

Abstract
Homology-based transferal remains the major approach to computational protein function annotations, but it becomes increasingly unreliable when the sequence identity between query and template decreases below 30%. We propose a novel pipeline, MetaGO, to deduce Gene Ontology attributes of proteins by combining sequence homology-based annotation with low-resolution structure prediction and comparison, and partner’s-homology based protein-protein network mapping. The pipeline was tested on a large-scale set of 1,000 non-redundant proteins from the CAFA3 experiment. Under the stringent benchmark conditions where templates with >30% sequence identity to the query are excluded, MetaGO achieves average F-measures of 0.487, 0.408, and 0.598, for Molecular Function, Biological Process, and Cellular Component, respectively, which are significantly higher than those achieved by other state-of-the-art function annotations methods. Detailed data analysis shows that the major advantage of the MetaGO lies in the new functional homolog detections from partner’s-homology based network mapping and structure-based local and global structure alignments, the confidence scores of which can be optimally combined through logistic regression. These data demonstrate the power of using a hybrid model incorporating protein structure and interaction networks to deduce new functional insights beyond traditional sequence-homology based referrals, especially for proteins that lack homologous function templates. The MetaGO pipeline is available at http://zhanglab.ccmb.med.umich.edu/MetaGO/.
Keywords: Protein function prediction, Gene Ontology, protein-protein interaction, sequence profiles, protein structure prediction
Graphical abstract
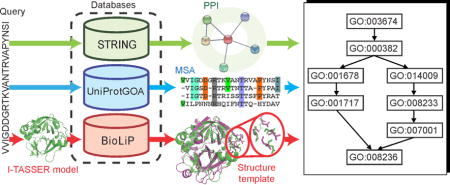
Introduction
The gap between the exponential growth of known protein sequences and the slow accumulation of manual curation of biological function annotation is continually increasing. As of November 2017 when the paper was submitted, the UniProt [1] database harbored ~90 million protein sequences, but only <1% of them were annotated with known Gene Ontology (GO) terms using experimental evidence [2]. The incomplete knowledge of protein function routinely impedes research progress, as un-annotated proteins are frequently implied to have functional roles in genetic screening [3], GWAS studies [4], and laboratory evolution experiments [5]. Development of efficient computational function annotation methods is highly urgent to fill the knowledge gap.
Many current function prediction methods depend on sequence homology [6], and transfer function annotations from inferred homologs [7–10] identified using tools such as BLAST and PSI-BLAST [11]. There are other approaches that combine data from multiple different sources and employ advanced machine learning techniques [12]; but it was found that most of these methods are not significantly better than the sequence-homology based detections in the community-wide CAFA experiments [6, 13] and that homology-based function prediction still constitutes the largest contribution to the best performing CAFA methods [12]. Nevertheless, high sequence similarity does not necessarily imply similar function. For enzymes, >70% of sequences have different enzyme commission numbers even at a sequence identity >50% [14]. Additionally, nearly 87% of protein sequences in UniProt do not have any function template with a sequence identity >50% (Figure S1), an observation significantly compromising the generalizability of purely sequence-homology approaches. To address this issue, tools have been developed to infer function from conserved local signatures [15] derived from Hidden Markov Models (HHMs) [16] or function pattern of sequence motifs [17]. While signature-based annotations have constituted the majority of electronic annotation in UniProt [18], such annotations depend on manual curation of the signatures, which is often laborious and slow to accumulate, and lack specificity in favor of general terms applicable to all members of the same protein family (e.g., “protein binding”).
To address the shortcomings of sequence-based approaches, various attempts have been made to utilize structure-based approaches to assist function annotations [19–23], in which functional templates are identified from structure-function databases through structure comparisons [24, 25]. As structural patterns are usually conserved over longer evolutionary differences than sequence patterns, structure-based predictors may generate more sensitive annotations for “hard” (or non-homology) targets. There are however several factors that can limit their usefulness. First, functionally distinct proteins can share similar global topologies. Typical examples include “TIM barrel” and “Rossmann” folds, each of which is adopted by diverse proteins performing >50 different biological functions. Second, structures are needed for both query and templates in the structure-based approaches, but most unannotated sequences have no experimental structures; meanwhile, ~80% of the proteins in function databases do not have experimentally determined structures, and thus cannot be directly used as functional templates in structure-based pipelines. Finally, many functionally important proteins do not adopt stable structure under physiological conditions, and thus cannot be modeled via structure-based approaches. While the first challenge can be partially mitigated by combining global structure search and local structure motif identification [26], the second and third challenges can only be met if structure-based function annotation is used in appropriate combination with non-structural approaches.
