

Abstract
Background
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a familial arrhythmogenic syndrome characterized by sudden death. There are several genetic forms of CPVT associated with mutations in genes encoding the cardiac ryanodine receptor (RyR2) and its auxiliary proteins including calsequestrin (CASQ2) and calmodulin (CaM). It has been suggested that impairment of the ability of RyR2 to stay closed (ie, refractory) during diastole may be a common mechanism for these diseases. Here, we explore the possibility of engineering CaM variants that normalize abbreviated RyR2 refractoriness for subsequent viral‐mediated delivery to alleviate arrhythmias in non–CaM‐related CPVT.
Methods and Results
To that end, we have designed a CaM protein (GSH‐M37Q; dubbed as therapeutic CaM or T‐CaM) that exhibited a slowed N‐terminal Ca dissociation rate and prolonged RyR2 refractoriness in permeabilized myocytes derived from CPVT mice carrying the CASQ2 mutation R33Q. This T‐CaM was introduced to the heart of R33Q mice through recombinant adeno‐associated viral vector serotype 9. Eight weeks postinfection, we performed confocal microscopy to assess Ca handling and recorded surface ECGs to assess susceptibility to arrhythmias in vivo. During catecholamine stimulation with isoproterenol, T‐CaM reduced isoproterenol‐promoted diastolic Ca waves in isolated CPVT cardiomyocytes. Importantly, T‐CaM exposure abolished ventricular tachycardia in CPVT mice challenged with catecholamines.
Conclusions
Our results suggest that gene transfer of T‐CaM by adeno‐associated viral vector serotype 9 improves myocyte Ca handling and alleviates arrhythmias in a calsequestrin‐associated CPVT model, thus supporting the potential of a CaM‐based antiarrhythmic approach as a therapeutic avenue for genetically distinct forms of CPVT.
Keywords: arrhythmia (mechanisms), calcium channel, calcium signaling, calmodulin, gene therapy
Subject Categories: Arrhythmias, Sudden Cardiac Death, Animal Models of Human Disease, Basic Science Research, Calcium Cycling/Excitation-Contraction Coupling
Clinical Perspective
What Is New?
Catecholaminergic polymorphic ventricular tachycardia is a life threatening inherited arrhythmia syndrome caused by mutations in the ryanodine receptor (RyR2) and its auxiliary proteins including calsequestrin 2 (CASQ2) and calmodulin (CaM).
The underlying cause of catecholaminergic polymorphic ventricular tachycardia involves the compromised ability of RyR2 to stay closed during diastole thereby resulting in aberrant arrhythmogenic intracellular Ca release.
Here we designed a CaM construct that combines enhanced Ca sensitivity with tighter RyR2 binding resulting in enhanced Ca‐dependent inhibition of RyR2.
What Are the Clinical Implications?
Gene transfer of this CaM construct (therapeutic CaM, TCaM) normalized myocyte Ca cycling and alleviated life‐threatening arrhythmias in vivo in mice affected by a mutation within another disease‐related protein (CASQ2).
Thus, TCaMs may provide a therapeutic strategy for multiple genetic forms of catecholaminergic polymorphic ventricular tachycardia.
Cardiac arrhythmia is a leading cause of mortality and morbidity worldwide.1, 2 Aberrant Ca handling, and alterations in the cardiac sarcoplasmic reticulum (SR) Ca release channel (ryanodine receptor, RyR2), in particular, is recognized as an important factor in the genesis of arrhythmia.3, 4 This link is especially evident in catecholaminergic polymorphic ventricular tachycardia (CPVT), an inherited arrhythmic syndrome caused by mutations in the RyR2 itself and its multiple accessory proteins including calsequestrin 2 (CASQ2) and calmodulin (CaM).5, 6
Interestingly, previous work from our and other laboratories have demonstrated that impairment of the ability of RyR2 to become refractory, ie, appropriately close after each systolic Ca release event, appears to be a common mechanism for various forms of genetic and acquired arrhythmias, including CPVT and post‐infarction ventricular fibrillation (VF).7, 8, 9 Specifically, shortened RyR2 refractoriness results in aberrant diastolic SR Ca release that synchronizes across the myocardium to induce triggered activity, thus, precipitating malignant arrhythmias.
Given its central role in Ca‐dependent arrhythmias, RyR2 is a logical target for the treatment of these arrhythmias.10, 11, 12, 13, 14 However, the development of effective and safe RyR2 based therapies is hindered by the complex nature of RyR2 regulation and myocyte Ca handling. For example, most of the current RyR2 targeting compounds act by reducing the overall RyR2 open probability (during both systole and diastole) making them inefficient or even detrimental in patient groups with impaired systolic function. The ideal RyR2 “blocker” would be one that allows normal Ca release during systole, but becomes inhibited only during diastole, ie, refractory. In other words, the blocker would specifically target the refractory window of RyR2 to prevent aberrant diastolic Ca release.
CaM is a cytosolic Ca binding protein that translates changes in Ca concentrations into changes in function of target proteins including RyR2.15, 16 In cardiac cells, most CaM is bound to RyR2 and inhibits its activity in a Ca‐dependent manner.17, 18, 19, 20 Such Ca dependent binding would be expected to inhibit RyR2 specifically after Ca release lasting late into the diastolic period. Consistent with this notion, mutations in CaM have recently been linked to CPVT, a syndrome associated with a shortened RyR2 refractory period.7, 8, 21 We reasoned that if indeed certain CPVT‐causing CaM mutations act by shortening RyR2 refractoriness, then CaM variants with slowed Ca dissociation could be engineered to prolong RyR2 refractoriness in cardiac disease settings when refractoriness is pathologically shortened. Such therapeutic CaMs potentially could be applied to alleviate aberrant Ca release and the resulting arrhythmias caused not only by mutations in CaM but possibly other genetic and acquired defects in the RyR2 complex.
In this study, we demonstrate that RyR2 refractoriness can indeed be directly modulated through altering the N‐terminal Ca exchange rates of CaM. Importantly, here we present a genetically engineered CaM that when applied via a viral delivery system, restores RyR2 refractoriness and relieves arrhythmia burden in vivo in a mouse model of CPVT caused by a mutation in another component of the RyR2 complex, CASQ2.
Methods
The data and analytic methods will be made available to other researchers for purposes of reproducing the results or replicating the procedure. The data that support the findings of this study are available from the corresponding author on reasonable request.
Ethical Approval
All animal procedures were approved by the Ohio State University Institutional Animal Care and Use Committee. The study conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 2011).
