

Abstract
Objective(s):
To reach an evidence-based knowledge in the context of the temporal-spatial pattern of neuronal death and find appropriate time of intervention in order to preserve spared neurons and promote regeneration after traumatic spinal cord injury (TSCI).
Materials and Methods:
The study design was based on Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA)-guided systematic review. PubMed and EMBASE were searched (24 October, 2015) with no temporal or linguistic restrictions. Hand-search was performed in the bibliographies of relevant articles. Non-interventional animal studies evaluating time-dependent neuronal death following acute mechanical trauma to the spinal cord were included. We separately evaluated the fate of various populations of neurons including propriospinal neurons, ventral motor neurons, Clarke’s column neurons, and supraspinal neurons.
Results:
We found 11,557 non-duplicated studies. Screening through the titles and abstracts led to 549 articles, 49 of which met the inclusion criteria. Both necrotic and apoptotic neuronal deaths occur after TSCI, though necrosis is the prominent mechanism. There are differences in the responses of intrinsic neurons of the spinal cord to the TSCI. Also, the extent of neuronal death in the supraspinal neurons depends on the anatomical location of their axons.
Conclusion:
In order to develop new therapies, selection of the injury model and time of intervention has a crucial role in the efficacy of therapy. In addition, examining the safety and efficacy of an intervention by reliable methods not confounded by the injury-related changes would promote translation of therapies to the clinical application.
Keywords: Apoptosis, Necrosis, Neuron, Pathophysiology, Spinal cord injury
Introduction
Traumatic spinal cord injury (TSCI) has a devastating effect on the patient’s quality of life, family, and society (1-3). Current clinical therapies attempt to prevent progression of secondary injuries that initiate after acute mechanical insult. However, the results of clinical trials suggest that current therapies are not sufficiently effective. In this regard, new therapeutic approaches, such as cell therapies, gene therapies, and tissue engineering have been proposed to compensate neuronal loss and inhibitory environment of the injured tissue. Detailed understanding of the time-dependent pathophysiological events after TSCI in animal studies may help scientists to accurately decide the time and type of intervention. However, despite abundant studies evaluating the TSCI pathophysiology, there is a controversy about neuronal death following mechanical insult. Some evidence suggests the immediate and immense neuronal death (4, 5), while there is other evidence for the preservation of spared neurons up to one year PI (6, 7). Additionally, there are some studies reviewing the occurrence of apoptosis after TSCI (8, 9), while in these studies the occurrence of apoptosis was evaluated in all cellular components of CNS including glial cells and neurons. In the current systematic review, we exclusively analyzed the available evidence regarding the occurrence of apoptosis in neurons. Additionally, the focus of the previous studies reviewing the fate of neurons after TSCI was limited to the apoptotic/necrotic neuronal death, while current study was designed to systematically analyze the survival potential of various populations of neurons after TSCI.
Materials and Methods
Search and selection of studies
We searched MEDLINE via PubMed and EMBASE via Ovid SP on 24 October, 2015. Keywords were collected through expert advice, literature review, controlled vocabulary (Medical Subject Headings = MeSH and Excerpta Medica Tree = EMTREE), and reviewing the results of pilot search. Search strategies were developed by assistance of a medical information specialist (see the Appendix S1). Search results were de-duplicated in EndNote X5 and then screened by two independent researchers. In addition, hand-search was performed in the bibliographies of relevant studies.
Selection criteria
Animal studies discussing time-dependent pathophy- siological changes of neurons following acute mechanical trauma to the spinal cord were included. No temporal or linguistic restrictions were considered. We included rat studies because: first, rats are the most commonly used animal models for investigating the pathophysiology of TSCI and design of therapeutic approaches (10, 11) and second, they show functional, electrophysiological, and morphological outcomes similar to humans following TSCI (12). We included following acute mechanical trauma models with no age, gender or strain restrictions: compression, contusion (the most common injury model which can be performed by weight drop using the NYU device, the Ohio State University device, or clips compression), hemisection, or transection. We excluded studies that 1) used transgenic rats, 2) reported cell death without identifying the cell type, 3) reported molecular events without any cellular examination, or 4) did not mention the exact time of the assessment after TSCI. Reviews and interventional studies were also excluded.
Outcome measures
We evaluated neuronal death (i.e., apoptosis and necrosis) in the spinal cord and brain after TSCI. We separately evaluated the fate of various populations of neurons including propriospinal neurons, ventral motor neurons, Clarke’s column neurons, and supraspinal neurons. Furthermore, in order to reduce the heterogeneity and to compare the effect of different injury models, the data were categorized according to the injury model into 3 groups of contusion, compression, and transection/hemisection/tractotomy.
Data extraction and quality assessment
Data were independently extracted into pre-developed forms by two reviewers. We used a consensus process to resolve the discrepancies. Quality assessment of the included papers was performed based on a 15-item checklist developed in our previous study (13). If two reviewers had different opinions about the quality of a study, the idea of a third reviewer (VRM) was asked. The authors of articles were contacted via email when we did not have access to the full-text or additional information required.
Results
Included studies
Search in electronic sources yielded 11,557 non-duplicated studies. Screening through titles and abstracts led to 549 articles, 49 of which met the inclusion criteria (4-7, 14-58). The search flowchart is presented in Figure 1. Characteristics of the included studies are presented in Table 1. The transection model, including complete transection (6, 17-18, 26, 30, 32, 36, 40-44, 47-49, 51, 56, 59), hemisection (7, 35, 50, 58), or tractotomy (23, 33, 54), was the most common used injury model performed on 25 experiments. Contusion was the next model used in 23 experiments (4-5, 7, 14-16, 19, 20, 22, 24, 25, 28-32, 34, 37, 38, 46, 51, 52, 55), and compression injury model was only used in 6 studies (21, 27, 39, 45, 53, 57).
Figure 1.
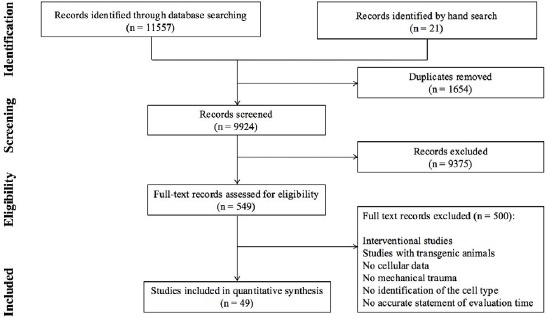
Flowchart of including studies
Table 1.
Characteristics of included studies
| No | Authror and Year | Number of animals in injury/control goup * | Methrod | Time of evaluation (post-injury) | |||
|---|---|---|---|---|---|---|---|
| Gender/ strain/ weighrt (g) or age | Methrod/level of injury | Severity of injury | Methrod of evaluation | ||||
| 1 | Andrade 2008 | at least 10/8 | M/ Wistar/ 220–250 | NYU impactor/ T9-T10 | 10 g*25 mm & 10 g*50 mm | Cresyl Violet staining | 7, 30 D |
| 2 | Barron 1988 | 3/3 | F/ Sprague–Dawley /6 W | Transection/ T7 | N.A. | HR&E & TEM | 10 W |
| 3 | Bose 2005 | 9/5 | F/ Sprague–Dawley /7-7.5 monthr-old | NYU impactor/ T8 | 10 g*25 mm | Retrogade labeling by CTB | 16 W |
| 4 | Chroo 2008 | 10 per goup/ 9 shram | M/ Sprague Dawley/ 347±28 | Contusion/ C4-C5 | 6 mm x 1.1±0.02 mm depthr | IHRC | 3 hr |
| 5 | Conta 2004 | at least 2/5 | F/ Long-Evans /Adult | NYU impactor/ T10 | 10 g*12.5 mm& 10 g*50 mm | Retrogade labeling by FG | 4, 6 D, 2 W |
