

Abstract
Background
Although transcription is the initial process of gene expression, posttranscriptional gene expression regulation has also played a critical role for fine‐tuning gene expression in a fast, precise, and cost‐effective manner. Although the regulation of sodium channel α‐subunit (SCN5A) mRNA expression has been studied at both transcriptional and pre‐mRNA splicing levels, the molecular mechanisms governing SCN5A mRNA expression are far from clear.
Methods and Results
Herein, we show that, as evidenced by ribonucleoprotein immunoprecipitation assay, RNA binding protein Hu antigen R/ELAV like RNA binding protein 1 (HuR/ELAVL1) and myocyte enhancer factor‐2C (MEF2C) transcription factor mRNA are associated. HuR positively regulated transcription factor MEF2C mRNA expression by protecting its mRNA from degradation. As demonstrated by both chromatin immunoprecipitation–quantitative polymerase chain reaction assay and an electrophoretic mobility shift assay, MEF2C enhanced SCN5A transcription by binding to a putative MEF2C binding site within SCN5A promoter region. Overexpression of HuR increased the expression of SCN5A mRNA, and this effect was attenuated by the presence of MEF2C small interfering RNA in cardiomyocytes.
Conclusions
In conclusion, our results suggested that HuR participates in a combined network at the DNA and RNA levels that regulates SCN5A mRNA expression. HuR upregulates MEF2C mRNA expression by protecting MEF2C mRNA from degradation, and consequently, the elevated MEF2C enhances SCN5A mRNA transcription.
Keywords: Mef2c, sodium channel α‐subunit, transcription factors, transcriptional regulation
Subject Categories: Basic Science Research, Arrhythmias, Ion Channels/Membrane Transport, Gene Expression & Regulation
Clinical Perspective
What Is New?
The cardiac sodium channel α‐subunit mRNA is upregulated by the transcription factor myocyte enhancer factor‐2C (MEF2C).
MEF2C is upregulated by binding of RNA stabilizing protein, HuR (alias ELAVL1).
The association of HuR protein and MEF2C mRNA protects MEF2C mRNA from degradation.
MEF2C and HuR participate in a combined network at the DNA and RNA levels to upregulate sodium channel α‐subunit mRNA expression.
What Are the Clinical Implications?
Because cardiac sodium channel levels are reduced in cardiomyopathy and reduced levels contribute to arrhythmic risk, increasing sodium channel α‐subunit mRNA by targeting MEF2C or HuR may be a useful strategy to reduce arrhythmic risk.
The precise regulation of gene expression is fundamental to the phenotype and normal function of cells. Although transcription is the initial process of gene expression, posttranscriptional gene expression regulation has also played a critical role in maintaining normal mRNA homeostasis. It allows the cell to fine‐tune gene expression in a fast, precise, and cost‐effective manner.1 RNA binding proteins (RBPs) participate in posttranscriptional regulation, which includes pre‐mRNA splicing, polyadenylation, mRNA stabilization, mRNA localization, and translation. The interaction of the cis‐acting RNA elements within mRNA and the RBPs determines the fate of the corresponding mRNA. More than 1000 RBPs have been identified, and many RBPs have links to heart disease and mitochondrial metabolism.2 The best‐characterized cis‐acting RNA elements commonly existing in the 3′‐untranslated region (3′‐UTR) of mRNAs are the adenylate‐uridylate‐rich elements (AREs), known to regulate mRNA stability, translation, localization, and splicing.3, 4 One of the RBPs, HuR/ELAVL1, plays a key role in the regulation of many aspects of RNA metabolism, from pre‐mRNA splicing to mRNA decay. HuR is well known for its ability to regulate mRNA stability through interacting with AREs within many mRNA 3′UTRs.5 Although the mRNA targets of HuR have been characterized in many types of cells, few studies have been conducted in cardiomyocytes.5, 6, 7
Myocyte enhancer factor‐2C (MEF2C) is a member of MEF2 family of transcription factors. In vertebrates, MEF2 members are regulators of many other core cardiac transcription factors that are required for heart development and maintaining normal cardiac function in adulthood. There are 4 MEF2 members (A–D) that play profound roles in the regulation of transcription in cardiomyocytes. Among them, MEF2C is the earliest expressed member during heart development. MEF2C cooperatively activates transcription of the Nppa gene with the basic helix‐loop‐helix (bHLH) proteins heart and neural crest derivatives expressed 1 (HAND1) and heart and neural crest derivatives expressed 2 (HAND2), which, like MEF2, are essential regulators of cardiac development.8, 9, 10 Global knockout of Mef2c is embryonic lethal because of impaired heart morphogenesis.11 In the postnatal heart, decreased expression of MEF2C has been associated with myotonic dystrophy type 1 heart disease.12
The cardiac sodium channel gene sodium channel α‐subunit (SCN5A) encodes the voltage‐gated sodium channel in cardiomyocytes, an ion channel essential for the electrical activation of the heart. The expression of SCN5A mRNA has been regulated through transcriptional and posttranscriptional pathways. Although T‐box 5 (TBX5), GATA binding protein 4 (GATA4), nuclear factor‐κB, and Snail have been shown to regulate SCN5A expression transcriptionally, abnormal splicing of SCN5A pre‐mRNA is one of the major posttranscriptional mechanisms governing SCN5A expression.13, 14, 15, 16, 17 Nevertheless, all of the mechanisms underlying mRNA expression of this critical gene are far from clear.
Although the regulation of mRNA stability for the expression of many short‐lived mRNAs, such as inflammatory factors and transcription factors, has been investigated, the regulation of MEF2C mRNA stability has not been studied. Bioinformatic prediction suggested that the 3′‐UTR of MEF2C contains multiple copies of AREs (http://nibiru.tbi.univie.ac.at/AREsite2/welcome).18 Therefore, we conducted this study to investigate whether HuR regulated the mRNA expression of MEF2C and, consequently, SCN5A.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. All supporting data are available within the article.
Cell Culture and Transfection
The human fetal cardiomyocyte cell line RL14 was purchased from ATCC (Manassas, VA). Because this cell line is publicly available and all information known about the cell line is also publically available, use of this human line was not considered to be human subjects research by the institutional review board of Lifespan or the University of Minnesota. Cells were cultured in a humidified 37°C, 5% CO2 environment in DMEM/F‐12 nutrient medium (GE Healthcare Life Sciences, Logan, UT) supplemented with 12.5% (v/v) fetal bovine serum (Sigma, St Louis, MO), streptomycin (100 μg/mL), and penicillin (100 U/mL). DNA plasmids were transfected into cultured cells using SuperFect (Qiagen, Valencia, CA), according to the manufacturer's protocol. In brief, cells were seeded in 6‐well plates the day before transfection. On the day of transfection, 2 μg of total plasmid was diluted in 150 μL of growth medium without serum or antibiotics. After mixing, 10 μL of SuperFect reagent was added and mixed well. Then, the DNA and SuperFect mixture was incubated at room temperature for 5 minutes before 600 μL of complete growth medium was added into the transfection complex. After washing the cells with PBS, the total volume of transfection complex was immediately transferred to the cells. The transfection complex medium was replaced with fresh complete medium after a 3‐hour incubation. Cells were collected for analysis 48 hours after transfection.
Plasmid Constructions
Hemagglutinin (HA)‐fusion plasmids were generated by amplifying the coding region of HuR using polymerase chain reaction (PCR) primers that included the HA sequences and subcloned into pcDNA3.1 to create an HA‐tag at its N‐terminus of HuR. SCN5A promoter region (2 kb upstream of exon 1) was amplified by PCR and was cloned into pGL3‐Basic vector at the upstream of Luciferase open reading frame to create pGL3‐SCN5A Promoter‐Luciferase vector. Doxycycline inducible MEF2C or HuR was constructed, as described.19 In brief, the AgeI‐MluI fragment containing the turboRFP tag and the 5′mir30/3′mir30 sequences of pTRIPZ (Thermo Fisher Scientific, Waltham, MA) was replaced by an AgeI‐HpaI‐XhoI‐EcoR1‐MluI polylinker sequence to create the inducible lentiviral vector, pTRIPO, under the tet‐on control system. MEF2C or HuR open reading frame fused with HA‐tag sequences at 3′ end was then cloned into pTRIPO at XhoI and EcoR1 sites using standard cloning procedure. Plasmid DNA sequence mutations were achieved using QuikChange II XL Site‐Directed Mutagenesis Kit (Foster City, CA), following the manufacturer's protocol.
