

Abstract
Background
We aimed to compare the associations of smoking exposure as assessed by self‐reports and urine cotinine with cardiovascular disease (CVD) risk and determine the potential utility of cotinine for CVD risk prediction.
Methods and Results
Smoking status by self‐reports and urine cotinine were assessed at baseline in 4737 participants (mean age, 53 years) of the PREVEND (Prevention of Renal and Vascular End‐Stage Disease) prospective study. Participants were classified as never, former, light current (≤10 cigarettes/day), and heavy current smokers (>10 cigarettes/day) according to self‐reports and analogous cutoffs for urine cotinine. During a median follow‐up of 8.5 years, 296 first CVD events were recorded. Compared with self‐reported never smokers, the hazard ratios (95% confidence interval) of CVD for former, light current, and heavy current smokers were 0.86 (0.64–1.17), 1.28 (0.83–1.97), and 1.80 (1.27–2.57) in multivariate analysis. Compared with urine cotinine–assessed never smokers, the corresponding hazard ratios of CVD for urine cotinine–assessed former, light current, and heavy current smokers were 1.70 (1.03–2.81), 1.62 (1.15–2.28), and 1.95 (1.39–2.73) respectively. The C‐index change on adding urine cotinine–assessed smoking status to a standard CVD risk prediction model (without self‐reported smoking status) was 0.0098 (0.0031–0.0164; P=0.004). The corresponding C‐index change for self‐reported smoking status was 0.0111 (0.0042–0.0179; P=0.002).
Conclusions
Smoking status as assessed by self‐reports and urine cotinine is associated with CVD risk; however, the nature of the association of urine cotinine with CVD is consistent with a dose‐response relationship. The ability of urine cotinine to improve CVD risk assessment is similar to that of self‐reported smoking status.
Keywords: cardiovascular disease, cohort study, cotinine, risk factor, risk prediction, smoking
Subject Categories: Cardiovascular Disease, Epidemiology, Lifestyle, Risk Factors
Clinical Perspective
What Is New?
In a population‐based prospective study of white men and women without a history of cardiovascular disease at baseline, smoking status as assessed by self‐reports and urine cotinine is associated with risk of cardiovascular disease.
Compared with self‐reports, the magnitude of the association using urine cotinine appears stronger and is consistent with a dose‐response relationship.
The ability of urine cotinine to improve cardiovascular disease risk assessment is similar to that provided by self‐reported smoking status.
What Are the Clinical Implications?
Urine cotinine may be equal to or better than self‐reported smoking for the assessment of smoking exposure.
In approaches that integrate smoking exposure in the primary prevention of cardiovascular disease, urine cotinine may serve as a reliable marker in instances where self‐reports are unreliable or cannot be ascertained.
Cardiovascular disease (CVD) is still the leading cause of global mortality; in 2015, there were an estimated 422.7 million CVD cases and 17.9 million deaths globally.1 It has been estimated that by 2030, over 23.6 million people will die from CVD.2 Major risk factors for CVD include a history of diabetes mellitus, blood pressure, and blood lipids, as well as smoking status.3 Cigarette smoking is highly prevalent globally, and its effect on CVD as well as all‐cause mortality is well established.4, 5, 6, 7 Indeed, the literature is a minefield of studies that have shown smoking to be an important cause of cardiovascular outcomes, which include coronary heart disease (CHD) and stroke. A strong dose‐response relationship has been demonstrated between cigarette smoking and CVD.7, 8 In fact, the strong epidemiological link suggests a causal link between smoking and CVD.
Notably, data on smoking exposure in these studies have mostly been dependent on self‐reports. There is, however, a challenge in the use of self‐reported smoking exposure; the assessment of smoking status by questionnaires may lead to inaccurate measures of smoking exposure due to smoking denial or difficulty in recalling the quantity and duration of smoking.9, 10 This misclassification potentially leads to the underestimation of the biological effects of smoking exposure. Cotinine is the major metabolite of nicotine and has a long biological half‐life of between 19 and 40 hours in the body compared with nicotine, which has a short half‐life of about 30 minutes to 2 hours.11 Cotinine is considered a highly sensitive and specific biomarker of cigarette smoking and is considered to be the gold standard measure of smoking exposure.11, 12 Cotinine concentrations can be accurately determined in serum or urine.13
A limited number of studies have evaluated the associations between cotinine‐assessed smoke exposure and the risk of cardiovascular outcomes and reported increased levels of blood cotinine to be associated with an increased risk of these outcomes.14, 15 However, there are uncertainties remaining regarding the nature, shape, and magnitude of the association between cotinine‐assessed cigarette smoking exposure and the risk of CVD because these previous reports were either cross sectional in design, were based on subclinical cardiovascular outcomes, or were insufficiently powerful to address these aspects of the association.14, 15 Whether a dose‐response relationship exists for the potential association is also not known. Furthermore, whether the assessment of cigarette smoking on the basis of self‐reports underestimates the risk between smoking status and CVD as a result of misclassification has not been previously investigated.
In this context, we aimed to compare in detail the associations of smoking exposure as assessed by self‐reports and urine cotinine with the risk of CVD. We also aimed to determine the potential utility of urine cotinine for CVD risk prediction.
Materials and Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure. We conducted this study in accordance with STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) guidelines for reporting observational studies in epidemiology (Table S1).16
Study Design and Population
The participants in this study were part of the PREVEND (Prevention of Renal and Vascular End‐Stage Disease) study, a general population–based prospective cohort study designed to investigate the natural course of urinary albumin excretion and its relationship to renal disease and CVD. The study design details and recruitment have been described in previous reports.17, 18 Participants in PREVEND consisted of a representative sample of inhabitants living in the city of Groningen in the Netherlands. The present cohort comprised 6894 individuals aged 32 to 80 years, who were invited for the second screening phase of the PREVEND study. Baseline examinations and measurements were performed between 2001 and 2003. In the present analysis, we used data of participants who had not experienced CVD, renal disease, or malignancy at baseline. This left a cohort of 4737 participants with nonmissing information on urine cotinine, smoking exposure on the basis of self‐reports, relevant covariates, and incident cardiovascular outcomes. The local ethics committee of the University Medical Center Groningen approved the PREVEND study, which was conducted in accordance with the Declaration of Helsinki. All participants provided written informed consent.
Assessment of Exposures and Risk Factors
Study participants completed 2 outpatient visits, during which baseline data on sociodemographics, anthropometric measurements, medical history, and use of medication were assessed or collected. Further information on medication use was complemented with data from all community pharmacies in the city of Groningen, which covers complete information on drug use in 95% of PREVEND participants.19 After an overnight fast and 15 minutes of rest, plasma and serum venous samples were taken from participants on which biomarker analyses were performed. Samples of 24‐hour urine collections were collected and stored at −80°C until assessment of cotinine. Cotinine concentrations were measured using the Immulite 2500 assay (Siemens, Los Angeles, CA) with the intra‐ and interassay coefficient of variation ranging from 2.2% to 5.7%. Smoking status was obtained by self‐reports. Participants provided details on their smoking habits, which included number of cigarettes smoked and duration of smoking. Smoking status was categorized as never smokers, former smokers, light current smokers, and heavy current smokers. Former smokers were those who were nonsmokers at the time of study inclusion but had ever smoked in their life, and current smokers were those who reported smoking at the time of inclusion. Light current smokers were current smokers who reported smoking ≤10 cigarettes per day, and heavy current smokers were current smokers who reported smoking >10 cigarettes per day. Blood pressure values were recorded as the mean of the last 2 readings of both visits.
Total cholesterol, high‐density lipoprotein cholesterol, high‐sensitivity C‐reactive protein (hsCRP), triglycerides, serum creatinine, and serum cystatin C were measured using standard laboratory protocols, which have been described in previous reports.20, 21, 22, 23, 24 Plasma glucose was measured by dry chemistry (Eastman Kodak, Rochester, NY). Estimated glomerular filtration rate was calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) combined creatinine–cystatin C equation.25 Hypertension was defined as systolic blood pressure of ≥140 mm Hg, a diastolic blood pressure of ≥90 mm Hg, and/or the use of antihypertensive medication, in accordance with recommendations from the Seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure.26
Ascertainment of Outcomes
The primary outcome for this study was first‐onset composite CVD, with incident CHD and stroke as secondary outcomes. Dates and causes of death were ascertained by record linkage with the Dutch Central Bureau of Statistics. Information on hospitalization for cardiovascular morbidity was retrieved from Prismant, the Dutch national registry of hospital discharge diagnoses.27 All outcome data were coded according to the International Classification of Diseases, Ninth Revision (ICD‐9) until January 1, 2009. After that date, the data were coded according to ICD, Tenth Revision (ICD‐10) codes. First‐onset CVD was defined as the combined end point of acute and subacute ischemic heart disease, acute myocardial infarction, coronary artery bypass grafting or percutaneous transluminal coronary angioplasty, subarachnoid hemorrhage, intracerebral hemorrhage, other intracranial hemorrhage, occlusion or stenosis of the precerebral or cerebral arteries, and other vascular interventions such as percutaneous transluminal angioplasty or bypass grafting of peripheral vessels and aorta. CHD events were defined as fatal or nonfatal ischemic heart disease, fatal or nonfatal myocardial infarction, coronary artery bypass graft, and percutaneous transluminal coronary angioplasty. Stroke events were defined as subarachnoid hemorrhage, intracerebral hemorrhage, other and unspecified intracranial hemorrhage, occlusion and stenosis of precerebral or cerebral arteries, and carotid obstruction.