| 6 | Dusart 1994 | at least 2/2 | F/ Lewis /4-5 W | Transection/ middle to lower throracic | two-thrirds of thre cord | IHRC | 1, 3, 6, 12 hrr, 1, 2, 4, 8, 15 21 D, 4, 8, 24 W |
| 7 | Eidelberg 1989 | 4/4 | M/ Sprague–Dawley /200-250 & 7 D | Transection/ T8-T10 | N.A. | TEM, HR&E staining, retrogade labeling by HRRP | 24 hrr, 2, 3, 4, 8, 12 D |
| 8 | Ek 2010 | 3/ 3 | M/ Sprague-Dawley/ 170–200 | Impactor/ T10 | 2 mm penetration depthr withr thre target velocity of 5 m/sec and dwell time 100 ms | TEM, IHRC, HR&E | 24 hrr, 1, 4, 10 W |
| 9 | Fehrlings 1995 | 25/ 5 control | F/ Wistar/ 220-280 | Compression/ T1 | 2 g, 18 g, 30 g, 50 g, 98 g | Tract tracing by HRRP | 44 D |
| 10 | Feringa 1984 | at least 3/6 | F/ Wistar/ 6 W | Transection/ T7-T8 | N.A. | Retrogade labeling by HRRP | 6, 8, 10, 12 W |
| 11 | Feringa 1985 a | 10/10 | F/ Wistar/ 6 W | Transection/ T9-T10 | N.A. | Cresyl violet staining/ Retrogade labeling by HRRP | 25 W |
| 12 | Feringa 1988 | at least 3/at least 3 | F/ Wistar/ 7-8 W | Transection/ T9 | N.A. | HR&E staining & Retrogade labeling by HRRP | 5, 10, 15, 25, 52 W |
| 13 | Feringa 1987 | at least 2/3 | F/ Wistar/ 7-8 W | Transection/ T9 | N.A. | HR&E staining | 1, 2, 3, 5, 7, 9, 12, 15 W |
| 14 | Feringa1985 b | 8/8 | F/ Wistar/ 6 W | Transection/ T9 | N.A. | Retrogade labeling by HRRP | 1 Y |
| 15 | Feringa 1983 | at least 3/ at least 3 | F/ Wistar/ 6 W | Transection/ T9 | N.A. | Retrogade labeling by HRRP | 5, 10, 25 W |
| 16 | Gossman 2000 | 5 /5 | F/ Sprague–Dawley /200-250 | Weighrt drop / T8 | 10 g*25 mm | HR&E staining, IHRC | 24 hrr, 4 W |
| 17 | Gossman 2001 | 5 /5 | F/ Sprague–Dawley / 200 –250 | Weighrt drop/ T8 | 10 g*25 mm | HR&E staining, IHRC, TEM | 15 min, 4, 8, 24 hr |
| 18 | HRains 2003 | 6 / 9 | M/ Sprague–Dawley / 150-175 | Transection/T9 | Complete | TUNEL & retrogade labeling by FG | 1, 2, 3, 4 W |
| 19 | HRoltz 1990 | 6 / 3 | M/ Sprague–Dawley / 330 g-380 | Compression by curved rectangular plate/ T7-8 | 35 g*5 min | HR&E, LFB, cresyl violet staining and TEM | 1 hr, 1, 4, 9, 21 D |
| 20 | HRoule 1999 | 3-4 for cell deathr, 36 for retrogade labeling/ N.A. | F/ Sprague–Dawley /225–250 | HRemisection/ C3 | N.A. | Retrogade labeling by TB and BDA | 1, 4. 8, 14 W |
| 21 | HRuang 2007 | 5 for eachr time point/5 for eachr time point | F/ Sprague–Dawley / 230–255 | Compression/ T12 | 50 g*5 min | IHRC | 1, 3, 7, 28 D |
| 22 | James 2011 | at least 4 per eachr time point /N.A. | F/ Sprague–Dawley /200–220 | IHR impactor/ T10 | 150 kdyn from 2–4 mm hreighrt | IHRC & TEM | 1 D, 1, 2, 4, 12, 24 W |
| 23 | Kim 2002 | at least 3/4 | F/ Long- Evans/ 220-270 | Weighrt drop/ T9-10 | 10 g * 6.25 mm 10 g * 12.5 mm 10 g * 25 mm |
Retrogade labeling by Fast Blue | 5, 6 D, 1 W |
| 24 | Lee 2004 | at least 9 /11 control | M/ Sprague–Dawley/ 300–350 | NYU impactor/ T9-T10 | 10 g*25 mm | TUNEL & IHRC | 12, 24, 48, 72 hr, 1 W |
| 25 | Li 1996 | 4 /4 control/4 shram | M /N.A./370 | Compression/ T7-T8 | 9 g, 35 g, 50 g for 5 min | TUNEL & IHRC | 4 hr. 1, 4, 9 D |
| 26 | Liu 2003 | at least 3 / at least 3 | F/ Wistar/ 90–105 | Rubrospinal tractotomy/ C2 | N.A. | Retrogade labeling by FB & TEM | 2 D, 1, 2, 4, 10 W |
| 27 | Liu 1997 | at least 4/ N.A. | F/ Long-Evans/ Adult | Weighrt drop/ T8-9 | 10 g * 6.25 mm 10 g * 12.5 mm |
TUNEL | 5 min & 4, 8, 24 hrr, 3, 7, 14, 30 D |
| 28 | Lou 1998 | Total number of 16 | N.A./Sprague- Dawley adult/ 400 | Allen weighrt-drop/ midthroracic | 35 g. 100 mm | HR&E staining & IHRC & TUNEL | 30 min & 4, 8, 12, 24, 72 hr |
| 29 | McBride 1992 | at least 3/ at least 4 | F/ Wistar/ 7 W | Transection/ T9 | N.A. | Retrogade labeling by FG | 10, 20, 52 W |
| 30 | McBride 1990 | at least 7/ at least 8 | F/ Wistar/ 7 W | Transection/ T9 | N.A. | Retrogade labeling by FG | 10, 20 W |
| 31 | McBride 1989 | at least 4/ at least 5 | F/ Wistar/ 7 W | Transection/ T9 | N.A. | Retrogade labeling by FG | 10, 20 W |
| 32 | McBride 1988 | at least 5/ at least 3 | F/ Wistar/ 135-170 | Transection/ T9 | N.A. | Intracerebellar injection of FG or true blue | 5, 10, 20 W |
| 33 | Morino 2003 | at least 5/37 | F/ Wistar/ 250 | Compression/ T11 | 20 g * 5, 10, 20, 30 or 40 min | TUNEL | 24, 48 hr |
| 34 | Naso 1993 | 15/ 6 | F/ Sprague-Dawley/ 250-350 | Contusion/ T8 | 200, 400, 600 g.mm: 5 g weighrt from various hreighrt | Retrogade labeling by FG | 3 D |
| 35 | Nielson 2010 | at least 5 except 3 w survival n=1/ 10 | M/ Sprague-Dawley/ 150–175 & F/ 225–250 | Dorsal funiculus lesion/ T9 & lateral hremisection/ C5 & Contusion/ T9 | 200 or 250 kdyn | Retrogade labeling by BDA | 1, 3 W, 1 Y |
| 36 | Prendergast 1976 | 5 / N.A. | N.A./Neonate (0-3 D) & adult | Midthroracic hremisection | N.A. | Cresyl violet staining | 90-120 D |
| 37 | Qiu, 2001 | 3/ 3 | M/ Sprague-Dawley / 250-260 | Contusion / T8 | 10 g.12.5 mm | Retrogade labeling by FG, Frag-EL Assay, ELISA, IHRC | 3 D |
| 38 | Rosenberg 1997 | 3/ 4 | F/ Sprague–Dawley/ 230-250 | Weighrt drop/ T8 | 10 g * 25 mm or 175 mm | TB staining & TEM | 15 min & 4, 24 hr |
| 39 | Siebert 2010 a | 4/4 | F/ Long-Evans/ 77 D | Weighrt drop/ T9 Transaction/ T9 |
10 g*25 mm Complete |
TUNEL | 3 D, 1, 2, 4 W |
| 40 | Siebert 2010 b | 4/12 | F/ Long-Evans/ 77 D | Weighrt drop/ T9 Transaction/ T9 |
10 g*25 mm Complete |
Microarray, qRT-PCR | 3 D & 1, 2, 5 W |
| 41 | Springer 1999 | 6 / 6 | Rat | NYU impactor/ T10 | 250 g.mm | IHRC | 1 hr |
| 42 | Steencken 2010 | at least 4/4 | F/ Long-Evans/ N.A. | Contusion / T9 | 25 mm | FG tracing | 2, 6, 16 W |
| 43 | Steencken 2011 | at least 3/ at least 2 | F/ Long-Evans/ N.A. | Contusion/ transection/ T9-T10 | 25 mm | FG tracing | 2, 4, 8, 16 W |
| 44 | Threriault 1994 | 5 /7 | F/ Wistar/ 280-350 g | Clip compression/ C8-T1 | 53 g*1 min | Tract tracing by HRRP & FG | 4, 8 W |
| 45 | Wang 2000 | at least 4/ at least 3 | F/ Wistar/ 90-105 | Rubrospinal tractotomy/ C2 | N.A. | Anterogade labeling by Dextran, Retrogade Labeling by FB | 8 W |
| 46 | Wang 2002 | 3/ 9 | F/ Wistar/ 100–150 | Unilateral tractotomy/ C2 or T10 | N.A. | Retrogade Labeling by FB, Intracellular Dye Injection of LY | 4, 8 W |
| 47 | Wang 2013 | 10/ 10 | M/ Sprague-Dawley/ N.A. | Contusion/ T10 | 500 g.mm | TUNEL, IHRC, | 12 hr |
| 48 | Yick 1998 | 4/ 4 | F/ Sprague–Dawley/ 200-250 | HRemisection/ C3, T1, or T11 | N.A. | IHRC | 3, 5, 10, 20, 30, 40, 50, 60 D |
| 49 | Yong 1998 | N.A. | F/ Sprague-Dawley/ 300-350 | NYU Impactor/ T9-T10 | 500 g.mm | HR&E, TUNEL, IHRC, TEM | 1, 3, 7, 14, 28 D |
The number of animals in each injury group (i.e., different time or severity of injury) and control/sham group
Abbreviations: N.A.: not applicable; min: minutes; Sce: seconds; hr: hours; D: days; W: weeks; M: months, IHC: immunohistochemistry; TEM: transmission electron microscopy; TB: True Blue; LY: Lucifer yellow; HRP: horseradish peroxidase; FG: Fluoro-Gold; BDA: Biotinylated dextran amines; CTB: Cholera toxin B, MRI: magnetic resonance imaging; WGA-HRP: wheat germ agglutinin-conjugated horseradish peroxidase
The quality assessment of included studies is presented in Table S2. All included studies used the appropriate method to study the objective. Severity and level of injury were defined in all studies. Age/weight of the animal, number of animals per group, type of strain, and genetic background were not stated in 6 (12.2%), 5 (10.2%), 2 (4.1%), and 38 (77.5%) studies, respectively. There was no statement about control group in 5 studies (10.2%). The methods used for statistical analysis and allocation to treatment groups were not stated in 6 (12.2%) and 45 (89.8%) studies, respectively. There was no statement about the compliance with regulations and ethical committees, bladder expression of injured animals, blindness of assessors, and exclusion of animals during the study in 29 (59.2%), 22 (44.9%), 38 (77.5%), and 44 (89.8%) studies, respectively. Also, in one study (2%) two different values were reported for the total number of animals used and considered as the source of risk of bias.