Lentivirus Packaging and Transduction
Pseudotyped lentiviral particles containing HuR or MEF2C were produced by transfection of pTRIPO‐HuR or pTRIPO‐MEF2C and lentiviral packaging plasmid mixture into Lenti‐X 293 cells (Clontech, Mountain View, CA), following the manufacturer's protocol. Briefly, 6×106 Lenti‐X cells were seeded in 10‐cm plates 24 hours before transfection. Then, cells were transfected using SuperFect (Qiagen) with pTRIPO‐HuR or pTRIPO‐MEF2C (vector plasmids), along with the lentiviral packaging helper plasmids psPAX and pMD2.G. Plasmids psPAX2 (plasmid no. 12260; Addgene, Cambridge, MA) and pMD2.G (plasmid no. 12259; Addgene) were gifts of Dr Didier Trono (School of Life Sciences, École Polytechnique Fédérale de Lausanne, Switzerland). Cell culture medium was changed after 24 hours, and then lentivirus containing medium was collected over 24‐hour periods for 2 days. After collection, PEG‐it Virus Precipitation Solution (System Biosciences, Palo Alto, CA) was added, and the mixture was stored at 4°C overnight, followed by centrifugation to concentrate the lentiviral particles. Concentrated lentiviral solution was stored at −80°C.
Real‐Time PCR Quantification
Total RNA from cells was extracted using the RNeasy Mini plus Kit (Qiagen), according to the manufacturer's instruction, and 1 μg of total RNA was reverse transcribed into cDNA using SuperScript VILO Master Mix (Thermo Fisher Scientific, Waltham, MA), following the manufacturer's protocol. Quantitative real‐time reverse transcription–PCR (RT‐qPCR) was performed using gene‐specific primers (Table), Fast SYBR Green Master Mix (Thermo Fisher Scientific, Waltham, MA), and 7500 Fast Real‐Time PCR System (Applied Biosystems, Foster City, CA). The RT‐qPCR was activated with an initial denaturation step at 95°C for 20 seconds, followed by cycles of denaturation at 95°C for 3 seconds, and annealing and extension at 60°C for 30 seconds. Samples were run in triplicate and averaged. Gene expression levels were normalized to the level of β‐actin.
Table 1.
Primers for Target Genes
| Gene | Primer | Sequences |
|---|---|---|
| SCN5A | Sense | 5′‐TGGTTGTCATCCTCTCCATCGT‐3′ |
| Antisense | 5′‐ATGAGGGCAAAGAGCAGCGT‐3′ | |
| SCN5A | ChIP sense | 5′‐TCTGAGTGAGAAACAGTGGGTG‐3′ |
| ChIP antisense | 5′‐CCACCCTCTTGAGCCACATAC‐3′ | |
| SCN5A | P2kb‐S‐Mlu I | 5′‐ATAACGCGTAGGAGAGTGAAAACCCCACTTCTG‐3′ |
| P2kb‐AS‐Bgl II | 5′‐ATAAGATCTACTCGCTCACCTGCTGGTCCC‐3′ | |
| HuR | Sense | 5′‐GACCATGACCCAGAAGGACG‐3′ |
| Antisense | 5′‐CGGATAAACGCAACCCCTCT‐3′ | |
| MEF2C | Sense | 5′‐TACAACGAGCCGCATGAGAG‐3′ |
| Antisense | 5′‐GGAATCGTCCGCATCGGG‐3′ | |
| MEF2C | RIP sense | 5′‐CATGTTGCAGGTTCAACGTTATTT‐3′ |
| RIP antisense | 5′‐CCATTACCGGGTCTGTCCA‐3′ | |
| 28S RNA | Sense | 5′‐GTGACGCGCATGAATGGA‐3′ |
| Antisense | 5′‐CCCTTGGCTGTGGTTTCG‐3′ | |
| ACTB | Sense | 5′‐AAGAGCTACGAGCTGCCTGAC‐3′ |
| Antisense | 5′‐ACAGGACTCCATGCCCAGGAA‐3′ | |
| Luciferase | Sense | 5′‐ACACCCGAGGGGGATGATAA‐3′ |
| Antisense | 5′‐TCTCACACACAGTTCGCCTC‐3′ | |
| EGFP | Sense | 5′‐GGACGACGGCAACTACAAGA‐3′ |
| Antisense | 5′‐TTGTACTCCAGCTTGTGCCC‐3′ |
ACTB indicates beta‐actin; ChIP, chromatin immunoprecipitation; EGFP, enhanced green fluorescent protein; HuR, Hu antigen R; MEF2C, myocyte enhancer factor‐2C; RIP, ribonucleoprotein immunoprecipitation.
Western Blotting
For Western blotting, whole‐cell lysates were prepared using radioimmunoprecipitation assay buffer (Boston Bio Products, Ashland, MA) with Halt proteinase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA). Protein concentration was measured using a BCA Protein Assay kit (Pierce, Rockford, MA) and a Nanodrop 8000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Equal amounts (30 μg) of proteins were separated onto 4% to 10% mini‐PROTEAN TGX gels (Biorad, Hercules, CA) under reducing and denaturing conditions and transferred to polyvinylidene difluoride membranes. The membranes were blotted with a 1:200 dilution of HuR antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight. An anti‐GAPDH (1:5000 dilution; Thermo Fisher Scientific, Waltham, MA) or an anti‐tubulin (Sigma) antibody was used as a loading control for all studies. Goat anti‐rabbit or anti‐mouse horseradish peroxidase antibodies (Santa Cruz Biotechnology) were used at a dilution of 1:5000. Proteins were visualized using Clarity Western ECL Blotting Substrate (Biorad). Band intensities were detected by ChemiDoc MP Imaging System and analyzed by Image Lab software (Biorad).
Small Interfering RNA and Transfection
Silencer select validated small interfering RNA (siRNA) for HuR (catalog no. s4608) and for MEF2C (catalog no. s8653) and negative control siRNA (catalog no. 4390843) were purchased from Thermo Fisher Scientific (Waltham, MA). To inhibit HuR or MEF2C, siRNA was transfected into RL14 cells using Lipofectamine RNAiMAX (Life Technologies, Carlsbad, CA), following the manufacturer's protocol.
Ribonucleoprotein Immunoprecipitation
To test if the endogenous HuR was associated with endogenous MEF2C mRNA, immunoprecipitation of ribonucleoprotein complexes was conducted using Ribocluster Profiler ribonucleoprotein immunoprecipitation assay kit (Medical & Biological Laboratories Co, Nagoya, Japan), following the manufacturer's instruction. In brief, 20 million cells were collected per sample. Cell lysates were incubated with anti‐HuR antibody–immobilized Protein A beads with rotation for 3 hours at 4°C after preclear up. RNA in immunoprecipitation materials was isolated and was converted in cDNA using SuperScript VILO Master Mix. RT‐PCR was then performed to detect MEF2C mRNA enrichment. 28S RNA was analyzed to evaluate nonspecific binding.
Chromatin Immunoprecipitation–qPCR
Chromatin immunoprecipitation (ChIP) experiments were performed in RL14 cells using the ab‐500 ChIP kit (Abcam, Cambridge, MA), following the manufacturer's protocol. Briefly, the cells were cross‐linked in 1% formaldehyde (Sigma‐Aldrich, St Louis, MO) for 10 minutes at room temperature and cross‐linking was stopped with 0.125 mol/L of glycine. Chromatin was fragmented by sonication using Fisher Scientific 50 Sonic Dismembrator Cell Disruptor (Fisher Scientific, Pittsburgh, PA). For immunoprecipitation, a 100‐μL aliquot of sonicated chromatin was incubated at 4°C overnight with ChIP grade anti‐MEF2C antibody (Abcam, Cambridge, MA). The antibody‐chromatin complex was then incubated with protein A beads at 4°C for 1 hour. The beads were washed, and 100 μL of DNA purifying slurry was added, mixed well, and incubated at 98°C for 10 minutes. After leaving the reaction at room temperature for 20 minutes, 1 μL of proteinase K was added and incubated at 55°C for 30 minutes. After incubation at 98°C for 10 minutes, DNA purifying slurry was pelleted by centrifugation. Supernatant was collected for qPCR analysis. qPCR was conducted using gene‐specific primers (Table), Fast SYBR Green Master Mix (Thermo Fisher Scientific, Waltham, MA), and 7500 Fast Real‐Time PCR System (Applied Biosystems). The enrichment of SCN5A DNA fragment was normalized with that of the input sample. We used the percentage input value method to calculate the relative abundance of SCN5A fragments in the ChIP assay. Fold enrichment was determined by dividing the enrichment of the immunoprecipitated chromatin with MEF2C antibody by that with control IgG.