Mouse Models
CASQ2 R33Q22 and knock out (KO)23 mice (3–6 months old, males) in the C57BL/6 genetic background were utilized in this study. All animal procedures were approved by The Ohio State University Institutional Animal Care and Use Committee. The study conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 2011).
Virus Production and Injection
Adeno‐ associated viral vector serotype 9 (AAV9) vectors were produced by co‐transfecting HEK293 cells with three plasmids as previously described,24 which were then expanded in 10% DMEM medium. Five days after transfection, the cells were harvested for downstream purification using PEG precipitation and an iodixanol gradient protocol.25 The 40% iodixanol gradient band was collected and subjected to further purification. The virus titer was obtained via TaqMan (LifeTechnologies) based quantification. AAV9 (100 μL, containing 1×1011 viral genomes) was injected into adult male mice (12–14 weeks old) through the intra‐thoracic cavity. After being lightly anaesthetized by ≈1% isoflurane, mice were kept in supine position to allow clear access to the chest area. A needle (29.5 gauge) was then inserted at an angle halfway between the ribs and ≈7.5 mm left of sternum. Care was taken to avoid insertion of the needle into the lungs or heart. This straightforward technique ensures efficient and highly reproducible viral transduction specifically to the adult heart, meanwhile minimizing the risk of damaging the beating heart.24
Measurement of the Rate of Ca Dissociation From CaM
Wild type (WT) and recombinant CaM were expressed in bacteria (DE3 BL21) and purified as previously described.26 Ca dissociation rates were measured using an Applied Photophysics Ltd. (Leatherhead, UK) model SX.18 MV stopped‐flow instrument with a dead time of 1.4 ms at 20°C. Each rate represents an average of at least five traces.
CaMF20W was utilized to record the rate of Ca dissociation from the N‐terminal domain of isolated CaM,26 the samples were excited using a 150 W xenon arc source. The tryptophan (Trp) emission was monitored through a UV‐transmitting black glass filter (UG1 from Oriel [Stanford, CT]). The data collected from CaMF20W were fit with a single exponential. Each koff represents an average of at least five traces. The fluorescent Ca chelator Quin‐2 was utilized to record the rate of Ca dissociation from CaM in the presence of the human RyR2 CaM‐binding domain (RSKKAVWHKLLSKQRKRAVVACFRMAPLYNL). Data were fit with a double exponential (variance less than 1.3×10−4) to account for the fact that Quin‐2 reports the rates of Ca dissociation from both the N‐ and C‐domains of CaM. The buffer used in all stopped‐flow experiments was 10 mmol/L MOPS, 150 mmol/L KCl, pH 7.0.
Electrocardiographic Recordings
After the mice were lightly anesthetized by isoflurane, baseline ECG was recorded for 5 minutes, followed by an additional 10 minutes after the administration of the α and β‐agonists epinephrine (EPI, 1.5 mg/kg) and caffeine (120 mg/kg) via intraperitoneal (IP) injection.27 ECG traces were analyzed using LabChart 7 Pro (AD Instruments). The mice utilized for the ECG recordings were not used for other experiments.
Cardiomyocyte Isolation and Confocal Ca Imaging
Myocyte isolation
Mice were fully anesthetized using 4% isoflurane in 95% oxygen, before surgically removing the heart. Mouse ventricular myocytes were isolated as previously described.28, 29 Briefly, the hearts were quickly excised and perfused on a Langendorff's apparatus at 37°C. After 5 minutes of perfusion with nominally Ca‐free perfusion solution (containing, in mmol/L: 140 NaCl, 5.4 KCl, 0.5 MgCl2, 10 Hepes, and 5.6 glucose [pH 7.3]), the perfusate was switched to perfusion solution containing Liberase TH (0.24 U; Roche) for the digestion of the connective tissue. After digestion, single ventricular myocytes were isolated from the dissected and triturated ventricles and stabilized in perfusion solution containing BSA (20 mg/mL).
Ca imaging in intact myocytes
The ventricular myocytes were loaded with 8 μmol/L Fluo‐3 AM (Invitrogen, Carlsbad, CA) for 25 minutes at room temperature, followed by 25 minutes of incubation in fresh perfusion solution (de‐esterification). Fluo‐3 was excited with the 488 nm line of an argon laser and emission was collected at 500 to 600 nm. Fluo‐3 fluorescence was recorded in the line‐scan mode of the confocal microscope (Olympus Fluoview 1000). The myocytes were field‐stimulated using extracellular platinum electrodes.
Patch‐clamp recordings
For the voltage‐clamp experiments the external solution contained in mmol/L: 140 NaCl, 5.4 CsCl, 2.0 CaCl2, 0.5 MgCl2, 10 HEPES, and 5.6 glucose (pH 7.4). Patch pipettes were filled with a solution that contained in mmol/L: 123 CsCl, 20 TEACl, 5 MgATP, 5 NaCl, 1 MgCl2, 0.1 Tris GTP, 10 HEPES, and 0.1 Rhod‐2 K‐salt (Molecular Probes, OR) (pH 7.2). Rhod‐2 dye was excited with 561 nm laser and fluorescence was collected at 570 to 620 nm wavelengths. Ca currents and corresponding Ca transients were evoked by 300 ms depolarizing steps from −50 to −40 … 60 mV in 10 mV intervals. The excitation‐contraction (EC) coupling gain was calculated as a ratio of Ca transient amplitude to the peak density of Ca current.
Ca imaging in permeabilized myocytes
The myocytes were permeabilized with saponin (0.01% for 50 seconds) dissolved in the internal solution, which contained (mmol/L): 120 potassium aspartate, 20 KCl, 0.81 MgCl2, 1 KH2PO4, 0.1 EGTA, (free [Ca] 120 nmol/L), 20 μmol/L cAMP, 3 MgATP, 10 phosphocreatine, 20 Hepes (pH 7.2) and 5 U/mL creatine phosphokinase. The cells were incubated with either WT or mutant CaM for 25 minutes to allow equilibration of CaM binding to its targets. Ca sparks were detected and analyzed using a custom MATLAB (2014b, The MathWorks, Inc, MA) script as described previously.30 Refractoriness factor (RF), calculated as the inverse of frequency of Ca sparks that occurred within 1 seconds following Ca wave, was used to characterize refractoriness of the SR Ca release.
Statistical Analysis
Results are expressed as Mean±SEM. Statistical significance was determined using either one‐way ANOVA or unpaired Student t test. For certain data sets with smaller sample size, The Wilcoxon rank sum or Kruskal–Wallis test was applied. A P<0.05 was considered statistically significant.