To explore the potential of such hybrid approaches, we propose in this study a new method, MetaGO, which generates automated Gene Ontology modeling by combining three complementary pipelines from global and local structure alignments, sequence and sequence-profile matches, and protein-protein interaction (PPI) network mapping (Figure 1A). We note that the ideas of deducing functional insights from sequence and structural comparisons and PPI mapping are not new, as these approaches have been implemented by several previous studies [19–22, 26–30]. The major new ideas that we focus on exploring include the novel combination of the global and local structural alignments on distant function-homology detection, and the possibility to increase the coverage of function deduction of the target proteins through the mapping of homologs of the PPI binding partners, instead of the direct PPI partners themselves as most previous studies used. To carefully examine the strengths and weaknesses of different component pipelines, we systematically tested MetaGO on a large set of benchmark proteins in comparison with state-of-the-art methods of the field. Since the pipeline is mainly designed for modeling the non- and distant-homology proteins, a stringent benchmark condition was applied to exclude homologous templates from all the structure and function prediction libraries.
Figure 1.
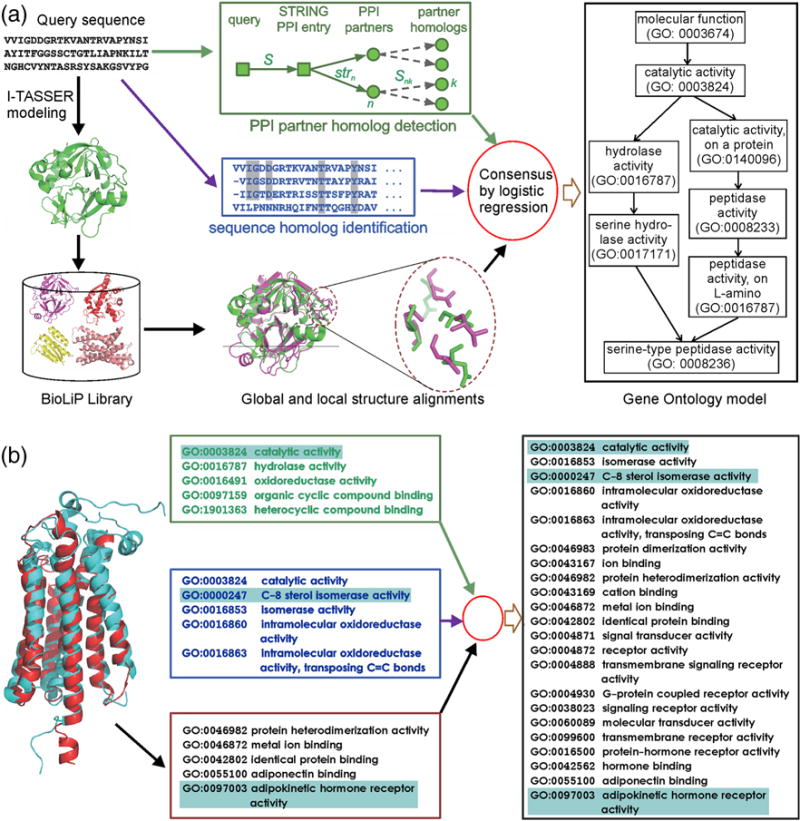
(A) The MetaGO algorithm for GO annotation, which contains three pipelines of global and local structure alignment (bottom red), PPI partner homolog detection (top green), and sequence homolog identification (center blue), followed by a logistic regression based combination. (B) An illustrative example of MetaGO being applied to human EBP protein (Q15125). Left panel is the superposition of I-TASSER model (red) and the PDB structure of the adiponectin receptor (cyan), which are combined with PPI-homolog (green box) and sequence-based predictions (blue box) to create the complete set of MetaGO predictions (right panel). Highlights are to illustrate how the representative function terms from individual pipelines are merged into the final MetaGO predictions.
Results and Discussion
Datasets
To test MetaGO, we curated a set of 1,000 non-redundant proteins randomly taken from the CAFA3 experiment (http://biofunctionprediction.org/cafa-targets/CAFA3_targets.tgz) but with the following criteria: (1) the target can be mapped to a UniProt entry with ≥90% sequence identity; (2) the length is between 30 and 300 amino acids; (3) it has experimental annotations in all three GO aspects: Molecular Function (MF), Biological Process (BP), and Cellular Component (CC), excluding the uninformative “protein binding” GO terms; (4) all proteins share ≤30% sequence identity to each other; (5) all test proteins have a sequence identity <30% to any protein used for training the MetaGO pipeline (see Methods). When structure models are constructed by I-TASSER [31], all structural templates with >30% sequence identity are removed from the LOMETS library [32]. While the benchmark test was mainly performed on small- and medium-size proteins (<300 residues), the prediction accuracy of MetaGO does not have apparent dependency on the length of target proteins (see Figure S5).
