

Abstract
Study Objective
Patients with schizophrenia are known to have higher rates of metabolic disease than the general population. Contributing factors likely include lifestyle and atypical antipsychotic (AAP) use, but the underlying mechanisms are unknown. The objective of this study was to identify metabolomic variability in adult patients with schizophrenia who were taking AAPs and grouped by fasting insulin concentration, our surrogate marker for metabolic risk.
Design
Metabolomics analysis.
Participants
Ninety-four adult patients with schizophrenia who were taking an AAP for at least 6 months, with no changes in their antipsychotic regimen for the previous 8 weeks, and who did not require treatment with insulin. Twenty age- and sex-matched nonobese (10 subjects) and obese (10 subjects) controls without cardiovascular disease or mental health diagnoses were used to match the body mass index range of the patients with schizophrenia to account for metabolite concentration differences attributable to body mass index.
Measurements and Main Results
Existing serum samples were used to identify aqueous metabolites (to differentiate fasting insulin concentration quartiles) and fatty acids with quantitative nuclear magnetic resonance (NMR) and gas chromatography (GC) methods, respectively. To exclude metabolites from our pathway mapping analysis that were due to variability in weight, we also subjected serum samples from the nonobese and obese controls to the same analyses. Patients with schizophrenia had a median age of 47.0 (interquartile range 41.0-52.0) years. Using a false discovery rate threshold of <25%, 10 metabolites, not attributable to weight, differentiated insulin concentration quartiles in patients with schizophrenia and identified variability in one-carbon metabolism between groups. Patients with higher fasting insulin concentrations (quartiles 3 and 4) also trended toward having higher levels of saturated fatty acids compared with patients with lower fasting insulin concentrations (quartiles 1 and 2).
Conclusion
These results illustrate the utility of metabolomics to identify pathways underlying variable fasting insulin concentration in patients with schizophrenia. Importantly, no significant difference in AAP exposure was observed among groups, suggesting that current antipsychotic use may not be a primary factor that differentiates middle-aged adult patients with schizophrenia by fasting insulin concentration.
Keywords: Adverse drug reactions, insulin, antipsychotics
Cardiovascular disease (CVD) is a major cause of premature mortality in patients with schizophrenia.1,2 It is not fully understood why these patients experience more CVD than the general population, but medication is likely an important contributing factor. Atypical antipsychotics (AAPs), in particular, have been identified as culprits because of their tendency to cause adverse metabolic effects such as weight gain, dyslipidemias, and diabetes mellitus. These adverse events are consistent with components of metabolic syndrome (MetS),3 a well-known risk factor for CVD. In 2004, the American Psychiatric Association collaborated with the American Diabetes Association and others to describe monitoring recommendations for the development of MetS in AAP users.4 Unfortunately, these recommendations are not often followed in practice,5 which stresses a need for improved understanding of the underlying mechanisms of MetS in order to develop more precise mechanisms for monitoring and preventing adverse metabolic side effects.
An alternative option for assessing CVD risk in AAP users is fasting insulin concentration, which has been associated with insulin resistance and future development of prediabetes and diabetes.6,7 The development of insulin resistance has also been observed prior to weight gain in response to AAPs,8,9 and it is considered an essential component of the definition of MetS.10 Therefore, we reasoned that a hypothesis-generating, metabolomics study would identify unique metabolite signatures associated with variable fasting insulin concentrations in AAP users with schizophrenia and provide added insight on the biological pathways differentiating patients with schizophrenia who have variable CVD risk. We also expected to see greater exposure to AAPs in general and increased use of more obesogenic AAPs (such as clozapine and olanzapine) in participants with relatively higher fasting insulin concentrations. Therefore, we conducted a metabolomics analysis of existing serum samples from adult patients with schizophrenia, grouped by log-transformed fasting insulin concentration quartiles, using quantitative 1D-1H-nuclear magnetic resonance (NMR) spectroscopy, and a gas chromatography (GC) assay for the detection of fatty acids (FA). Since the median body mass indexes (BMIs) of patients in each fasting insulin quartile were different, we also assayed existing serum samples from obese (BMI ≥30 kg/m2) and nonobese (BMI <30 kg/m2) subjects without CVD or mental health diagnoses that were matched to the schizophrenia cohort by sex, age, and BMI range to account for metabolite concentration differences attributable to BMI. AAP use was compared across quartiles with two approaches: (1) normalizing exposure using chlorpromazine equivalents, and (2) comparing percentages of patients using AAPs divided into three groups based on their propensity to cause weight gain.
Methods
Study Design, Patients, and Data Collection
For this metabolomics pilot study, existing serum samples from patients with schizophrenia were selected from a large, ongoing cross-sectional study on CVD risk in schizophrenia. Inclusion criteria for the parent study are age 18-90 years old; Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition11 diagnosis of schizophrenia, schizophreniform disorder, schizoaffective disorder, or psychotic disorder not otherwise specified; and taking an antipsychotic for at least 6 months with no changes in antipsychotic regimen for the previous 8 weeks. Exclusion criteria were inability to give informed consent, presence of diabetes mellitus type 2 diagnosis prior to initiation of antipsychotic therapy, and an active substance abuse diagnosis. For inclusion in this pilot study, patients had to be taking an AAP and not require treatment with insulin.
For the parent study, anthropometric measurements and venipuncture were performed at the University of Michigan’s Clinical Research Unit (MCRU; http://www.michr.umich.edu/services/mcru). A dietician collected information on dietary recall from all participants, and information on past and current medication use was collected by the study team. The parent study protocol was approved by the University of Michigan’s Institutional Review Board (IRBMED HUM00017774) and the IRBs of the Washtenaw County Health Organization (WCHO), the Detroit-Wayne County Community Mental Health Agency (DWCCMHA), and the Ann Arbor Veterans Affairs Medical Center. The study was performed in accordance with the ethical standards of the Helsinki Declaration of 1975 (as revised in 1983).
AAP Exposure Standardization and Risk Categories
AAP exposure was standardized between groups using the formulas described by Andreasen et al.12 to calculate chlorpromazine equivalents for aripiprazole, clozapine, olanzapine, quetiapine, risperidone, and ziprasidone. The equations for calculating chlorpromazine equivalents for paliperidone, iloperidone, and lurasidone were obtained from Woods.13 As AAPs are known to have a varying propensity to cause adverse metabolic side effects, medication use was further described by risk group, as follows: high (olanzapine or clozapine), moderate (quetiapine, risperidone, iloperidone, or paliperidone), and low (aripiprazole, ziprasidone, and lurasidone).14–17 When participants were taking more than one AAP, the medication with the highest risk was used to group the participant into medication risk category. Antipsychotic polypharmacy was also described as use of 2-3 antipsychotics (typical or atypical).