It should be noted that we did not receive any explanation about the reason of this discrepancy when we contacted the authors of this article.
Methods used to assess the neuronal cell loss following injury were light microscopy using the cresyl violet staining method (14, 40, 50), hematoxylin and eosin (H&E) staining method (17, 19, 20, 25, 36, 41, 42, 55), transmission electron microscopy (TEM) (17, 19, 29, 36, 55), retrograde labeling by Cholera toxin B (CTB) (11), Fluoro-Gold (FG) (5, 16, 26, 28, 32, 44, 47-49, 52, 53), horseradish peroxidase (HRP) (13-15, 40, 41, 43), Fast blue (FB) (4, 23, 33, 54), True Blue (TB) (58) and Biotinylated dextran amines (BDA) (7, 58), anterograde labeling by dextran (33), immunohistochemistry (IHC) (19, 20, 25, 28, 31, 35, 45, 46, 55-57), Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) (24, 25, 30, 34, 44, 46, 55, 57), quantitative rearl-time polymerase chain reaction (qRT-PCR) (34), and microarray (51).
Apoptotic and necrotic neuronal death at the epicenter contusion injury model
An immediate consequence of SCI, as early as five minutes (min) PI, was the disappearance of Nissl bodies, an indication of neural degeneration. However, up to 30 min, no apoptotic cell death was detected at the injury site (24, 25). As the first evidence, immunoreactivity of DFF40/CAD (DNA Fragmentation Factor 40/Caspase-activated DNase) at the injury site was detected in neurons one hour PI, as one of the nucleases primarily responsible for genomic DNA fragmentation during apoptosis (31).
Furthermore, the occurrence of apoptosis was reported 3 hr PI in the gray matter (GM) of the lesion epicenter by detection of the cytosolic presence of cytochrome c released from neuronal mitochondria (37). Also, TUNEL staining of the sections from the ventral horn revealed the presence of apoptotic neurons at 4 hr PI (24). The number of apoptotic neurons was maximized 8 hr PI and subsequently decreased (24). By 24 hr apoptotic neurons were no longer detectable in the lesion area (defined by the breakdown of axons and myelin as well as the invasion of blood cells to the white matter) (24). Similarly, following a severe contusion injury, the highest quantity of apoptotic cell staining, which included both neuronal and glial cells, was observed 4-8 hr PI at the epicenter (25). Assessments at 24 and 72 hr after the injury showed no discernable apoptotic activity in the immediate zone of the SCI. In another study, the time-dependent response of both neurons and glial cells to a severe injury was evaluated during 1 to 14 days PI. One day PI the normal structure of most neurons was destroyed and only a few spared neurons were observed in the peripheral GM and about 13% of these neurons had apoptotic characteristics (55). The maximum apoptosis in neurons was detected 3 days PI and gradually decreased afterward (55). It is worth noting that in this study the amount of apoptosis was measured within the limit of the necrotic region, determined approximately 7-8 mm rostral and caudal to the epicenter. At the lesion center, the neurons were completely wiped out, while significantly more neurons remained intact at 4 mm and longer distances (55).
Coexistence ultrastructural changes owing to apoptotic and necrotic cell death was also detected inside the lesion area within 4 to 24 hr PI (24, 55).
At 12 hr PI, TUNEL-positive neurons were still detectable in the GM of the injury site (34). Also, expression of C/EBP homologous protein (CHOP), a prominent marker of the endoplasmic reticulum stress-induced apoptosis, was detected by IHC and qRT-PCR. The number of CHOP-positive neurons was significantly increased in the injury site (34).
NeuN (neuron-specific nuclear protein) immunostaining showed that 2 hr PI the ratio of neurons at the lesion epicenter was 79% of the sham-operated animals (558±41 vs. 707±35, respectively) (38). Subsequently, at 24 hr, neurons had almost completely disappeared (38). Also, it has been shown that only neurons at the very peripheral region of the dorsal horns looked normal as long as 24 hr PI (29). There is another evidence indicating the complete disappearance of NeuN-positive neurons at the epicenter from 2 weeks PI (22).
Compression injury model
Based on optical microscopic results at one hour PI, neurons were hyperchromatic, irregular, and shrunken (a marker of apoptosis). In addition, cytoplasmic microvacuolation (a marker of necrosis) was observed in some neurons located in the ventral horn at the epicenter (21).
Assessment of neuroprotective activities through estimating the expression of Bcl-2, an endogenous inhibitor of apoptosis, showed no sign of Bcl-2 expression in neurons of GM one day after the moderate injury. However, very intense immunoreactivity for Bcl-2 was observed in the axons of the dorsal, ventral and lateral tracts, but not in the CST (57). Four days PI the staining intensity and the number of Bcl-2 positive axons were decreased. At day 9, Bcl-2 immunoreactivity was no longer evident in the injured axons (57). In the animals with mild compression, the Bcl-2 positive axons were confined to the injured T8-T9 segment. In the animals with moderate and severe compression, Bcl-2 immunoreactivity was observed in the compressed segment as well as in the caudal T10 segment. The Bcl-2 positive axons were more frequent in rats with moderate injury than those with mild injury. In rats with severe compression, the Bcl-2 positive axons were observed in the spared subpial region of the cord (57). These results reveal the anti-apoptotic activities of the supraspinal neurons at least up to 4 days PI.
After dorsal compression, the neuronal loss was greatest in the ventral horn and intermediate GM, while the lateral dorsal horn relatively remained spared. This heterogeneous regional distribution was evaluated through immunostaining of activating transcription factor-3 (ATF3), a member of the activating transcription factor that only expresses in injured neurons. The ATF3-positive neurons were abundant in the ventral horn and intermediate GM and only a few ATF3-positive neurons were detected in the dorsal horn (45). Furthermore, it has been shown that the expression of ATF3 in injured neurons has a transient pattern; ATF3 expression peaked at 3 days and diminished at 7 days PI (45). The reduction in the ATF3 expression may be a consequence of neuronal loss, since NeuN immunostaining showed that 44% of neurons were lost from the injury epicenter by one day PI. The loss of neurons increased to 73% in 3 days and this reduction continued for 1 month. Neuronal loss was also detected ±5 mm away from the epicenter but was not statistically significant until day 3 (45).
The reduction trend of neurons that had started previously continued (21) and by one month PI 81% of neurons were lost and similar to previous time points this reduction was more abundant in the ventral horn and intermediate GM while some neurons were still preserved in the lateral dorsal horn (45).
Transection injury model
One hr after transection, the area of neuronal loss was confined to the primary lesion site (56). Between 3 and 6 hr PI, within an area about 800 µm away from the transected site, severely damaged neurons appeared. Histological evaluation showed that most of the dying neurons had characteristics of ischaemic necrosis (i.e., swollen cytoplasm) (56). Subsequently, 12 hr PI, the area of cell loss was increased and areas with morphologically changed cells were not observable anymore (56). Additionally, the expression of pro-apoptotic genes was detected during the first days after transection (51).
Summary of results of apoptotic and necrotic neuronal death at the epicenter
Both necrotic and apoptotic neuronal deaths occur after TSCI, though necrosis is the prominent cell death mechanism. Necrosis is almost immediately initiated following injury and continues up to months PI. However, apoptotic neuronal death is a time-limited pathophysiological event that initiates about one hour PI at the lesion epicenter and up to 2 weeks PI extends as far as 8 mm rostral and caudal to the injury site. During this period two peaks of apoptosis incidence have been reported; first at 4-8 hr PI at the lesion epicenter and the second at 3 days PI about 7-8 mm rostral and caudal to the epicenter (Figure 2).
Figure 2.
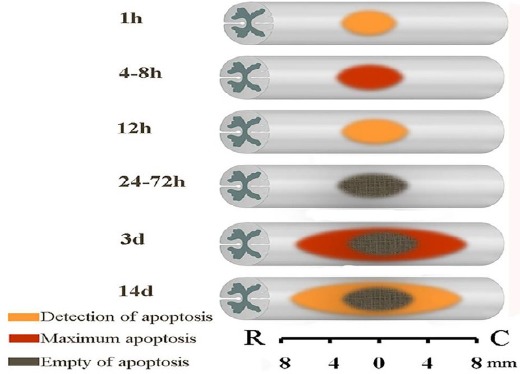
Evidence-based representation of neuronal apoptotic cell death extension after
Fate of Clarke’s column neurons after SCI
The effect of the proximity of injury site to the cell body on the death of neurons in Clarke’s nucleus, the secondary ascending spinocerebellar neurons, was assessed following hemisection at either thoracic or cervical levels (35). A severe neuronal death was detected in Clarke’s nucleus, at segment L1 following hemisection at either T11, T1, or C3. Significant cell loss was first observed 5-10 days PI when hemisection was at T11 and T1 (35). However, after cervical transection the significant decrease in the cell numbers was observed on day 20. In all transected groups the number of surviving neurons compared to the total number of Clarke’s neurons in the intact contralateral side of the spinal segment reached a minimum by 40 days PI (35).