Statistical Analyses
Skewed variables (eg, hsCRP, creatinine, and urinary albumin excretion) were natural logarithm (loge) transformed to achieve approximately normal distributions. We summarized baseline characteristics of participants using descriptive statistics. Normally distributed variables are presented as means (standard deviation) and variables with a skewed distribution are given as median (interquartile range). Continuous and categorical variables were compared between groups by ANOVA and chi‐square testing, respectively. ρ coefficient was estimated to measure the degree of association between self‐reported smoking status and urine cotinine–measured smoking status. To assess the measure of agreement of the classification of smoking exposure on the basis of self‐report and urine cotinine, we calculated Cohen's kappa (κ). A κ <0.21 is considered poor, a κ between 0.21 and 0.40 is considered weak; a κ between 0.41 and 0.60 is considered moderate; a κ between 0.61 and 0.80 is considered strong; and a κ >0.80 is considered very strong.28 Time‐to‐event Cox proportional hazards models were used to assess the associations of smoking exposure as assessed by self‐report and cotinine concentrations with risk of CVD, after confirmation of no major departure from the proportionality‐of‐hazards assumptions.29 We categorized cotinine‐assessed smoking exposure as never smokers, former smokers, light current smokers, and heavy current smokers on the basis of cutoffs for urine cotinine reported in the literature. The cutoffs for urine cotinine were <100 ng/mL, 100 to 500 ng/mL, and >500 ng/mL for the categories of never smokers, former smokers, and current smokers, respectively, as employed in several previous reports.30, 31, 32, 33 Current smokers were then subdivided into light and heavy current smokers on the basis of the median cotinine level in current smokers, as reported in a previous study.31 We plotted cumulative Kaplan‐Meier curves for CVD during follow‐up according to categories of smoking status as assessed by self‐report and urine cotinine. To assess the independence of the association between smoking exposure and CVD risk, hazard ratios were calculated with progressive adjustment for age and sex, other established CVD risk factors (history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol), other potential confounders (body mass index, alcohol consumption, fasting glucose, and estimated glomerular filtration rate), and hsCRP. Given that assuming a linear relationship between a continuous variable (urine cotinine) and an outcome (CVD) can yield misleading analyses, we employed a multivariate fractional polynomials model,34 which allows for flexible modeling of the relationship between urine cotinine and risk of CVD. We used interaction tests to assess statistical evidence of effect modification by relevant clinical characteristics. To minimize bias due to reverse causation, we performed sensitivity analyses that excluded the first 2 years of follow‐up, participants with a history of diabetes mellitus at baseline, or participants on regular statin medication.
To assess whether adding information on urine cotinine assessed smoking exposure to conventional cardiovascular risk factors35 is associated with an improvement in the prediction of CVD risk, we calculated measures of discrimination for censored time‐to‐event data (Harrell's C‐index36) and reclassification. To investigate the change in C‐index, we added smoking status to a model on the basis of traditional risk factors included in the Framingham CVD Risk Score (ie, age, sex, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol).37 Second, we evaluated whether urine cotinine–assessed smoking exposure helps to correctly classify participants into categories of predicted CVD risk. Using the cardiovascular risk categories of low (<5%), intermediate (5 to <7.5%), and high (≥7.5%) risk,38 reclassification was assessed using the categorical net reclassification improvement.39 Reclassification analysis was based on the 9 years of follow time for this study. Finally, we calculated the integrated discrimination improvement (IDI), which integrates the net reclassification improvement over all possible cutoffs and is equivalent to the difference in discrimination slopes.39 Risk prediction analysis was restricted to participants without a known history of diabetes mellitus or CVD at baseline. All statistical analyses were conducted using Stata version 14 (Stata Corp, College Station, TX).
Results
Baseline Characteristics
Baseline characteristics of all 4737 participants overall and according to their self‐reported smoking status are reported in Table 1. The mean (standard deviation) age of participants at baseline was 53 (12) years, and 45.5% were men. The mean (standard deviation) of urine cotinine was 370 (721) ng/mL. Former smokers were older, heavier, and more likely to have preexisting disease such as diabetes mellitus and hypertension compared with other categories. Heavy current smokers had higher levels of total cholesterol and hsCRP and lower levels of high‐density lipoprotein cholesterol compared with other categories. There was a strong correlation between self‐reported smoking status and urine cotinine–measured smoking status (ρ=0.76, P<0.001). However, the classification of self‐report corresponded weakly with that of urine cotinine on the basis of a Cohen's κ of 0.24 (interrater agreement of 45%). Table 2 shows a cross tabulation of self‐reported smoking status and urine cotinine–measured smoking status. Of the 1458 self‐reported never smokers, 8 (0.5%) had urine cotinine concentrations consistent with active smoking; and of the 1997 self‐reported former smokers, 53 (2.7%) had urine cotinine concentrations consistent with active smoking. Hence, the misclassification rate of active smokers (the number of misclassified active smokers divided by the number of self‐reported active smokers40) was 4.8%. Furthermore, of the 3407 never smokers as assessed by urine cotinine concentrations, a majority (1887, 55.4%) were classified as former smokers by self‐reports.
Table 1.
Baseline Participant Characteristics Overall and According to Self‐Reported Smoking Status
| Overall (N=4737) Mean (SD) or Median (IQR) or n (%) | Never Smokers (N=1458) Mean (SD) Median (IQR) or n (%) | Former Smokers (N=1997) Mean (SD) or Median (IQR) or n (%) | Light Current Smokers (N=495) Mean (SD) or Median (IQR) or n (%) | Heavy Current Smokers (N=787) Mean (SD) or Median (IQR) or n (%) | P Value for ANOVA | |
|---|---|---|---|---|---|---|
| Urine cotinine (ng/mL)a | 370 (721) | 11 (109) | 47 (256) | 805 (681) | 1580 (753) | <0.001 |
| Questionnaire | ||||||
| Male | 2156 (45.5) | 578 (39.6) | 992 (49.7) | 214 (43.2) | 372 (47.3) | <0.001 |
| Age at survey, y | 53 (12) | 52 (12) | 55 (12) | 52 (11) | 50 (10) | <0.001 |
| History of diabetes mellitus | 236 (5.0) | 69 (4.7) | 107 (5.4) | 28 (5.7) | 32 (4.1) | 0.447 |
| Alcohol consumers | 3570 (75.4) | 1007 (69.1) | 1577 (79.0) | 391 (79.0) | 595 (75.6) | <0.001 |
| Regular use of antihypertensive medication | 742 (16.6) | 197 (14.5) | 393 (20.8) | 68 (14.5) | 84 (11.2) | <0.001 |
| Regular use of lipid‐lowering medication | 126 (3.2) | 37 (3.2) | 60 (3.6) | 14 (3.4) | 15 (2.3) | 0.498 |
| Physical measurements | ||||||
| BMI, kg/m2 | 26.5 (4.3) | 26.5 (4.4) | 27.1 (4.2) | 25.6 (4.2) | 25.8 (4.1) | <0.001 |
| SBP, mm Hg | 125 (19) | 125 (19) | 127 (19) | 122 (17) | 123 (18) | <0.001 |
| DBP, mm Hg | 73 (9) | 72 (9) | 74 (9) | 72 (9) | 73 (9) | <0.001 |
| Lipid markers | ||||||
| Total cholesterol, mmol/L | 5.47 (1.05) | 5.34 (1.04) | 5.51 (1.03) | 5.42 (1.03) | 5.63 (1.09) | <0.001 |
| HDL‐C, mmol/L | 1.28 (0.31) | 1.29 (0.29) | 1.29 (0.32) | 1.28 (0.33) | 1.21 (0.31) | <0.001 |
| Metabolic, inflammatory, and renal function markers | ||||||
| hsCRP, mg/L | 1.30 (0.60–2.89) | 1.05 (0.50–2.49) | 1.35 (0.65–2.87) | 1.21 (0.53–2.91) | 1.81 (0.77–3.78) | <0.001 |
| Fasting plasma glucose, mmol/L | 4.98 (1.08) | 4.94 (1.10) | 5.05 (1.10) | 4.92 (0.96) | 4.93 (1.07) | 0.002 |
| Creatinine, μmol/L | 71 (62–80) | 70 (62–79) | 72 (64–82) | 69 (62–78) | 67 (60–76) | <0.001 |
| Cystatine C, mg/dL | 0.90 (0.20) | 0.87 (0.19) | 0.91 (0.22) | 0.92 (0.21) | 0.92 (0.16) | <0.001 |
| eGFR, mL/min per 1.73 m2 | 92.4 (16.8) | 94.1 (16.9) | 90.7 (17.3) | 92.3 (17.0) | 93.8 (14.6) | <0.001 |
Continuous variables are reported as mean±SD or median (interquartile range) and categorical variables are reported as n (%). BMI indicates body mass index; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate (as calculated using the Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation); HDL‐C, high‐density lipoprotein cholesterol; hsCRP, high‐sensitivity C‐reactive protein; IQR, interquartile range; SBP, systolic blood pressure; SD, standard deviation.