Controls
Existing serum samples from a cohort of patients who were enrolled in the University of Michigan’s Weight Management Program (NCT02043457)18,19 and technical replicate serum samples from healthy subjects who were recruited from the Claude D. Pepper Older Americans Independence Center (OAIC) Research Participant Program at the University of Michigan’s Geriatric Center18–20 were used as controls. To be included in the weight management program, participants were required to meet a minimum BMI of 32 kg/m2 with at least one comorbidity, or a BMI of at least 35 kg/m2.18,19 For this pilot study, the additional exclusion criteria were applied: present cardiovascular disease or diabetes, psychiatric disorder, and use of weight-potentiating medication such as steroids. From these cohorts, two control groups (without CVD or mental health diagnoses) were formed to match the range of BMI of the patients with schizophrenia. The first group was selected to have a mean BMI <30 kg/m2 (nonobese) and the second >30 kg/m2 (obese). All were matched to the schizophrenia cohort for sex and age range. The protocols under which the control data were acquired were also approved by IRBMED (obese controls: HUM00030088, nonobese controls: HUM00038122), and all participants in the schizophrenia and BMI control groups provided informed consent.
Serum Sample Preparation and Fasting Insulin Assay
Whole blood from fasting patients with schizophrenia was collected by direct venipuncture into additive-free collection tubes, allowed to coagulate for at least 30 minutes, and then centrifuged (2500 × g for 10 min) to generate serum. Serum was aliquoted into vials for storage (-80°C) until time of assay except for exposure to one freeze-thaw cycle for an analysis related to the parent study. The blood collections from obese and nonobese participants were similar and have previously been described.18–20 For this pilot study, an additional aliquot (minimum of 125 μL) of frozen serum from participants with schizophrenia and negative controls was sent to the Michigan Diabetes Research Center to assess fasting insulin concentration by radioimmunoassay.
Prior to the time of assay, serum samples were randomized into batches of 10 for extraction and NMR analysis. At the time of assay, samples were subjected to a water (H2O)/methanol (MeOH):chloroform (CHCl3) extraction as previously described.21,22 The dried CHCl3 fraction was transported to the Michigan Comprehensive Regional Metabolomics Core (MRC)2 for FA analysis.
Quantitative 1D-1H-NMR Metabolomics and GC to Detect Free FA
The 1D-1H-NMR spectra of the H2O/MeOH fraction of each sample were acquired at the University of Michigan’s NMR Biochemical Core Laboratory on a Varian (now Agilent Inc., Santa Clara, CA, USA) 11.74 Tesla (500 MHz) NMR spectrometer. This process has been previously described in detail,23 and further description of the methods are provided in the supplementary information in addition to a representative NMR spectrum (Supplementary Figure 1). The CHCl3 fractions containing the lipids of the serum samples were assayed for FA content, utilizing an Agilent 5890 gas chromatograph with an Agilent HP 88 column, by the MRC2 using a modified technique previously described by Das and Hajra.24 A representative gas chromatogram can also be found in the supporting information (Supplementary Figure 2).
Statistical and Pathway Analysis
For the patients with schizophrenia, groups were created by separating their log-transformed fasting insulin concentrations into quartiles. The quantified metabolomics data sets were transformed and scaled in Metaboanalyst 3.0 (http://www.metaboanalyst.ca/) to achieve normal distribution in preparation for parametric statistical analyses.27,28 The FA data were analyzed as relative percentage of total to account for different initial sample volumes. Normalized data were analyzed by analysis of variance (ANOVA) with a Tukey-Kramer post hoc test, and demographic and clinical data were compared with Wilcoxon, ANOVA, or χ2 tests, as appropriate in JMP Pro 11 (SAS Institute, Inc. Cary, NC). For the control participants, metabolite mean concentrations were compared between BMI groups with an unpaired Student t test. For all normalized mean metabolite comparisons, Welch’s corrections were applied when variance between groups was not similar. Daily AAP exposure was calculated by translating total AAP medication dose into chlorpromazine equivalents.12,13 To control for multiple comparisons, a false discovery rate (FDR) of the resulting post hoc P values was calculated for each metabolite using the method described by Storey et al.29 Metabolites with an FDR <25% were used in pathway analysis by Metscape (http://metscape.ncibi.org/)30, a plugin for Cytoscape (http://www.cytoscape.org/). Figures were constructed using “R” (https://www.r-project.org/).31
Results
Participant Characteristics
Patients with Schizophrenia
Ninety-four patients with schizophrenia were included in this study. Patients were grouped by log-transformed insulin concentration quartile, which was different across all four quartiles (Figure 1A). Of note, several metabolic parameters were significantly different among schizophrenia groups (p<0.05), including BMI (Figure 1C) and MetS diagnosis (Table 1). Cumulative average AAP dose exposure was available for 96.8% (91/94 patients) and was not significantly different among groups, and neither was the variability among quartiles when examining AAP risk groups. With respect to polypharmacy, the rates were not significantly different among quartiles (Table 1). A total of six patients were using a typical and atypical antipsychotic medication combination regimen: five were using fluphenazine and one was using haloperidol, in addition to an AAP. The participant taking haloperidol was also using olanzapine, and the participants taking fluphenazine were using moderate-risk AAPs (n=4) or ziprasidone (n=1). Risk category was not modified for participants based on typical antipsychotic use. Time since schizophrenia diagnosis was assessed by group as a surrogate marker for approximate length of exposure to antipsychotics. This measure was also not significantly different between groups.
Figure 1.