Electrophoretic Mobility Shift Assay
Nuclear extracts were isolated from RL14 cells using NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham, MA) containing Halt proteinase inhibitor cocktail, following the manufacturer's instruction. As before, protein concentration was determined with a BCA Protein Assay kit using a Nanodrop 8000c. Electrophoretic mobility shift assays (EMSAs) were performed using biotin 5′‐end–labelled oligonucleotides synthesized by Life Technologies (Carlsbad, CA), as described before with modification.20 The biotinylated complementary oligonucleotides were then annealed to generate the double‐stranded oligonucleotides as probes. The sequences of SCN5A oligonucleotides were as follows: wild‐type (WT) oligo, 5′‐TGGAAGTATATATATAAAGAGGT‐3′ (sense) and 5′‐ACCTCTTTATATATATATCTTCCA‐3′ (antisense); and mutant oligo, 5′‐TGGAAGTCTCTCTCTGGCGAGG‐3′ (sense) and 5′‐ACCTCGCCAGAGAGAGACTTCCA‐3′ (antisense). Binding reactions were performed by using nuclear extract and probe for 30 minutes on ice in a 25‐μL total volume, according to the instructions for the LightShift Chemiluminescent EMS assay kit (Thermo Fisher Scientific, Waltham, MA). Where indicated, binding reactions contained 1 μg of anti‐MEF2C antibody (Santa Cruz Biotechnology). The binding reactions were resolved onto a 5% nondenaturing polyacrylamide gel at 100 V for 2.5 hours at 4°C. Supershift EMSAs were performed by first preincubating nuclear extract with 1 μg of either normal rabbit IgG or the corresponding antibody for 30 minutes at room temperature, followed by the addition of the biotin‐labeled DNA probe. For competition experiments, unlabeled WT and mutated SCN5A oligonucleotides were added in a 200‐fold molar excess before the addition of the biotinylated probes. After electrophoresis, the DNA‐protein complex was transferred to nylon membrane. To detect DNA protein complexes, the membrane was incubated with streptavidin‐conjugated horseradish peroxidase and then visualized with chemiluminescent substrate, according to the manufacturer's instructions.
Statistical Analysis
Continuous variables are expressed as mean+SD. Mann‐Whitney test was performed for statistical analysis using IBM SPSS Statistics, version 24.0 (International Business Machines Co, Armonk, NY). P<0.05 was considered statistically significant. The Bonferroni correction was used for multiple‐comparison correction.
Results
HuR‐Regulated MEF2C mRNA Expression
MEF2C mRNA carries multiple putative ARE sites within its 3′UTR (Figure 1A), and HuR is known to regulate the fate of target mRNAs through binding to ARE sites.21 Therefore, we performed RT‐qPCR to compare MEF2C mRNA expression levels with or without HuR overexpression to understand whether HuR regulates MEF2C mRNA expression in cardiomyocytes. The results indicated that overexpression of HuR increased MEF2C mRNA levels by 47.3% compared with that in the control group (P<0.05; Figure 1B). The overexpression of HuR tagged with HA was confirmed by Western blot using anti‐HA antibody (Figure 1C). Positive regulation of MEF2C mRNA expression by HuR was further confirmed by the HuR knockdown experiments. The MEF2C mRNA decreased by 24.5% (P<0.05, Figure 1D) in cardiomyocytes transfected with HuR siRNA compared with those in the cells transfected with control scrambled siRNA. Reduced expression of HuR by siRNA against HuR was confirmed by Western blot analysis (Figure 1E).
Figure 1.
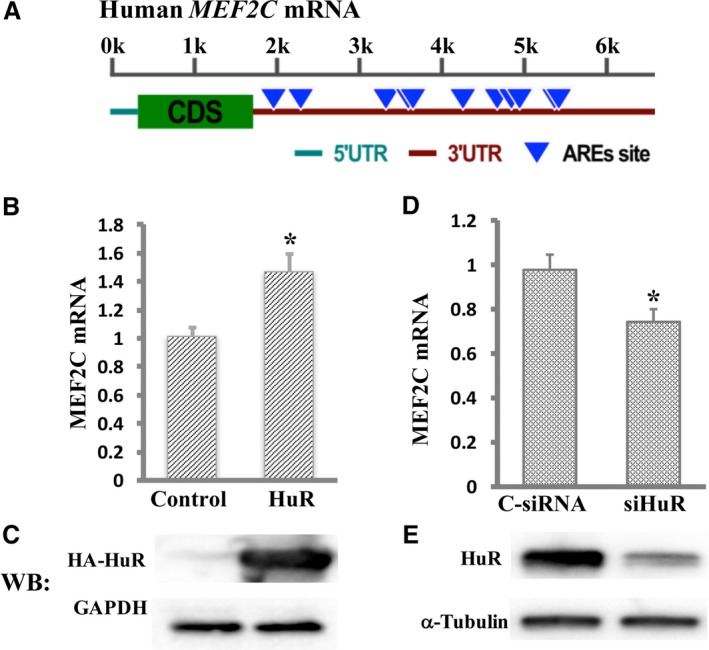
Hu Antigen R (HuR) positively regulates myocyte enhancer factor‐2C (MEF2C) mRNA expression in cardiomyocytes. A, Schematic representation of the human MEF2C adenylate‐uridylate–rich element (ARE) sites in its 3′‐untranslated region (UTR). The length of the mRNA is indicated by a kilobase (k) scale. Multiple putative ARE sites are predicted in the 3′‐UTR of MEF2C. B, The abundance of MEF2C mRNA on overexpression of HuR was measured by real‐time quantitative reverse transcription–polymerase chain reaction (RT‐qPCR). C, Western blot analysis shows overexpressed HA‐tagged HuR using anti‐HA antibody. D, Knockdown of HuR by small interfering RNA (siRNA) decreased MEF2C mRNA expression, as measured by RT‐qPCR. E, Reduced expression of HuR in cells transfected with siHuR was confirmed by immunoblot using anti‐HuR antibody. Levels of all mRNAs are normalized to β‐actin and were shown as fold changes. Error bars represent mean+SD (n=3). C‐siRNA indicates control siRNA; siHuR, HuR‐specific siRNA; CDS, Coding sequences; and WB, Western blot. *P<0.05.
To study whether the effect of HuR on MEF2C mRNA expression was mediated by the AREs within the 3′UTR of MEF2C, we cloned partial 3′UTR sequences containing the first 2 copies of putative AREs of MEF2C into pGL3‐Promoter vector downstream of the luciferase gene because these ARE sequences are conserved in multiple species (Figure 2A and 2B). Then, the plasmids were transfected into cardiomyocytes with either overexpression or knockdown of HuR. Enhanced green fluorescent protein (EGFP) plasmids were also cotransfected to determine the transfection efficiency. The abundance of luciferase mRNA was measured by RT‐qPCR and normalized with EGFP mRNA. Our data indicated that overexpression of HuR in cardiomyocytes increased the luciferase mRNA by 48.0% (P<0.05; Figure 2C), whereas knockdown of HuR decreased the luciferase mRNA by 63.3% (P<0.05; Figure 2D). Then, we deleted the first 2 copies of the AREs from the WT sequences to form a mutation construct and cotransfected these 2 plasmids into cardiomyocytes bearing TetOn‐HuR with EGFP plasmid, in the presence of HuR overexpression achieved by doxycycline induction for 48 hours. Luciferase mRNA quantification revealed that the luciferase mRNA levels were 38.0% lower in cardiomyocytes transfected with ARE‐deleted plasmids compared with those transfected with WT plasmids (P<0.05, Figure 2E). Taken together, our data suggest that the regulation of MEF2C mRNA expression by HuR is mediated by AREs within MEF2C 3′UTR.
Figure 2.
