 16 Research automated nucleic acid extraction system (Promega, Madison, WI) from whole blood samples collected on the study day, prior to MEB administration. The samples were genotyped for the SLCO1B1 rs2306283 (c.388A>G, p.N130D), rs11045819 (c.463C>A, p.P155T), rs4149056 (c.521T>C, p.V174A), and rs71581941 (c.1738C>T, p.R580Stop) single nucleotide polymorphisms by allelic discrimination using Taqman® 5′-nuclease assays on a QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA).
Pharmacokinetic Data Analysis
Blood concentration-time and the liver counts/sec-time profiles were used to conduct a non-compartmental analysis (NCA) using Phoenix® WinNonlin® version 7 (Certara USA, Inc., Princeton, NJ). The area under the blood concentration-time curve and the liver activity-time curve from time zero to the last time point (AUC0-300,blood and AUC0-180,liver, respectively) were determined using a linear-up log-down trapezoidal algorithm. The terminal half-life (t1/2) was calculated as 0.693/λz, where λz was estimated from the terminal elimination phase. The percentage of the dose excreted in urine was calculated by dividing the total mass of MEB in urine after 180min (the product of urine concentration and total volume collected) by the administered dose.
In addition to the NCA, a PopPK model was developed using ordinary differential equations to describe the disposition of MEB in the blood and liver using the Quasi-random Parametric Expectation Maximization (QRPEM) estimation method in Phoenix® WinNonlin® version 7. Various structural models were evaluated, including models without a peripheral compartment, and with two liver sub-compartments. The final model was selected based on the objective function values, overall model fit, and diagnostic plots. Urinary excretion of MEB was not included in the model because it was not a major route of elimination (<1% of the dose was recovered in urine after 180min). Liver concentrations were obtained by converting liver counts to amount using an instrument-specific conversion factor of 193CPM/μCi and dividing by estimated liver volume calculated for each individual subject using a previously published formula based on body surface area.40 The volume of the peripheral compartment was estimated to be approximately 1.5L in the initial iterations of the model, and was fixed at this value for subsequent runs. A multiplicative error model was used for blood and liver concentrations, and an exponential random effects model was used for interindividual variability on the model parameters.
The effects of potential covariates (body weight and NASH disease status) on Vcentral, CLuptake, CLbile, and CLefflux were evaluated using a forward-inclusion, backward-elimination, stepwise algorithm. A visual predictive check was performed with 100 replicates using the parameter and interindividual variability estimates from the final model to evaluate the performance of the model (Figure S1). The final version of the PopPK model was used to compute parameter estimates for each NASH patient and each healthy participant to evaluate the impact of NASH.
Statistical Analysis Strategy
Individual-specific estimates of MEB blood and liver pharmacokinetic parameters from the NCA and from the PopPK model were used to characterize and compare the two cohorts. Median differences between the two cohorts were estimated using the Hodges-Lehmann estimator (HLE) of location shift for two-sample data. The HLE was tabulated along with the corresponding 95% confidence interval for the median difference. The Mann-Whitney-Wilcoxon test was used to detect differences in pharmacokinetic parameters between the healthy and NASH cohorts. All statistical analyses were conducted using GraphPad Prism version 7.00 for Windows (GraphPad Software, La Jolla, CA, USA).
Supplementary Material
Figure S1: Visual Predictive Checks for 99mTc-Mebrofenin in Blood and Liver.
Table S1: SLCO1B1 Genotype of study participants stratified by group.
Study Highlights.
What is the current knowledge on the topic?
Alterations in the expression of some hepatic uptake and efflux transporters in NASH have been reported previously. However functional data in patients with NASH are lacking.
What question did this study address?
This study was designed to evaluate NASH-mediated changes in OATP1B1/1B3 and MRP2 function using MEB, a clinical probe for transporter function assessment. A population pharmacokinetic model was developed to quantify changes in systemic and hepatic disposition of MEB in NASH patients compared to healthy subjects.
What does this study add to our knowledge?
Patients with NASH exhibited increased systemic and hepatic exposure to MEB, likely due to decreased hepatic uptake and impaired biliary excretion of MEB.
How might this change clinical pharmacology or translational science?
OATPs and MRP2 are key drug transporters that play an important role in the disposition of many medications, including those commonly used in patients with NASH (e.g., statins, angiotensin receptor antagonists). Therefore, disease-mediated impairment in the function of these transporters, as shown here, may have important clinical implications for the treatment of patients with NASH.