Overall performance of MetaGO and the comparison to control methods
In Figure 2, we present a summary of MetaGO predictions on the three aspects of Gene Ontologies, where the detailed Fmax values are listed in Table S1 in the Supplementary Information (SI). In general, the Cellular Component prediction has on average the highest accuracy, followed by Molecular Function and finally Biological Process. Since MetaGO generated the GO models mainly by functional template inference, we used three different sequence identity cutoffs to filter out the templates, i.e., to consider templates only with a sequence identity below 20%, 30% and 50% respectively. As expected, the overall performance has a slight dependence on the level of cutoffs. For example, the average Fmax scores are 0.454, 0.391 and 0.589 for MF, BP and CC at the 20% cutoff, and gradually increased to 0.487, 0.408 and 0.598, and 0.518, 0.428 and 0.605, when the sequence identity cutoffs are increased to 30% and 50%, respectively.
Figure 2.
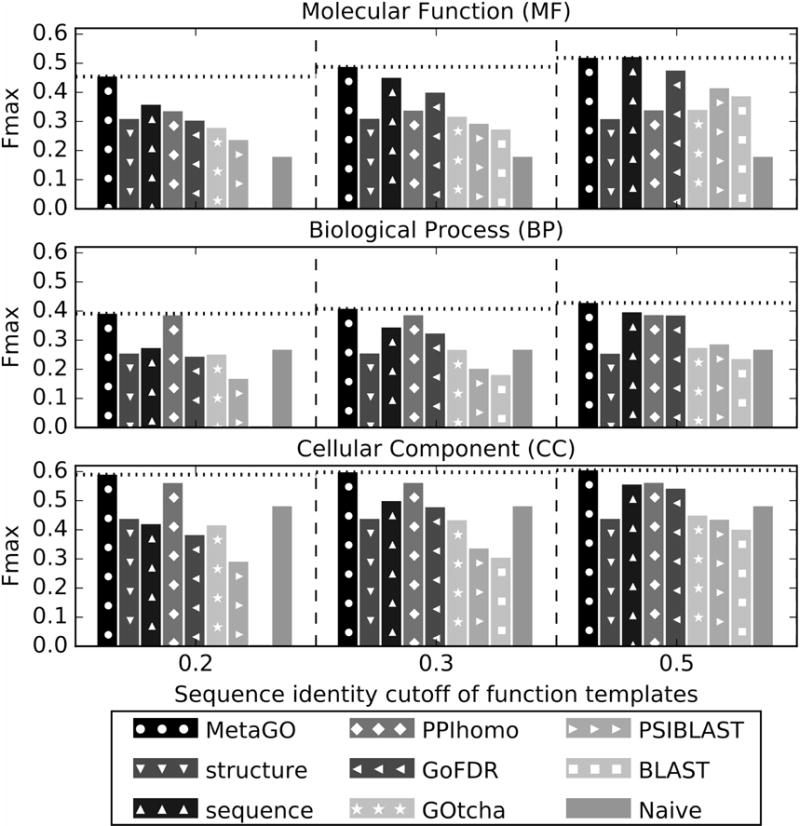
Fmax score of the GO predictions by MetaGO, compared to that by the three component pipelines (structure, sequence, and PPI-homolog), and four control methods (GoFDR, GOtcha, BLAST, PSI-BLAST, and Naïve) at different sequence identity cut-offs for filtering functional templates. The dotted lines label the performance of MetaGO. A color version of this figure is provided as Figure S2 in Supplemental Material.
As a control, we also list the results from four other methods, including GoFDR [7], GOtcha [10], Naïve, BLAST, and PSI-BLAST [11]. Here, GoFDR is one of the best GO predictors in the CAFA2, while “Naïve”, “BLAST”, and “PSI-BLAST” are three standard baseline methods implemented in the CAFA experiments (see Text S1)[12, 33]. The data in Figure 2 shows that MetaGO consistently outperforms the control methods in all GO aspects. In particular, although MetaGO showed some dependence of the performance on the sequence identity cutoffs for templates, it is much more robust to low template similarity than the control methods that generate GO predictions mainly on sequence homologous transferals [7, 11]. For example, the average Fmax values on MF by BLAST, PSI-BLAST, GoFDR and GOtcha decrease by 11267%, 75%, 56%, and 22%, respectively, when the sequence identity cutoff goes from 50% to 20%, while that by MetaGO decreases only by 14%. Similar patterns are also seen in BP and CC predictions. The robustness of MetaGO on the GO prediction is mainly due to the introduction of the two additional pipelines from structural alignments and protein-protein interactions, which show essentially no dependence on the sequence similarity levels of the functional templates; this is particularly important for the uses in annotating ‘hard’ targets that do not have closely homologous templates.