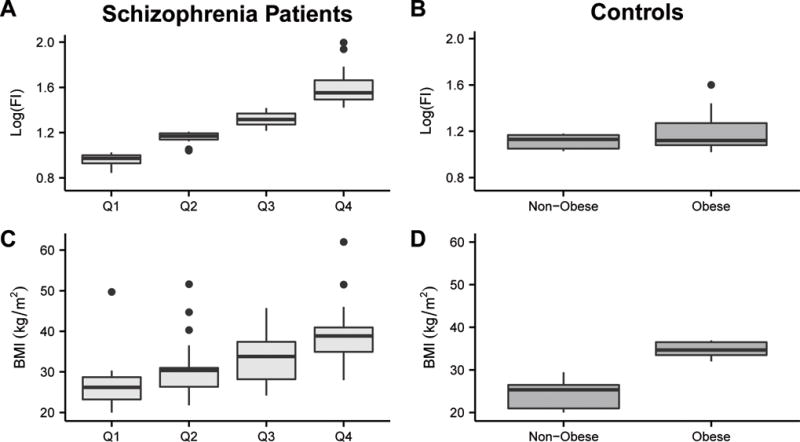
Box and whisker plots of log-transformed fasting insulin concentration (Log(FI), measured as mU/mL) and body mass index (BMI) by quartile for the patients with schizophrenia (A and C, respectively) and by BMI group for the controls (B and D, respectively). The crossbar of the box indicates the median, the box spans the interquartile range, and the whiskers indicate the entire data range, with outliers identified as individual points. The schizophrenia quartiles (Q1-4) are designated by log(FI). BMI was significantly different among all of the schizophrenia groups (P<0.01) with the exceptions of Q2 and Q3. Log(FI) was significantly different among all schizophrenia groups (P<0.05). BMI was significantly different between the two obese and nonobese control groups (P<0.001). The BMI of the obese control group was significantly greater than the BMI of schizophrenia quartiles Q1 (P<0.001) and Q2 (P=0.003). The BMI of the nonobese controls was significantly less than that of Q2 (P=0.003), Q3 (P<0.001), and Q4 (P<0.001). Log(FI) was significantly different between the BMI control groups and all the schizophrenia quartiles, with the exception of the obese controls and Q2. Log(FI) was not significantly different between the nonobese and obese controls. Wilcoxon tests were used to compare BMI and fasting insulin concentrations amongst groups, and post hoc Wilcoxon each pair analyses were completed to identify differences within groups. For example, a Wilcoxon test noted that fasting insulin was significantly different between schizophrenia quartiles, and the post hoc tests identified that all quartiles varied significantly from each other by fasting insulin concentration.
Table 1.
Demographic and Clinical Characteristics of the Patients with Schizophrenia
| Insulin Concentration Quartilea | |||||
|---|---|---|---|---|---|
|
| |||||
| Characteristic | Q1 (n=25) |
Q2 (n=22) |
Q3 (n=25) |
Q4 (n=22) |
p Valueb |
| BMI (kg/m2) | 26.2 (23.2-29) |
30.4 (26.2-31.2) |
33.8 (28.0-37.9) |
38.9 (34.3-41.6) |
<0.001 |
| Age (yrs) | 45.0 (34.5-50.5) |
46.5 (41.0-50.5) |
49.0 (42-53) |
47.5 (40.8-53) |
0.486 |
| Metabolic syndrome diagnosis | 6 (24.0) | 13 (59.1) | 14 (56.0) | 19 (86.4) | 0.002 |
| Use of antidiabetic medication | 3 (12.0) | 5 (22.7) | 2 (8.0) | 4 (18.2) | 0.502 |
| Female sex | 11 (44.0) | 8 (36.4) | 12 (48.0) | 9 (40.9) | 0.875 |
| Average daily calorie intake (kcal) (mean ± SD) | 1986.0 ± 652.3 | 1873.2 ± 563.9 | 1882.3 ± 628.2 | 1933.5 ± 499.5 | 0.890 |
| Caucasian race | 16 (64.0) | 15 (68.2) | 14 (56.0) | 13 (59.1) | 0.837 |
| Smoker | 15 (60.0) | 12 (54.6) | 9 (36.0) | 11 (50.0) | 0.370 |
| Log-transformed fasting insulin concentration | 0.973 (0.922-1.002) |
1.169 (1.136-1.197) |
1.316 (1.272-1.369) |
1.552 (1.485-1.674) |
<0.001 |
| Time since schizophrenia diagnosis (yrs) | 17.0 (9.0-30.5) |
20.0 (14.3-27.8) |
26.0 (15.5-32.5) |
16.0 (9.5-21.8) |
0.342 |
| Daily exposure to AAPs in chlorpromazine equivalents (mg) | 516.0 (247.2-795.8) |
456.6 (334.7-669.6) |
692.1 (379.5-1069.0) |
516.8 (352.0-639.4) |
0.092 |
| AAP Risk Groupc | |||||
| High | 6 (24.0) | 11 (50.0) | 11 (44.0) | 7 (31.8) | |
| Moderate | 16 (64.0) | 7 (31.8) | 10 (40.0) | 12 (54.6) | 0.420 |
| Low | 3 (12.0) | 4 (18.2) | 4 (16.0) | 3 (13.6) | |
| Antipsychotic polypharmacyd | 8 (32.0) | 2 (9.1) | 10 (40.0) | 6 (27.3) | 0.115 |
Data are median (interquartile range) or no. (%) of patients unless otherwise specified.
BMI = body mass index; AAP = atypical antipsychotic.
Insulin concentration quartiles were based on log-transformed fasting insulin values, with Q1 corresponding to the lowest quartile and Q4 corresponding to the highest quartile.
χ2 or analysis of variance p value as appropriate.
AAP risk groups were defined as follows: high = olanzapine or clozapine; moderate = risperidone, quetiapine, paliperidone, or iloperidone; low = aripiprazole, lurasidone, or ziprasidone.
Antipsychotic polypharmacy was defined as use of 2-3 total antipsychotics, including first-generation (typical) and second-generation (atypical) antipsychotic medications.
Obese and Nonobese Control Subjects
The median age of the 10 obese control participants was 46.5 (IQR 45.0-49.0) years and 46.5 (IQR 41.8-50.3) years for the 10 nonobese participants (p=0.704). Median log-transformed fasting insulin values were 1.13 (IQR 1.05-1.17) μU/mL and 1.12 (IQR 1.02-1.32) μU/mL for the nonobese and obese controls, respectively (p=0.500) (Figure 1B). The median BMI for the nonobese and obese controls was 25.3 (IQR 20.9-27.0) kg/m2, and 34.7 (IQR 33.4-36.7) kg/m2, respectively (p<0.001) (Figure 1D).
Quantitative 1H-NMR Metabolomics and Pathway Analysis
Thirty-six metabolites were identified and quantified by 1H-NMR in all serum samples. Of these, 13 1H-NMR detected metabolites differentiated (FDR < 25%) the insulin quartiles based on the ANOVA P value (Table 2). To identify associated metabolic pathways, these compounds were entered into Metscape (http://metscape.ncibi.org/), an open-source bioinformatics platform that permits the visualization and identification of associated metabolic pathways.30 Among resulting pathways, one-carbon metabolism, and glycine, serine, alanine, and threonine metabolism were represented by at least three metabolites.
Table 2.