Acknowledgments
WinNonlin software was generously provided to the Division of Pharmacotherapy and Experimental Therapeutics, UNC Eshelman School of Pharmacy, by Certara as a member of the Pharsight Academic Center of Excellence Program. We gratefully acknowledge Dr. Daniel Gonzalez for his advice in development of the population pharmacokinetic model, and Krista Sherrell for nuclear medicine technical assistance.
This work, in part, was submitted to the UNC-CH Graduate School in partial fulfillment of requirements for the Doctor of Philosophy degree in Pharmaceutical Sciences (J.R.S.), and was presented at the 2016 American Association of Pharmaceutical Scientists Annual Meeting, and the 2017 American Society of Clinical Pharmacology and Therapeutics Annual Meeting.
Funding: This research was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under Award Numbers R01GM041935 and R35GM122576 (K.L.R.B.). I.A. was supported by an NIH NIGMS Clinical Pharmacology Training Grant (Award Number T32GM086330). The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH. J.D.K. was supported by a Clinical Pharmacokinetics/Pharmacodynamics fellowship sponsored by Quintiles.
Footnotes
Conflict of Interest: The authors declared no competing interests for this work.
Author Contributions: I.A., J.R.S., J.D.K., M.I., M.N., P.W.S., A.S.B., and K.L.R.B. wrote the manuscript; J.R.S., P.W.S., A.S.B., and K.L.R.B. designed the research; I.A., J.R.S., J.D.K., M.I., M.N., and A.S.B. performed the research; I.A., J.R.S., and J.D.K. analyzed the data.
References
- 1.Bellentani S. The Epidemiology of Non-Alcoholic Fatty Liver Disease. Liver Int. 2017;37:81–84. doi: 10.1111/liv.13299. [DOI] [PubMed] [Google Scholar]
- 2.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global Epidemiology of Nonalcoholic Fatty Liver Disease. Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 3.Zezos P, Renner EL. Liver Transplantation and Non-Alcoholic Fatty Liver Disease. World J Gastroenterol. 2014;20:15532–8. doi: 10.3748/wjg.v20.i42.15532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chalasani N, et al. The Diagnosis and Management of Non-alcoholic Fatty Liver Disease: Practice Guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and Outcomes of Liver Transplantation for Nonalcoholic Steatohepatitis in the United States. Gastroenterology. 2011;141:1249–1253. doi: 10.1053/j.gastro.2011.06.061. [DOI] [PubMed] [Google Scholar]
- 6.Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The Natural History of Nonalcoholic Steatohepatitis: a Follow-up Study of Forty-Two Patients for up to 21 Years. Hepatology. 1990;11:74–80. doi: 10.1002/hep.1840110114. [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto E, et al. Hepatocellular Carcinoma in Patients with Nonalcoholic Steatohepatitis. J Gastroenterol. 2009;44:89–95. doi: 10.1007/s00535-008-2262-x. [DOI] [PubMed] [Google Scholar]
- 8.Hui J, et al. Long-Term Outcomes of Cirrhosis in Nonalcoholic Steatohepatitis Compared with Hepatitis C. Hepatology. 2003;38:420–427. doi: 10.1053/jhep.2003.50320. [DOI] [PubMed] [Google Scholar]
- 9.Thakkar N, Slizgi JR, Brouwer KLR. Effect of Liver Disease on Hepatic Transporter Expression and Function. J Pharm Sci. 2017;106:2282–2294. doi: 10.1016/j.xphs.2017.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.König J, Müller F, Fromm MF. Transporters and Drug-Drug Interactions: Important Determinants of Drug Disposition and Effects. Pharmacol Rev. 2013;65:944–966. doi: 10.1124/pr.113.007518. [DOI] [PubMed] [Google Scholar]
- 11.Lai Y, et al. Impact of Drug Transporter Pharmacogenomics on Pharmacokinetic and Pharmacodynamic Variability – Considerations for Drug Development. Expert Opin Drug Metab Toxicol. 2012;8:723–743. doi: 10.1517/17425255.2012.678048. [DOI] [PubMed] [Google Scholar]