As the function prediction accuracy usually relies on the confidence score cutoffs, we show in Figure 3 the precision-recall curves of MetaGO and the control methods at the 30% sequence identity cutoff. As expected, a higher confidence score cutoff will result in a higher accuracy (precision) but with a lower recall rate. Overall, MetaGO has the highest precision at all different recall rates.
Figure 3.
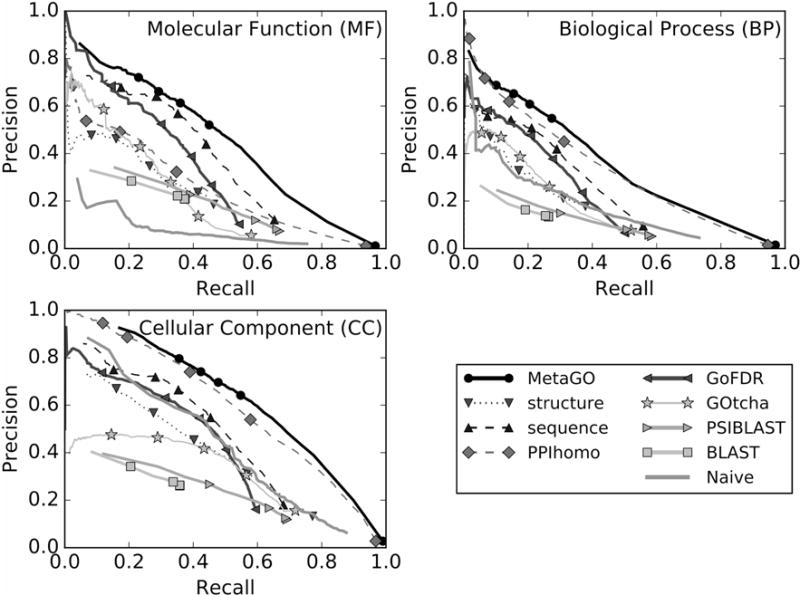
Precision-recall curves of GO predictions by MetaGO, compared to that by the three component pipelines (structure, PPIhomo, and sequence), and five control methods (GoFDR, GOtcha, BLAST, PSI-BLAST, and Naïve) at 30% sequence identity cut-off of functional templates. A color version of this figure is provided as Figure S3 in Supplemental Material.
To examine the contribution of different resources of information, we also list in Figures 2 and 3 the performance of three component pipelines of MetaGO. The combined MetaGO model has a higher accuracy than the components through all the recall regions, demonstrating the importance of appropriate combination of different sources of information. In Figure 1B, we present an illustrative example from the human EBP protein (Q15125), which is both a steroid delta-isomerase (GO:0004769) and a transmembrane signaling receptor (GO:0004888) for anti-ischemic drugs. As shown in Figure 1B, while the PPI-homolog based approach simply predicts the protein to be an enzyme (top green box), sequence-based pipeline predicts it as an isomerase for steroid (central blue box). The structure-based approach, on the other hand, found significant structural similarity between the I-TASSER model (red cartoon) and a transmembrane adiponectin receptor, ADIPOR1 (PDBID: 3wxvA), with TM-score =0.88, based on which it infers the transmembrane signaling receptor activity (bottom red box). The final MetaGO prediction generated the complete set of annotations (right panel), by combining the enzymatic activity prediction from the PPI-homolog and sequence-based pipelines and the transmembrane signaling receptors prediction from the structure-based pipeline, which well match with the experimental annotations in UniProt-GOA [18], despite the fact that all templates with a sequence identity >30% have been excluded. This example illustrates how the various pipelines of MetaGO complement each other to yield a comprehensive annotation of protein targets.
Performance of structure-based GO prediction
In MetaGO, structure templates with GO annotations are mainly identified from the BioLiP database [24] using global structure alignment by TM-align [34]. To tackle structural promiscuity, each query-template pair is re-aligned by combining the global structure alignment with both evolutionary score and local functional site comparison. This combinatory pipeline achieves F-measures of 0.309, 0.254, and 0.438 for MF, BP, and CC, respectively, if functional templates sharing >30% sequence identity are removed.
In contrast, under the same sequence identity cutoff, if the TM-scores of the templates are directly used, the F-measures will be reduced to 0.204, 0.161, and 0.290 for the three GO aspects, representing a drop by 51%, 60% and 51% compared to the full version structure-based pipeline. This result affirms that functional-site-evolution-aware global and local structure alignments are useful post-processing steps contributing to MetaGO’s performance.