Serum Metabolites and Fatty Acids Identified and Quantified
| KEGG ID | ANOVA P value |
False Discovery Rate (%) | Post Hoc Resultsa | |
|---|---|---|---|---|
| Metabolites | ||||
| Glutamate | C00302 | 0.0001 | 0.36 | Q3>Q1; Q3>Q2; Q4>Q1; Q4>Q2 |
| Acetate | C00033 | 0.0057 | 10.26 | Q2>Q4; Q3>Q4; Q1>Q4 |
| Tyrosine | C00082 | 0.0129 | 15.48 | Q4>Q1 |
| 3-hydroxybutyrate | C01089 | 0.0269 | 16.87 | Q1>Q4 |
| Carnitine | C00487 | 0.0269 | 16.87 | Q2>Q4 |
| Glycine | C00037 | 0.0286 | 16.87 | Q1>Q4 |
| Alanine | C00133 | 0.0328 | 16.87 | Q3>Q1 |
| Serine | C00065 | 0.0536 | 22.76 | Q3>Q4 |
| Creatine phosphate | C02305 | 0.0690 | 23.84 | Q3>Q4 |
| Valine | C00183 | 0.0842 | 23.84 | |
| Taurine | C00245 | 0.0862 | 23.84 | |
| Proline | C00148 | 0.0891 | 23.84 | |
| Leucine | C00123 | 0.0927 | 23.84 | |
|
| ||||
| Fatty Acids | ||||
|
| ||||
| 14:0 | C06424 | <0.0001 | 0.02 | Q3>Q1; Q4>Q1; Q2>Q1 |
| 16:1 | C08362 | <0.0001 | 0.02 | Q3>Q1; Q4>Q1; CQ3>Q2; Q4>Q2 |
| 18:2 | C01595 | 0.0008 | 0.61 | Q1>Q4; Q1>Q3; Q2>Q4 |
| 16:0 | C00249 | 0.0025 | 1.44 | Q4>Q1 |
| 24:0 | C08320 | 0.0349 | 16.05 | |
Serum metabolites detected by 1H-nuclear magnetic resonance and fatty acids detected with gas chromatography were identified as differentiating patients with schizophrenia across insulin concentration quartiles (false discovery rate of <25%). Metabolites in italics and boldface were those that were determined to be associated with body mass index (see text).
ANOVA = analysis of variance; Q = quartile.
Post hoc results were determined to be significant when Tukey-Kramer p values were <0.05.
If metabolites were different (FDR < 25%) between obese and nonobese controls, they were considered to be attributable to BMI and were removed from the Metscape analysis. However, if differentiating metabolites between the obese and nonobese controls were also represented as a differentiating metabolite in the schizophrenia analysis, but their concentrations trended in the opposite direction as weight (e.g., glycine), they were not removed from the Metscape analysis (Table 2). After this adjustment, we determined that differences in the concentrations of the remaining seven metabolites were influenced by insulin concentration (Figures 2A–N). Metscape mapping of these compounds generated a one-carbon metabolism pathway that involved three of these metabolites: serine, glycine, and taurine (Figure 3). Supporting information includes all differentiating metabolites (FDR < 25%) from the control analyses (Supplementary Table 1) and all pathways that were mapped with Metscape (Supplementary Table 2). The 1H-NMR and FA data sets are freely accessible via the Metabolomics Workbench (http://www.metabolomicsworkbench.org/).
Figure 2.
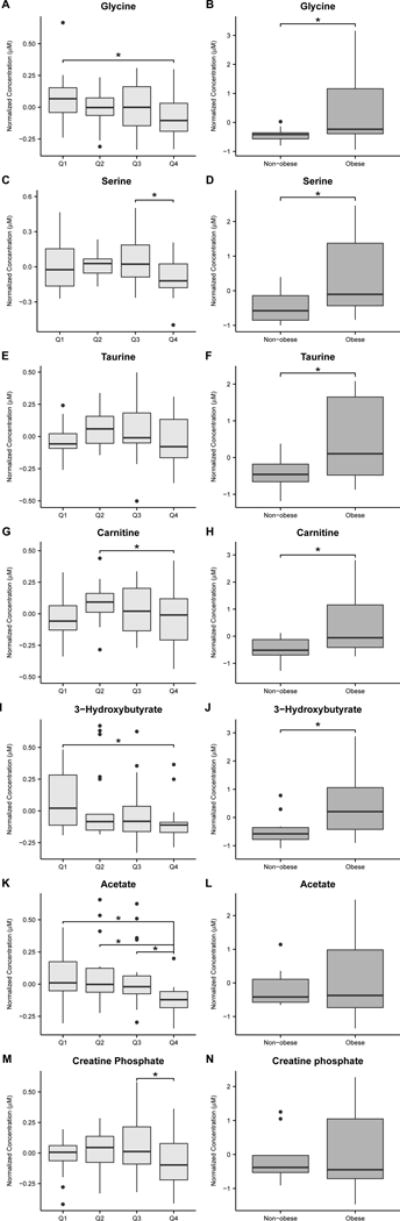
Box and whisker plots of normalized serum metabolite concentrations detected and quantified by 1H-nuclear magnetic resonance (A-N) in the schizophrenia quartiles (A, C, E, G, I, K, and M) and BMI controls (B, D, F, H, J, L, and N). Metabolites shown in the schizophrenia column are those that differentiated insulin quartiles (ANOVA FDR <25%) that were determined to be independent of BMI (see text). This was determined if the Student t-test results comparing the same normalized metabolite concentrations between the BMI control groups was not significant, or the metabolite concentrations trended in opposite directions with weight in the BMI controls and schizophrenia groups (e.g., glycine). In the patients with schizophrenia, glycine (A) concentration trended lower but was increased in obese BMI controls compared with nonobese BMI controls (B). Serine (C and D), taurine (E and F), carnitine (G and H) and 3-hydroxybutyrate (I and J) concentrations followed the same trends. Acetate and creatine phosphate concentrations trended lower in patients with schizophrenia (K and M) but were not significantly different between nonobese and obese controls (L and N). Asterisks indicate metabolite concentrations in the schizophrenia group that were significantly different between quartiles (Tukey-Kramer post hoc P<0.05) or between BMI controls (Student t-test P<0.05).
Figure 3.