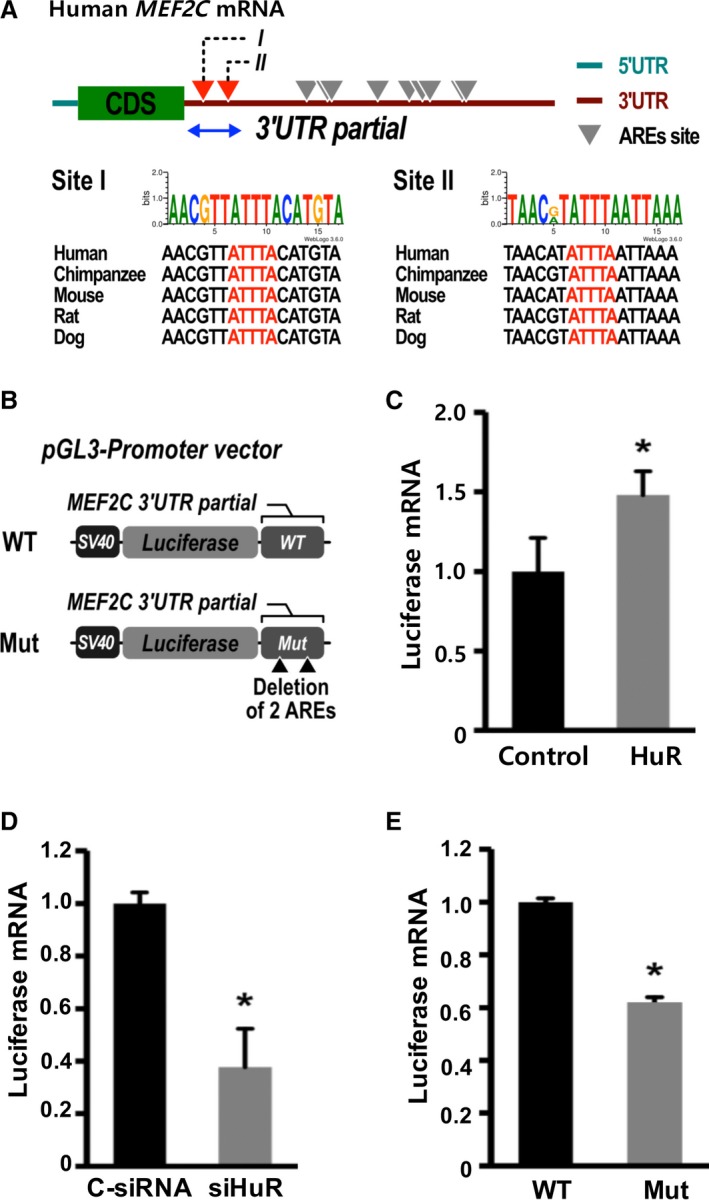
Adenylate‐uridylate–rich elements (AREs) within the 3′‐untranslated region (UTR) of myocyte enhancer factor‐2C (MEF2C) mediate the effects of Hu Antigen R (HuR) on MEF2C mRNA expression. A, Conservation of the first 2 copies of AREs (red) in the MEF2C mRNA 3′UTR among species. B, Schemes of luciferase reporter plasmids harboring partial MEF2C 3′ UTRs. Wild‐type (WT) or deletion mutants of 2 ARE sites (Mut) were generated by cloning MEF2C partial 3′UTR DNA fragments downstream of luciferase in the pGL3‐SV40 promoter vector. C, Cardiomyocytes with TetOn‐HuR were cotransfected with the WT MEF2C 3′UTR plasmid, and enhanced green fluorescent protein (EGFP) plasmids followed by overexpression of HuR. D, Cardiomyocytes were transfected with WT MEF2C 3′UTR plasmid DNA in the absence or presence of HuR small interfering RNA (siRNA). E, The cardiomyocytes with TetOn‐HuR were transfected with either WT or Mut plasmid and EGFP plasmid, followed by overexpression of HuR. Overexpression of HuR was achieved by induction with doxycycline for 48 hours. Total RNA was isolated 48 hours after transfection, and real‐time quantitative reverse transcription–polymerase chain reaction was performed to measure mRNA abundance. The luciferase mRNA levels were normalized by EGFP mRNA and were shown as fold changes. Error bars represent mean+SD (n=3). C‐siRNA indicates control siRNA; CDS, coding sequences; siHuR, HuR‐specific siRNA. *P<0.05.
HuR Was Associated With and Protected MEF2C mRNA From Decay
It is well known that HuR interacts with mRNA directly, and MEF2C contains AREs.7 Therefore, it seemed possible that HuR was associated with MEF2C mRNA. As shown in the ribonucleoprotein immunoprecipitation assay, MEF2C PCR products were highly enriched in samples incubated with anti‐HuR antibody (Figure 3A, lane 2), whereas no MEF2C PCR product was detected in control IgG pellets (Figure 3A, lane 4). There was no MEF2C PCR product detected in samples precipitated with anti‐HuR antibody or with IgG without RT (Figure 3A, lanes 3 and 5). These findings indicate that HuR specifically associates with MEF2C mRNA. Next, we investigated whether the association of HuR and MEF2C mRNA altered the fate of MEF2C mRNA. We first established a doxycycline‐inducible HuR overexpression system, as described in the Methods. After 48 hours of doxycycline induction, de novo transcription was blocked by adding actinomycin D. Cells were collected at different time points to isolate total RNA. MEF2C mRNA abundance was detected by RT‐qPCR and normalized by β‐actin mRNA. The initial MEF2C mRNA levels measured immediately before adding actinomycin D to block de novo transcription were set to 100%. The percentage of remaining MEF2C mRNA in cardiomyocytes with or without HuR overexpression was calculated by comparing the MEF2C mRNA abundance at the given time point after blocking de novo transcription to the initial MEF2C mRNA level in each group. As shown in Figure 3B, the percentages of remaining MEF2C mRNA were higher in cardiomyocytes at 30, 60, 90, 120, and 150 minutes after transcription blocking with HuR overexpression compared with that in cardiomyocytes without HuR overexpression (n=3, P<0.05). These results indicate that MEF2C mRNA degraded more slowly in cells with HuR overexpression compared with those in cells without exogenous HuR expression. Overexpression of HuR was confirmed by Western blot analysis using anti‐HuR antibody (Figure 3C).
Figure 3.
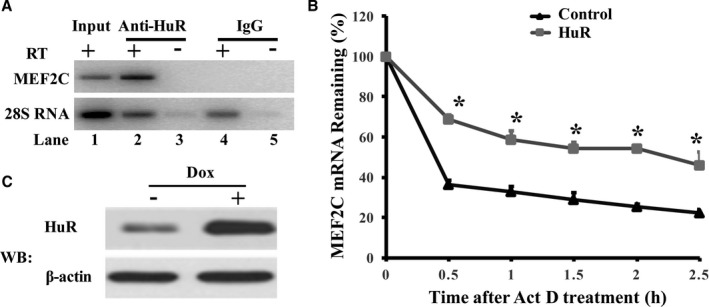
Hu Antigen R (HuR) is associated with myocyte enhancer factor‐2C (MEF2C) and protects MEF2C mRNA from decay. A, HuR was associated with MEF2C mRNA. RNA was isolated and converted into cDNA using SuperScript VILO Master Mix after immunoprecipitation of RNA‐protein complexes from cardiomyocytes using either anti‐HuR antibody (Anti‐HuR) or control IgG. The reverse transcription–polymerase chain reaction (RT‐PCR) product was separated and visualized in ethidium bromide–stained agarose gel. B, Overexpression of HuR protected MEF2C mRNA from degradation. De novo transcription of the cells was blocked by addition of actinomycin D (ActD; 5 μg/mL) after 48 hours of doxycycline (Dox) induction. Total RNA was isolated from cells harvested at different time points after ActD addition. The abundance of mRNA was determined by real‐time quantitative RT‐PCR. The graph shows the percentage of the remaining MEF2C mRNA compared with the mRNA of MEF2C measured immediately before addition of ActD. The MEF2C mRNA was normalized by β‐actin mRNA measured at the same time point. The initial mRNA levels measured immediately before adding ActD were set to 100%. The percentage of remaining MEF2C mRNA in cardiomyocytes with or without HuR overexpression was calculated by comparing the MEF2C mRNA abundance at the given time point after blocking de novo transcription to the initial MEF2C mRNA level. The percentages of remaining MEF2C mRNA after transcription blocking are shown in the graph (n=4). C, Overexpressed HuR on Dox induction was confirmed by Western blot (WB) analysis using anti‐HuR antibody. *P<0.05.