Results
CPVT CaM Mutants Shorten RyR2 Refractoriness
Genetic causes of certain forms of CPVT (eg, mutations in the luminal RyR2 accessory protein CASQ2) involve premature reactivation of RyR2 via abbreviation of the time RyR2 remains refractory.7 Recently, mutations in CaM have also been shown to cause CPVT.31, 32, 33 Considering CaM binding to the cytosolic surface of RyR2 has been reported to contribute to inactivation of RyR2,17, 18, 19, 20 we tested whether arrhythmogenic mutations of CaM also act by shortening RyR2 refractoriness. Ca imaging was performed in permeabilized cardiac myocytes (cytosolic Ca buffered at 120 nmol/L) supplemented with different CaM protein variants.34, 35 Consistent with previous studies,31 the addition of the CPVT CaM N98S (100 nmol/L), to permeabilized myocytes isolated from WT mice significantly increased the frequency of Ca waves (as compared with WT CaM; Figure 1A and 1C). RyR2 refractoriness in permeabilized myocytes was assessed by examining sparks restitution following Ca waves.9, 36 Ca sparks restitution was accelerated by CaM N98S, as indicated by the increased Ca spark frequency following a Ca wave (Figure 1B and 1C). Refractoriness factor (RF) was calculated as inverse to spark frequency occurring within 1‐second following Ca wave. RyR2 refractoriness was significantly shortened by CaM N98S compared with WT CaM (Figure 1C). Similar results were obtained with the CPVT CaM N54I (Figure S1). Together with previous results on CASQ2 and RyR2‐mediated CPVT, these data suggests that shortened RyR2 refractoriness may be a common mechanism that promotes arrhythmias caused by both CASQ2 and CaM mutants.7, 8
Figure 1.
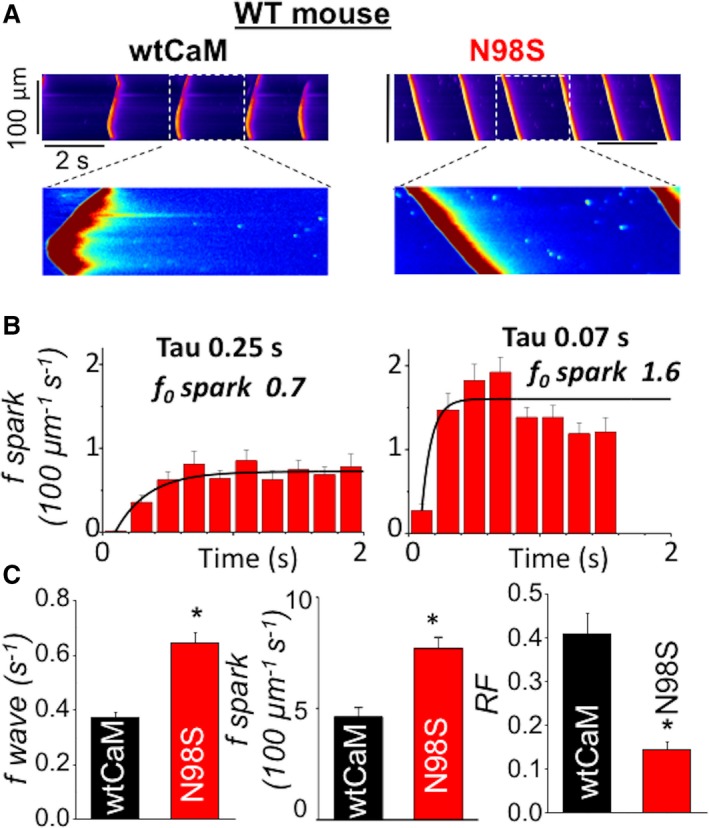
CPVT CaM N98S increased Ca wave frequency and shortened RyR2 refractoriness. A, representative line‐scan images of spontaneous Ca waves (SCWs) in permeabilized WT myocytes exposed to 100 nmol/L wtCaM or 100 nmol/L CPVT CaM N98S. Cytosolic Ca was clamped at ≈120 nmol/L with the slow Ca buffer EGTA. B, time‐dependent recovery of Ca sparks after occurrence of SCWs. Frequency of Ca sparks (f sparks) was calculated at 200 ms intervals. Recovery of Ca sparks was fitted with mono‐exponential curve with tau of 0.25 seconds for wtCaM and 0.07 seconds for N98S CaM, respectively. Ca sparks steady state levels (f0) were 0.7 (per 100 μm per second) for wtCaM and 1.6 (per 100 μm per second) for N98S CaM, respectively. C, Average data for SCW frequency, Ca spark frequency and refractoriness factor (RF) obtained from WT mouse ventricular myocytes permeabilized with wild‐type (wt) and CPVT (N98S) CaMs, respectively, n=38 to 58 cells, *P<0.05 vs wtCaM. Ca indicates calcium; CaM, calmodulin; CPVT, catecholaminergic polymorphic ventricular tachycardia; EGTA, ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid; RF, refractoriness factor; RyR2, ryanodine receptor 2; SCW, spontaneous Ca waves; WT, wild type.
To determine whether these two different regulators of RyR2 are additive or if they operate independently, we examined the impact of arrhythmogenic CaMs in myocytes derived from CPVT mice devoid of CASQ2 (CASQ2‐KO).23 As expected, in the presence of WT CaM permeabilzed CASQ2‐KO myocytes displayed significantly higher frequency of Ca waves when compared with myocytes from WT animals (0.54±0.01 versus 0.37±0.02 per seconds, P<0.001). Notably, supplementation of permeabilized CPVT myocytes with CaM N98S further increased the frequency of Ca waves and shortened the refractory period in these cells (Figure 2). Thus, CPVT mutations in CaM and CASQ2 act in parallel to modulate RyR2 refractoriness. These results suggest that it may be possible to modulate the composite RyR2 refractoriness by targeting either one of these proteins. We examined the possibility of modulating RyR2 refractoriness through modifying the Ca‐binding properties of CaM.
Figure 2.