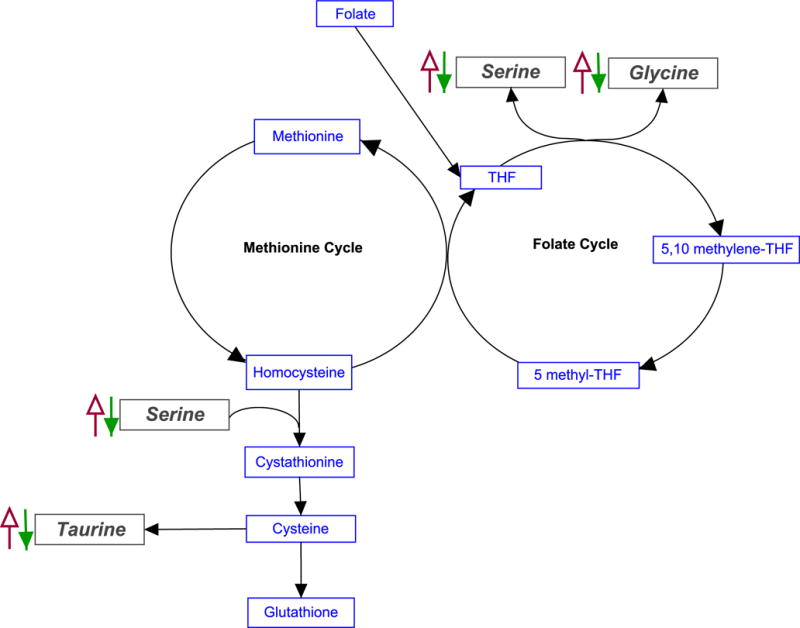
Simplified schematic diagrams of one-carbon metabolism. Arrows depict trend of metabolite concentration with increasing BMI. Solid (green) arrows indicate differentiating metabolites whose concentrations trended inversely with weight among the schizophrenia quartiles. Open (red) arrows indicate differentiating metabolites whose concentrations increased with weight among the BMI controls. The inverse relationship between the concentration of these metabolites with BMI indicates that variability in one-carbon metabolism is not solely due to weight.
Quantitative Fatty Acid Analysis
A total of 23 serum FA were identified and quantified. In the schizophrenia cohort, significant differences were apparent in several saturated FA, such as 16:0 (palmitic acid), 14:0 (myristic acid), 24:0 (lignoceric acid), and the unsaturated essential FA 18:2 (linoleic acid). Three FA—16:0, 18:2, and 24:0—were not associated with BMI and differentiated the four insulin quartiles of the schizophrenia cohort (Table 2 and Figures 4A–F).
Figure 4.
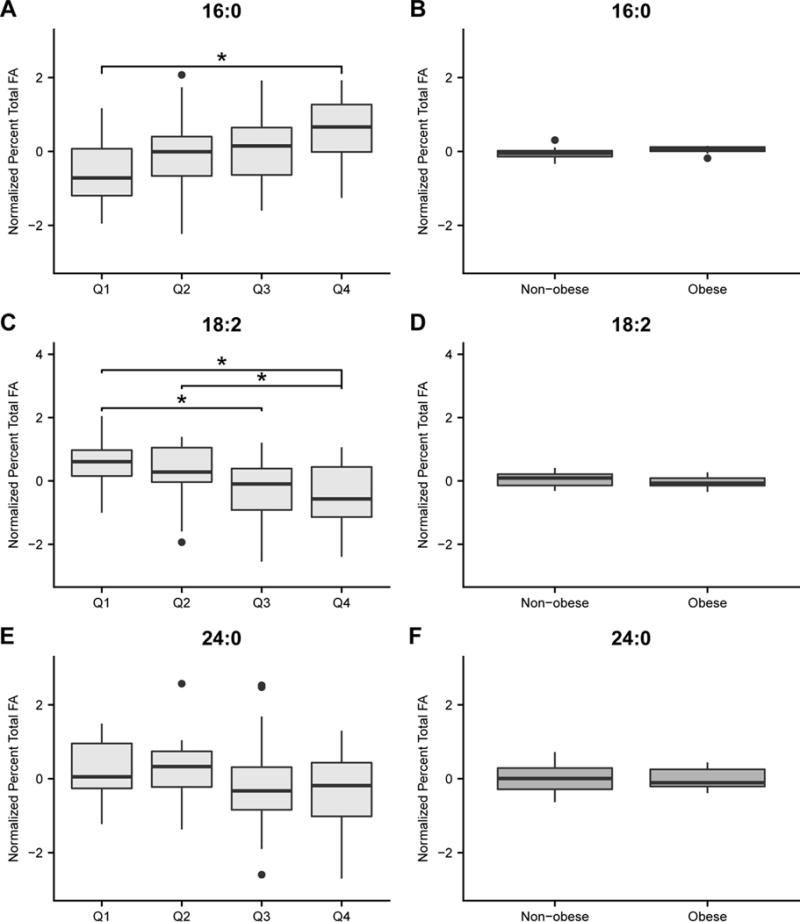
Box and whisker plots of normalized concentrations of individual fatty acids (FA) by percent total that differentiated insulin quartiles in patients wih schizophrenia (ANOVA FDR <25%; A, C, and E) but not BMI (B, D, and F). This was determined if the Student t-test p values comparing the same normalized FA concentrations between the BMI control groups were not significant, or if the FA concentrations were not associated with weight in the BMI schizophrenia groups. Asterisks indicate FA that were significantly different (Tukey Kramer post hoc P<0.05) between quartiles.
Discussion
We conducted a metabolomics analysis of serum samples from patients with schizophrenia who were taking AAPs and analyzed the results based on their fasting insulin concentration quartiles. This approach permitted the detection of unique metabolic signatures associated with variable fasting insulin concentrations and demonstrated the utility of fasting insulin concentration as a biochemical anchor for the metabolic phenotyping of this population. We discuss the importance of perturbed one-carbon metabolism as a contributing factor to cardiovascular risk in patients with schizophrenia. We suggest that analyzing CVD risk by fasting insulin concentration in this population provides insight into metabolic variances between AAP users because there were no significant differences in average daily AAP exposure or higher metabolic risk AAP use between the insulin quartiles. Furthermore, these findings were not solely attributable to differences in BMI.
Among the affected pathways identified from differentiating metabolites by Metscape, the folate cycle and methionine and cysteine metabolism intersect through their involvement in one-carbon metabolism, which is essential for DNA methylation, purine synthesis, and homocysteine metabolism (Figure 3). In our cohort, concentrations of the methyl donors glycine and serine trended higher in the lower insulin quartiles. This suggests increased availability of one-carbon groups available for the folate cycle. Additionally, serine is used in the formation of cystathionine from homocysteine, ultimately leading to production of the endogenous antioxidant glutathione. This is important because elevations in homocysteine have been repeatedly linked to CVD through mechanisms that are not yet understood.32,33
Perturbed one-carbon metabolism has also been implicated in CVD risk in patients with schizophrenia.34 Our group has observed decreased methylation capacity and worsening endothelial function in schizophrenia patients with genetic polymorphisms in folate cycle enzymes associated with hyperhomocysteinemia.35,36 We also found that supplemental folate administration improved methylation capability and endothelial function in schizophrenia patients taking AAPs.37 Additionally, the folate cycle is intimately tied to FA metabolism, as it is needed to contribute methyl groups for carnitine methylation and FA chain elongation and desaturation.38 Assies et al 39 recently proposed that the complex relationship between FA and the folate cycle in CVD and psychiatric disease could be described as oxidative stress leading to the one-carbon cycle spending proportionally more time on transsulfation compared to methylation. This could decrease available methyl groups, resulting in less FA chain elongation and desaturation. We observed metabolite concentration changes that may agree with this hypothesis: saturated FA were elevated in schizophrenia patients with higher fasting insulin concentrations (e.g., Q3-4), whereas patients with lower fasting insulin concentrations (e.g., Q1-2) had higher concentrations of methyl-donating amino acids. This pattern of elevation in saturated FA has also been observed in settings of CVD40 in patients without mental health disorders.