In another study, a significant loss of large neurons of Clarke’s column after axotomy at T9 level began at 5 weeks and continued to the last time point evaluated in this study, i.e., the 15th week (42). The number of surviving neurons 15 weeks PI reduced by almost 40% compared to the age-matched controls. Indeed, examination of the somatic size of the axotomized neurons at this time point showed that larger neurons, with the somatic cross-sectional area > 100 µm2, were more vulnerable to injury than smaller neurons (42). In a separate experiment, at 25 weeks PI the number of neurons in Clarke’s column reduced by 93% relative to age-matched control animals (40)
The fate of pre-labeled Clarke’s column neurons at the L1 level was studied from 5 to 20 weeks following complete transection at T9 (49). The neurons were labeled by intra-cerebellar injection of either FG or TB 4 days before injury and 5 weeks PI, the total number of Clarke’s column neurons decreased but were not statistically significant compared to the control animals (49). The decrease in the number of labeled neurons was continued for 10 and 20 weeks PI. At these time points, the extent of surviving neurons was significantly lower than age-matched controls (49).
Summary of results on the fate of Clarke’s column neurons after SCI
Based on one study the proximity of the injury site to the cell body can affect the neuron survival. Following thoracic injury, the loss of Clarke’s column neurons at the L1 level began one week PI. However, neuron loss was statistically significant after 10 weeks PI and by 25 weeks PI a small number of neurons survived the injury.
Fate of propriospinal neurons after SCI
Contusion injury model
Propriospinal neurons, which are intrinsic neurons of the spinal cord, can be divided into two subpopulations. Short thoracic propriospinal (TPS) neurons are located in the thoracic levels and their axons project for a few segments in either rostral or caudal directions. Long projection propriospinal neurons include long ascending propriospinal tract (LAPT) neurons found in the lumbosacral enlargement that projects rostrally to the cervical enlargement, and long descending propriospinal tract (LDPT) neurons found in the cervical enlargement projecting mainly caudally to the lumbosacral enlargement. LDPT neurons are thought to be important in mediating reflex control and in coordination during locomotion. TPS neurons are postulated to be involved in regulating axial musculature and postural mechanisms. Therefore, functional recovery might be expected from protection of propriospinal neurons after injury.
The amount of damage to propriospinal neurons after different severities of injury has been reported (16, 30, 32, 52). It has been shown that propriospinal neurons have differential vulnerabilities to contusion injury. At one week PI, although apoptosis was detected in TPS neurons, no TUNEL-positive cell was observed in LDPT neurons (30, 32). Similarly, in another study apoptosis was only detected in a subset of TPS neurons at one week PI, even though at longer time points TPS neurons were TUNEL-negative (32).
However, loss of TPS neurons continued through necrosis and a significant decrease in the number of TPS neurons was detected at 2 weeks PI (32). At the same time point, an overall examination of the surviving neurons through tract tracing showed that 87% of LDPT (located in C3-C6 spinal levels) and 90% of TPS neurons (located in T5-T7 spinal levels) disappear after injury (52). The same research group also reported that about 90% of LDPT neurons and 99% of TPS neurons found in all segments rostral to and at the injury level (T9-T10), disappear after injury and there was no significant difference in the number of the neurons when comparing cell counts after mild or severe injuries (16). No significant decrease in the number of propriospinal neurons was found at the later times, indicating that the maximum cell loss in these two subpopulations of neuron occurs within the first two weeks PI (52) and this period can be a golden time for therapeutic interventions which aim to preserve spinal cord intrinsic neurons.
Caudal to the lesion, labeling of TPS neurons was intensive and comparable to the uninjured animals (16). In a separate experiment, pre-labeling of TPS neurons 4 days prior to contusion showed that while TPS neurons appeared to be severely damaged at the epicenter, their cell bodies remained intact at least for 12 days PI (16).
The total number of neurons in the GM of the entire spinal cord, from cervical to sacral including epicenter, was measured by cresyl violet staining. No difference was found in the number of neurons in the rostral regions. However, the number of neurons decreased in the caudal regions by 7 days after either moderate or severe injuries. A trend for decrease was observed in the number of surviving neurons after moderate and severe injuries (14). Contrary to differential response of the spinal cord neurons to moderate and severe contusion at the T1 level at 7 days PI, no difference was observed in the estimated total number of spinal cord neurons, from cervical to sacral at 30 days PI regardless of the injury severity, and caudal to the lesion after both moderate and severe contusion lesions the number reduced by about 57% (14).
Cell death after the injury is a major factor responsible for the loss of TPS neurons during the acute phase (by 2 weeks PI). However, cell death may not be the only reason for the decrease in the number of labeled LDPT neurons. It may be attributed to the impaired retrograde transport of tracer at long PI survival time points (32). To confirm this hypothesis, it has been shown that deficit in the retrograde transport due to chronic axotomy could be reinitiated by reaxotomy (32). In the same study, no TUNEL-positive profile was evident in LDPT neurons 2, 4 or 8 weeks PI.
Transection injury model
Expression of many pro-apoptotic genes in TPS neurons confirmed the occurrence of apoptosis following low thoracic transection in TPS neurons. These pro-apoptotic genes were Bax, Casp3, Bard, Dap, and Pycard, which were highly upregulated during the first 3 days and subsequently returned to the baseline level in the later times (51). It is worth noting that a decrease in gene expression after 3 days PI may be due to the sample selection error, in which after early cell death the sampling was from the surviving axotomized short propriospinal neurons (51).
Furthermore, after a low thoracic injury, gene expression evaluation showed immediate changes in the expression of apoptosis-related genes (Casp2 and Pycard) in LDPN. Both Casp2 and Pycard genes were downregulated. While Pycard remained downregulated up to one month, the level of Caps2 showed a gradual increase in expression, approaching the baseline by one month PI. These suggest that LDPT neurons do not represent an apoptotic response up to one month PI in contrast with TPS neurons (30). However, in the longer times up to 1 month, there was no change in the expression of pro-apoptotic genes in the TPS neurons compared to the healthy spinal cord tissue (51).
Summary of results on the fate of propriospinal neurons after SCI
There are differences in the response of intrinsic neurons of the spinal cord to the TSCI. Although TPS neurons represent an apoptotic response during the acute phase, LDPT neurons do not undergo apoptosis for at least one month PI (4 studies, 91 rats: 28 rats in the injury group and 63 rats in the control group) (16, 30, 32, 52).
Fate of ventral motor neurons after SCI
Contusion injury model
Investigation of ventral motor neurons (VMNs) following injury revealed a time-dependent symmetrical loss of VMNs at specified distances rostral and caudal to the lesion center. Fifteen min PI, tight network of cells seen in control animals had disappeared at the impact site. Quantitative assessment revealed that only 33% of the VNMs were present at the epicenter and this number increased to approximately 60% at ± 3 mm and reached to the level of the control tissue at ± 4 mm from the epicenter (19). However, neurons in most of the peripheral area of the dorsal horns generally appeared normal (29). It has been shown that increasing the severity of injury did not change the microscopic appearance of VMNs during 15 min PI (29).
Further evaluation revealed complete VMN loss at the lesion center at 4 hr PI (19). The neuron loss progressed and at 24 hr PI nearly no VMNs were detected at distances up to ± 2 mm from the epicenter (19, 20). However, at ± 4 mm from the epicenter 44% of VMNs were present and the tissue further away was morphologically normal (19, 20). Measuring the number of surviving VMNs at various distances from the lesion center up to one month PI showed no significant additional cell deaths after 24 hr (20). Using the similar injury model, the number of soleus motoneurons, located in the central and dorsolateral parts of the ventral horn, was also measured four months PI and 16% reduction was observed at the distance of 8–9 segments caudal to the epicenter (15).
A variety of pathologies were also seen at 4 and 24 hr PI at 2–4 mm distal from the epicenter; many of VMNs looked pyknotic, with vacuolization either in the nucleus or in the cytoplasm. Some neurons appeared swollen. Some nucleoli looked small and condensed; some appeared swollen and large while others were fragmented. Most of the neuronal pathologies were more consistent with a necrotic phenotype rather than apoptotic (19).
Compression injury model
One day PI very few motor neurons with shrunken morphology remained (21, 45). At 4 days PI, there were a few hyperchromatic and shrunken motor neurons at the L2 level, where the injury site was at T7-8 (21). Seven days after a mild injury, most of the VMNs disappeared. However, TUNEL staining showed no apoptotic cells in the compressed area (27).
Transection injury model
There are two opposite reports about the death of VMNs after transection (17, 47). Eidelberg et. al. showed that after transection of T8-T9 the death of VMNs occurs at the lumbar level and similar to severe compression injury prominent cell death occurs by the first day PI when the number of motoneurons reduces to about 75% (17).