Majority of participants had urine cotinine concentrations below the assay's detection limit.
Table 2.
Cross‐Tabulation of Participants by Self‐Reported Smoking Status and Urine Cotinine Measured Smoking Status
| Self‐Reported Smoking Status | Urine Cotinine–Assessed Smoking Status | Total | |||
|---|---|---|---|---|---|
| Never Smokers | Former Smokers | Light Current Smokers | Heavy Current Smokers | ||
| Never smokers | 1441 (98.8) | 9 (0.6) | 4 (0.3) | 4 (0.3) | 1458 (100.0) |
| Former smokers | 1887 (94.5) | 57 (2.9) | 31 (1.6) | 22 (1.1) | 1997 (100.0) |
| Light current smokers | 66 (13.3) | 135 (27.3) | 202 (40.8) | 92 (18.6) | 495 (100.0) |
| Heavy current smokers | 13 (1.7) | 26 (3.3) | 315 (40.0) | 433 (55.0) | 787 (100.0) |
| Total | 3407 (71.9) | 227 (4.8) | 552 (11.7) | 551 (11.6) | 4737 (100.0) |
Data are n (%).
Smoking Exposures and Risk of Incident CVD
During a median follow‐up of 8.5 (interquartile range, 7.8–8.9) years (37 392 person‐years at risk), 296 incident CVD events (annual rate 7.92/1000 person‐years at risk; 95% confidence interval [CI], 7.06–8.87) were recorded. Cumulative hazard curves showed increased risks of CVD among heavy current smokers (as assessed by self‐reports and urine cotinine) compared with other categories of smoking exposure (P value for log‐rank test <0.05 for all; Figure 1). Table 3 shows the associations of smoking exposure categories assessed by self‐reports and urine cotinine with the risk of CVD. Compared with self‐reported never smokers, the hazard ratio (95% CI) of CVD for heavy current smokers was 1.93 (1.37–2.73) in the analysis adjusted for established cardiovascular risk factors. The association remained consistent on additional adjustment for body mass index, alcohol consumption, glucose, and estimated glomerular filtration rate 1.96 (1.39–2.78). The association was minimally attenuated by further adjustment for loge hsCRP 1.80 (1.27–2.57). The associations of self‐reported former and light current smoking exposures with CVD were not significant. Fitting of a fractional polynomial model suggested a linear dose‐response relationship between urine cotinine and CVD risk (Figure 2). Compared with urine cotinine–assessed never smokers in analysis adjusted for established cardiovascular risk factors, the hazard ratios (95% CI) of CVD for the following urine cotinine assessed smoking groups: former smokers, light current smokers, and heavy current smokers were 1.65 (1.00–2.72), 1.68 (1.20–2.36), and 2.04 (1.47–2.83), respectively. The hazard ratios were 1.70 (1.03–2.81), 1.62 (1.15–2.28), and 1.95, (1.39–2.73), respectively, after further adjustment for other potential confounders and loge hsCRP. In subsidiary analyses that modeled urine cotinine as a continuous variable (per 1000 ng/mL), significant positive associations were observed in all models (Table 3). In separate analyses for other cardiovascular outcomes, the associations of both exposures were generally similar for CHD and stroke; except for less robust associations of cotinine‐assessed former smoking with risk of CHD and stroke (Tables 4, 5 and 5). In sensitivity analyses, the hazard ratios remained similar on exclusion of the first 2 years of follow‐up, people with diabetes mellitus at baseline, or people on cholesterol‐lowering medication (Tables 6, 7, 8). In further sensitivity analyses, we assessed the associations of urinary cotinine multiplied by urinary volume and urinary cotinine/urine creatinine ratio with the risk of CVD. In multivariate analyses that compared the top to the bottom tertiles of urinary cotinine*urinary volume and urinary cotinine/urine creatinine ratio, there was an increased risk of CVD (Tables 9, 10and 10). The associations of self‐reported and urine cotinine–assessed smoking status with incident CVD were not significantly modified by several clinically relevant characteristics such as age and sex, except for evidence of effect modification by total cholesterol on the association between self‐reported smoking and CVD risk (P for interaction=0.044). A strong association was observed in those with total cholesterol levels ≥5.41 mmol/L compared to a modest association in participants with cholesterol levels <5.41 mmol/L (Figures 3 and 4).
Figure 1.
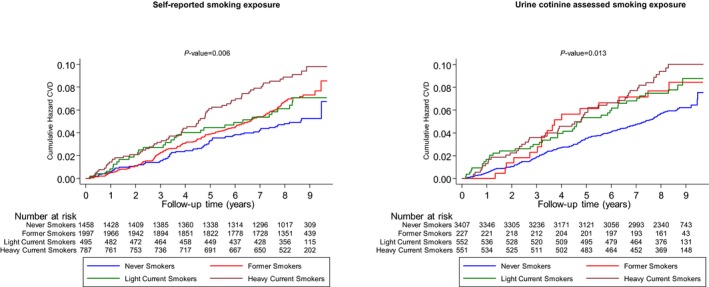
Cumulative Kaplan‐Meier curves for cardiovascular disease during follow‐up according to smoking exposure categories as assessed by self‐reports and urine cotinine. CVD indicates cardiovascular disease.
Table 3.
Prospective Associations of Smoking Exposure With Development of Cardiovascular Disease
| Smoking Exposure | Events/Total | Model 1 | Model 2 | Model 3 | Model 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | ||
| Self‐reported smoking | |||||||||
| Never smokers | 69/1458 | ref | ref | ref | ref | ||||
| Former smokers | 130/1997 | 0.89 (0.66–1.19) | 0.425 | 0.87 (0.64–1.17) | 0.359 | 0.87 (0.64–1.18) | 0.383 | 0.86 (0.64–1.17) | 0.337 |
| Light current smokers | 31/495 | 1.27 (0.83–1.94) | 0.274 | 1.31 (0.85–2.00) | 0.218 | 1.34 (0.87–2.06) | 0.180 | 1.28 (0.83–1.97) | 0.259 |
| Heavy current smokers | 66/787 | 2.10 (1.49–2.96) | <0.001 | 1.93 (1.37–2.73) | <0.001 | 1.96 (1.39–2.78) | <0.001 | 1.80 (1.27–2.57) | 0.001 |
| Urine cotinine | |||||||||
| Per 1000 ng/mL | 296/4737 | 1.47 (1.29–1.68) | <0.001 | 1.44 (1.26–1.65) | <0.001 | 1.46 (1.27–1.67) | <0.001 | 1.40 (1.22–1.61) | <0.001 |
| Never smokers | 190/3407 | ref | ref | ref | ref | ||||
| Former smokers | 17/227 | 1.70 (1.04–2.80) | 0.036 | 1.65 (1.00–2.72) | 0.050 | 1.73 (1.05–2.86) | 0.032 | 1.70 (1.03–2.81) | 0.038 |
| Light current smokers | 41/552 | 1.67 (1.19–2.35) | 0.003 | 1.68 (1.20–2.36) | 0.003 | 1.69 (1.20–2.38) | 0.003 | 1.62 (1.15–2.28) | 0.006 |
| Heavy current smokers | 48/551 | 2.12 (1.54–2.93) | <0.001 | 2.04 (1.47–2.83) | <0.001 | 2.12 (1.52–2.96) | <0.001 | 1.95 (1.39–2.73) | <0.001 |
Model 1: Age and sex. Model 2: Model 1 plus history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol. Model 3: Model 2 plus body mass index, alcohol consumption, glucose, and estimated glomerular filtration rate (as calculated using the Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation). Model 4: Model 3 plus loge high‐sensitivity C‐reactive protein. CI indicates confidence interval; HR, hazard ratio.
Figure 2.
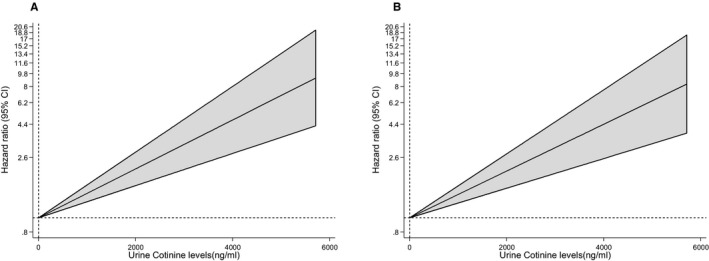
Hazard ratios for incident cardiovascular disease, by baseline concentrations of urine cotinine using multivariate fractional polynomial models. A, Hazard ratios were adjusted for age and sex; B, adjustment in A plus history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol. A fractional polynomial was used to model the relationship between urine cotinine as a continuous risk factor and cardiovascular disease. The shaded regions denote the 95% confidence interval for the fractional polynomial model.
Table 4.