Comparison between PPI-based and PPI-homolog-based GO prediction
In developing MetaGO, we tested two PPI approaches, one deducing function from the direct PPI partners similar to previous approaches [28, 29] and another combining multiple homologs of the PPI partners. At the 30% sequence identity cutoff, the direct PPI-based approach has F-measures of 0.222, 0.333 and 0.560 for MF, BP and CC, which is 52%, 16% and 0.2% lower than the respective F-measures of 0.337, 0.386 and 0.561 from the PPI-homolog-based approaches. This is mainly due to the extension of the functional template entries through the additional homologous search, which helps increasing the coverage of functional referrals.
It is of interest to note that both pipelines perform relatively poorly on the MF relative to the BP and CC aspects, while most of other methods/pipelines have a higher performance in MF than BP. This is understandable, as proteins that physically interact with each other do not necessarily perform the same molecular task (MF), even though they generally co-localize and involve in the same pathway (related to BP and CC).
Confidence score for BLAST and PSI-BLAST baseline methods
As implemented in the CAFA experiments [33], we have used localID (i.e. the sequence identity normalized by number of aligned residues) as the confidence score for the “BLAST” and “PSI-BLAST” in the control studies (Text S1). However, we note that such a score might not be the best choice for these baseline methods. In Figure S4, we compare the performance of BLAST and PSI-BLAST using different ‘confidence scores’, including localID, globalID (sequence identity normalized by the query length), evalue (lowest E-value), and frequency (the number of homologs annotated with a GO term of interest among all identified homologs). It shows that frequency consistently has the highest Fmax at all cutoffs through all GO aspects, indicating that a consensus of multiple template hits is probably a more robust indicator than the score of the best individual template. We therefore recommend the use of the frequency of (PSI−)BLAST hits as a more reliable and challenging baseline method in future assessment experiments. We also find that sequence identity is more indicative of GO annotation similarity than E-value, where both globalID and localID have consistently a higher Fmax than evalue.
Based on these observations, in Eq. (6) we have combined the homologous templates from both BLAST and PSI-BLAST, with the sequence identity as the weight of the combinations. The result shows that the combination outperforms the simple counting of the frequency in each individual program (Figure S4). Thus, even the poorly performing BLAST and PSI-BLAST methods may be substantially improved by careful consideration of the applied scoring schemes.
Performance of MetaGO in blind test
As a blind test, an early version of MetaGO participated in the community-wide CAFA3 experiment as “Zhang-Freddolino Lab”. Since MetaGO was not finalized by the time of CAFA3, we were using PPI-based prediction instead of PPI-homolog-based prediction, and simple confidence score averaging instead of logistic regression. Due to the limit of computational resource, we were only able to complete structure-based function prediction for 5,000 targets. Nevertheless, MetaGO model was ranked at the first place in BP prediction among the 148 models submitted by 80 registered teams (-although the performance of MetaGO in other categories was relatively worse), according to preliminary results from the CAFA3 assessors (N. Zhou and I. Friedberg, personal communication, see also https://www.synapse.org/#!Synapse:syn11587254).
As a case study, we inspected the MetaGO prediction for target T96060009790 (Human Lysozyme-like protein 6, O75951). According to the experimental annotation obtained after our CAFA3 model was submitted, this is a sperm protein exhibiting bacteriolytic activity against gram-positive bacteria (GO:0042742 defense response to bacterium). While the structure of this protein is known, the I-TASSER model shows remarkable similarity to human lysozyme C (PDBID: 1di3A) with TM-score 0.85. Therefore, the structure-based pipeline of MetaGO asserts that the BP of this protein is involved in defense response to bacterium (GO:0042742) with a high confidence score of 0.98. Interestingly, the sequence-based methods only give a low confidence score of 0.3 for the BP term GO:0042742; but these methods identify the bacteriolytic MF (GO:003796) with a high confidence (0.98). Thus, the correct BP and MF annotations are both assigned with confidence scores >0.9, when structure, sequence, and PPI information are combined, showing the advantage of multisource information in the annotation of highly specific functions.
Conclusion
Protein function is a multifaceted and complex phenomenon, and there is currently no single algorithm that can generate models which cover all aspects of functions. To explore the potential of multi-source approaches, we developed a new hybrid method, MetaGO, to predict Gene Ontology of proteins by combining information from evolutionary homology, structural analogous comparison, and protein-protein interaction mapping.
The method was tested on a large-scale set of 1,000 non-redundant proteins, which demonstrated significant advantages on GO prediction compared to the traditional sequence-homology based approaches. Detailed data analysis showed that the major advantage of MetaGO comes from the introduction of additional pipelines from global and local structure alignments, and the protein-protein interaction based functional referrals. In contrast with sequence-only methods, whose performance decrease rapidly when the sequence identity level of functional templates is reduced below 30%, the structure- and PPI-based pipelines have essentially no dependence on the sequence similarity level of the functional templates, demonstrating the potential of MetaGO for function annotation of distant- and non-homologous protein targets, a long-standing problem in the field of computational biology [12, 35].