The authors also confirmed the neuronal damage by TEM (17). On the other hand, McBride and Feringa showed that thoracic transection does not result in the transneuronal degeneration of VMNs at the lumbar enlargement up to 1 year PI (47). This difference can be attributed to the different methods used (Table 1) and further assessments are needed to reach a consensus.
Summary of results about the fate of ventral motor neurons after SCI
Death of VMNs had a time-dependent symmetrical pattern at specified distances rostral and caudal to the lesion center (3 studies, 142 rats: 95 rats in the injury group and 47 rats in the control group) (19, 20, 27). After a severe injury, regardless of the injury model, prominent cell death occurs by the first day PI (17, 19-21, 45).
Nevertheless, after a mild compression, at the lesion epicenter VMNs survived by 3 days PI and more cell death occurred by 7th day through necrosis mechanism (27).
Fate of descending & ascending supraspinal neurons
Contusion injury model
Traumatic injury in the spinal cord may also cause cell death in the brain. In this regard, hindlimb area in the brain sensorimotor cortex was examined for detecting apoptosis after moderate contusion at T9 level (46). The number of TUNEL-positive cells significantly increased between 12 hr and 48 hr PI compared with control animals. Double labeling showed that most of the TUNEL-positive cells were neurons (46).
On the other hand, a proportion of cortical neurons were immunoreactive for Calbindin D28K, a calcium-binding protein which has a neuroprotective effect (60). The number of Calbindin D28K-positive neurons peaked at 24 hr PI and was significantly more than control animals by 48 hr PI. These results reveal some intrinsic neuroprotective activity which may initiate to restrict apoptosis after injury.
The degree of neuronal apoptosis in the spinothalamic tract (STT), a central pain-signaling pathway, has also been evaluated (28). STT neurons were identified through retrograde transport of FG tracer and the occurrence of apoptosis in the FG-labeled neurons detected morphologically by nucleus staining with Hoechst 33342. There was a 15.2% decrease in the number of FG-labeled STT neurons at two vertebral segments rostral to the injury site 3 days PI and 9.7% of the remaining STT neurons displayed apoptotic properties. At the lesion epicenter, 65.4% neuron loss with 6.3% apoptotic neurons was detected in comparison to the sham-operated animals (28). Similarly, the percentage of STT neurons with immunoreactivity for Bcl-xL, an anti-apoptotic protein, significantly decreased 3 days after contusion compared to uninjured animals (28).
The proportion of spared neurons of a particular spinal cord tract was closely correlated to the severity of the injury as well as the location of that pathway. Nine days after contusion the number of retrogradely labeled neurons by FB tracer were counted in the following supraspinal nuclei: raphe nuclei (RaN), the lateral vestibular nucleus (LVN), the locus coeruleus (LC), the subcoeruleus alpha (SubCA), the caudal pontine reticular nucleus (PnC), the red nucleus (RN), and the hindlimb area of motor cortex (4). The number of neurons decreased in all supraspinal nuclei as the severity of injury increased. There were statistical differences in the number of surviving neurons of all nuclei between control and either moderately or severely injured groups with an exception of CST. Ninety-nine percent of CST neurons were lost following a mild injury up to 9 days PI (4).
Similarly, the effect of the severity of injury on the survival of RN neurons was evaluated (5). The number of rubrospinal neurons did correlate well with the trauma force delivered to the spinal cord, so that by increasing the injury force from 200 to 400 and 600 g.mm (by dropping a 5 g weight from various heights), the mean number of neurons 3 days PI decreased to 151±48, 43±6, and 3±7, respectively, compared to 579±78 neurons in sham-operated animals (5). These results show that the rat’s rubrospinal neurons are relatively spared at a low trauma force and may offer an opportunity to develop effective therapeutics in this situation.
In one study the survived corticospinal neurons were assessed through estimation of Wallerian degeneration and axon counts in the medullary pyramid (7). No sign of degenerating axons in the medullary pyramid was detected up to 1 year after moderate contusion injury at either thoracic T9 or cervical C5 levels; thus the authors concluded that SCI does not result in the significant death of CST neurons.
Compression injury model
One and 2 months following severe clip compression, RN neurons of both sides of midbrain were counted. One month PI the number of retrogradely labeled neurons significantly reduced to 88% (5,425 compared to 6,176 in uninjured animals) and subsequently 2 months PI 73% (4,531 compared to 6,176 in uninjured animals) of the RN neurons retrogradely labeled with FG (53). These results indicate that following severe clip compression a considerable number of neurons are functionally intact and capable of retrograde transport, at least up to 2 months PI. However, there was considerable cellular atrophy at this time point (53).
By 44 days after graded injury, the number of spared neurons was counted in the motor cortex and brain stem. Injection of HRP caudal to the injury site revealed numerous labeled neurons in the brain stem nuclei, particularly in the RN. Labeled neurons were largely confined to the layer V of the sensorimotor cortex, magnocellular red nucleus, and the raphe nuclei (39). These are the origins of main descending tracts in the thoracic spinal cord of rats that are involved in locomotor control. Some labeled neurons were also seen in the hypothalamic nuclei (39). With clip compression injuries of 18 g for 1 min or more, there were virtually no retrogradely labeled corticospinal neurons. In contrast, counts of rubrospinal, vestibulospinal, raphespinal, and reticulospinal neurons decreased in an approximately linear fashion with increased severity of injury (39).
Transection injury model
The survival rate after unilateral rubrospinal tractotomy at C2 was calculated from the number of retrogradely labeled neurons in the injured red nucleus divided by that of the corresponding control red nuclei of the opposite side. A relatively minor neuron death (~ 7%) occurred during the first 2 weeks PI (23).
The number of TUNEL-positive CST neurons maximized at one week PI and approximately 40% of CST neurons were TUNEL-positive (44). Corticospinal neurons showed an increase in the expression of activated caspase-3 and Box, markers of apoptosis, at only one week PI. Caspase-3 staining was evident in 32.1±5.8% of neurons and 66.2±0.87% of neurons were Box-positive (44).
The loss of brainstem neurons after cervical hemisection was evaluated in the RN and LVN during a period up to 8 weeks PI (58). At 4 weeks PI, no detectable decrease was observed in the number of surviving neurons in the RN compared to one week PI. However, the number of surviving neurons reduced to75% between 4 and 8 weeks PI in comparison with one week PI (58). The response of LVN neurons was different, no significant reduction in the number of surviving LVN neurons was found at 8 weeks compared to one week PI (58).
In another study, the surviving neurons after unilateral RS tractotomy was quantified from 2nd to 10th week PI, 8% reduction of the surviving neurons was observed during this period and at the end of the 10th survival week, 85% of the RS neurons survived the axotomy (23). These results show that a considerable proportion of RS neurons survive spinal axotomy at least during the first 10 weeks PI.
Similarly, the number of cortical motor neurons at 2 and 4 weeks after transection of dorsal funiculus was decreased compared with one week PI and cell loss was between 35 and 42%. However, during this period the number of TUNEL-positive corticospinal neurons was negligible (44). Comparably, at 10 weeks PI the percent of cell loss in corticospinal neurons was about 80% (43). H&E staining also revealed a significant number of dying neurons at 10 weeks PI: the number of dying neurons in injured animals was 33.8±14.3 in each coronal section compared with 3.2±1.9 in the same cortical area of control animals (18).
Assessment of magnocellular portion of RN 90–120 days after midthoracic hemisection revealed a decrease in cell numbers contralateral to the hemisection side (50). In addition, cell loss of the magnocellular portion of the RN was significantly more in the 0–3 day old neonates than in adult animals (50). The decrease in the number of RN neurons continued by 10 weeks PI and the prominent reduction was observed in the caudal portion of RN (41, 50).
In contrast, there is evidence of no reduction in the number of surviving RN neurons by 8 weeks PI (33). Similarly, McBride et al. showed that complete transection does not affect the number of retrogradely FG-labeled corticospinal and rubrospinal neurons 10 and 20 weeks PI (26). In spite of detection of no changes in the number of rubrospinal neurons, a significant shrinkage of soma size of these neurons was detected at 10 weeks PI (26, 36) and remained shrunken up to 20 weeks (26). There is also similar evidence showing the survival of neurons in layer Vb of the sensorimotor cortex by 10 weeks PI (36).
Twenty-five weeks PI, approximately 7% of corticospinal neurons survived the injury, measured through tract tracing by HRP (6). These surviving corticospinal neurons preserved up to one year PI (6). Furthermore, evaluation of RS neurons 52 weeks following complete transection showed 29% decrease in the total number of HRP-labeled neurons in RN (41).
Summary of results about the fate of supraspinal neurons after SCI
During the first week PI, neurons in the sensorimotor cortex of the brain undergo apoptosis (2 studies, 129 rats: 104 rats in the injury group and 25 rats in the control group) (44, 46).