MEF2C Regulated SCN5A Expression
MEF2C is the earliest expressed MEF2 member during heart development. In vertebrates, MEF2 members are regulators of many other core cardiac genes that are required for the differentiation and maintenance of normal cellular function of cardiomyocytes.22 Bioinformatic predictions revealed an AT‐rich sequence, which might be a putative MEF2C binding site within in the SCN5A promoter region (Figure 4A), and these E‐box sequences have been shown to mediate the transcription activation of mygenic regulatory factor 4 (MRF4) by interacting with MEF2 proteins.23 We therefore tested the ability of MEF2C to regulate expression of SCN5A. We first constructed a doxycycline‐inducible MEF2C overexpression lentiviral plasmid. Cardiomyocytes were infected with lentiviral particles carrying MEF2C. The expression of SCN5A mRNA increased on doxycycline induction in a dose‐dependent manner (P<0.05, Figure 4B). Overexpressed HA‐tagged MEF2C was detected by Western blot analysis using anti‐HA antibody (Figure 4C). We next compared the mRNA levels of SCN5A in cardiomyocytes with reduced MEF2C expression. As expected, knockdown of MEF2C dramatically decreased SCN5A mRNA expression by 22.3% (P<0.05, Figure 5A). Reduced expression of MEF2C was confirmed by RT‐qPCR (Figure 5B, P<0.05).
Figure 4.
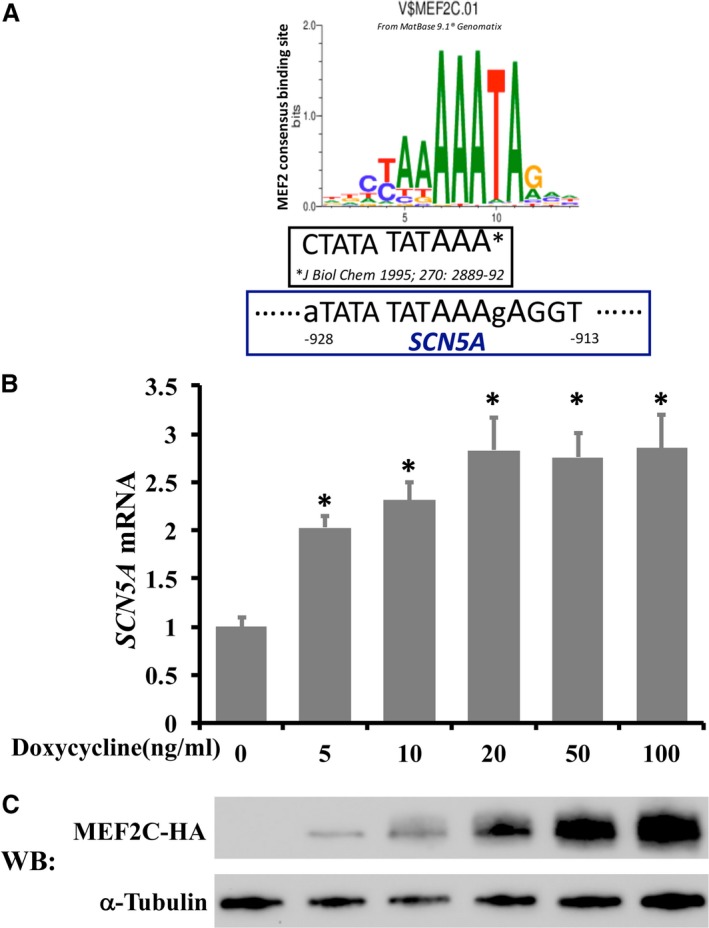
Myocyte enhancer factor‐2C (MEF2C) increases SCN5A mRNA expression in a dose‐dependent manner. A, An MEF2 consensus binding site is predicted in the SCN5A promoter region using Matbase of Genomatix. MEF2C binding sequences in MRF423 are also shown. SCN5A sequences in lowercase indicate the nucleotide at this position is different from the consensus sequences from the Matbase database. B, Cardiomyocytes were infected with lentiviral particles carrying MEF2C, and the overexpression of MEF2C was achieved by addition of doxycycline. Cells were harvested and extracted for total cellular RNA 48 hours after doxycycline induction. Changes in SCN5A mRNA levels were determined by real‐time quantitative reverse transcription–polymerase chain reaction were normalized to β‐actin mRNA, and were shown as fold changes. C, Control of exogenous MEF2C expression by addition of different amount of doxycycline is confirmed by Western blot (WB) analysis using anti‐HA antibody. Error bars represent mean+SD (n=3). *P<0.05 compared with those without doxycycline induction.
Figure 5.
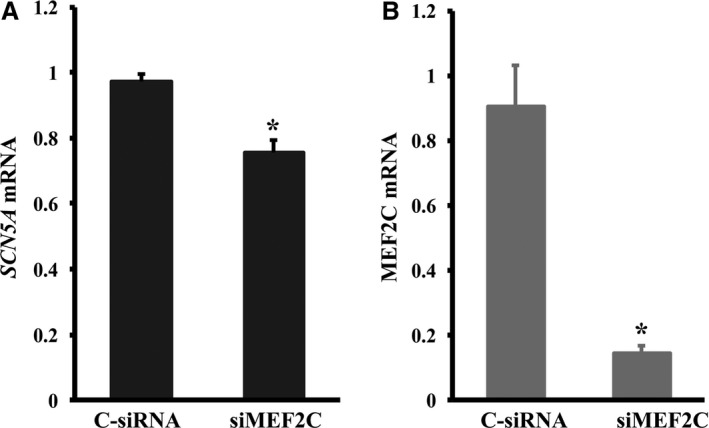
Knockdown of myocyte enhancer factor‐2C (MEF2C) decreases SCN5A mRNA expression. A, Silencing of MEF2C by small interfering RNA (siRNA) decreases SCN5A mRNA expression, as determined by real‐time quantitative reverse transcription–polymerase chain reaction (RT‐qPCR). B, Decreased MEF2C mRNA expression in cells transfected with siRNA against MEF2C by RT‐qPCR. Levels of all mRNAs are normalized to β‐actin. Error bars represent mean+SD (n=3). C‐siRNA indicates control siRNA; siMEF2C, MEF2C‐specific siRNA. *P<0.05.
MEF2C Enhances SCN5A Transcription
To determine whether MEF2C binds to SCN5A genomic DNA in live cells, we performed ChIP‐qPCR experiments in RL14 cells. Anti‐MEF2C antibody showed 3.2‐fold higher enrichment of SCN5A than control IgG (P<0.05, Figure 6A). We then tested if the association of MEF2C and the SCN5A gene regulated SCN5A transcription. A 2‐kb DNA fragment containing the SCN5A promoter region was generated from human genomic DNA (Clontech) by PCR. The fragment was cloned into a pGL3‐Basic vector at the upstream of the luciferase gene to create a pGL3 SCN5A WT promoter plasmid (Figure 6C). The plasmid was cotransfected into RL14 cardiomyocytes with the Mef2C construct or a control vector. pEGFP‐C1 plasmid (Clontech) was also included into the transfection to determine the transfection efficiency. The abundance of luciferase mRNA was measured by RT‐qPCR and normalized with EGFP mRNA. A 3.9‐fold increase of luciferase mRNA driven by the SCN5A WT promoter was observed with the Mef2C overexpression in RL14 cells compared with those in cells without Mef2C plasmid transfected (Figure 6B, P<0.05). Then, the putative MEF2C binding site within the SCN5A promoter was altered to create a mutated construct (Figure 6C) by performing site‐directed mutagenesis using the pGL3 SCN5A WT promoter plasmid as a template. The WT and mutated plasmids were then cotransfected with EGFP plasmid into cardiomyocytes with HuR overexpression. The luciferase mRNA levels were measured by RT‐qPCR and normalized by the EGFP mRNA. The results showed lower luciferase mRNA levels in cardiomyocytes transfected with mutated SCN5A promoter plasmids than in those transfected with WT SCN5A promoter plasmids (P<0.05, Figure 6D). These results suggest that MEF2C enhances the transcription of SCN5A through interacting with the MEF2C binding site within SCN5A promoter.
Figure 6.