As insulin concentration is known to be associated with weight, this complicated our analysis because both weight and fasting insulin concentration were significantly different between quartiles. To account for weight variability, we used two groups of control subjects: obese and nonobese participants without CVD or any psychiatric diagnosis, and completed the same metabolomics assays for this cohort. Importantly, fasting insulin concentration was not significantly different between groups. As such, we attributed differences in metabolite and FA concentrations between the BMI control groups as those that were primarily due to differences in weight. We used these results to interpret the metabolomics data from the schizophrenia cohort’s insulin concentration quartiles. With respect to FA, palmitic acid (16:0), linoleic acid (18:2), and lignoceric acid (24:0) remained uniquely altered in schizophrenia patients. Palmitic acid trended upward in Q3-4, whereas linoleic and lignoceric acids trended lower. Of these FA, linoleic acid (18:2) is an essential FA and reflects variable diet. This suggests that linoleic acid intake trended inversely with fasting insulin in the schizophrenia cohort.
To test the rigor of our analysis, we separated the schizophrenia participants into two groups (obese and nonobese) to act as their own controls for BMI variability and completed the same analyses that were performed for the BMI controls without psychiatric diagnoses or cardiovascular disease. The table of differentiating metabolites from this analysis is provided in the supplement (Supplementary Table 3). There were no differentiating 1H-NMR metabolites that were common to both the fasting insulin quartile analysis and the BMI analysis that were not already identified as associated with BMI by the control analysis (and indicated in Table 2). After a similar comparison of the FA differentiating the schizophrenia insulin quartiles and BMI groups, palmitic acid was the only remaining FA that was associated with fasting insulin concentration and not BMI. Unlike the BMI controls used for the primary analysis, log-transformed fasting insulin concentration differentiated the schizophrenia groups separated by obese or nonobese BMI classification (Wilcoxon test P <0.001), making interpretation of these results less straightforward. Although not confirmatory, these results indicate that the metabolite variability of the schizophrenia cohort are neither entirely due to differences in BMI nor to AAP exposure between the insulin quartiles.
We chose to describe AAP use among insulin quartiles in the schizophrenia cohort with two different approaches: (1) comparing the proportion of patients using medications at three different levels of metabolic risk, and 2) normalizing average daily AAP exposure using chlorpromazine equivalents. This was done because AAPs have variable propensity to cause metabolic adverse effects, and no AAP is likely free from this risk.41 Neither measure was significantly different among schizophrenia quartiles (Table 1). This was an interesting finding because it suggests that current AAP use may not impact fasting insulin concentration in this population. The lack of difference in AAP exposure among quartiles may be due to the similar length of time since schizophrenia spectrum diagnosis (Table 1). This could be reflective of previous exposure to a variety of antipsychotic treatment trials. Since many patients could not recall an in-depth account of past antipsychotic exposure, it was not possible to assess the impact of the duration of treatment on the metabolome. Previous research suggests that duration of exposure to AAP influences the metabolome early in schizophrenia treatment, but it is still unclear how drug use influences the metabolome and metabolic risk after several antipsychotic trials. For example, a lipidomics study by McEvoy et al42 noted significant changes in lipid profiles following only 2 weeks of drug therapy in patients experiencing their first episode of schizophrenia, and Suvitival et al43 found metabolites associated with future weight gain in patients with first-episode psychosis followed over one year.
The lack of a relationship between fasting insulin concentration and current AAP exposure is interesting, particularly given the association between AAP use and the development of adverse metabolic events.44 This leaves unanswered an important question regarding how essential the timing and duration of AAP exposure is to encouraging the disruption of glucose-insulin homeostasis. In the future, it will be important to capture changes in the metabolome before and for an extended period of time after AAP initiation and medication switches. It will also be necessary to have studies adequately powered to allow for further group separation by sex, as recent research shows this to be an important factor in determining changes in folate cycle metabolites following antipsychotic administration.45
Despite these intriguing findings, we acknowledge that this was an exploratory study with several limitations. First, this was a retrospective investigation that used existing blood samples from a relatively small sample size. This prevents us from making conclusions about AAP-induced changes on the metabolome. Additionally, the use of NMR-based metabolomics results in significantly fewer metabolites than approaches using mass spectrometry–based detection methods. It is possible that we could have identified additional biochemical pathways associated with variability in fasting insulin concentration with the use of a more sensitive platform like liquid chromatography–mass spectrometry. However, for this preliminary study, the advantages of quantitative NMR metabolomics outweighed this option because NMR is routinely quantitative, highly reproducible, and nondestructive to the sample. Use of this approach will also direct the selection of more sensitive targeted assays for future studies. Another limitation of the study stems from the liberal FDR of <25%, which is consistent with exploratory genetics analyses,46 but increases the likelihood of type 1 error.
Conclusion
Our metabolomics analyses identified differences in concentrations of metabolites that are associated with variable one-carbon metabolism (glycine, serine, and taurine) among fasting insulin concentration quartiles of a schizophrenic cohort—specifically, that fasting insulin concentration trended inversely with increased methylation capability. We also found that patients with schizophrenia who had higher insulin concentrations (e.g., Q3-4) had higher levels of the saturated FA 16:0 (palmitic acid). These findings appeared to be independent of current AAP use and demonstrated the utility of anchoring metabolomics studies to fasting insulin concentration as a measure of metabolic risk in this population, as compared to MetS diagnosis that is observable after fasting insulin concentration elevations. In aggregate, these results have implications for furthering knowledge about the mechanisms involved in the development of CVD in schizophrenic patients since loss of, or a decline in, methylation capability may be associated with endothelial dysfunction. Future studies will need to include a targeted, prospective, metabolomics approach with a more stringent FDR. Ultimately, understanding the metabolic consequences of AAPs across all phases of schizophrenia and how AAPs impact variable baseline risk for CVD will allow for safer medication use through improved metabolic monitoring and the design of more precise treatment regimens.
Supplementary Material
Acknowledgments
We would like to thank all participants for their involvement in this study and the nurses from the Michigan Clinical Research Unit (MCRU) for their assistance with study visits.