Despite the encouraging performance, MetaGO can be further improved in several aspects. For instance, as shown in CAFA3, one issue in MetaGO is that the accuracy of Molecular Function is relatively low compared to other GO aspects. This is partly because MF, especially the enzymatic activity, usually depends only on a small number of critical residues, while the current sequence and PPI-homolog pipelines consider only global template comparison. Implementing local similarity comparisons of the critical residues in the pipelines should help improving the MF prediction. Another issue is the relatively slow process of the structure-based pipeline, as it needs to do alignment search through the entire BioLiP database which can take a few hours for a single protein (Figure S5). The process can be significantly accelerated by a hierarchical search, in which the BioLiP proteins are pre-clustered on the structural similarity, where an initial scan can be quickly completed on a representative member of each cluster, followed by detailed alignment search only on the interested clusters. Work along these lines is in progress.
Methods
MetaGO consists of three separate GO prediction pipelines, which detect functional homologies based on structure and sequence comparisons, and protein-protein interaction networks. A consensus is then deduced by logistic regression, using the confidence scores of different pipelines as features. The pipeline of MetaGO is illustrated in Figure 1A.
Structure-based GO prediction
The structure-based GO prediction protocol in MetaGO is extended from the COFACTOR algorithm designed for ligand binding site prediction [26] (see bottom of Fig. 1A).
Global structure search
The pipeline starts from a structure model predicted by I-TASSER [31]. TM-align [34] is then used to match the query structure against the BioLiP [24], which contains a non-redundant set of 35,238 proteins with known GO, where 20 templates with the highest TM-scores [36] are returned. Next, each of the templates is re-aligned to the query by a modified TM-align program using a new Gsim score:
| (1) |
where L is the length of the query, Lali is the number of aligned residues, di is Cα atom distance between ith pair of aligned residues, and is a distance scale, as defined in TM-score [36]. F(i, a) is the frequency of amino acid a at the ith position of the sequence profile generated by searching the query against the NCBI non-redundant (NR) database using PSI-BLAST with an E-value cutoff 0.001. P(i, a) is the log-odds value of a at the ith position in the template sequence profile, pre-generated for all templates in the BioLiP. δi equals 1 when amino acid type at the ith aligned position is identical in query and template, and 0 otherwise.
The same heuristic iterative algorithm from TM-align is used to search for the alignment with the highest Gsim. Since additional mutation terms are included, this re-alignment process on Gsim considers both structural and evolutionary information, where the latter helps enhance the functional connections between the proteins.
Local structure alignment
To evaluate the local structure similarity between query and template, their active/binding sites are superposed to each other according to
| (2) |
where Nt is the total number of residues in the active/binding site, Nali is the number of aligned residues, and Mi is the normalized BLOSUM62 substitution score [37] between the ith pair of aligned residues.
This local structure alignment is performed by an iterative process similar to that in TM-align. Briefly, starting from the superposition from the global structure alignment, a dynamic programming is performed to create a new alignment using lsimij as the alignment score and −1 as the gap penalty. Based on this alignment, the structures of active/binding residues are re-superposed by TM-score, where the new superposition will result in a new lsimij matrix, which in turn is used to create a newer alignment and newer superposition by dynamic programming. This process is repeated iteratively until the alignment is converged.
Confidence score of GO terms from structure templates
The confidence score of a structural template hit is calculated as a combination of the global and local structure similarities:
| (3) |
with Sbs being the sequence identity at the active/binding site. The confidence score of a GO term q, which is transferred from N(q) functional templates, is calculated by
| (4) |
where GLi(q) is the GL value of the ith template associated with q. We note that if q is assigned to the query, its direct parent GO term p should also be considered annotated to the query. To enforce this hierarchy relation, we compute the confidence score for p by
| (5) |
where Np and Nq are the numbers of proteins in the UniProt with GO terms p and q, respectively. Eq. (5) is iteratively applied to all parent terms toward the root until Cscorestructure(p) > 0.3, as upstream parent terms with a higher Cscore are automatically included.
Sequence and sequence-profile based GO predictions
In the sequence-based pipeline, a query is searched against the UniProt-GOA by BLAST with an E-value cutoff 0.01 to identify sequence homologs, where unreviewed annotations with “IEA” or “ND” evidences are excluded. Similarly, a three-iteration PSI-BLAST search is performed for the query through the UniRef90 database to create a sequence profile, which is used to jump-start a one-iteration PSI-BLAST search through the UniProt-GOA. The confidence score of a GO term q transferred from the sequence templates is
| (6) |
where and are the sequence identity of the nth homolog identified by BLAST and PSI-BLAST, respectively, and Nblast and Npsiblast are the total numbers of homologs identified by the two programs. , (q), Nblast(q), and Npsiblast(q) are the corresponding values for the proteins with GO term q in UniProt-GOA. To balance the BLAST and PSI-BLAST terms, the weight is introduced as the maximum sequence identity for all PSI-BLAST hits, so that BLAST has stronger weight than PSI-BLAST when close homologs are found.