In the majority of studies discussing the fate of supraspinal neurons, the injury model was transection (12 out of 16 studies). Only 2 studies used contusion to cause TSCI, 2 applied compression and one used both transection and contusion injury models. The proportion of spared neurons of a particular supraspinal tract is closely correlated to the severity of injury (2 studies, 37 rats: 27 rats in the injury group and 10 rats in the control group) (4, 5) as well as the location of that pathway due to heterogeneity of the spinal cord tissue (10 studies, 243 rats: 179 rats in the injury group and 34 rats in the control group) (16, 26, 5, 29, 37, 39, 58, 61-63). Corticospinal neurons are more susceptible to the contusion and compression injuries than other supraspinal tracts (4, 39).
This exceptional response can be attributed to the course of the supraspinal tract (Figure 3a), as a major proportion of CST in the rat occupy the dorsal funiculus close to the central canal and it has been shown that the mechanical loads during trauma are the highest in this anatomical region (64).
Figure 3.
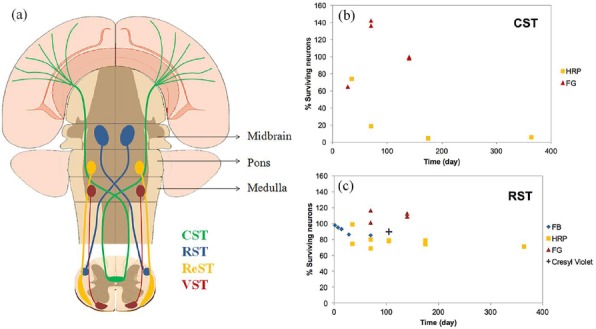
(a) Schematic representation of the course of 4 most studied descending tracts including corticospinal tract (CST), rubrospinal tract (RST), reticulospinal tract (ReST), and vestibulospinal tract (VST). The percent of surviving neurons of (b) corticospinal tract (CST) and (c) rubrospinal tract (RST) following transection injury. The results are categorized based on the evaluation methods including tract tracing by FG, HRP, FB () and cresyl violet staining (+)
Following transection, some studies revealed a significant degree of corticospinal neuron death by 2 hr PI (3 studies, 153 rats: 123 rats in the injury group and 30 rats in the control group) (6, 43, 44).
However, some studies showed no neuronal death in the sensorimotor cortex by 2 hr PI (3 studies, 71 rats: 35 rats in the injury group and 36 rats in the control group) (26, 36, 48) and chronic phase of injury (> 6 months PI) (1 study, 52 rats: 42 rats in the injury group and 10 rats in the control group) (7). Quantitative results of these studies are summarized in Figure 3b. As depicted in this figure, the aforementioned controversy can be attributed to different evaluation methods used in these studies. Further studies are required for a detailed understanding of the fate of CST neurons following transection injury.
Regardless of the model and level of injury, a proportion of RST neurons can survive the injury at least up to one year PI (Figure 3c) (9 studies, 190 rats: 167 rats in the injury group and 123 rats in the control group) (23, 26, 33, 39, 41, 48, 50, 53, 58).
Similar to CST, the extent of neuronal loss in the RST depends on the severity of the injury. In addition, cell loss of the magnocellular portion of the RN was significantly more in the 0–3 day old neonates compared with adult animals (50).
Based on one study, 11 rats in the injury group with no control animal, LVN neurons can survive the injury better than RN neurons, as depicted no significant decrease in the number of surviving neurons in LVN was detected by 8 weeks PI (58).
Discussion
The present study was designed to comprehensively review evidence from animal studies discussing the fate of neurons after TSCI in rats. Despite abundant studies evaluating the TSCI pathophysiology, there is a controversy about the neuronal death following mechanical insult; some evidence suggests the immediate and immense neuronal death (4-5), while there is other evidence for the preservation of spared neurons up to one year PI (6-7). In the current study, we found that different populations of neurons differently survive the TSCI, and neuronal loss occurs through both mechanisms of apoptosis and necrosis.
Although the intrinsic and extrinsic factors contributing to the apoptotic and necrotic cell death have been extensively reviewed (65-68), we found no report discussing the mechanism of neuronal death after TSCI. Also, previous reviews have not evaluated the rostro-caudal extension of apoptosis after injury. In addition, the focus of the studies reviewing the fate of neurons after TSCI was limited to the apoptotic/necrotic neuronal death, while as revealed in this systematic review, a considerable proportion of supraspinal neurons can survive the injury at least up to one year PI. This population of neurons which remain in a non-regenerative state can be a worthwhile therapeutic target for scientists working on nerve tissue regeneration.
Based on the articles included in our study necrosis is almost immediately initiated following injury and continues up to months PI. However, apoptotic neuronal death is a time-limited pathophysiological event which initiates about one hour PI at the lesion epicenter and up to 2 weeks PI extends as far as 8 mm rostral and caudal to the injury site. Accordingly, if the aim of a therapy is reduction of the apoptotic neuronal death, the first 2 weeks PI would be a golden time for this kind of therapeutic interventions.
SCI can also result in the apoptotic neuronal death of the supraspinal neurons. Accordingly, cell transplantation and/or neurotrophic factor delivery to the brain stem/brain after TSCI may be a promising intervention to preserve injured neurons and improve locomotor recovery.
Various factors are found in this systematic review which affect the extent of neuronal death after TSCI including the type of injury model, severity of injury, the proximity of the injury site to the cell body, and the anatomical location of the supraspinal tracts. It was found that neuronal death after compression and contusion injuries commences with a slight delay when compared to transection injury (5 studies, 155 rats: 122 rats in the injury group and 33 rats in the control group) (14, 22, 38, 45, 56).
Limitations
Identification of the mode of cell loss as apoptotic rather than necrotic relies on several morphological and biochemical criteria. Condensation of chromatin in the nucleus, nuclear shrinkage, preservation of membrane, and organelle structure are morphological hallmarks of apoptosis. Furthermore, DNA fragmentation can occur at a late stage of apoptosis. DNA fragmentation during apoptosis has been widely identified through the TUNEL technique, while internucleosomal DNA fragmentation may occur during necrosis, so necrotic neurons can be TUNEL-positive, as well (69).
Furthermore, in vivo evaluation of cells undergoing apoptosis is intricate, as the phagocytosis of apoptotic cells and their fragments (apoptotic bodies) by activated microglia/macrophages, with subsequent lysosomal degradation, occurs almost immediately after their formation. This rapid uptake and elimination of apoptotic cells can result in underestimation of the extent of apoptotic cell death, especially using TUNEL assay, since the free DNA strand ends are fabricated at a late stage of apoptosis (70). These problems associated with in vivo measurements make the interpretation of apoptosis extensively difficult.
The other problem with detection of apoptosis using the TUNEL is that the preservation methods to prepare the tissue sections can affect the detection of free DNA strand ends (71).
The morphological appearance of cells undergoing apoptosis is both distinct and specific. Thus, the ultrastructural observations to assess the characteristics change in nuclear morphology is the most accurate indicator of apoptosis and is still the gold standard for identification of apoptosis. Meanwhile, application of more than one method, each based on a different principal (i.e. detecting a different cellular feature of apoptosis), offers a better chance of detecting apoptosis than does any single method.
In the current review among the articles reporting the occurrence of apoptosis after TSCI (i.e. 14 articles) (21, 24, 25, 27, 28, 30-32, 34, 37, 44, 46, 51, 55), TUNEL assay was the only method used for identification of apoptosis in 5 articles (24, 25, 27, 32, 46), TUNEL was in combination with other tests (i.e. either TEM or gene expression) in 5 articles (21, 30, 34, 44, 55) and in the rest of the articles one test other than TUNEL was used for identification of apoptosis (28, 31, 37, 51).
Conclusions and future perspectives
As the secondary injury is a progressive phenomenon, in order to develop new therapies to preserve the spared neurons, selection of the injury model and time of intervention has a crucial role in the efficacy of therapy. In addition, examining the safety and efficacy of the intervention by reliable methods not confounded by the injury-related changes would promote translation of therapies to clinical application. To reach this goal further studies are needed to detail understanding of TSCI pathophysiology in rats.
Acknowledgment
This study was supported by Sina Trauma and Surgery Research Center, Tehran University of Medical Sciences, Tehran, Iran (No. 93-02-38-25620).