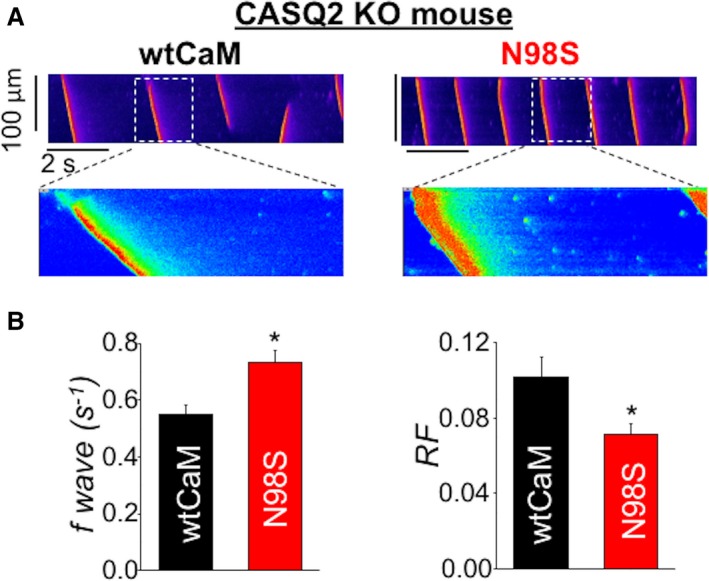
CPVT CaM N98S increased Ca waves frequency and shortened RyR2 refractoriness in CASQ2 KO myocytes. A, representative line‐scan images of SCWs in permeabilized CASQ2 KO myocytes exposed to either 100 nmol/L of wtCaM or 100 nmol/L of N98S CaM. Cytosolic Ca was clamped at ≈120 nmol/L with the slow Ca buffer EGTA. B, Average data for SCW frequency and refractoriness factor obtained from CASQ2 KO ventricular myocytes permeabilized with 100 nmol/L wtCaM and 100 nmol/L of N98S CaM, respectively, n=31 to 38 cells, *P<0.05 vs wtCaM. Ca indicates calcium; CaM, calmodulin; CASQ2, calsequestrin; CPVT, catecholaminergic polymorphic ventricular tachycardia; EGTA, ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid; KO, knock out; RyR2, ryanodine receptor 2; SCW, spontaneous Ca waves.
Modulating RyR2 Refractoriness Via Tuning Ca Binding to CaM
Structurally CaM is thought to be composed of a C‐terminal domain that controls the docking of CaM to RyR2 and an N‐terminal domain that governs the Ca‐dependent regulation of RyR2 by CaM (Figure 3A).37, 38, 39 Based on this model, we hypothesized that altering (ie, accelerating or slowing) the release of Ca from the N‐terminal domain of CaM should accordingly change CaM's ability to help keep RyR2 refractory during diastole. To test this hypothesis we measured CaM Ca dissociation in vitro in WT and mutant recombinant CaM protein (Figure 3B and 3C). Since association with target proteins is known to influence CaM's Ca binding kinetics,40 we performed experiments with the CaMs alone (Figure 3B) and CaMs complexed with a peptide corresponding to the CaM binding domain of RyR241 (CaM‐pRyR2) (Figure 3C). In accordance with our prediction, the CPVT associated mutation N54I accelerated the rate of Ca dissociation from the N‐domain of isolated CaM (Figure 3B). Furthermore, the N54I mutation, in addition to shortening RyR2 refractoriness (Figure S1) accelerated the N‐terminal rate of Ca dissociation from CaM when complexed with the RyR2 peptide (Figure 3C). In the presence of the target peptide, the values of the Ca dissociation rates (ranging from 10.6 to 19.0/second) were now comparable to Ca spark restitution rates in myocytes in the presence of the corresponding CaM variants (≈3/second).
Figure 3.
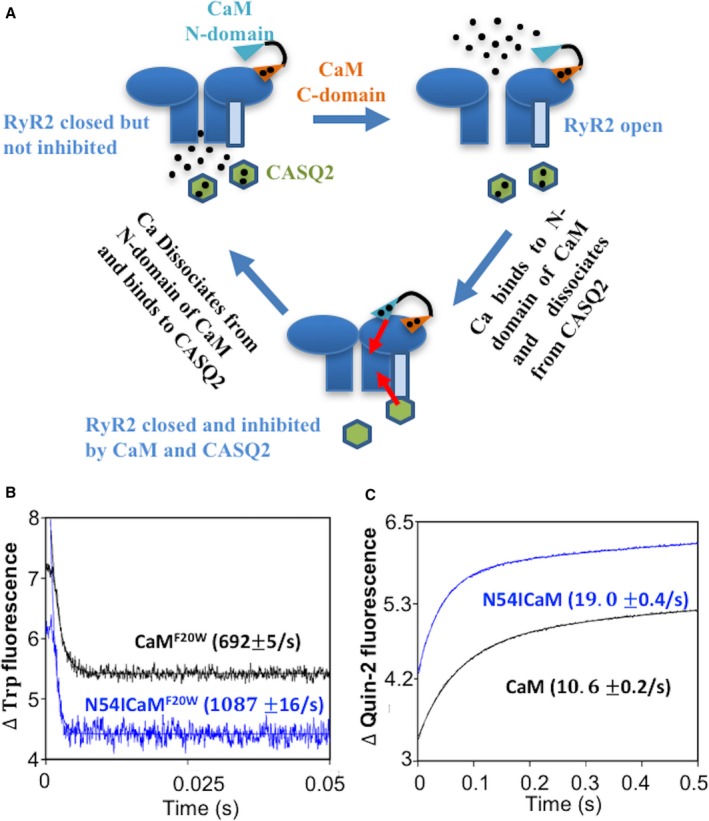
The effect of CPVT CaM N54I on the rate of Ca dissociation from N‐domain of CaM. A, Ca bound N‐domain of CaM binds RyR2 and keeps it refractory following CICR. Meanwhile, CASQ2 senses luminal (Ca) decrease and contributes to RyR2 refractoriness. B, the time course of the decrease in Trp fluorescence as Ca dissociated from CaMF20W constructs. The data traces have been staggered and normalized for clarity. C, the time course of the increase in Quin‐2 fluorescence as Ca dissociated from CaM‐pRyR2 complex. The data traces have been staggered and normalized for clarity. Ca indicates, calcium; CaM, calmodulin; CASQ2, calsequestrin; CICR, calcium induced calcium release; CPVT, catecholaminergic polymorphic ventricular tachycardia; RyR2, ryanodine receptor 2; Trp, tryptophan.
To test further the notion that the rate of Ca dissociation from CaM's N‐domain contributes to CaM regulation of RyR2 refractoriness, a CaM construct (CaM D57A) with hindered ability to bind Ca at the N‐terminal was engineered and its effect on SR Ca release was examined. The D to A point mutation in CaM at position 57 (CaM D57A) greatly accelerated the Ca dissociation from the N‐terminal domain of CaM (Figure 4A). Similar to the CPVT CaMs, supplementation of CaM D57A to WT myocytes significantly increased the frequency of Ca waves, and shortened RyR2 refractoriness compared with WT CaM (Figure 4B and 4C). Thus, RyR2 refractoriness can be modulated by altering the N‐terminal Ca dissociation rate of CaM. This property of CaM makes it an excellent therapeutic target for resetting CPVT associated abbreviated RyR2 refractoriness.