- 12.Patel M, Taskar KS, Zamek-Gliszczynski MJ. Importance of Hepatic Transporters in Clinical Disposition of Drugs and Their Metabolites. J Clin Pharmacol. 2016;56:S23–S39. doi: 10.1002/jcph.671. [DOI] [PubMed] [Google Scholar]
- 13.Clarke JD, et al. Synergistic Interaction between Genetics and Disease on Pravastatin Disposition. J Hepatol. 2014;61:139–47. doi: 10.1016/j.jhep.2014.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferslew B, et al. Altered Morphine Glucuronide and Bile Acid Disposition in Patients with Nonalcoholic Steatohepatitis. Clin Pharmacol Ther. 2015;97:419–427. doi: 10.1002/cpt.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardwick RN, Fisher CD, Canet MJ, Scheffer GL, Cherrington NJ. Variations in ATP-binding Cassette Transporter Regulation During the Progression of Human Nonalcoholic Fatty Liver Disease. Drug Metab Dispos. 2011;39:2395–402. doi: 10.1124/dmd.111.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardwick RN, et al. Increased Susceptibility to Methotrexate-Induced Toxicity in Nonalcoholic Steatohepatitis. Toxicol Sci. 2014;142:45–55. doi: 10.1093/toxsci/kfu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke JD, Novak P, Lake AD, Hardwick RN, Cherrington NJ. Impaired N-linked Glycosylation of Uptake and Efflux Transporters in Human Non-alcoholic Fatty Liver Disease. Liver Int. 2017;37:1074–1081. doi: 10.1111/liv.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang P, Tian X, Chandra P, Brouwer KLR. Role of Glycosylation in Trafficking of Mrp2 in Sandwich-cultured Rat Hepatocytes. Mol Pharmacol. 2005;67:1334–41. doi: 10.1124/mol.104.004481. [DOI] [PubMed] [Google Scholar]
- 19.Hashimoto K, et al. Trafficking and Functional Defects by Mutations of the ATP-binding Domains in MRP2 in Patients with Dubin-Johnson Syndrome. Hepatology. 2002;36:1236–1245. doi: 10.1053/jhep.2002.36368. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. Physiologically Based Pharmacokinetic Modeling to Predict Transporter-Mediated Clearance and Distribution of Pravastatin in Humans. J Pharmacol Exp Ther. 2009;328:652–662. doi: 10.1124/jpet.108.146647. [DOI] [PubMed] [Google Scholar]
- 21.Krishnamurthy GT, Turner FE. Pharmacokinetics and Clinical Application of Technetium 99m-labeled Hepatobiliary Agents. Semin Nucl Med. 1990;20:130–49. doi: 10.1016/s0001-2998(05)80166-7. [DOI] [PubMed] [Google Scholar]
- 22.De Graaf W, et al. Transporters Involved in the Hepatic Uptake of 99mTc-Mebrofenin and Indocyanine Green. J Hepatol. 2011;54:738–745. doi: 10.1016/j.jhep.2010.07.047. [DOI] [PubMed] [Google Scholar]
- 23.Ghibellini G, Leslie EM, Pollack GM, Brouwer KLR. Use of Tc-99m Mebrofenin as a Clinical Probe to Assess Altered Hepatobiliary Transport: Integration of In Vitro, Pharmacokinetic Modeling, and Simulation Studies. Pharm Res. 2008;25:1851–1860. doi: 10.1007/s11095-008-9597-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swift B, Yue W, Brouwer KLR. Evaluation of (99m)Technetium-mebrofenin and (99m)Technetium-sestamibi as Specific Probes for Hepatic Transport Protein Function in Rat and Human Hepatocytes. Pharm Res. 2010;27:1987–98. doi: 10.1007/s11095-010-0203-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfeifer ND, et al. Effect of Ritonavir on (99m)Technetium-Mebrofenin Disposition in Humans: A Semi-PBPK Modeling and In Vitro Approach to Predict Transporter-Mediated DDIs. CPT pharmacometrics Syst Pharmacol. 2013;2:e20. doi: 10.1038/psp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhargava KK, et al. Adenosine Triphosphate-Binding Cassette Subfamily C Member 2 is the Major Transporter of the Hepatobiliary Imaging Agent (99m)Tc-mebrofenin. J Nucl Med. 2009;50:1140–6. doi: 10.2967/jnumed.109.062448. [DOI] [PubMed] [Google Scholar]