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- 1.Saadat S, Javadi M, Divshali BS, Tavakoli AH, Ghodsi SM, Montazeri A, et al. Health-related quality of life among individuals with long-standing spinal cord injury: a comparative study of veterans and non-veterans. BMC Public Health. 2010;10:1–7. doi: 10.1186/1471-2458-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eslami V, Saadat S, Habibi Arejan R, Vaccaro AR, Ghodsi SM, Rahimi-Movaghar V. Health-related quality of life among individuals with long-standing spinal cord injury. Spinal Cord. 2012;50:899–903. doi: 10.1038/sc.2012.75. [DOI] [PubMed] [Google Scholar]
- 3.Rahimi-Movaghar V, Moradi-Lakeh M, Rasouli MR, Vaccaro AR. Burden of spinal cord injury in Tehran, Iran. Spinal Cord. 2010;48:492–497. doi: 10.1038/sc.2009.158. [DOI] [PubMed] [Google Scholar]
- 4.Kim ES, Kim GM, Lu X, Hsu CY, Xu XM. Neural circuitry of the adult rat central nervous system after spinal cord injury: a study using fast blue and the Bartha strain of pseudorabies virus. J Neurotrauma. 2002;19:787–800. doi: 10.1089/08977150260139156. [DOI] [PubMed] [Google Scholar]
- 5.Naso WB, Cox RD, McBryde JP, Perot PL., Jr Rubrospinal neurons and retrograde transport of fluoro-gold in acute spinal cord injury--a dose-response curve. Neurosci Lett. 1993;155:125–127. doi: 10.1016/0304-3940(93)90688-h. [DOI] [PubMed] [Google Scholar]
- 6.Feringa ER, Vahlsing HL. Labeled corticospinal neurons one year after spinal cord transection. Neurosci lett. 1985;58:283–286. doi: 10.1016/0304-3940(85)90067-9. [DOI] [PubMed] [Google Scholar]
- 7.Nielson JL, Sears-Kraxberger I, Strong MK, Wong JK, Willenberg R, Steward O. Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. J Neurosci. 2010;30:11516–11528. doi: 10.1523/JNEUROSCI.1433-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beattie MS, Farooqui AA, Bresnahan JC. Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma. 2000;17:915–925. doi: 10.1089/neu.2000.17.915. [DOI] [PubMed] [Google Scholar]
- 9.Chu D, Qiu J, Grafe M, Fabian R, Kent TA, Rassin D, et al. Delayed cell death signaling in traumatized central nervous system: hypoxia. Neurochem Res. 2002;27:97–106. doi: 10.1023/a:1014858707218. [DOI] [PubMed] [Google Scholar]
- 10.Cheriyan T, Ryan DJ, Weinreb JH, Cheriyan J, Paul JC, Lafage V, et al. Spinal cord injury models: a review. Spinal Cord. 2014;52:588–595. doi: 10.1038/sc.2014.91. [DOI] [PubMed] [Google Scholar]
- 11.Sharif-Alhoseini M, Khormali M, Rezaei M, Safdarian M, Hajighadery A, Khalatbari MM, et al. Animal models of spinal cord injury: a systematic review. Spinal Cord. 2017;55:714–721. doi: 10.1038/sc.2016.187. [DOI] [PubMed] [Google Scholar]
- 12.Metz GA, Curt A, van de Meent H, Klusman I, Schwab ME, Dietz V. Validation of the weight-drop contusion model in rats: a comparative study of human spinal cord injury. J Neurotrauma. 2000;17:1–17. doi: 10.1089/neu.2000.17.1. [DOI] [PubMed] [Google Scholar]
- 13.Hassannejad Z, Sharif-Alhoseini M, Shakouri-Motlagh A, Vahedi F, Zadegan SA, Mokhatab M, et al. Potential variables affecting the quality of animal studies regarding pathophysiology of traumatic spinal cord injuries. Spinal Cord. 2016;54:579–583. doi: 10.1038/sc.2015.215. [DOI] [PubMed] [Google Scholar]
- 14.Andrade M, Hanania F, Daci K, Leme R, Chadi G. Contuse lesion of the rat spinal cord of moderate intensity leads to a higher time-dependent secondary neurodegeneration than severe one: An open-window for experimental neuroprotective interventions. Tissue Cell. 2008;40:143–156. doi: 10.1016/j.tice.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Bose P, Parmer R, Reier PJ, Thompson FJ. Morphological changes of the soleus motoneuron pool in chronic midthoracic contused rats. Exp Neurol. 2005;191:13–23. doi: 10.1016/j.expneurol.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 16.Conta AC, Stelzner DJ. Differential vulnerability of propriospinal tract neurons to spinal cord contusion injury. J Comp Neurol. 2004;479:347–359. doi: 10.1002/cne.20319. [DOI] [PubMed] [Google Scholar]
- 17.Eidelberg E, Nguyen LH, Polich R, Walden JG. Transsynaptic degeneration of motoneurones caudal to spinal cord lesions. Brain Res Bull. 1989;22:39–45. doi: 10.1016/0361-9230(89)90125-1. [DOI] [PubMed] [Google Scholar]
- 18.Feringa ER, Gilbertie WJ, Vahlsing HL. Histologic evidence for death of cortical neurons after spinal cord transection. Neurology. 1984;34:1002–1006. doi: 10.1212/wnl.34.8.1002. [DOI] [PubMed] [Google Scholar]
- 19.Grossman S, Rosenberg L, Wrathall J. Temporal–spatial pattern of acute neuronal and glial loss after spinal cord contusion. Exp Neurol. 2001;168:273–282. doi: 10.1006/exnr.2001.7628. [DOI] [PubMed] [Google Scholar]
- 20.Grossman SD, Wolfe BB, Yasuda RP, Wrathall JR. Changes in NMDA receptor subunit expression in response to contusive spinal cord injury. J Neurochem. 2000;75:174–184. doi: 10.1046/j.1471-4159.2000.0750174.x. [DOI] [PubMed] [Google Scholar]
- 21.Holtz A, Nyström B, Gerdin B, Olsson Y. Neuropathological changes and neurological function after spinal cord compression in the rat. J Neurotrauma. 1990;7:155–167. doi: 10.1089/neu.1990.7.155. [DOI] [PubMed] [Google Scholar]
- 22.James ND, Bartus K, Grist J, Bennett DL, McMahon SB, Bradbury EJ. Conduction failure following spinal cord injury: functional and anatomical changes from acute to chronic stages. J Neurosci. 2011;31:18543–18555. doi: 10.1523/JNEUROSCI.4306-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu PH, Wang YJ, Tseng GF. Close axonal injury of rubrospinal neurons induced transient perineuronal astrocytic and microglial reaction that coincided with their massive degeneration. Exp Neurol. 2003;179:111–126. doi: 10.1006/exnr.2002.8057. [DOI] [PubMed] [Google Scholar]
- 24.Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, et al. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lou J, Lenke LG, Ludwig FJ, O'Brien MF. Apoptosis as a mechanism of neuronal cell death following acute experimental spinal cord injury. Spinal Cord. 1998;36:683–690. doi: 10.1038/sj.sc.3100632. [DOI] [PubMed] [Google Scholar]
- 26.McBride RL, Feringa ER, Garver MK, Williams JJ. Retrograde transport of fluoro-gold in corticospinal and rubrospinal neurons 10 and 20 weeks after T-9 spinal cord transection. Exp Neurol. 1990;108:83–85. doi: 10.1016/0014-4886(90)90011-g. [DOI] [PubMed] [Google Scholar]
- 27.Morino T, Ogata T, Horiuchi H, Takeba J, Okumura H, Miyazaki T, et al. Delayed neuronal damage related to microglia proliferation after mild spinal cord compression injury. Neurosci Res. 2003;46:309–318. doi: 10.1016/s0168-0102(03)00095-6. [DOI] [PubMed] [Google Scholar]
- 28.Qiu J, Nesic O, Ye Z, Rea H, Westlund KN, Xu GY, et al. Bcl-xL expression after contusion to the rat spinal cord. J Neurotrauma. 2001;18:1267–1278. doi: 10.1089/089771501317095304. [DOI] [PubMed] [Google Scholar]
- 29.Rosenberg LJ, Wrathall JR. Quantitative analysis of acute axonal pathology in experimental spinal cord contusion. J Neurotrauma. 1997;14:823–838. doi: 10.1089/neu.1997.14.823. [DOI] [PubMed] [Google Scholar]
- 30.Siebert JR, Middleton FA, Stelzner DJ. Long descending cervical propriospinal neurons differ from thoracic propriospinal neurons in response to low thoracic spinal injury. BMC Neurosci. 2010;11:148–165. doi: 10.1186/1471-2202-11-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Springer JE, Azbill RD, Knapp PE. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med. 1999;5:943–946. doi: 10.1038/11387. [DOI] [PubMed] [Google Scholar]
- 32.Steencken AC, Smirnov I, Stelzner DJ. Cell survival or cell death: differential vulnerability of long descending and thoracic propriospinal neurons to low thoracic axotomy in the adult rat. Neuroscience. 2011;194:359–371. doi: 10.1016/j.neuroscience.2011.05.052. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y-J, HO H-WT, Seng G-F. Fate of the supraspinal collaterals of cord-projection neurons following upper spinal axonal injury. J Neurotrauma. 2000;17:231–241. doi: 10.1089/neu.2000.17.231. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Zhang C, Hong Z, Chen H, Chen W, Chen G. C/EBP homologous protein (CHOP) mediates neuronal apoptosis in rats with spinal cord injury. Exp Ther Med. 2013;5:107–111. doi: 10.3892/etm.2012.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yick LW, Wu W, So KF, Wong S-Y. Time course of NOS expression and neuronal death in Clarke's nucleus following traumatic injury in adult rat spinal cord. Neurosci Lett. 1998;241:156–158. doi: 10.1016/s0304-3940(98)00020-2. [DOI] [PubMed] [Google Scholar]