Figure 4.
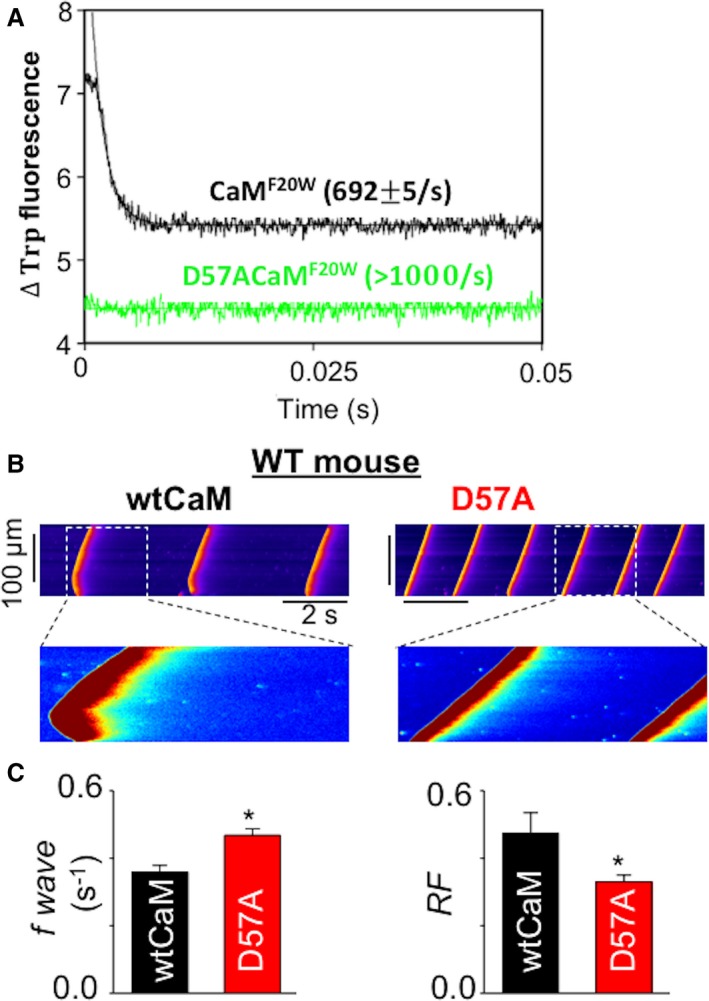
Engineered CaM variant D57A exhibited a faster rate of Ca dissociation and mimicked the effects of CPVT CaMs. A, the time course of the decrease in Trp fluorescence as Ca dissociated from CaMF20W constructs. The data traces have been staggered and normalized for clarity. B, representative line‐scan images of SCWs in permeabilized WT myocytes exposed to 100 nmol/L wtCaM or 100 nmol/L D57A CaM. Cytosolic Ca was clamped at ≈120 nmol/L with the slow Ca buffer EGTA. C, Average data for SCW frequency and refractoriness factor obtained from WT mouse ventricular myocytes permeabilized with wtCaM and D57A CaM, respectively, n=40 cells, *P<0.05 vs wtCaM. Ca indicates calcium; CaM, calmodulin; cAMP, cyclic adenosine monophosphate; EGTA, ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid; SCW, spontaneous Ca waves; TCaM, therapeutic calmodulin; Trp, tryptophan; WT, wild type.
Engineered Therapeutic CaMs
Based on the above mentioned results, stabilizing Ca binding to CaM by slowing its rate of dissociation should prolong refractoriness and may potentially be therapeutic against arrhythmias. To slow CaM's N‐terminal rate of Ca dissociation, we stabilized its Ca binding induced open conformation by mutating a key hydrophobic residue, Met 37, into a polar residue Gln. Based on our previous studies on CaM's evolutionary relative troponin C, a similar mutation in troponin C facilitates the Ca‐induced structural transition of the protein, thus stabilizing Ca binding.42, 43 As shown in Figure 5A, mutating Met 37 to Gln (CaM M37Q) drastically slowed the rate of N‐terminal Ca dissociation from CaM. As predicted, addition of CaM M37Q to permeabilized CPVT myocytes (CASQ2 R33Q44) reduced Ca waves (Figure 5B and 5C), and prolonged the refractory period of RyR2 (Figure 5C) relative to WT CaM.
Figure 5.
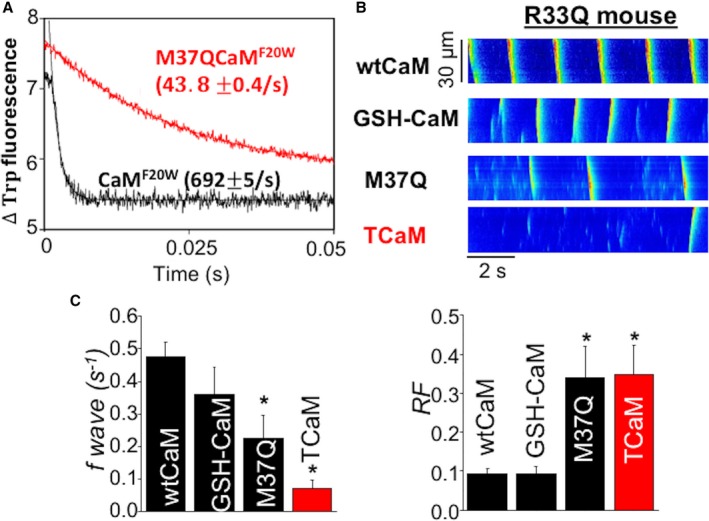
TCaM significantly reduced spontaneous Ca waves in permeabilized R33Q myocytes and prolonged refractoriness. A, the time course of the decrease in Trp fluorescence as Ca dissociated from CaMF20W constructs. The data traces have been staggered and normalized for clarity. B, representative line‐scan images of SCWs in permeabilized R33Q myocytes exposed to cAMP. Cytosolic Ca was clamped at ≈120 nmol/L with the slow Ca buffer EGTA. C, Average data for SCW frequency (n=18–39 cells) and refractoriness factor (n=11–15 cells) obtained from R33Q ventricular myocytes permeabilized with wtCaM, GSH‐CaM, M37Q CaM, and GSH‐M37Q CaM (TCaM), *P<0.05 vs wtCaM. Ca indicates calcium; CaM, calmodulin; cAMP, cyclic adenosine monophosphate; EGTA, ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid; SCW, spontaneous Ca waves; TCaM, therapeutic calmodulin; Trp, tryptophan; WT, wild type.