Prospective Associations of Smoking Exposure With Development of Coronary Heart Disease
| Smoking Exposure | Events/Total | Model 1 | Model 2 | Model 3 | Model 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | ||
| Self‐reported smoking | |||||||||
| Never smokers | 46/1458 | ref | ref | ref | ref | ||||
| Former smokers | 98/1997 | 1.00 (0.70–1.43) | 0.988 | 0.97 (0.67–1.39) | 0.863 | 0.97 (0.68–1.40) | 0.879 | 0.96 (0.67–1.39) | 0.840 |
| Light current smokers | 23/495 | 1.37 (0.83–2.26) | 0.223 | 1.37 (0.83–2.27) | 0.219 | 1.40 (0.85–2.33) | 0.189 | 1.36 (0.82–2.27) | 0.231 |
| Heavy current smokers | 45/787 | 1.98 (1.30–2.99) | 0.001 | 1.76 (1.16–2.68) | 0.008 | 1.79 (1.17–2.73) | 0.007 | 1.70 (1.11–2.61) | 0.016 |
| Urine cotinine | |||||||||
| Per 1000 ng/mL | 212/4737 | 1.46 (1.25–1.71) | <0.001 | 1.41 (1.20–1.65) | <0.001 | 1.42 (1.21–1.66) | <0.001 | 1.39 (1.18–1.64) | <0.001 |
| Never smokers | 137/3407 | ref | ref | ref | ref | ||||
| Former smokers | 10/227 | 1.33 (0.70–2.54) | 0.381 | 1.24 (0.65–2.37) | 0.518 | 1.31 (0.68–2.50) | 0.418 | 1.30 (0.68–2.48) | 0.433 |
| Light current smokers | 29/552 | 1.56 (1.05–2.34) | 0.029 | 1.57 (1.05–2.35) | 0.028 | 1.58 (1.05–2.38) | 0.027 | 1.54 (1.02–2.32) | 0.038 |
| Heavy current smokers | 36/551 | 2.09 (1.43–3.03) | <0.001 | 1.90 (1.30–2.79) | 0.001 | 1.98 (1.35–2.92) | 0.001 | 1.90 (1.28–2.81) | 0.001 |
Model 1: Age and sex. Model 2: Model 1 plus history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol. Model 3: Model 2 plus body mass index, alcohol consumption, glucose, and estimated glomerular filtration rate (as calculated using the Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation). Model 4: Model 3 plus loge high‐sensitivity C‐reactive protein. CI indicates confidence interval; HR, hazard ratio.
Table 5.
Prospective Associations of Smoking Exposure With Development of Stroke
| Smoking Exposure | Events/Total | Model 1 | Model 2 | Model 3 | Model 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | ||
| Self‐reported smoking | |||||||||
| Never smokers | 23/1458 | ref | ref | ref | ref | ||||
| Former smokers | 31/1997 | 0.68 (0.39–1.19) | 0.178 | 0.68 (0.39–1.19) | 0.172 | 0.71 (0.40–1.24) | 0.227 | 0.68 (0.39–1.20) | 0.184 |
| Light current smokers | 7/495 | 0.94 (0.40–2.19) | 0.878 | 1.00 (0.43–2.34) | 0.995 | 1.09 (0.46–2.57) | 0.851 | 1.02 (0.43–2.41) | 0.965 |
| Heavy current smokers | 22/787 | 2.39 (1.31–4.37) | 0.005 | 2.33 (1.27–4.26) | 0.006 | 2.49 (1.35–4.59) | 0.003 | 2.15 (1.16–4.00) | 0.015 |
| Urine cotinine | |||||||||
| Per 1000 ng/mL | 83/4737 | 1.51 (1.17–1.96) | 0.002 | 1.56 (1.20–2.03) | 0.001 | 1.59 (1.22–2.07) | 0.001 | 1.50 (1.14–1.96) | 0.003 |
| Never smokers | 53/3407 | ref | ref | ref | ref | ||||
| Former smokers | 4/227 | 1.45 (0.52–4.02) | 0.474 | 1.53 (0.55–4.24) | 0.416 | 1.66 (0.60–4.61) | 0.334 | 1.58 (0.57–4.41) | 0.380 |
| Light current smokers | 13/552 | 2.02 (1.09–3.72) | 0.024 | 2.02 (1.09–3.72) | 0.025 | 2.12 (1.14–3.93) | 0.017 | 1.99 (1.07–3.70) | 0.029 |
| Heavy current smokers | 13/551 | 2.25 (1.21–4.18) | 0.010 | 2.41 (1.29–4.52) | 0.006 | 2.56 (1.36–4.83) | 0.004 | 2.21 (1.16–4.20) | 0.016 |
Model 1: Age and sex. Model 2: Model 1 plus history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol. Model 3: Model 2 plus body mass index, alcohol consumption, glucose, and estimated glomerular filtration rate (as calculated using the Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation). Model 4: Model 3 plus loge high‐sensitivity C‐reactive protein. CI indicates confidence interval; HR, hazard ratio.
Table 6.
Prospective Associations of Smoking Exposure With Development of Cardiovascular Disease on Exclusion of First 2 Years of Follow‐Up
| Smoking Exposure | Events/Total | Model 1 | Model 2 | Model 3 | Model 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | ||
| Self‐reported smoking | |||||||||
| Never smokers | 53/1409 | ref | ref | ref | ref | ||||
| Former smokers | 109/1942 | 0.98 (0.70–1.37) | 0.908 | 0.96 (0.69–1.35) | 0.836 | 0.98 (0.70–1.38) | 0.917 | 0.97 (0.69–1.37) | 0.870 |
| Light current smokers | 21/472 | 1.13 (0.68–1.87) | 0.646 | 1.15 (0.69–1.91) | 0.591 | 1.18 (0.71–1.97) | 0.528 | 1.13 (0.68–1.89) | 0.633 |
| Heavy current smokers | 50/753 | 2.14 (1.45–3.17) | <0.001 | 1.99 (1.34–2.96) | 0.001 | 2.02 (1.35–3.01) | 0.001 | 1.88 (1.25–2.82) | 0.002 |
| Urine cotinine | |||||||||
| Per 1000 ng/mL | 233/4576 | 1.40 (1.20–1.64) | <0.001 | 1.38 (1.17–1.62) | <0.001 | 1.38 (1.17–1.62) | <0.001 | 1.34 (1.13–1.58) | <0.001 |
| Never smokers | 155/3305 | ref | ref | ref | ref | ||||
| Former smokers | 14/218 | 1.75 (1.01–3.02) | 0.046 | 1.67 (0.96–2.91) | 0.068 | 1.74 (1.00–3.03) | 0.049 | 1.71 (0.99–2.98) | 0.056 |
| Light current smokers | 28/528 | 1.41 (0.94–2.12) | 0.094 | 1.41 (0.94–2.12) | 0.093 | 1.42 (0.95–2.14) | 0.091 | 1.37 (0.91–2.06) | 0.134 |
| Heavy current smokers | 36/525 | 2.00 (1.38–2.89) | <0.001 | 1.94 (1.33–2.82) | 0.001 | 1.98 (1.35–2.90) | <0.001 | 1.84 (1.25–2.71) | 0.002 |
Model 1: Age and sex. Model 2: Model 1 plus history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol. Model 3: Model 2 plus body mass index, alcohol consumption, glucose, and estimated glomerular filtration rate (as calculated using the Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation). Model 4: Model 3 plus loge high‐sensitivity C‐reactive protein. CI indicates confidence interval; HR, hazard ratio.
Table 7.
Prospective Associations of Smoking Exposure With Development of Cardiovascular Disease on Exclusion of Participants With a History of Diabetes Mellitus at Baseline
| Smoking Exposure | Events/Total | Model 1 | Model 2 | Model 3 | Model 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | ||
| Self‐reported smoking | |||||||||
| Never smokers | 60/1389 | ref | ref | ref | ref | ||||
| Former smokers | 114/1890 | 0.88 (0.64–1.22) | 0.452 | 0.88 (0.64–1.22) | 0.456 | 0.88 (0.63–1.21) | 0.424 | 0.86 (0.62–1.20) | 0.378 |
| Light current smokers | 28/467 | 1.33 (0.85–2.08) | 0.220 | 1.39 (0.88–2.19) | 0.153 | 1.39 (0.88–2.18) | 0.160 | 1.33 (0.84–2.10) | 0.221 |
| Heavy current smokers | 62/755 | 2.16 (1.51–3.10) | <0.001 | 1.98 (1.37–2.84) | <0.001 | 1.98 (1.37–2.85) | <0.001 | 1.80 (1.24–2.61) | 0.002 |
| Urine cotinine | |||||||||
| Per 1000 ng/mL | 264/4501 | 1.50 (1.31–1.72) | <0.001 | 1.47 (1.27–1.69) | <0.001 | 1.47 (1.28–1.70) | <0.001 | 1.41 (1.22–1.64) | <0.001 |
| Never smokers | 165/3235 | ref | ref | ref | ref | ||||
| Former smokers | 16/218 | 1.82 (1.09–3.05) | 0.022 | 1.82 (1.09–3.05) | 0.023 | 1.87 (1.12–3.15) | 0.017 | 1.84 (1.10–3.09) | 0.021 |
| Light current smokers | 38/521 | 1.75 (1.23–2.49) | 0.002 | 1.73 (1.22–2.47) | 0.002 | 1.73 (1.21–2.47) | 0.003 | 1.65 (1.15–2.36) | 0.006 |
| Heavy current smokers | 45/527 | 2.26 (1.62–3.17) | <0.001 | 2.19 (1.56–3.09) | <0.001 | 2.24 (1.58–3.17) | <0.001 | 2.03 (1.43–2.89) | <0.001 |
Model 1: Age and sex. Model 2: Model 1 plus systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol. Model 3: Model 2 plus body mass index, alcohol consumption, glucose, and estimated glomerular filtration rate (as calculated using the Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation). Model 4: Model 3 plus loge high‐sensitivity C‐reactive protein. CI indicates confidence interval; HR, hazard ratio.