- 36.Barron KD, Dentinger MP, Popp AJ, Mankes R. Neurons of layer Vb of rat sensorimotor cortex atrophy but do not die after thoracic cord transection. J Neuropathol Exp Neurol. 1988;47:62–74. doi: 10.1097/00005072-198801000-00008. [DOI] [PubMed] [Google Scholar]
- 37.Choo AM, Liu J, Dvorak M, Tetzlaff W, Oxland TR. Secondary pathology following contusion, dislocation, and distraction spinal cord injuries. Exp Neurol. 2008;212:490–506. doi: 10.1016/j.expneurol.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 38.Ek CJ, Habgood MD, Callaway JK, Dennis R, Dziegielewska KM, Johansson PA, et al. Spatio-temporal progression of grey and white matter damage following contusion injury in rat spinal cord. PLoS One. 2010;5:12021–12036. doi: 10.1371/journal.pone.0012021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fehlings MG, Tator CH. The relationships among the severity of spinal cord injury, residual neurological function, axon counts, and counts of retrogradely labeled neurons after experimental spinal cord injury. Exp Neurol. 1995;132:220–228. doi: 10.1016/0014-4886(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 40.Feringa ER, Lee GW, Vahlsing HL. Cell death in Clarke's column after spinal cord transection. J Neuropathol Exp Neurol. 1985;44:156–164. doi: 10.1097/00005072-198503000-00004. [DOI] [PubMed] [Google Scholar]
- 41.Feringa ER, McBride RL, Pruitt JN. Loss of neurons in the red nucleus after spinal cord transection. Exp Neurol. 1988;100:112–120. doi: 10.1016/0014-4886(88)90205-1. [DOI] [PubMed] [Google Scholar]
- 42.Feringa ER, Pruitt JN, 2nd, McBride RL, Vahlsing HL. Changes in number and size of Clarke's column neurons after cord transection. J Neuropathol Exp Neurol. 1987;46:695–702. doi: 10.1097/00005072-198711000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Feringa ER, Vahlsing HL, Smith BE. Retrograde transport in corticospinal neurons after spinal cord transection. Neurology. 1983;33:478–482. doi: 10.1212/wnl.33.4.478. [DOI] [PubMed] [Google Scholar]
- 44.Hains BC, Black JA, Waxman SG. Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. J Comp Neurol. 2003;462:328–341. doi: 10.1002/cne.10733. [DOI] [PubMed] [Google Scholar]
- 45.Huang WL, George KJ, Ibba V, Liu MC, Averill S, Quartu M, et al. The characteristics of neuronal injury in a static compression model of spinal cord injury in adult rats. Eur J Neurosci. 2007;25:362–372. doi: 10.1111/j.1460-9568.2006.05284.x. [DOI] [PubMed] [Google Scholar]
- 46.Lee BH, Lee KH, Kim UJ, Yoon DH, Sohn JH, Choi SS, et al. Injury in the spinal cord may produce cell death in the brain. Brain Res. 2004;1020:37–44. doi: 10.1016/j.brainres.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 47.McBride RL, Feringa ER. Ventral horn motoneurons 10, 20 and 52 weeks after T-9 spinal cord transection. Brain Res Bull. 1992;28:57–60. doi: 10.1016/0361-9230(92)90230-u. [DOI] [PubMed] [Google Scholar]
- 48.McBride RL, Feringa ER, Garver MK, Williams JK., Jr Prelabeled red nucleus and sensorimotor cortex neurons of the rat survive 10 and 20 weeks after spinal cord transection. J Neuropathol Exp Neurol. 1989;48:568–576. doi: 10.1097/00005072-198909000-00007. [DOI] [PubMed] [Google Scholar]
- 49.McBride RL, Feringa ER, Smith BE. The fate of prelabeled Clarke's column neurons after axotomy. Exp Neurol. 1988;102:236–243. doi: 10.1016/0014-4886(88)90099-4. [DOI] [PubMed] [Google Scholar]
- 50.Prendergast J, Stelzner DJ. Changes in the magnocellular portion of the red nucleus following thoracic hemisection in the neonatal and adult rat. J Comp Neurol. 1976;166:163–171. doi: 10.1002/cne.901660204. [DOI] [PubMed] [Google Scholar]
- 51.Siebert JR, Middelton FA, Stelzner DJ. Intrinsic response of thoracic propriospinal neurons to axotomy. BMC Neurosci. 2010;11:69–92. doi: 10.1186/1471-2202-11-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steencken AC, Stelzner D. Loss of propriospinal neurons after spinal contusion injury as assessed by retrograde labeling. Neuroscience. 2010;170:971–980. doi: 10.1016/j.neuroscience.2010.06.064. [DOI] [PubMed] [Google Scholar]
- 53.Theriault E, Tator CH. Persistence of rubrospinal projections following spinal cord injury in the rat. J Comp Neurol. 1994;342:249–258. doi: 10.1002/cne.903420208. [DOI] [PubMed] [Google Scholar]
- 54.Wang YJ, Chen JR, Tseng GF. Fate of the soma and dendrites of cord-projection central neurons after proximal and distal spinal axotomy: an intracellular dye injection study. J Neurotrauma. 2002;19:1487–1502. doi: 10.1089/089771502320914714. [DOI] [PubMed] [Google Scholar]
- 55.Yong C, Arnold PM, Zoubine MN, Citron BA, Watanabe I, Berman NE, et al. Apoptosis in cellular compartments of rat spinal cord after severe contusion injury. J Neurotrauma. 1998;15:459–472. doi: 10.1089/neu.1998.15.459. [DOI] [PubMed] [Google Scholar]
- 56.Dusart I, Schwab M. Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur J Neurosci. 1994;6:712–724. doi: 10.1111/j.1460-9568.1994.tb00983.x. [DOI] [PubMed] [Google Scholar]
- 57.Li GL, Brodin G, Farooque M, Funa K, Holtz A, Wang WL, et al. Apoptosis and expression of Bcl-2 after compression trauma to rat spinal cord. J Neuropathol Exp Neurol. 1996;55:280–289. doi: 10.1097/00005072-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 58.Houle JD, Ye JH. Survival of chronically-injured neurons can be prolonged by treatment with neurotrophic factors. Neuroscience. 1999;94:929–936. doi: 10.1016/s0306-4522(99)00359-0. [DOI] [PubMed] [Google Scholar]
- 59.Wang L, Hu B, Wong WM, Lu P, Wu W, Xu XM. Glial and axonal responses in areas of Wallerian degeneration of the corticospinal and dorsal ascending tracts after spinal cord dorsal funiculotomy. Neuropathology. 2009;29:230–241. doi: 10.1111/j.1440-1789.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 60.Sun S, Li F, Gao X, Zhu Y, Chen J, Zhu X, et al. Calbindin-D28K inhibits apoptosis in dopaminergic neurons by activation of the PI3-kinase-Akt signaling pathway. Neuroscience. 2011;199:359–367. doi: 10.1016/j.neuroscience.2011.09.054. [DOI] [PubMed] [Google Scholar]
- 61.Brook GA, Plate D, Franzen R, Martin D, Moonen G, Schoenen J, et al. Spontaneous longitudinally orientated axonal regeneration is associated with the Schwann cell framework within the lesion site following spinal cord compression injury of the rat. J Neurosci Res. 1998;53:51–65. doi: 10.1002/(SICI)1097-4547(19980701)53:1<51::AID-JNR6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 62.Nashmi R, Fehlings MG. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience. 2001;104:235–251. doi: 10.1016/s0306-4522(01)00009-4. [DOI] [PubMed] [Google Scholar]
- 63.Frei E, Klusman I, Schnell L, Schwab ME. Reactions of oligodendrocytes to spinal cord injury: cell survival and myelin repair. Exp Neurol. 2000;163:373–380. doi: 10.1006/exnr.2000.7379. [DOI] [PubMed] [Google Scholar]
- 64.LaPlaca MC, Simon CM, Prado GR, Cullen DK. CNS injury biomechanics and experimental models. Prog Brain Res. 2007;161:13–26. doi: 10.1016/S0079-6123(06)61002-9. [DOI] [PubMed] [Google Scholar]
- 65.Artal-Sanz M, Tavernarakis N. Proteolytic mechanisms in necrotic cell death and neurodegeneration. FEBS letters. 2005;579:3287–3296. doi: 10.1016/j.febslet.2005.03.052. [DOI] [PubMed] [Google Scholar]
- 66.Barres BA, Barde Y. Neuronal and glial cell biology. Curr opin neurobiol. 2000;10:642–648. doi: 10.1016/s0959-4388(00)00134-3. [DOI] [PubMed] [Google Scholar]
- 67.Frade JM, Ovejero-Benito MC. Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle. 2015;14:712–720. doi: 10.1080/15384101.2015.1004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, Wang W, Li Z, Hao S, Wang B. A novel perspective on neuron study: damaging and promoting effects in different neurons induced by mechanical stress. Biomech Model Mechanobiol. 2016;15:1019–1027. doi: 10.1007/s10237-015-0743-4. [DOI] [PubMed] [Google Scholar]
- 69.Lu J, Ashwell KW, Waite P. Advances in secondary spinal cord injury: role of apoptosis. Spine (Phila Pa 1976) 2000;25:1859–1866. doi: 10.1097/00007632-200007150-00022. [DOI] [PubMed] [Google Scholar]
- 70.Allen RT, Hunter WJ, 3rd, Agrawal DK. Morphological and biochemical characterization and analysis of apoptosis. J Pharmacol Toxicol Methods. 1997;37:215–228. doi: 10.1016/s1056-8719(97)00033-6. [DOI] [PubMed] [Google Scholar]
- 71.Willingham MC. Cytochemical methods for the detection of apoptosis. J Histochem Cytochem. 1999;47:1101–1110. doi: 10.1177/002215549904700901. [DOI] [PubMed] [Google Scholar]