Previous reports have suggested that aberrant RyR2 Ca release in myocytes derived from failing hearts can be blunted by addition of a CaM with enhanced binding affinity to RyR2, GSH‐CaM.45, 46 Unexpectedly, the addition of GSH‐CaM to the CPVT myocytes had no effect at preventing the adverse Ca waves and accordingly had no significant effect at prolonging the pathologically abbreviated refractory period in these myocytes (Figure 5B and 5C). However, the addition of GSH to the N‐terminus of CaM M37Q to help target our engineered CaM to RyR2 markedly enhanced its potentially beneficial effects in the CASQ2 R33Q myocytes (Figure 5B and 5C). One could surmise that prolonging refractoriness with this GSH‐M37Q‐CaM (termed therapeutic CaM [TCaM]) in CPVT mice harboring the arrhythmogenic CASQ2 R33Q mutation would be anti‐arrhythmic in vivo. This hypothesis can be tested in mice harboring the CPVT CASQ2 R33Q mutation.
Viral‐Mediated Gene Delivery of TCaM Alleviates Arrhythmia Burden in CPVT
In order to incorporate TCaM into the murine heart, we generated an AAV9‐TCaM viral construct which was injected transthoracically into CPVT R33Q mice. The construct also contained a sequence encoding mCherry for transfection verification.24 Figure 6A demonstrates that ≈40% of the ventricular myocytes could be transduced with our viral construct based on mCherry fluorescence. Consistent with the CPVT phenotype, myocytes isolated from R33Q mice transfected with our control mCherry AAV9 virus displayed frequent diastolic Ca waves, when stimulated at 0.5 Hz in the presence of 100 nmol/L isoproterenol, a β‐adrenergic agonist (Figure 6B). Notably, the frequency of these arrhythmogenic events was markedly reduced (≈6‐fold) in AAV9‐TCaM myocytes (Figure 6B and 6C). Additionally, RyR2 refractoriness was significantly prolonged in AAV9‐TCaM myocytes consistent with the results obtained in permeabilized myocytes (Figure 6C). Therefore, TCaM alleviated cellular arrhythmogenesis in intact myocytes isolated from AAV9 infected R33Q mice. Of note, TCaM did not alter Ca transient amplitude, baseline cytosolic Ca (Figure 6D) or the extent of shortening in the same myocytes (Figure S2). Moreover TCaM had no significant impact on inward Ca currents, Ca transients and EC coupling gain in patch clamped R33Q myocytes (Figure S3). Thus, while inhibiting arrhythmogenic diastolic Ca waves, TCaM did not significantly alter EC coupling, systolic Ca release or contractile function in R33Q myocytes.
Figure 6.
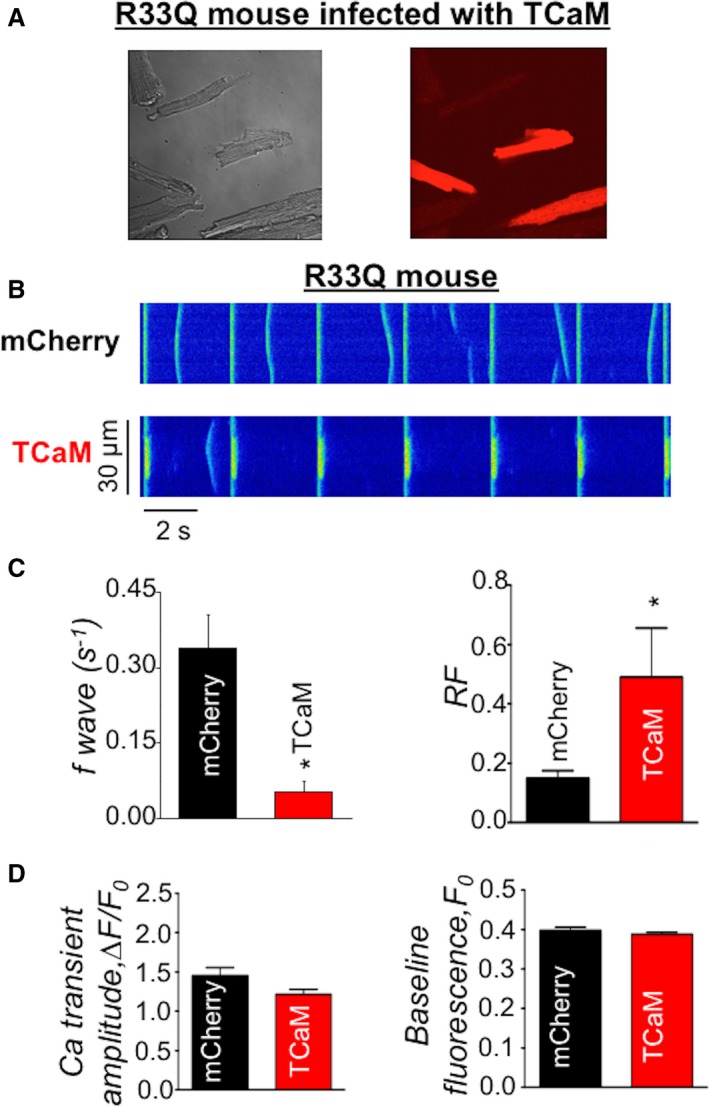
TCaM infected R33Q myocytes had improved Ca handling. A, evaluation of the infection efficiency of TCaM: phase contrast and mCherry fluorescence of TCaM injected R33Q myocytes. B, representative line‐scan images of SCWs in intact myocytes isolated from mCherry or TCaM virus infected R33Q mouse. Myocytes were field‐stimulated at 0.3 Hz in the presence of 1 μmol/L isoproterenol. C, Average data for SCW frequency (n=45–50 cells) and refractoriness factor (n=15–17 cells) obtained from R33Q myocytes infected with control (mCherry) and TCaM virus, respectively, *P<0.05 vs mCherry. D, Average of Ca transient amplitude and baseline fluorescence obtained from R33Q myocytes infected with control (mCherry) and TCaM virus, respectively, *P<0.05 vs mCherry. Ca indicates calcium; TCaM, therapeutic calmodulin; SCW, spontaneous Ca waves.
To test the ability of AAV9‐TCaM to reduce arrhythmia propensity in vivo, we performed surface ECG measurements in anesthetized CASQ2 R33Q mice challenged with epinephrine (Epi)+caffeine (Caf).27 Following IP injection of Epi and Caf, only ≈10% of WT control mice developed episodes of VT, but nearly all untransfected CASQ2 R33Q mice exhibited CPVT (Figure 7A and 7B). Additionally, 7 out of 8 (88%) of these mice died following arrhythmia induction. Notably, the AAV9‐TCaM infected mice had markedly reduced incidence of VT following epinephrine and caffeine challenge. Moreover, the majority of the mice, 7 out of 8 (88%), survived following the Epi/Caf injection (Figure 7C). Thus, our rationally engineered therapeutic protein, TCaM, alleviated life threatening ventricular arrhythmias in the CASQ2 R33Q mice in vivo.