Table 8.
Prospective Associations of Smoking Exposure With Development of Cardiovascular Disease on Exclusion of Participants on Cholesterol‐Lowering Medication
| Smoking Exposure | Events/Total | Model 1 | Model 2 | Model 3 | Model 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | ||
| Self‐reported smoking | |||||||||
| Never smokers | 62/1421 | ref | ref | ref | ref | ||||
| Former smokers | 121/1937 | 0.91 (0.66–1.24) | 0.542 | 0.90 (0.66–1.23) | 0.513 | 0.91 (0.66–1.25) | 0.549 | 0.90 (0.65–1.23) | 0.498 |
| Light current smokers | 28/481 | 1.27 (0.81–1.99) | 0.291 | 1.33 (0.85–2.08) | 0.219 | 1.35 (0.86–2.13) | 0.189 | 1.29 (0.82–2.03) | 0.272 |
| Heavy current smokers | 64/772 | 2.22 (1.56–3.17) | <0.001 | 2.06 (1.44–2.95) | <0.001 | 2.10 (1.46–3.01) | <0.001 | 1.92 (1.33–2.76) | <0.001 |
| Urine cotinine | |||||||||
| Per 1000 ng/mL | 275/4611 | 1.47 (1.29–1.69) | <0.001 | 1.45 (1.26–1.66) | <0.001 | 1.46 (1.27–1.68) | <0.001 | 1.40 (1.21–1.62) | <0.001 |
| Never smokers | 175/3313 | ref | ref | ref | ref | ||||
| Former smokers | 16/219 | 1.79 (1.07–2.99) | 0.027 | 1.73 (1.03–2.91) | 0.037 | 1.80 (1.07–3.03) | 0.026 | 1.76 (1.05–2.96) | 0.031 |
| Light current smokers | 39/540 | 1.69 (1.19–2.40) | 0.003 | 1.70 (1.20–2.41) | 0.003 | 1.71 (1.20–2.43) | 0.003 | 1.63 (1.14–2.32) | 0.007 |
| Heavy current smokers | 45/539 | 2.12 (1.52–2.96) | <0.001 | 2.06 (1.47–2.90) | <0.001 | 2.13 (1.51–3.00) | <0.001 | 1.94 (1.37–2.75) | <0.001 |
Model 1: Age and sex. Model 2: Model 1 plus history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol. Model 3: Model 2 plus body mass index, alcohol consumption, glucose, and estimated glomerular filtration rate (as calculated using the Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation). Model 4: Model 3 plus loge high‐sensitivity C‐reactive protein. CI indicates confidence interval; HR, hazard ratio.
Table 9.
Prospective Associations of Smoking Exposure (Urine Cotinine Multiplied by Urinary Volume) With Development of Cardiovascular Disease
| Smoking Exposure (ng) | Events/Total | Model 1 | Model 2 | Model 3 | Model 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | ||
| Tertile 1 | 162/2837 | ref | ref | ref | ref | ||||
| Tertile 2 | 16/318 | 1.02 (0.61–1.70) | 0.945 | 1.00 (0.60–1.67) | 0.998 | 0.99 (0.59–1.66) | 0.969 | 0.96 (0.57–1.60) | 0.866 |
| Tertile 3 | 118/1577 | 1.68 (1.32–2.13) | <0.001 | 1.63 (1.28–2.08) | <0.001 | 1.68 (1.31–2.14) | <0.001 | 1.59 (1.24–2.03) | <0.001 |
Model 1: Age and sex. Model 2: Model 1 plus history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol. Model 3: Model 2 plus body mass index, alcohol consumption, glucose, and estimated glomerular filtration rate (as calculated using the Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation). Model 4: Model 3 plus loge high‐sensitivity C‐reactive protein. CI indicates confidence interval; HR, hazard ratio.
Table 10.
Prospective Associations of Smoking Exposure (Urine Cotinine/Urine Creatinine Ratio) With Development of Cardiovascular Disease
| Smoking Exposure, ng/mmol per L | Events/Total | Model 1 | Model 2 | Model 3 | Model 4 | ||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | HR (95% CI) | P Value | ||
| Tertile 1 | 159/2785 | ref | ref | ref | ref | ||||
| Tertile 2 | 16/320 | 0.96 (0.57–1.61) | 0.880 | 0.97 (0.58–1.62) | 0.907 | 0.96 (0.57–1.61) | 0.871 | 0.93 (0.55–1.55) | 0.771 |
| Tertile 3 | 118/1552 | 1.72 (1.35–2.19) | <0.001 | 1.67 (1.31–2.13) | <0.001 | 1.72 (1.34–2.19) | <0.001 | 1.63 (1.27–2.09) | <0.001 |
Model 1: Age and sex. Model 2: Model 1 plus history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol. Model 3: Model 2 plus body mass index, alcohol consumption, glucose, and estimated glomerular filtration rate (as calculated using the Chronic Kidney Disease Epidemiology Collaboration combined creatinine–cystatin C equation). Model 4: Model 3 plus loge high‐sensitivity C‐reactive protein. CI indicates confidence interval; HR, hazard ratio.
Figure 3.
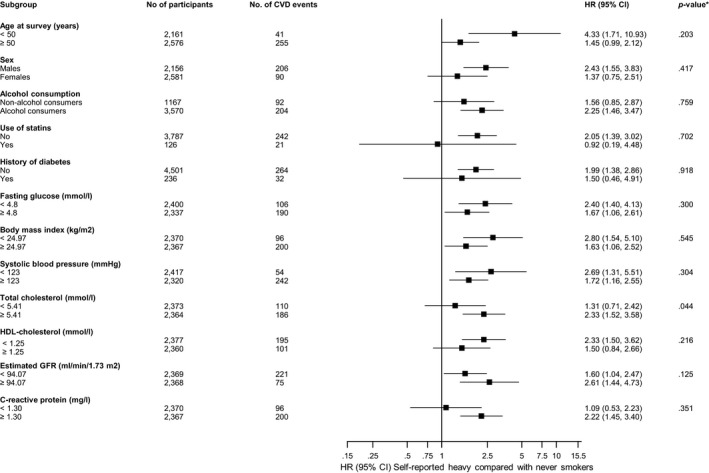
Hazard ratios for self‐reported smoking and cardiovascular disease risk by several participant‐level characteristics. Hazard ratios were adjusted for age, sex, history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein cholesterol; CI indicates confidence interval (bars); CVD, cardiovascular disease; HDL, high‐density lipoprotein; HR, hazard ratio. *P value for interaction; cutoffs used for fasting glucose, body mass index, systolic blood pressure, total cholesterol, HDL cholesterol, estimated glomerular filtration rate (GFR), and C‐reactive protein are median values.
Figure 4.
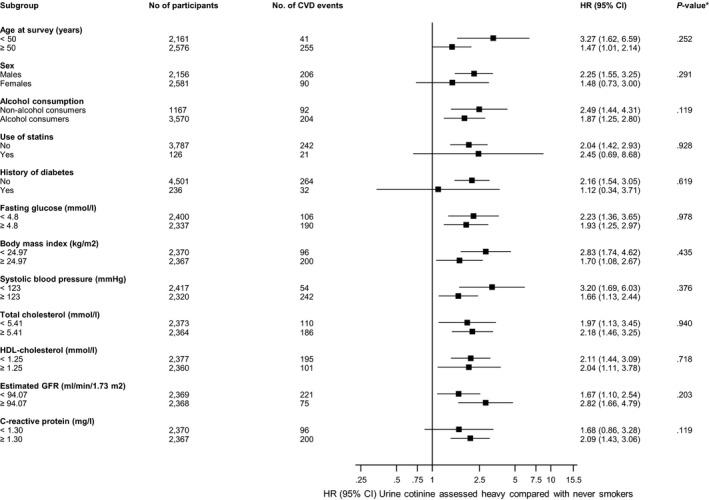
Hazard ratios for urine cotinine assessed smoking and cardiovascular disease risk by several participant level characteristics. Hazard ratios were adjusted for age, sex, history of diabetes mellitus, systolic blood pressure, total cholesterol, and high‐density lipoprotein‐cholesterol; CI indicates confidence interval (bars); CVD, cardiovascular disease; HDL, high‐density lipoprotein; HR, hazard ratio. *P value for interaction; cutoffs used for fasting glucose, body mass index, systolic blood pressure, total cholesterol, HDL cholesterol, estimated glomerular filtration rate (GFR), and C‐reactive protein are median values.