Funding
Funding for this work was supported in part by grants from the following centers: the University of Michigan Claude D. Pepper Older Americans Independence Center (National Institute on Aging [NIA] grant AGA024824); Drs. Amy Rothberg and Charles Burant, and the University of Michigan’s Nutrition Obesity Research Center (grant DK089503) and Weight Management Program, and the Michigan Regional Comprehensive Metabolomics Resource Coure (grant DK097153), the Michigan Center for Diabetes Translational Research (grant P30DK092926), and the A. Alfred Taubman Medical Institute and the Robert C. And Veronica Atkins Foundation. This work was also supported in part by a metabolomics supplement to a grant from the National Institute of Mental Health (NIMH; grant MH082784; Dr. Ellingrod). Dr. Stringer’s effort is supported in part by a grant from the National Institute of General Medical Sciences (NIGMS; grant GM111400).
Footnotes
DR KRISTEN MARIE WARD (Orcid ID : 0000-0002-5881-3456)
DR KATHLEEN A. STRINGER (Orcid ID : 0000-0003-0238-7774)
Disclaimer
The content is solely the responsibility of the authors and does not necessarily present the official views of the NIA, National Institute of Diabetes and Digestive and Kidney Diseases, NIMH, NIGMS, or the National Institutes of Health.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Contributor Information
Kristen M. Ward, Department of Clinical Pharmacy, College of Pharmacy, University of Michigan, Ann Arbor, MI
Larisa Yeomans, NMR Metabolomics Laboratory, College of Pharmacy, University of Michigan, Ann Arbor, MI.
Cora McHugh, NMR Metabolomics Laboratory, College of Pharmacy, University of Michigan, Ann Arbor, MI.
A. Zarina Kraal, Psychology Department, College of Literature, Science, and the Arts, University of Michigan, Ann Arbor, MI.
Stephanie A. Flowers, Department of Pharmacy Practice, College of Pharmacy, University of Illinois, Chicago, IL
Amy E. Rothberg, Department of Internal Medicine, Division of Metabolism, Endocrinology and Diabetes, School of Medicine, University of Michigan, Ann Arbor, MI
Alla Karnovsky, Department of Bioinformatics and Computational Medicine, School of Medicine and the Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan, Ann Arbor, MI.
Arun Das, Department of Internal Medicine, Division of Metabolism, Endocrinology and Diabetes, School of Medicine and the Michigan Regional Comprehensive Metabolomics Resource Core, University of Michigan, Ann Arbor, MI.
Vicki L. Ellingrod, Department of Clinical Pharmacy, College of Pharmacy, and the Department of Psychiatry, School of Medicine, University of Michigan, Ann Arbor, MI
Kathleen A. Stringer, Department of Clinical Pharmacy, College of Pharmacy; Division of Pulmonary and Critical Care Medicine, School of Medicine, and the NMR Metabolomics Laboratory, University of Michigan, Ann Arbor, MI
References
- 1.Hennekens CH, Hennekens AR, Hollar D, Casey DE. Schizophrenia and increased risks of cardiovascular disease. Am Heart J. 2005;150:1115–1121. doi: 10.1016/j.ahj.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Ringen PA, Engh JA, Birkenaes AB, Dieset I, Andreassen OA. Increased mortality in schizophrenia due to cardiovascular disease – a non-systematic review of epidemiology, possible causes, and interventions. Front psychiatry. 2014;5:137. doi: 10.3389/fpsyt.2014.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grundy SM, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–52. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- 4.Consensus Development Conference on Antipsychotic Drugs and Obesity and Diabetes AMERICAN DIABETES ASSOCIATION AMERICAN PSYCHIATRIC ASSOCIATION AMERICAN ASSOCIATION OF CLINICAL ENDOCRINOLOGISTS NORTH AMERICAN ASSOCIATION FOR THE STUDY OF OBESITY
- 5.Mitchell AJ, Delaffon V, Vancampfort D, Correll CU, De Hert M. Guideline concordant monitoring of metabolic risk in people treated with antipsychotic medication: systematic review and meta-analysis of screening practices. Psychol Med. 2012;42:125–147. doi: 10.1017/S003329171100105X. [DOI] [PubMed] [Google Scholar]
- 6.Weyer C, Hanson RL, Tataranni PA, Bogardus C, Pratley RE. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: evidence for a pathogenic role of relative hyperinsulinemia. Diabetes. 2000;49:2094–101. doi: 10.2337/diabetes.49.12.2094. [DOI] [PubMed] [Google Scholar]
- 7.Johnson J, Duick D, Chui M, Aldasouqi S. Identifying Prediabetes Using Fasting Insulin Levels. Endocr Pract. 2010;16:47–52. doi: 10.4158/EP09031.OR. [DOI] [PubMed] [Google Scholar]
- 8.Henderson DC, et al. Glucose Metabolism in Patients With Schizophrenia Treated With Atypical Antipsychotic Agents. Arch Gen Psychiatry. 2005;62:19. doi: 10.1001/archpsyc.62.1.19. [DOI] [PubMed] [Google Scholar]
- 9.Houseknecht KL, et al. Acute Effects of Atypical Antipsychotics on Whole-Body Insulin Resistance in Rats: Implications for Adverse Metabolic Effects. Neuropsychopharmacology. 2007;32:289–297. doi: 10.1038/sj.npp.1301209. [DOI] [PubMed] [Google Scholar]
- 10.Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. 2009;2:231–7. doi: 10.1242/dmm.001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.First MB, Spitzer RL, Miriam G, Williams JB. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version. New York: Biometrics Research, New York State Psychiatric Institute; 2002. ((SCID-I/P) [Google Scholar]
- 12.Andreasen NC, Pressler M, Nopoulos P, Miller D, Ho BC. Antipsychotic dose equivalents and dose-years: a standardized method for comparing exposure to different drugs. Biol Psychiatry. 2010;67:255–62. doi: 10.1016/j.biopsych.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woods SW. Chlorpromazine Equivalent Doses for Atypical Antipsychotics: An Update 2003-2010. 2011 doi: 10.4088/jcp.v64n0607. at < http://scottwilliamwoods.com/files/WoodsEquivUpdate.doc>. [DOI] [PubMed]