Protein-protein interaction based GO predictions
We tested two different approaches to utilize the PPI network for GO prediction (green pipeline in Fig. 1A). In the first approach, the query sequence is mapped to its closest BLAST hit in STRING PPI database [38]. Since each protein in STRING can have multiple interaction partners, the GO terms q of the PPI partners, as annotated in the STRING database, are transferred to the query with a confidence score of
| (7) |
where S is the sequence identity between the query and the STRING entry that it is mapped to, N is the total number of PPI partners for this entry, and strn is the score assigned by STRING as confidence of interaction. N(q) and strn(q) are the corresponding partner numbers and STRING score for PPI partners annotated with q.
In the second PPI-homolog based approach, the PPI partners are identified similarly as in the first approach. Next, these PPI partners are searched through the UniProt-GOA by BLAST to identify homologs of the PPI partners. The GO terms (e.g., q) of the BLAST homologs are then transferred to the query with a confidence score calculated by
| (8) |
where Kn and Sn,k are the total number of the BLAST hits for nth PPI partner and the sequence identity between nth PPI partner and its kth homolog, while Kn(q) and Sn,k(q) are those for the PPI partner homologs annotated with q.
Consensus MetaGO prediction
The final GO prediction in MetaGO is a combination of the three pipelines, where the confidence score of a GO term q is calculated through logistic regression on their weights:
| (9) |
Here, mε{structure, sequence, PPIhomo, Naive}, and Cscorem(q)ε[0,1] are the confidence score for q by the mth feature. The first three features in the regression, “structure”, “sequence”, and “PPIhomo”, are the confidence score from the three structure, sequence, and PPI-homolog based pipelines in Eqs. (4–8). The fourth feature “Naïve” is the background probability of q being annotated in UniProt-GOA (see Text S1).
Here, we note that w0 and wm are the only free parameters in the MetaGO pipelines, where all other parameters in the structure pipeline in Eq. (1–3) are inherited from the COFACTOR program without further optimization, and the parameters in Eq. (4–8) have been uniquely determined from the UniProt-GOA database, the sequence identity of function templates, or the confidence score of protein-protein interaction assigned by STRING database. The five free parameters are trained by gradient descend on the 1,224 training proteins that are selected from E. coli genome and have a sequence identity <30% to any of the test proteins used in this study (see Table S2).
Assessment criteria of GO prediction
Following the CAFA experiments [12, 33], only GO terms with experimental evidence codes (EXP, IDA, IMP, IGI, IEP, TAS or IC) are considered as “gold standards”. To explicitly consider the hierarchical nature of GO terms, if a child term is annotated to a protein, all its direct and indirect parents, as defined by the “is_a” relation, are also considered gold standards. Similarly, for each prediction, the confidence scores for predicted GO terms are recursively propagated towards the root of the ontology such that each parent term receives the highest score among its children.
To evaluate the predictions, the maximum F1-score, i.e. F-measure, is calculated as [12]
| (10) |
where pr(t) and rc(t) are the precision and recall for the GO predictions with confidence score ≥ t, defined by
| (11) |
Here, tp(t) is the number of GO terms correctly predicted, tp(t) + fp(t) the number of all predicted GO terms for the query, and tp(t) + fn(t) are all the GO terms annotated to the query in the “gold standard”.
Supplementary Material
Highlights.
Detect functional templates using homology of protein interaction partners.
Low-resolution structure prediction is found useful for function annotation.
The new pipeline is particularly robust for non-homologous proteins.
Webserver and source codes are freely available to the community.