Smoking Exposure and CVD Risk Prediction
A CVD risk prediction model comprising traditional risk factors (excluding self‐reported smoking) yielded a C‐index of 0.8002 (95% CI, 0.7799–0.8205). On addition of information on urine cotinine concentration–assessed smoking status to this prognostic model, the C‐index was 0.8100 (0.7905–0.8294), representing a small significant increase of 0.0098 (0.0031–0.0164; P=0.004). There was no improvement in the classification of participants into predicted CVD risk categories (net reclassification improvement, 0.28%, −4.50 to 5.06%; P=0.908), whereas there was significant improvement if reclassification was assessed as continuous variable, with an integrated discrimination improvement of 0.0101 (95% CI, 0.0042–0.0161; P=0.001).
To compare the predictive ability of urine cotinine assessed smoking status with that of self‐reported smoking status in the same sample, information on self‐reported smoking status was added to the model containing conventional risk factors. There was a C‐index change of 0.0111 (95% CI, 0.0042–0.0179 P=0.002). After taking into account inappropriate reclassification, there was no significant improvement in the classification of participants into the predicted CVD risk categories (net reclassification improvement, 1.86%, −2.92 to 6.64%; P=0.447), whereas there was significant improvement if reclassification was assessed as a continuous variable, with an integrated discrimination improvement of 0.0102 (0.0040–0.0164; P=0.001).
Discussion
Summary of Main Findings
We have evaluated the associations of smoking exposure (as assessed by self‐reports and urine cotinine) with the risk of CVD in a population‐based prospective cohort study comprising white men and women without a history of CVD at baseline. By using cotinine‐based measurements of smoking exposure, 61 (1.8%) of 3455 self‐reported never and former smokers could be reclassified as active smokers (misclassification rate of 4.8%). More than half of urine cotinine–assessed never smokers were classified as former smokers by self‐reports, which reflects evidence questioning the reliability of cotinine in distinguishing between never smoking and former smoking.31 In addition, we observed a strong correlation between self‐reported smoking and urine cotinine–assessed smoking status. However, the kappa value suggested weak agreement between self‐report and urine cotinine in smoking status classification. Compared with self‐reported never smokers, self‐reported heavy current smokers had an increased risk of CVD, and this association was independent of several established cardiovascular risk factors and other potential confounders. However, the associations of self‐reported former and light current smoking exposures with CVD were not significant. On evaluation of the association between smoking status as assessed by urine cotinine and risk of CVD, former smokers, light current smokers, and heavy current smokers were each independently associated with an increased risk of CVD, and this was consistent with a linear dose‐response relationship. The associations were similar in several sensitivity analyses. The magnitudes of the associations were generally similar for the specific end points of CHD and stroke, except for modest associations of cotinine‐assessed former smoking with risk of CHD and stroke, which could be attributed to the low event rate in that smoking exposure category. Though the association between self‐reported smoking and CVD risk was significantly modified by total cholesterol, the associations remained generally consistent across several clinically relevant subgroups such as age and sex for both exposures. The stronger association between smoking status and CVD risk in participants with high cholesterol levels (≥5.41 mmol/L) may be consistent with established evidence that shows that smoking is associated with a more atherogenic lipid profile (higher total cholesterol and triglyceride with lower high‐density lipoprotein cholesterol levels) and increases the risk of CHD in people with high cholesterol levels and other risk factors that increase the risk of CVD.41, 42 Though there was no significant evidence of effect modification by sex on the associations, the associations were more extreme for men compared with women, which may reflect evidence that the attributable risk of CHD as a result of smoking is generally lower in women than in men.43 However, evidence suggests that smoking has a much larger relative detrimental impact on CHD in women, although the detrimental effect of smoking on CVD in women with respect to men has mostly been conflicting in studies and may be related to factors such as differences in smoking habits in populations and cessation during follow‐up.43 Given the absence of significant evidence of effect modification by sex in our analyses and the low event rate in women, the current results should be interpreted with caution. Finally, addition of urine cotinine assessed smoking status was not associated with a clinically meaningful improvement in assessment of CVD risk, although the change was statistically significant, which could be attributed to the relatively large sample size we employed. Additional analyses in the same set of participants showed the improvement provided by self‐reported smoking exposure in prediction of CVD risk was similar to that of urine cotinine, with no obvious superiority of urine cotinine. Both exposures did not improve the reclassification of participants across clinical risk categories currently recommended to inform decisions about the initiation of preventive treatment.38
Comparison With Previous Work
We are unable to directly compare the current findings with previous work, as our search of the literature did not identify any prospective study that has assessed and also compared the associations of smoking exposure as assessed by self‐reports and urine cotinine with the risk of CVD. Of note, Delgado and colleagues compared the association of plasma cotinine and cigarette smoking in pack‐years with cardiovascular and all‐cause mortality.15 Both exposures were significantly associated with both outcomes, and the magnitude of the associations were higher for cotinine compared to pack‐years, findings that were consistent with the results of our study. In a recent cross‐sectional analysis, increased serum cotinine levels were demonstrated to be associated with an increased risk of subclinical myocardial injury.14 In our study, we found a misclassification rate of ≈5%. In comparison, previous studies have reported misclassification rates for current smokers reporting themselves to be nonsmokers to range from 0.8% to 15.3%.31, 40 The strong correlation observed between the 2 exposures is consistent with that of a previous study.31 Consistent with our findings, a previous study demonstrated serum cotinine, pack‐years, or self‐reported smoking to significantly improve mortality risk prediction beyond traditional risk factors.15 The majority of previously published studies have evaluated the associations between cotinine‐assessed passive smoke exposure and the risk of CVD and have suggested dose‐response relationships.44, 45, 46
Possible Explanations for Findings
Our findings indicate that smoking exposure is associated with an increased risk of CVD, which is consistent with established evidence.4, 5, 6, 7 Although there was no marked superiority of urine cotinine over self‐reported smoking status in the associations, the stronger magnitude of the associations using cotinine‐assessed smoking exposure and the dose‐dependent nature of the relationship suggest smoking exposure using urine cotinine may be a more reliable indicator than self‐reported smoking. Indeed, it has been shown that serum cotinine is better than self‐report when quantifying the risks with several outcomes.47 Cotinine measurements could be a more reliable way of quantifying risks than are self‐reports for the following reasons: (1) potential for individuals to underreport smoking exposure due to actual difficulty in recalling or deliberate denial—misclassification rates for current smokers reporting themselves to be nonsmokers have been reported to range from 0.8% to 15.3%40; and (2) differences in cigarettes smoked and smoke inhalation, which result in differences in smoking exposure among individuals. Furthermore, it has been shown in a recent study that quantification of smoking exposure using cotinine measurements led to significant reclassification compared with self‐report.31 Another study reported absence of a correlation between self‐reported cigarette smoking and measured cotinine concentrations.15 The overall findings suggest that it may be more reliable to assess smoking exposure using objective measures such as cotinine assessed in the saliva, hair, urine, or blood. Although cotinine, a major metabolite of nicotine, has long been used as a marker of smoking exposure, its use has some drawbacks. First, there is variability in the amount of nicotine that is converted to cotinine, which ranges between 55% and 92%.48 Second, there is between‐person variation in rates of metabolism and excretion of cotinine. Third, cotinine cannot be used to reliably distinguish between never smoking and former smoking.31 Fourth, cotinine may not be useful for distinguishing between passive smoking and nonsmoking exposure groups.49 Fifth, cotinine concentrations reflect smoking exposure of several days and may not provide accurate estimates if there is a break in smoking. Sixth, genetic factors that control nicotine metabolism may influence cotinine concentrations.50 The interrater reliability between self‐report and urine cotinine in smoking status classification was weak, which may reflect (1) some of the limitations of urine cotinine in distinguishing between some smoking exposure categories, or (2) that urine cotinine may indeed be a more reliable measure of smoking status than is self‐reported smoking. However, further investigation is needed. Findings from our risk prediction analysis showed that urine cotinine–assessed smoking exposure augmented CVD risk prediction, which was comparable to that of self‐reported smoking status; this and the observation of a graded association between urine cotinine and CVD risk suggests that urine cotinine–assessed smoking status is potentially suitable for population‐level risk assessment.