- 14.Meyer JM, Ng-Mak DS, Chuang CC, Rajagopalan K, Loebel A. Weight changes before and after lurasidone treatment: a real-world analysis using electronic health records. Ann Gen Psychiatry. 2017;16:36. doi: 10.1186/s12991-017-0159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Hert M, et al. Body Weight and Metabolic Adverse Effects of Asenapine, Iloperidone, Lurasidone and Paliperidone in the Treatment of Schizophrenia and Bipolar Disorder. CNS Drugs. 2012;26:733–759. doi: 10.2165/11634500-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 16.Maayan L, Correll CU. Management of antipsychotic-related weight gain. Expert Rev Neurother. 2010;10:1175–200. doi: 10.1586/ern.10.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parsons B, et al. Weight effects associated with antipsychotics: A comprehensive database analysis. Schizophr Res. 2009;110:103–110. doi: 10.1016/j.schres.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 18.Rothberg AE, et al. The impact of weight loss on health-related quality-of-life: implications for cost-effectiveness analyses. Qual Life Res. 2014;23:1371–6. doi: 10.1007/s11136-013-0557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothberg AE, McEwen LN, Kraftson AT, Fowler CE, Herman WH. Very-low-energy diet for type 2 diabetes: an underutilized therapy? J Diabetes Complications. 28:506–10. doi: 10.1016/j.jdiacomp.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stringer KA, et al. Whole Blood Reveals More Metabolic Detail of the Human Metabolome than Serum as Measured by 1H-NMR Spectroscopy. Shock. 2015;44:200–208. doi: 10.1097/SHK.0000000000000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obi AT, et al. 1D-1H-nuclear magnetic resonance metabolomics reveals age-related changes in metabolites associated with experimental venous thrombosis. J Vasc Surg Venous Lymphat Disord. 2016;4:221–230. doi: 10.1016/j.jvsv.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Stringer KA, et al. Metabolic consequences of sepsis-induced acute lung injury revealed by plasma 1H-nuclear magnetic resonance quantitative metabolomics and computational analysis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L4–L11. doi: 10.1152/ajplung.00231.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lacy P, et al. Signal intensities derived from different NMR probes and parameters contribute to variations in quantification of metabolites. PLoS One. 2014;9:e85732. doi: 10.1371/journal.pone.0085732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das AK, Hajra AK. Quantification, characterization and fatty acid composition of lysophosphatidic acid in different rat tissues. Lipids. 1989;24:329–33. doi: 10.1007/BF02535172. [DOI] [PubMed] [Google Scholar]
- 25.MORRISON WR, SMITH LM. PREPARATION OF FATTY ACID METHYL ESTERS AND DIMETHYLACETALS FROM LIPIDS WITH BORON FLUORIDE–METHANOL. J Lipid Res. 1964;5:600–8. [PubMed] [Google Scholar]
- 26.Mangold HK. Thin-Layer Chromatography. Springer; New York: p. 1969. [Google Scholar]
- 27.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 2015;43:W251–W257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van den Berg RA, Hoefsloot HCJ, Westerhuis JA, Smilde AK, van der Werf MJ. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics. 2006;7:142. doi: 10.1186/1471-2164-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Storey JD. THE POSITIVE FALSE DISCOVERY RATE: A BAYESIAN INTERPRETATION AND THE q-VALUE 1. Ann Stat. 2003;31:2013–2035. [Google Scholar]
- 30.Gao J, et al. Metscape: a Cytoscape plug-in for visualizing and interpreting metabolomic data in the context of human metabolic networks. Bioinformatics. 2010;26:971–3. doi: 10.1093/bioinformatics/btq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.R Core Team. R: A language and environment for statistical computing. 2014 [Google Scholar]
- 32.Mayer EL, Jacobsen DW, Robinson K. Homocysteine and coronary atherosclerosis. J Am Coll Cardiol. 1996;27:517–27. doi: 10.1016/0735-1097(95)00508-0. [DOI] [PubMed] [Google Scholar]
- 33.Humphrey LL, Fu R, Rogers K, Freeman M, Helfand M. Homocysteine Level and Coronary Heart Disease Incidence: A Systematic Review and Meta-analysis. Mayo Clin Proc. 2008;83:1203–1212. doi: 10.4065/83.11.1203. [DOI] [PubMed] [Google Scholar]
- 34.Assies J, et al. Effects of oxidative stress on fatty acid- and one-carbon-metabolism in psychiatric and cardiovascular disease comorbidity. Acta Psychiatr Scand. 2014;130:163–180. doi: 10.1111/acps.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellingrod VL, et al. Dietary, lifestyle and pharmacogenetic factors associated with arteriole endothelial-dependent vasodilatation in schizophrenia patients treated with atypical antipsychotics (AAPs) Schizophr Res. 2011;130:20–26. doi: 10.1016/j.schres.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ellingrod VL, et al. Metabolic syndrome and insulin resistance in schizophrenia patients receiving antipsychotics genotyped for the methylenetetrahydrofolate reductase (MTHFR) 677C/T and 1298A/C variants. Schizophr Res. 2008;98:47–54. doi: 10.1016/j.schres.2007.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellingrod VL, Grove TB, Burghardt KJ, Taylor SF, Dalack G. The effect of folate supplementation and genotype on cardiovascular and epigenetic measures in schizophrenia subjects. npj Schizophr. 2015;1:15046. doi: 10.1038/npjschz.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.da Silva RP, Kelly KB, Al Rajabi A, Jacobs RL. Novel insights on interactions between folate and lipid metabolism. Biofactors. 2014;40:277–83. doi: 10.1002/biof.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Assies J, et al. Effects of oxidative stress on fatty acid- and one-carbon-metabolism in psychiatric and cardiovascular disease comorbidity. Acta Psychiatr Scand. 2014;130:163–80. doi: 10.1111/acps.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khaw KT, Friesen MD, Riboli E, Luben R, Wareham N. Plasma phospholipid fatty acid concentration and incident coronary heart disease in men and women: the EPIC-Norfolk prospective study. PLoS Med. 2012;9:e1001255. doi: 10.1371/journal.pmed.1001255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kahn RS, et al. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet (London, England) 2008;371:1085–97. doi: 10.1016/S0140-6736(08)60486-9. [DOI] [PubMed] [Google Scholar]
- 42.McEvoy J, et al. Lipidomics Reveals Early Metabolic Changes in Subjects with Schizophrenia: Effects of Atypical Antipsychotics. PLoS One. 2013;8:e68717. doi: 10.1371/journal.pone.0068717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suvitaival T, et al. Serum metabolite profile associates with the development of metabolic co-morbidities in first-episode psychosis. Transl Psychiatry. 2016;6:e951. doi: 10.1038/tp.2016.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Correll CU, et al. Cardiometabolic Risk in Patients With First-Episode Schizophrenia Spectrum Disorders. JAMA Psychiatry. 2014;71:1350. doi: 10.1001/jamapsychiatry.2014.1314. [DOI] [PubMed] [Google Scholar]
- 45.Misiak B, Frydecka D, Łaczmański Ł, Ślęzak R, Kiejna A. Effects of second-generation antipsychotics on selected markers of one-carbon metabolism metabolic syndrome components in first-episode schizophrenia patients. Eur J Clin Pharmacol. 2014;70:1433–41. doi: 10.1007/s00228-014-1762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chesler EJ, et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet. 2005;37:233–42. doi: 10.1038/ng1518. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.