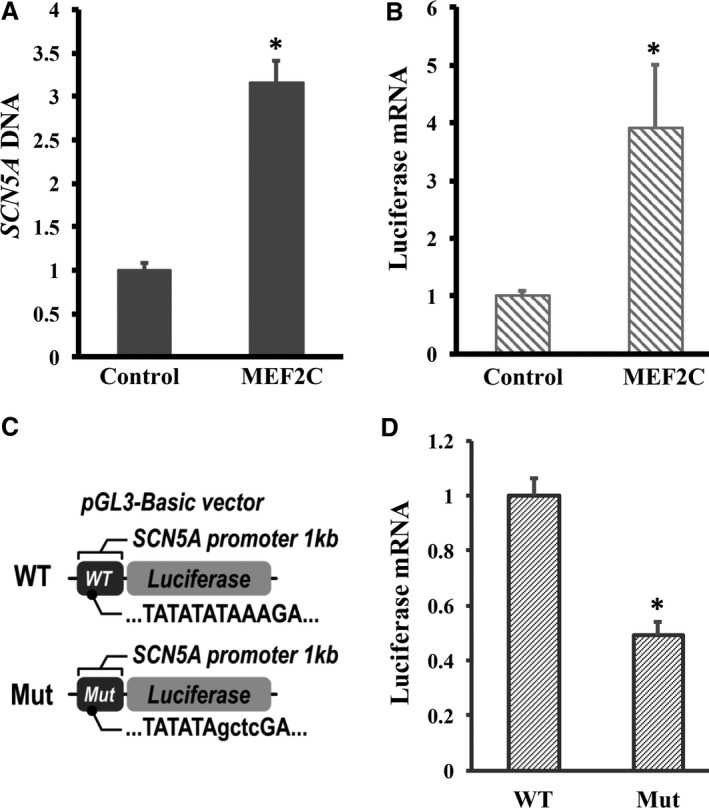
Myocyte enhancer factor‐2C (MEF2C) is associated with the SCN5A gene and enhances SCN5A promoter transcription in cardiomyocytes. A, The MEF2C protein and SCN5A DNA complex was pulled down by anti‐MEF2C antibody using ab‐500 chromatin immunoprecipitation kit. DNA fragments were purified and gene‐specific primers and real‐time quantitative PCR (qPCR) were used to determine the enrichment of SCN5A DNA in anti‐MEF2C antibody or control IgG pellets, respectively. n=6. B, SCN5A promoter (2 kb upstream of exon 1) was cloned into pGL3‐Basic vector and transfected into cardiomyocytes with or without MEF2C overexpression. pGEFP vector was cotransfected to determine the transfection efficiency. Cells were harvested and extracted for total cellular RNA 48 hours after transfection. Changes in luciferase mRNA levels were determined by reverse transcription (RT)–qPCR using enhanced green fluorescent protein (EGFP) mRNA for normalization. n=3. C, Schemes of luciferase reporter plasmids harboring wild‐type (WT) or mutated (Mut) SCN5A promoter. Mut plasmids were constructed by replacing the putative MEF2C binding sequences TAAA with sequences GCTC. D, Cardiomyocytes with MEF2C overexpression by doxycycline induction were transfected with either WT or Mut plasmids. The expression of luciferase or EGFP mRNA was measured by RT‐qPCR. Levels of luciferase mRNA were normalized by EGFP mRNA. n=3. Error bars represent mean+SD. *P<0.05.
MEF2C Bound to the SCN5A Promoter Region
To test if MEF2C bound to its putative binding site in SCN5A promoter region, EMSA was performed. A shift band was detected when the WT sequences were incubated with cardiomyocyte nuclear extract, whereas no band shift was detected in the reaction using mutated oligonucleotide (Figure 7A, lanes 2 and 7). The binding of MEF2C and SCN5A WT oligonucleotides could be competed off with unlabeled WT oligonucleotide but not with unlabeled mutant oligonucleotide (Figure 7A, lanes 4 and 5). We also performed antibody EMSAs using monoclonal antibodies that recognize MEF2C to determine whether MEF2C was one of the components of this complex. The addition of the MEF2C antibody to binding reactions partially supershifted the band (Figure 7A, lane 3), which indicated that MEF2C was one of the proteins of this protein DNA complex. The sequences of the oligonucleotides used in the experiment are shown in Figure 7B. This result further confirmed that MEF2C bound to the SCN5A gene.
Figure 7.
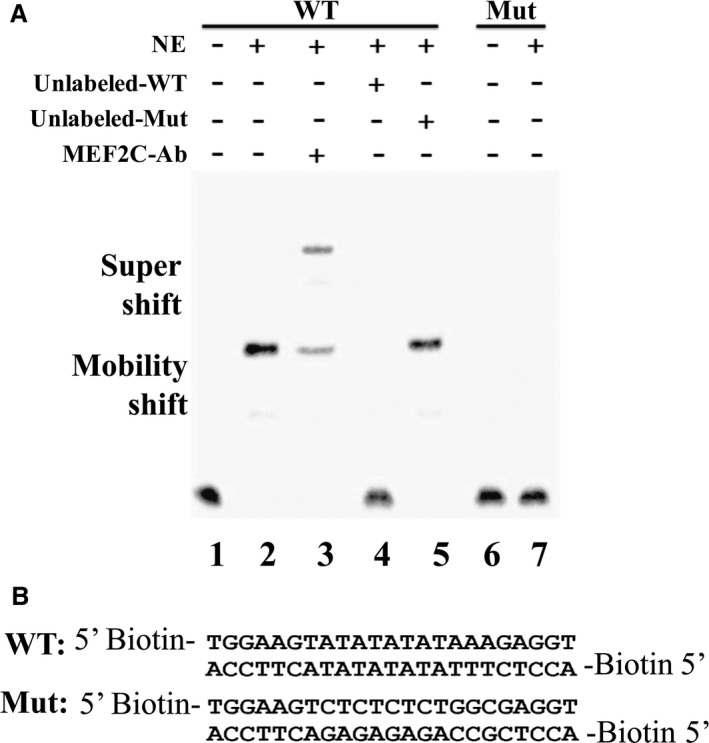
Myocyte enhancer factor‐2C (MEF2C) binds to the SCN5A promoter region. A, Electrophoretic mobility shift assay (EMSA) was performed using nuclear extracts (NEs) from cardiomyocyte and biotin‐labelled wild‐type (WT) or mutant (Mut) SCN5A oligonucleotides, using the LightShift Chemiluminescent EMSA kit. Shift of DNA oligonucleotides was detected with NEs added (lane 2), and this shift band was eliminated by excess of WT unlabeled DNA (lane 4) but not by Mut oligonucleotides (lane 5). A supershift assay with a MEF2C‐specific antibodies (Abs) revealed a corresponding shift (lane 3). No shift was detected with biotin‐labeled Mut oligonucleotides (lane 7). B, Sequences of biotin‐labeled WT and Mut oligonucleotides used for EMSA.
MEF2C Mediated the Increase of SCN5A mRNA Expression in the Presence of HuR Expression
Our data indicated that HuR positively regulated MEF2C mRNA expression and that MEF2C enhanced cardiac sodium channel α subunit SCN5A mRNA expression, so it is of great interest to know whether overexpression of HuR increases the SCN5A mRNA expression. Therefore, we investigated the expression of SCN5A mRNA with or without overexpression of HuR in cardiomyocytes. First, we overexpressed HuR in cardiomyocytes and measured SCN5A mRNA expression. As expected, the SCN5A mRNA expression levels were 41.0% higher in cardiomyocytes with HuR overexpression compared with that in cardiomyocytes without HuR overexpression (P<0.05, Figure 8A). More interesting, the increase of SCN5A mRNA expression by overexpression of HuR was attenuated by the presence of MEF2C siRNA (P<0.05, Figure 8A), whereas the expression of HuR was not affected by MEF2C siRNA (Figure 8B). Our data suggest that the increased expression of SCN5A mRNA in the presence of HuR is mediated by MEF2C in cardiomyocytes.
Figure 8.