Strengths and Limitations
This is the first comparative prospective assessment of the associations of smoking exposure as measured by self‐reports and urine cotinine with the risk of composite CVD as well as specific end points of CHD and stroke. We also compared the potential utility of both exposures for CVD risk prediction. Other strengths include the relatively large sample size, which was also representative of the general population; the extended follow‐up enabling time‐to‐event analyses; exclusion of individuals with a baseline history of CVD; and measurements on a comprehensive panel of cardiovascular risk markers that enabled adequate adjustment for potential confounding. The cutoffs we employed to distinguish between no smoking and active smoking were appropriate (have high sensitivity and specificity values), conservative, and have been used in several previous studies.30, 31, 32, 33, 49 To enhance the validity of the findings, we restricted analyses to people with complete information on exposures, risk factors, and outcomes. The findings were robust to exclusion of the first 2 years of follow‐up, participants with a history of diabetes mellitus at baseline, or participants on regular statin medication. In addition to the previously mentioned drawbacks to the use of cotinine as a measure of smoking exposure, there were some other limitations to our study. First, our analyses were based on a single measure of cotinine, introducing the possibility of regression dilution bias and underestimation of the association between cotinine assessed smoking exposure and CVD risk. Second, there was a potential for residual confounding due to other unmeasured covariates and errors in measurements of risk markers. Third, we classified urine cotinine–assessed light and current smokers on the basis of the median urine cotinine values of current smokers as reported in a previous study.31 Given the limited evidence on this approach, there is a possibility of misclassification. Finally, the findings may not be generalizable to individuals of different ethnicities. Irrespective of the limitations, our overall findings suggest that urine cotinine may be equal to or better than self‐reported smoking for the assessment of smoking exposure.
Conclusion
Smoking status as assessed by self‐reports and urine cotinine is associated with risk of CVD. However, the nature of the association of urine cotinine with CVD is consistent with a dose‐response relationship. The ability of urine cotinine to improve CVD risk assessment is similar to that provided by self‐reported smoking status.
Sources of Funding
The cotinine measurements were supported by the Food Biomarkers Alliance (FoodBAll) project, which is funded by the BIO‐NH call under the Joint Programming Initiative, “a Healthy Diet for a Healthy Life” (grant number 529051002). The FoodBAll project is funded nationally by the respective Research Councils. The Dutch Kidney Foundation supported the infrastructure of the PREVEND program from 1997 to 2003 (grant E.033). The University Medical Center Groningen supported the infrastructure from 2003 to 2006. Dade Behring, Ausam, Roche, and Abbott financed laboratory equipment and reagents by which various laboratory determinations could be performed. The Dutch Heart Foundation supported studies on lipid metabolism (grant 2001‐005). The contents of this article are solely the responsibility of the authors and do not represent the views of the sponsors. The funder had no role in design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the article. Dr Setor K. Kunutsor acknowledges support from the National Institute for Health Research Biomedical Research Centre at the University Hospitals Bristol NHS Foundation Trust and the University of Bristol. The views expressed in this publication are those of the author and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
Disclosures
None.
Supporting information
Table S1. STROBE 2007 Statement—Checklist of Items That Should Be Included in Reports of Cohort Studies
(J Am Heart Assoc. 2018;7:e008726 DOI: 10.1161/JAHA.118.008726.)29720504
References
- 1. Roth GA, Johnson C, Abajobir A, Abd‐Allah F, Abera SF, Abyu G, Ahmed M, Aksut B, Alam T, Alam K, Alla F, Alvis‐Guzman N, Amrock S, Ansari H, Arnlov J, Asayesh H, Atey TM, Avila‐Burgos L, Awasthi A, Banerjee A, Barac A, Barnighausen T, Barregard L, Bedi N, Belay Ketema E, Bennett D, Berhe G, Bhutta Z, Bitew S, Carapetis J, Carrero JJ, Malta DC, Castaneda‐Orjuela CA, Castillo‐Rivas J, Catala‐Lopez F, Choi JY, Christensen H, Cirillo M, Cooper L Jr, Criqui M, Cundiff D, Damasceno A, Dandona L, Dandona R, Davletov K, Dharmaratne S, Dorairaj P, Dubey M, Ehrenkranz R, El Sayed Zaki M, Faraon EJA, Esteghamati A, Farid T, Farvid M, Feigin V, Ding EL, Fowkes G, Gebrehiwot T, Gillum R, Gold A, Gona P, Gupta R, Habtewold TD, Hafezi‐Nejad N, Hailu T, Hailu GB, Hankey G, Hassen HY, Abate KH, Havmoeller R, Hay SI, Horino M, Hotez PJ, Jacobsen K, James S, Javanbakht M, Jeemon P, John D, Jonas J, Kalkonde Y, Karimkhani C, Kasaeian A, Khader Y, Khan A, Khang YH, Khera S, Khoja AT, Khubchandani J, Kim D, Kolte D, Kosen S, Krohn KJ, Kumar GA, Kwan GF, Lal DK, Larsson A, Linn S, Lopez A, Lotufo PA, El Razek HMA, Malekzadeh R, Mazidi M, Meier T, Meles KG, Mensah G, Meretoja A, Mezgebe H, Miller T, Mirrakhimov E, Mohammed S, Moran AE, Musa KI, Narula J, Neal B, Ngalesoni F, Nguyen G, Obermeyer CM, Owolabi M, Patton G, Pedro J, Qato D, Qorbani M, Rahimi K, Rai RK, Rawaf S, Ribeiro A, Safiri S, Salomon JA, Santos I, Santric Milicevic M, Sartorius B, Schutte A, Sepanlou S, Shaikh MA, Shin MJ, Shishehbor M, Shore H, Silva DAS, Sobngwi E, Stranges S, Swaminathan S, Tabares‐Seisdedos R, Tadele Atnafu N, Tesfay F, Thakur JS, Thrift A, Topor‐Madry R, Truelsen T, Tyrovolas S, Ukwaja KN, Uthman O, Vasankari T, Vlassov V, Vollset SE, Wakayo T, Watkins D, Weintraub R, Werdecker A, Westerman R, Wiysonge CS, Wolfe C, Workicho A, Xu G, Yano Y, Yip P, Yonemoto N, Younis M, Yu C, Vos T, Naghavi M, Murray C. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Global status report on noncommunicable diseases 2010. Geneva, Switzerland: World Health Organization; 2011. http://www.Who.Int/nmh/publications/ncd_report_full_en.pdf. Accessed January 23, 2018. [Google Scholar]
- 3. Wood D. Established and emerging cardiovascular risk factors. Am Heart J. 2001;141:S49–S57. [DOI] [PubMed] [Google Scholar]
- 4. Danaei G, Ding EL, Mozaffarian D, Taylor B, Rehm J, Murray CJ, Ezzati M. The preventable causes of death in the United States: comparative risk assessment of dietary, lifestyle, and metabolic risk factors. PLoS Med. 2009;6:e1000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Samet JM. Tobacco smoking: the leading cause of preventable disease worldwide. Thorac Surg Clin. 2013;23:103–112. [DOI] [PubMed] [Google Scholar]
- 6. Benowitz NL. Cigarette smoking and cardiovascular disease: pathophysiology and implications for treatment. Prog Cardiovasc Dis. 2003;46:91–111. [DOI] [PubMed] [Google Scholar]
- 7. Mons U, Muezzinler A, Gellert C, Schottker B, Abnet CC, Bobak M, de Groot L, Freedman ND, Jansen E, Kee F, Kromhout D, Kuulasmaa K, Laatikainen T, O'Doherty MG, Bueno‐de‐Mesquita B, Orfanos P, Peters A, van der Schouw YT, Wilsgaard T, Wolk A, Trichopoulou A, Boffetta P, Brenner H; CHANCES Consortium . Impact of smoking and smoking cessation on cardiovascular events and mortality among older adults: meta‐analysis of individual participant data from prospective cohort studies of the CHANCES consortium. BMJ. 2015;350:h1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhat VM, Cole JW, Sorkin JD, Wozniak MA, Malarcher AM, Giles WH, Stern BJ, Kittner SJ. Dose‐response relationship between cigarette smoking and risk of ischemic stroke in young women. Stroke. 2008;39:2439–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Connor Gorber S, Schofield‐Hurwitz S, Hardt J, Levasseur G, Tremblay M. The accuracy of self‐reported smoking: a systematic review of the relationship between self‐reported and cotinine‐assessed smoking status. Nicotine Tob Res. 2009;11:12–24. [DOI] [PubMed] [Google Scholar]
- 10. Kang HG, Kwon KH, Lee IW, Jung B, Park EC, Jang SI. Biochemically‐verified smoking rate trends and factors associated with inaccurate self‐reporting of smoking habits in Korean women. Asian Pac J Cancer Prev. 2013;14:6807–6812. [DOI] [PubMed] [Google Scholar]
- 11. Benowitz NL. Cotinine as a biomarker of environmental tobacco smoke exposure. Epidemiol Rev. 1996;18:188–204. [DOI] [PubMed] [Google Scholar]
- 12. Florescu A, Ferrence R, Einarson T, Selby P, Soldin O, Koren G. Methods for quantification of exposure to cigarette smoking and environmental tobacco smoke: focus on developmental toxicology. Ther Drug Monit. 2009;31:14–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verification SSoB . Biochemical verification of tobacco use and cessation. Nicotine Tob Res. 2002;4:149–159. [DOI] [PubMed] [Google Scholar]