Figure 7.
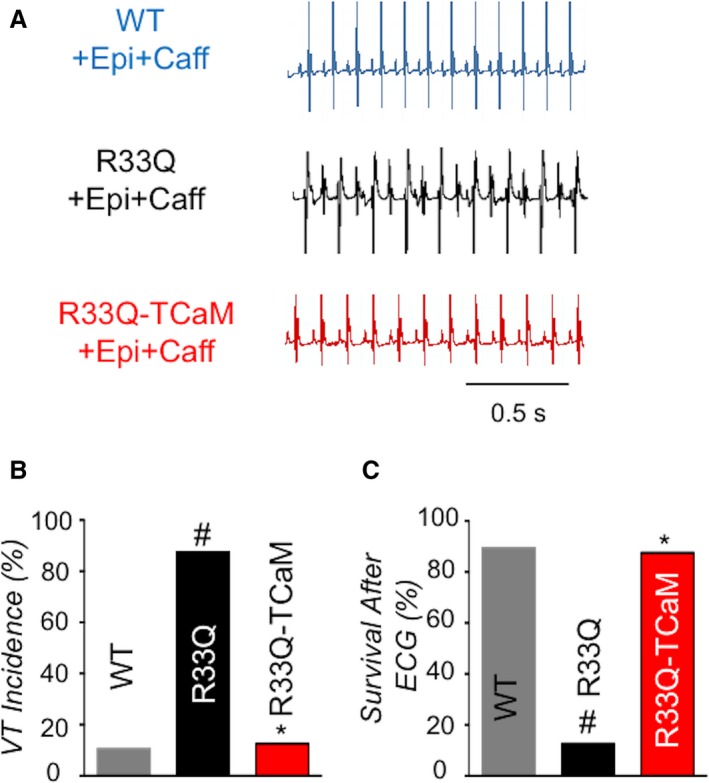
TCaM alleviated ventricular tachycardia in vivo in CPVT mice R33Q. A, Representative surface ECG traces from WT mice, R33Q mice or R33Q mice treated with TCaM. B, Average of VT incidence, n=8 to 19, *P<0.05 vs R33Q, #P<0.05 vs WT. C, Percentage of survival after ECG n=8 to 19, *P<0.05 vs R33Q, #P<0.05 vs WT. CPVT indicates catecholaminergic polymorphic ventricular tachycardia; ECG, electrocardiogram; TCaM, therapeutic calmodulin; VT, ventricular tachycardia.
Discussion
In this study, we provided evidence that rationally engineered CaM can be delivered to CPVT mice to treat arrhythmias. In particular, we demonstrated that therapeutic CaM proteins can be designed that interact with RyR2 to prolong its refractoriness (ie, ability to stay appropriately closed during diastole) which is abnormally shortened in CPVT. Importantly, application of such a TCaM normalized myocyte Ca cycling and reduced in vivo arrhythmia in mice affected by a mutation that occurs in another disease‐related protein (CASQ2). Thus, TCaMs may provide a therapeutic strategy for multiple genetic forms of CPVT and potentially other forms of Ca‐dependent arrhythmias associated with altered RyR2 function.
Following a Ca transient Ca release channels become inactivated and remain unresponsive for a certain time interval termed mechanical/Ca refractory period.36, 47 Refractoriness is modulated by a number of different factors, including luminal Ca, CASQ2,21, 48, 49, 50 RyR2 phosphorylation and oxidation states.51, 52, 53 Here we demonstrated that this process is also influenced by CaM and cytosolic Ca (Figure 8). Moreover, we showed that a CaM protein, ie, TCaM, designed to enhance RyR2 refractoriness can normalize pathologically abbreviated refractoriness to inhibit arrhythmogenesis in CPVT.
Figure 8.
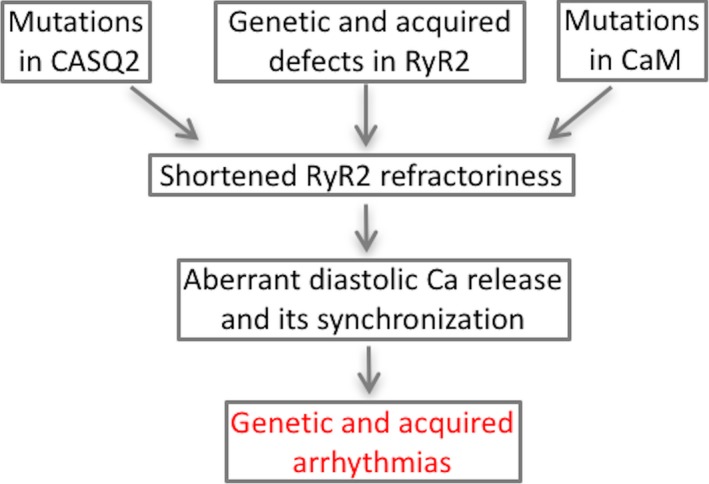
Multiple influences converge to impair RyR2 refractoriness and contribute to ryanopathies.57 CAM indicates calmodulin; CASQ2, calsequestrin; RyR2, ryanodine receptor 2.