- 27.Doo E, Krishnamurthy GT, Eklem MJ, Gilbert S, Brown PH. Quantification of Hepatobiliary Function as an Integral Part of Imaging with Technetium-99m-mebrofenin in Health and Disease. J Nucl Med. 1991;32:48–57. [PubMed] [Google Scholar]
- 28.Hendrikse NH, et al. In vivo imaging of hepatobiliary transport function mediated by multidrug resistance associated protein and P-glycoprotein. Cancer Chemother Pharmacol. 2004;54:131–8. doi: 10.1007/s00280-004-0793-2. [DOI] [PubMed] [Google Scholar]
- 29.Bujanover Y, Bar-Meir S, Hayman I, Baron J. 99mTc-HIDA Cholescintigraphy in Children with Dubin-Johnson Syndrome. J Pediatr Gastroenterol Nutr. 1983;2:311–2. [PubMed] [Google Scholar]
- 30.Wilke RA, et al. The Clinical Pharmacogenomics Implementation Consortium: CPIC Guideline for SLCO1B1 and Simvastatin-induced Myopathy. Clin Pharmacol Ther. 2012;92:112–7. doi: 10.1038/clpt.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niemi M, Pasanen MK, Neuvonen PJ. Organic Anion Transporting Polypeptide 1B1: a Genetically Polymorphic Transporter of Major Importance for Hepatic Drug Uptake. Pharmacol Rev. 2011;63:157–181. doi: 10.1124/pr.110.002857. [DOI] [PubMed] [Google Scholar]
- 32.Grover A, Benet LZ. Effects of Drug Transporters on Volume of Distribution. AAPS J. 2009;11:250–61. doi: 10.1208/s12248-009-9102-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.König J, Cui Y, Nies AT, Keppler D. Localization and Genomic Organization of a New Hepatocellular Organic Anion Transporting Polypeptide. J Biol Chem. 2000;275:23161–8. doi: 10.1074/jbc.M001448200. [DOI] [PubMed] [Google Scholar]
- 34.Shigefuku R, et al. Correlations of Hepatic Hemodynamics, Liver Function, and Fibrosis Markers in Nonalcoholic Fatty Liver Disease: Comparison with Chronic Hepatitis Related to Hepatitis C Virus. Int J Mol Sci. 2016;17:1545. doi: 10.3390/ijms17091545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Köck K, Brouwer KLR. A Perspective on Efflux Transport Proteins in the Liver. Clin Pharmacol Ther. 2012;92:599–612. doi: 10.1038/clpt.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel CG, Ogasawara K, Akhlaghi F. Mycophenolic Acid Glucuronide is Transported by Multidrug Resistance-Associated Protein 2 and this Transport is not Inhibited by Cyclosporine, Tacrolimus or Sirolimus. Xenobiotica. 2013;43:229–35. doi: 10.3109/00498254.2012.713531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Schoor LW, Verkade HJ, Kuipers F, Jonker JW. New Insights in the Biology of ABC Transporters ABCC2 and ABCC3: Impact on Drug Disposition. Expert Opin Drug Metab Toxicol. 2015;11:273–293. doi: 10.1517/17425255.2015.981152. [DOI] [PubMed] [Google Scholar]
- 38.Corpechot C, Ping C, Wendum D, Matsuda F, Barbu V, Poupon R. Identification of a novel 974C–>G Nonsense Mutation of the MRP2/ABCC2 Gene in a Patient with Dubin-Johnson Syndrome and Analysis of the Effects of Rifampicin and Ursodeoxycholic Acid on Serum Bilirubin and Bile Acids. Am J Gastroenterol. 2006;101:2427–32. doi: 10.1111/j.1572-0241.2006.00695.x. [DOI] [PubMed] [Google Scholar]
- 39.Zollner G, et al. Adaptive Changes in Hepatobiliary Transporter Expression in Primary Biliary Cirrhosis. J Hepatol. 2003;38:717–27. doi: 10.1016/s0168-8278(03)00096-5. [DOI] [PubMed] [Google Scholar]
- 40.Heinemann A, Wischhusen F, Püschel K, Rogiers X. Standard Liver Volume in the Caucasian Population. Liver Transplant Surg. 1999;5:366–368. doi: 10.1002/lt.500050516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Visual Predictive Checks for 99mTc-Mebrofenin in Blood and Liver.
Table S1: SLCO1B1 Genotype of study participants stratified by group.