- 14. Ali M, Li Y, O'Neal WT, Soliman EZ. Tobacco exposure as determined by serum cotinine and subclinical myocardial injury in individuals free from cardiovascular disease. Am J Cardiol. 2017;120:1114–1117. [DOI] [PubMed] [Google Scholar]
- 15. Delgado G, Siekmeier R, Kramer BK, Marz W, Kleber ME. Cotinine as a marker for risk prediction in the Ludwigshafen Risk and Cardiovascular Health Study. Respir Physiol Neurobiol. 2015;209:17–22. [DOI] [PubMed] [Google Scholar]
- 16. von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: guidelines for reporting observational studies. J Clin Epidemiol. 2008;61:344–349. [DOI] [PubMed] [Google Scholar]
- 17. Lambers Heerspink HJ, Brantsma AH, de Zeeuw D, Bakker SJ, de Jong PE, Gansevoort RT; PREVEND Study Group . Albuminuria assessed from first‐morning‐void urine samples versus 24‐hour urine collections as a predictor of cardiovascular morbidity and mortality. Am J Epidemiol. 2008;168:897–905. [DOI] [PubMed] [Google Scholar]
- 18. Kunutsor SK, Bakker SJ, Kootstra‐Ros JE, Gansevoort RT, Dullaart RP. Circulating gamma glutamyltransferase and prediction of cardiovascular disease. Atherosclerosis. 2014;238:356–364. [DOI] [PubMed] [Google Scholar]
- 19. Visser ST, Schuiling‐Veninga CC, Bos JH, van de Jong‐ den Berg LT, Postma MJ. The population‐based prescription database IADB.nl: its development, usefulness in outcomes research and challenges. Expert Rev Pharmacoecon Outcomes Res. 2013;13:285–292. [DOI] [PubMed] [Google Scholar]
- 20. Borggreve SE, Hillege HL, Dallinga‐Thie GM, de Jong PE, Wolffenbuttel BH, Grobbee DE, van Tol A, Dullaart RP; Group PS . High plasma cholesteryl ester transfer protein levels may favour reduced incidence of cardiovascular events in men with low triglycerides. Eur Heart J. 2007;28:1012–1018. [DOI] [PubMed] [Google Scholar]
- 21. Dullaart RP, Perton F, van der Klauw MM, Hillege HL, Sluiter WJ; Group PS . High plasma lecithin:cholesterol acyltransferase activity does not predict low incidence of cardiovascular events: possible attenuation of cardioprotection associated with high HDL cholesterol. Atherosclerosis. 2010;208:537–542. [DOI] [PubMed] [Google Scholar]
- 22. Corsetti JP, Bakker SJ, Sparks CE, Dullaart RP. Apolipoprotein A‐II influences apolipoprotein E‐linked cardiovascular disease risk in women with high levels of HDL cholesterol and C‐reactive protein. PLoS One. 2012;7:e39110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kunutsor SK, Bakker SJ, Kootstra‐Ros JE, Blokzijl H, Gansevoort RT, Dullaart RP. Inverse linear associations between liver aminotransferases and incident cardiovascular disease risk: the PREVEND study. Atherosclerosis. 2015;243:138–147. [DOI] [PubMed] [Google Scholar]
- 24. Kunutsor SK, Bakker SJ, Kootstra‐Ros JE, Gansevoort RT, Gregson J, Dullaart RP. Serum alkaline phosphatase and risk of incident cardiovascular disease: interrelationship with high sensitivity C‐reactive protein. PLoS One. 2015;10:e0132822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Inker LA, Schmid CH, Tighiouart H, Eckfeldt JH, Feldman HI, Greene T, Kusek JW, Manzi J, Van Lente F, Zhang YL, Coresh J, Levey AS; Investigators C‐E . Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012;367:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults . Executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III). JAMA. 2001;285:2486–2497. [DOI] [PubMed] [Google Scholar]
- 27. Stricker BH, Herings RM. [Plea for the retention of the Dutch National Medical Registration (LMR) to provide reliable information regarding public health and healthcare]. Ned Tijdschr Geneeskd. 2006;150:1916–1917. [PubMed] [Google Scholar]
- 28. Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33:159–174. [PubMed] [Google Scholar]
- 29. Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. New York, NY: Springer; 2000:39–77. [Google Scholar]
- 30. Hobbs SD, Wilmink AB, Adam DJ, Bradbury AW. Assessment of smoking status in patients with peripheral arterial disease. J Vasc Surg. 2005;41:451–456. [DOI] [PubMed] [Google Scholar]
- 31. Hellemons ME, Sanders JS, Seelen MA, Gans RO, Muller Kobold AC, van Son WJ, Postmus D, Navis GJ, Bakker SJ. Assessment of cotinine reveals a dose‐dependent effect of smoking exposure on long‐term outcomes after renal transplantation. Transplantation. 2015;99:1926–1932. [DOI] [PubMed] [Google Scholar]
- 32. Holl RW, Grabert M, Heinze E, Debatin KM. Objective assessment of smoking habits by urinary cotinine measurement in adolescents and young adults with type 1 diabetes. Reliability of reported cigarette consumption and relationship to urinary albumin excretion. Diabetes Care. 1998;21:787–791. [DOI] [PubMed] [Google Scholar]
- 33. Zielinska‐Danch W, Wardas W, Sobczak A, Szoltysek‐Boldys I. Estimation of urinary cotinine cut‐off points distinguishing non‐smokers, passive and active smokers. Biomarkers. 2007;12:484–496. [DOI] [PubMed] [Google Scholar]
- 34. Royston P, Ambler G, Sauerbrei W. The use of fractional polynomials to model continuous risk variables in epidemiology. Int J Epidemiol. 1999;28:964–974. [DOI] [PubMed] [Google Scholar]
- 35. Greenland P, Alpert JS, Beller GA, Benjamin EJ, Budoff MJ, Fayad ZA, Foster E, Hlatky MA, Hodgson JM, Kushner FG, Lauer MS, Shaw LJ, Smith SC Jr, Taylor AJ, Weintraub WS, Wenger NK, Jacobs AK; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines . 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2010;122:e584–e636. [DOI] [PubMed] [Google Scholar]
- 36. Harrell FE Jr, Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med. 1996;15:361–387. [DOI] [PubMed] [Google Scholar]
- 37. D'Agostino RB Sr, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, Kannel WB. General cardiovascular risk profile for use in primary care: the Framingham heart study. Circulation. 2008;117:743–753. [DOI] [PubMed] [Google Scholar]
- 38. Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd‐Jones DM, McBride P, Schwartz JS, Shero ST, Smith SC Jr, Watson K, Wilson PW; American College of Cardiology/American Heart Association Task Force on Practice Guidelines . 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2889–2934. [DOI] [PubMed] [Google Scholar]
- 39. Pencina MJ, D'Agostino RB Sr, D'Agostino RB Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172; discussion 207–112. [DOI] [PubMed] [Google Scholar]
- 40. Wells AJ, English PB, Posner SF, Wagenknecht LE, Perez‐Stable EJ. Misclassification rates for current smokers misclassified as nonsmokers. Am J Public Health. 1998;88:1503–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gossett LK, Johnson HM, Piper ME, Fiore MC, Baker TB, Stein JH. Smoking intensity and lipoprotein abnormalities in active smokers. J Clin Lipidol. 2009;3:372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chelland Campbell S, Moffatt RJ, Stamford BA. Smoking and smoking cessation—the relationship between cardiovascular disease and lipoprotein metabolism: a review. Atherosclerosis. 2008;201:225–235. [DOI] [PubMed] [Google Scholar]
- 43. Bolego C, Poli A, Paoletti R. Smoking and gender. Cardiovasc Res. 2002;53:568–576. [DOI] [PubMed] [Google Scholar]
- 44. Hamer M, Stamatakis E, Kivimaki M, Lowe GD, Batty GD. Objectively measured secondhand smoke exposure and risk of cardiovascular disease: what is the mediating role of inflammatory and hemostatic factors? J Am Coll Cardiol. 2010;56:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Whincup PH, Gilg JA, Emberson JR, Jarvis MJ, Feyerabend C, Bryant A, Walker M, Cook DG. Passive smoking and risk of coronary heart disease and stroke: prospective study with cotinine measurement. BMJ. 2004;329:200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tunstall‐Pedoe H, Brown CA, Woodward M, Tavendale R. Passive smoking by self report and serum cotinine and the prevalence of respiratory and coronary heart disease in the Scottish heart health study. J Epidemiol Community Health. 1995;49:139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perez‐Stable EJ, Benowitz NL, Marin G. Is serum cotinine a better measure of cigarette smoking than self‐report? Prev Med. 1995;24:171–179. [DOI] [PubMed] [Google Scholar]
- 48. Benowitz NL, Jacob P III. Metabolism of nicotine to cotinine studied by a dual stable isotope method. Clin Pharmacol Ther. 1994;56:483–493. [DOI] [PubMed] [Google Scholar]
- 49. Jung S, Lee IS, Kim SB, Moon CS, Jung JY, Kang YA, Park MS, Kim YS, Kim SK, Chang J, Kim EY. Urine cotinine for assessing tobacco smoke exposure in Korean: analysis of the Korea National Health and Nutrition Examination Survey (KNHANES). Tuberc Respir Dis (Seoul). 2012;73:210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Benowitz NL. Clinical pharmacology of nicotine: implications for understanding, preventing, and treating tobacco addiction. Clin Pharmacol Ther. 2008;83:531–541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. STROBE 2007 Statement—Checklist of Items That Should Be Included in Reports of Cohort Studies