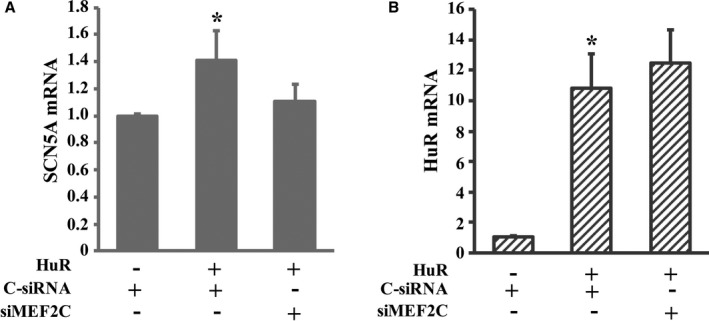
Myocyte enhancer factor‐2C (MEF2C) mediated the increase of sodium channel α‐subunit (SCN5A) mRNA expression in the presence of HuR expression. A, Knockdown of MEF2C attenuated the effect of Hu Antigen R (HuR) on the expression of SCN5A. Cardiomyocytes with overexpressed HuR were transfected with either scrambled control small interfering RNA (C‐siRNA) or MEF2C siRNA (siMEF2C). Cardiomyocytes without HuR overexpression transfected with C‐siRNA were used as a control to study the effect of HuR on the expression of SCN5A. Total RNA was isolated from the cells 48 hours after siRNA transfection. The expression of mRNA was determined by real‐time quantitative reverse transcription polymerase chain reaction and normalized by GAPDH. *P<0.05 compared with those without HuR overexpression or those with HuR overexpression and the presence of siMEF2C. B, The expression of HuR was not statistically altered in the presence of siMEF2C. n=3. Error bars represent mean+SD. *P<0.05 compared with those without HuR overexpression only.
Discussion
RBPs have been shown to regulate RNA metabolism at all stages of RNA life cycle, from the synthesis of RNA to its decay.24 In the present study, we identified that the RBP, HuR, regulated MEF2C mRNA levels by stabilizing its mRNA and ultimately increased cardiac sodium channel α subunit mRNA transcription (Figure 9). Our data indicated a combined regulatory network for the expression of the cardiac sodium channel gene SCN5A at the DNA and RNA levels.
Figure 9.
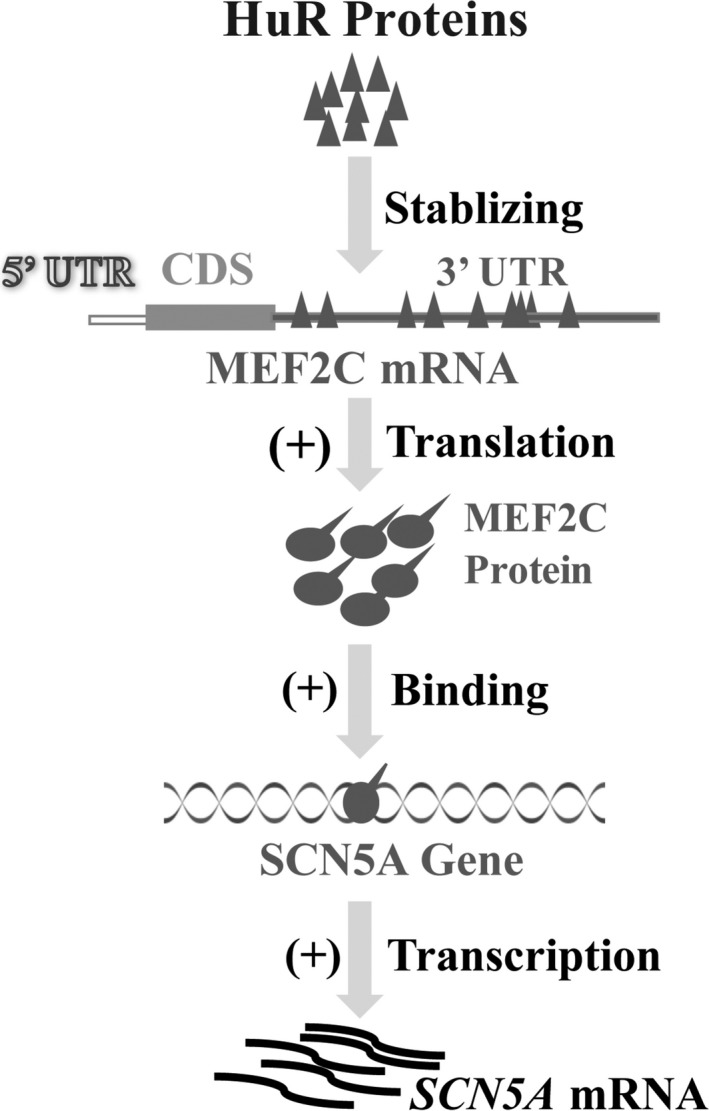
Schematic representation of a combined regulatory network for the expression of the cardiac sodium channel gene SCN5A. RNA binding protein, Hu Antigen R (HuR), is associated with myocyte enhancer factor‐2C (MEF2C) mRNA, and the association protects MEF2C mRNA from degradation. The elevated MEF2C transcriptionally enhances SCN5A mRNA expression. SCN5A indicates sodium channel α‐subunit; CDS, coding sequences; UTR, untranslated region.
The study of MEF2C expression regulation has traditionally focused on the transcription level, and significant efforts have been made on the identification and characterization of MEF2C enhancer or silencer sequences.22 MEF2C is the direct transcriptional target of ISL1 and GATA factors during mouse embryonic development.25 In contrast, TBX1 suppresses MEF2C expression through direct interaction with MEF2C and through regulating GATA4 expression.26
We are now aware that posttranscriptional regulation plays an important role in gene expression regulation. All mRNAs are ultimately degraded, and the steady‐state mRNA abundance is determined by the balance between transcription and mRNA decay. mRNA decay can influence timing, quantity, and perhaps even location of encoded proteins.24 In this study, in vitro gain‐of‐function and loss‐of‐function results suggested that the mRNA expression of MEF2C was regulated by HuR. Overexpression of HuR upregulated, whereas knockdown of HuR downregulated, MEF2C mRNA expression. The positive effect of HuR on the regulation of MEF2C mRNA expression appeared to be achieved through interacting with AREs within MEF2C 3′UTR. We further asked how HuR regulated MEF2C mRNA expression. The mRNA degradation study demonstrated that HuR upregulated MEF2C mRNA expression through stabilizing MEF2C mRNA because MEF2C mRNA degraded more slowly with HuR overexpression than without HuR overexpression. This is the first demonstration that HuR is involved in the regulation of MEF2C expression. The association of HuR protein and MEF2C mRNA by ribonucleoprotein study further confirmed this concept. Therefore, the interaction of HuR and MEF2C mRNA may serve to fine‐tune MEF2C expression.
MEF2C has been shown to be involved in many different developmental processes, especially in cardiac muscle differentiation and cardiogenesis.11 In addition to its important roles in development, MEF2C plays a critical role in postnatal adaptation to a wide array of physiological and pathological signals. MEF2C is first member expressed during cardiogenesis, and its expression in the heart is maintained at a basic level during the lifecycle of the animal, which indicates MEF2C may play a critical role in maintaining basic cardiac function. This idea is evidenced by the fact that decreased expression of MEF2C has been associated with myotonic dystrophy type 1 heart disease.12 Furthermore, both MEF2C and SCN5A have been shown to be expressed in the heart and circulating white blood cells.15, 27 Therefore, a transcriptional effect of MEF2C on SCN5A might be anticipated.
Normal function of voltage‐gated cardiac sodium channels encoded by SCN5A is essential for the initiation and propagation of action potentials in cardiomyocytes.28 Dysregulated expression of SCN5A because of common pathological conditions, such as heart failure or genetic mutation of SCN5A, has been associated with a wide range of arrhythmias.29, 30 To date, TBX5 and GATA4 are major transcription factors studied to promote SCN5A transcription, whereas nuclear factor‐κB, TBX3, and Snail function as repressors of SCN5A expression transcriptionally.13, 16, 17, 31, 32, 33
In this article, we identified MEF2C is one of the transcription factors positively regulating SCN5A expression in cardiomyocytes. The regulatory role of MEF2C on SCN5A expression was evidenced by the increased expression of SCN5A mRNA on MEF2C overexpression. Moreover, reduced SCN5A mRNA expression was also observed in cardiomyocytes with siRNA‐mediated reduced MEF2C expression. A putative MEF2C binding site located in the promoter region of SCN5A suggests that MEF2C regulates SCN5A by binding to this site.23 Our results showing that MEF2C was associated with the SCN5A gene and that overexpression of MEF2C enhanced the luciferase mRNA expression driven by the WT SCN5A promoter support this idea. On the other hand, on MEF2C overexpression, mutation of the MEF2C binding sequences in the SCN5A promoter region lowered the luciferase mRNA abundance compared with the WT SCN5A promoter. The fact revealed by the EMSA experiment that MEF2C was associated with SCN5A sequences confirmed that MEF2C directly interacts with SCN5A gene promoter.