Acknowledgments
We like to thank Dr. Xiaoqiong Wei for insightful discussion. The work was supported in part by the NIGMS [GM083107, GM116960], and the National Science Foundation [DBI1564756]. The benchmark test was performed on the Extreme Science and Engineering Discovery Environment (XSEDE) clusters [39].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bateman A, Martin MJ, O’Donovan C, Magrane M, Apweiler R, Alpi E, et al. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D12. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nature Genetics. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, et al. Phenotypic landscape of a bacterial cell. Cell. 2011;144:143–56. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirschhorn JN. Genomewide Association Studies - Illuminating Biologic Pathways. New Engl J Med. 2009;360:1699–701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 5.Good BH, McDonald MJ, Barrick JE, Lenski RE, Desai MM. The dynamics of molecular evolution over 60,000 generations. Nature. 2017 doi: 10.1038/nature24287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillis J, Pavlidis P. Characterizing the state of the art in the computational assignment of gene function: lessons from the first critical assessment of functional annotation (CAFA) Bmc Bioinformatics: BioMed Central. 2013:S15. doi: 10.1186/1471-2105-14-S3-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong QT, Ning W, Tian WD. GoFDR: A sequence alignment based method for predicting protein functions. Methods. 2016;93:3–14. doi: 10.1016/j.ymeth.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 8.Profiti G, Martelli PL, Casadio R. The Bologna Annotation Resource (BAR 3.0): improving protein functional annotation. Nucleic Acids Res. 2017;45:W285–W90. doi: 10.1093/nar/gkx330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hawkins T, Chitale M, Luban S, Kihara D. PFP: Automated prediction of gene ontology functional annotations with confidence scores using protein sequence data. Proteins. 2009;74:566–82. doi: 10.1002/prot.22172. [DOI] [PubMed] [Google Scholar]
- 10.Martin DMA, Berriman M, Barton GJ. GOtcha: a new method for prediction of protein function assessed by the annotation of seven genomes. Bmc Bioinformatics. 2004;5 doi: 10.1186/1471-2105-5-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, Sokolov A, et al. A large-scale evaluation of computational protein function prediction. Nat Methods. 2013;10:221–7. doi: 10.1038/nmeth.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamp T, Kassner R, Seemayer S, Vicedo E, Schaefer C, Achten D, et al. Homology-based inference sets the bar high for protein function prediction. BMC Bioinformatics. 2013;14(Suppl 3):S7. doi: 10.1186/1471-2105-14-S3-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rost B. Enzyme function less conserved than anticipated. Journal of molecular biology. 2002;318:595–608. doi: 10.1016/S0022-2836(02)00016-5. [DOI] [PubMed] [Google Scholar]
- 15.Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017;45:D190–D9. doi: 10.1093/nar/gkw1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D85. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sigrist CJA, de Castro E, Cerutti L, Cuche BA, Hulo N, Bridge A, et al. New and continuing developments at PROSITE. Nucleic Acids Res. 2013;41:E344–E7. doi: 10.1093/nar/gks1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huntley RP, Sawford T, Mutowo-Meullenet P, Shypitsyna A, Bonilla C, Martin MJ, et al. The GOA database: Gene Ontology annotation updates for 2015. Nucleic Acids Res. 2015;43:D1057–D63. doi: 10.1093/nar/gku1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy A, Yang JY, Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012;40:W471–W7. doi: 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res. 2005;33:W89–W93. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pazos F, Sternberg MJE. Automated prediction of protein function and detection of functional sites from structure. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:14754–9. doi: 10.1073/pnas.0404569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pal D, Eisenberg D. Inference of protein function from protein structure. Structure. 2005;13:121–30. doi: 10.1016/j.str.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 23.Xin F, Radivojac P. Computational methods for identification of functional residues in protein structures. Curr Protein Pept Sci. 2011;12:456–69. doi: 10.2174/138920311796957685. [DOI] [PubMed] [Google Scholar]
- 24.Yang JY, Roy A, Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013;41:D1096–D103. doi: 10.1093/nar/gks966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Beer TAP, Berka K, Thornton JM, Laskowski RA. PDBsum additions. Nucleic Acids Res. 2014;42:D292–D6. doi: 10.1093/nar/gkt940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy A, Zhang Y. Recognizing protein-ligand binding sites by global structural alignment and local geometry refinement. Structure. 2012;20:987–97. doi: 10.1016/j.str.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lan L, Djuric N, Guo YH, Vucetic S. MS-kNN: protein function prediction by integrating multiple data sources. Bmc Bioinformatics. 2013;14 doi: 10.1186/1471-2105-14-S3-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piovesan D, Giollo M, Leonardi E, Ferrari C, Tosatto SCE. INGA: protein function prediction combining interaction networks, domain assignments and sequence similarity. Nucleic Acids Res. 2015;43:W134–W40. doi: 10.1093/nar/gkv523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wass MN, Barton G, Sternberg MJE. CombFunc: predicting protein function using heterogeneous data sources. Nucleic Acids Res. 2012;40:W466–W70. doi: 10.1093/nar/gks489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol. 2007;3:88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nature Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu ST, Zhang Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007;35:3375–82. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang Y, Oron TR, Clark WT, Bankapur AR, D’Andrea D, Lepore R, et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016;17:184. doi: 10.1186/s13059-016-1037-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–9. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y. Protein structure prediction: when is it useful? Curr Opin Struct Biol. 2009;19:145–55. doi: 10.1016/j.sbi.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57:702–10. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 37.Yang JY, Roy A, Zhang Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29:2588–95. doi: 10.1093/bioinformatics/btt447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D52. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, et al. XSEDE: Accelerating Scientific Discovery. Computing in Science & Engineering. 2014;16:62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.