Furthermore, overexpression of HuR also increased SCN5A mRNA expression, and this increase was attenuated by the presence of MEF2C siRNA without altering HuR expression. Therefore, it is highly likely that MEF2C at least partially mediates the increase of SCN5A mRNA expression in the presence of HuR overexpression. Because we did not measure HuR or MEF2C in cardiac pathological conditions, the overall significance of these findings remains to be seen.
In summary, our results suggested a combined regulatory network at the DNA and RNA levels that regulates SCN5A mRNA expression. HuR upregulates MEF2C mRNA expression by protecting MEF2C mRNA from degradation, and consequently, the elevated MEF2C enhances SCN5A mRNA transcription.
Sources of Funding
The present work was supported in part by National Institutes of Health grant R01 HL104025.
Disclosures
Dudley is the inventor of PCT/US2015/046162, Assessment of Heart Failure and Arrhythmic Risk Stratification by Measuring Circulating Hu Proteins. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2018;7:e007802 DOI: 10.1161/JAHA.117.007802.)29678826
References
- 1. Keene JD. RNA regulons: coordination of post‐transcriptional events. Nat Rev Genet. 2007;8:533–543. [DOI] [PubMed] [Google Scholar]
- 2. Liao Y, Castello A, Fischer B, Leicht S, Foehr S, Frese CK, Ragan C, Kurscheid S, Pagler E, Yang H, Krijgsveld J, Hentze MW, Preiss T. The cardiomyocyte RNA‐binding proteome: links to intermediary metabolism and heart disease. Cell Rep. 2016;16:1456–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barreau C, Paillard L, Osborne HB. AU‐rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matoulkova E, Michalova E, Vojtesek B, Hrstka R. The role of the 3′ untranslated region in post‐transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012;9:563–576. [DOI] [PubMed] [Google Scholar]
- 5. Blackinton JG, Keene JD. Functional coordination and HuR‐mediated regulation of mRNA stability during T cell activation. Nucleic Acids Res. 2016;44:426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lebedeva S, Jens M, Theil K, Schwanhausser B, Selbach M, Landthaler M, Rajewsky N. Transcriptome‐wide analysis of regulatory interactions of the RNA‐binding protein HuR. Mol Cell. 2011;43:340–352. [DOI] [PubMed] [Google Scholar]
- 7. Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M Jr, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA‐binding protein HuR couples pre‐mRNA processing and mRNA stability. Mol Cell. 2011;43:327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morin S, Pozzulo G, Robitaille L, Cross J, Nemer M. MEF2‐dependent recruitment of the HAND1 transcription factor results in synergistic activation of target promoters. J Biol Chem. 2005;280:32272–32278. [DOI] [PubMed] [Google Scholar]
- 9. Srivastava D, Cserjesi P, Olson EN. A subclass of bHLH proteins required for cardiac morphogenesis. Science. 1995;270:1995–1999. [DOI] [PubMed] [Google Scholar]
- 10. Zang MX, Li Y, Xue LX, Jia HT, Jing H. Cooperative activation of atrial naturetic peptide promoter by dHAND and MEF2C. J Cell Biochem. 2004;93:1255–1266. [DOI] [PubMed] [Google Scholar]
- 11. Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ, Cooper TA. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep. 2014;6:336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Atack TC, Stroud DM, Watanabe H, Yang T, Hall L, Hipkens SB, Lowe JS, Leake B, Magnuson MA, Yang P, Roden DM. Informatic and functional approaches to identifying a regulatory region for the cardiac sodium channel. Circ Res. 2011;109:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao G, Xie A, Huang SC, Zhou A, Zhang J, Herman AM, Ghassemzadeh S, Jeong EM, Kasturirangan S, Raicu M, Sobieski MA, Bhat G, Tatooles A, Benz EJ Jr, Kamp TJ, Dudley SC Jr. Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure. Circulation. 2011;124:1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shang LL, Pfahnl AE, Sanyal S, Jiao Z, Allen J, Banach K, Fahrenbach J, Weiss D, Taylor WR, Zafari AM, Dudley SC Jr. Human heart failure is associated with abnormal C‐terminal splicing variants in the cardiac sodium channel. Circ Res. 2007;101:1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, Dudley SC Jr. NF‐kB‐dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am J Physiol Cell Physiol. 2008;294:C372–C379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, Bezzina CR, ‘t Hoen PA, Bakkers J, Barnett P, Christoffels VM. Genetic variation in T‐box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest. 2012;122:2519–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fallmann J, Sedlyarov V, Tanzer A, Kovarik P, Hofacker IL. AREsite2: an enhanced database for the comprehensive investigation of AU/GU/U‐rich elements. Nucleic Acids Res. 2016;44:D90–D95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Ree JH, Jeganathan KB, Malureanu L, van Deursen JM. Overexpression of the E2 ubiquitin‐conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol. 2010;188:83–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang H, Chen C, Song X, Chen J, Zhen Y, Sun K, Hui R. Mef2c is an essential regulatory element required for unique expression of the cardiac‐specific CARK gene. J Cell Mol Med. 2008;12:304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hinman MN, Lou H. Diverse molecular functions of Hu proteins. Cell Mol Life Sci. 2008;65:3168–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Black BC, Cripps RM. Myocyte enhancer factor 2 transcription factors in heart development and disease In: Rosenthal N, Harvey RP, eds. Heart Development and Regeneration. Oxford: Academic Press; 2010:673–699. [Google Scholar]
- 23. Black BL, Martin JF, Olson EN. The mouse MRF4 promoter is trans‐activated directly and indirectly by muscle‐specific transcription factors. J Biol Chem. 1995;270:2889–2892. [DOI] [PubMed] [Google Scholar]
- 24. Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nat Rev Genet. 2012;13:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dodou E, Verzi MP, Anderson JP, Xu SM, Black BL. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–3942. [DOI] [PubMed] [Google Scholar]
- 26. Pane LS, Zhang Z, Ferrentino R, Huynh T, Cutillo L, Baldini A. Tbx1 is a negative modulator of Mef2c. Hum Mol Genet. 2012;21:2485–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cante‐Barrett K, Pieters R, Meijerink JP. Myocyte enhancer factor 2C in hematopoiesis and leukemia. Oncogene. 2014;33:403–410. [DOI] [PubMed] [Google Scholar]
- 28. Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ. Voltage‐gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation. 2015;132:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu M, Gu L, Sulkin MS, Liu H, Jeong EM, Greener I, Xie A, Efimov IR, Dudley SC Jr. Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol. 2013;54:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Remme CA, Verkerk AO, Hoogaars WM, Aanhaanen WT, Scicluna BP, Annink C, van den Hoff MJ, Wilde AA, van Veen TA, Veldkamp MW, de Bakker JM, Christoffels VM, Bezzina CR. The cardiac sodium channel displays differential distribution in the conduction system and transmural heterogeneity in the murine ventricular myocardium. Basic Res Cardiol. 2009;104:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arnolds DE, Liu F, Fahrenbach JP, Kim GH, Schillinger KJ, Smemo S, McNally EM, Nobrega MA, Patel VV, Moskowitz IP. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J Clin Invest. 2012;122:2509–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hesse M, Kondo CS, Clark RB, Su L, Allen FL, Geary‐Joo CT, Kunnathu S, Severson DL, Nygren A, Giles WR, Cross JC. Dilated cardiomyopathy is associated with reduced expression of the cardiac sodium channel Scn5a. Cardiovasc Res. 2007;75:498–509. [DOI] [PubMed] [Google Scholar]
- 33. van den Boogaard M, Smemo S, Burnicka‐Turek O, Arnolds DE, van de Werken HJ, Klous P, McKean D, Muehlschlegel JD, Moosmann J, Toka O, Yang XH, Koopmann TT, Adriaens ME, Bezzina CR, de Laat W, Seidman C, Seidman JG, Christoffels VM, Nobrega MA, Barnett P, Moskowitz IP. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J Clin Invest. 2014;124:1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]