The potential role of CaM in regulating RyR2 has long been recognized.17, 18, 19, 20 CaM is associated with RyR2 at its cytosolic regulatory domain and inhibits RyR2 activity at elevated Ca.18 Recently several mutations of CaM have been linked to cardiac arrhythmia and specifically to CPVT.31 However, precisely how and through what molecular steps CaM regulates RyR2 mediated SR Ca release in cardiomyocytes and how these processes are altered to cause arrhythmia remains to be elucidated. Here, we demonstrated that CaM contributes to RyR2 refractoriness through binding of Ca to CaM's N‐terminal domain and subsequent inhibition of RyR2 following SR Ca release. In support of this conclusion engineered mutations that accelerated Ca dissociation from the N‐terminal domain of CaM shortened RyR2 refractoriness whereas engineered mutations that slowed Ca dissociation prolonged RyR2 refractoriness in permeabilized myocytes (Figures 4 and 5). Moreover, we also demonstrated that impairment of this regulatory mechanism accounts for arrhythmogenesis in CaM‐dependent CPVT. In particular, our experiments showed that CPVT mutations in CaM accelerated Ca dissociation from the N‐terminal domain of CaM and shortened RyR2 refractoriness (Figures 1 and 3; Figure S1). These findings are supported also by previous results of Sondergaard et al who showed that CPVT CaM N54I increased the rate of Ca dissociation from N‐domain of CaM.54
We and others have previously demonstrated that the generation of arrhythmogenic Ca release in CPVT involves shortened refractoriness of RyR2 in CPVT models associated with mutations in CASQ2.7, 21 CPVT mutations in CaM and CASQ2 produced additive effects on RyR2 refractoriness and cellular arrhythmogenesis, supporting the notion that shortened refractoriness is a common mechanistic step in CPVT (Figure 2). Given the general role of RyR2 refractoriness in arrhythmogenesis, we reasoned that normalizing shortened RyR2 refractoriness by a rationally designed CaM with enhanced ability to induce RyR2 refractoriness may provide a therapeutic strategy for genetically distinct forms of CPVT. We were able to test this hypothesis by designing a therapeutic CaM that possessed a slowed N‐terminal domain Ca dissociation rate and whose binding to RyR2 was enhanced by the GSH tag attached to the N‐terminus of the protein. Introduction of this TCaM into permeabilized myocytes derived from mice affected by CASQ2‐dependent CPVT normalized shortened refractoriness and reduced the frequency of diastolic Ca waves in these cells. Moreover viral‐mediated transthoracic delivery of TCaM to adult CASQ2 R33Q mice reduced myocyte arrhythmic potential, in vivo susceptibility to arrhythmias and death in these mice.
Recent studies from Priori and coworkers55, 56 demonstrated that wild type CASQ2 gene replacement utilizing AAV can rescue a CPVT phenotype associated with a mutated or absent CASQ2. However, such wild type replacement‐gene transfer has the limitation that it can tackle only the subset of CPVT disorders dependent on aberrant CASQ2. Here we demonstrated that an engineered gene transfer of the RyR2 regulatory protein CaM can counter a non‐CaM modulated arrhythmia model, suggesting that this therapeutic TCaM approach may serve as a general therapeutic avenue for CPVT mutations that result in aberrant Ca release. We have previously shown that abnormally shortened RyR2 refractoriness contributes to arrhythmogenesis associated with altered posttranslational modifications of RyR2 in settings of acquired cardiac disease, including post‐infarction VF and non‐ischemic heart failure.9, 53, 57 TCaMs could potentially serve as a general therapeutic approach to alleviate arrhythmia caused by various genetic and acquired molecular defects in the RyR2 channel complex.
It should be noted that TCaM proved highly effective at preventing arrhythmia despite a moderate transfection efficacy (≈40% myocytes transfected). Priori et al noted a similar unexpectedly high antiarrhythmic potential in the face of low transfection efficacy of virally transferred WT CASQ2.55, 56 The unexpectedly strong antiarrhythmic effects of this delivery method could be explained by considering the role of synchronization of aberrant diastolic SR Ca release in the genesis of Ca‐dependent arrhythmia. In order to initiate triggered activity in the form of triggered action potentials, aberrant Ca release has to occur synchronously across a critical mass of myocardium. We recently demonstrated that diastolic Ca release is indeed highly synchronized in CPVT ventricular myocytes7 and in cardiac tissue, due in part to abnormally abbreviated Ca release refractoriness.7 Therefore, slowing Ca release refractoriness with TCaMs is expected to reduce the incidence of synchronized aberrant Ca release events on the cellular level, thus reducing triggered activity in tissue and arrhythmias in vivo.
In summary, this study demonstrates that a smartly formulated CaM variant designed to prolong RyR2 refractoriness inhibits arrhythmogenic aberrant Ca release in myocytes derived from CPVT hearts. Importantly, adenoviral‐mediated thoracic cavity delivery of this CaM alleviated CPVT episodes and death in vivo. Our study points to a new therapeutic strategy for CPVT and possibly other forms of Ca‐dependent arrhythmias using rationally engineered CaMs. Although we are discovering common aberrant mechanisms for arrhythmias, not all diseased hearts have the same etiology, genetic background or co‐morbidities. Designer proteins open the doors for unprecedented personalized, and potentially, even generalized medicines as gene therapy or protein delivery techniques come to fruition.
Sources of Funding
This work was supported by the National Institutes of Health (RO1 HL132213, HL138579, and R21 AG051913 to Davis and RO1 HL074045, HL063043, and HL138579 to Györke,); and American Heart Association (SDG 17SDG33410716 to Liu).
Disclosures
None.
Supporting information
Figure S1. CPTV CaM N541 increased Ca waves frequency and shortened RyR2 refractoriness. A, Representative line‐scan images of SCWs in permeabilized WT myocytes exposed to cAMP. B, Average frequency of SCWs (n=46–51 cells) and refractoriness (n=21–22 cells), *P<0.05 vs wtCaM. Ca indicates calcium; CaM, calmodulin; cAMP, cyclic adenosine monophosphate; CPTV, catecholaminergic polymorphic ventricular tachycardia; RyR2, Ry00anodine receptor 2; SCW, spontaneous Ca waves; WT, wild type.
Figure S2. TCaM did not alter the extent of myocyte shortening in R33Q myocytes. A, Representative shortening traces of R33Q myocytes infected with control (mCherry) or TCaM virus. B, Average percentage of shortening (n=45–47 cells), *P<0.05 vs mCherry. TCaM indicates therapeutic calmodulin.
Figure S3. EC‐coupling in R33Q myocytes noninfected (control) and infected with TCaM. A, Voltage‐dependence of Ca currents (ICa) and corresponding Ca transients recorded in control R33Q myocytes (n=6) and in R33Q myocyte expressing TCaM (n=4). B, Representative traces of Ca transients and ICa evoked by depolarizing steps from −50 to −20, 0, and 20 mV in control and TCaM myocytes. *P<0.05 vs control. Ca indicates calcium; EC, excitation contraction; ICa, Calcium currents; TCaM, therapeutic calmodulin.
Acknowledgments
We thank Dr Jianchao Zhang for the purification of AAV9.
(J Am Heart Assoc. 2018;7:e008155 DOI: 10.1161/JAHA.117.008155.)29720499
Contributor Information
Jonathan P. Davis, Email: davis.812@osu.edu.
Sándor Györke, Email: sandor.gyorke@osumc.edu.
References
- 1. Chugh SS, Reinier K, Teodorescu C, Evanado A, Kehr E, Al Samara M, Mariani R, Gunson K, Jui J. Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis. 2008;51:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehra R. Global public health problem of sudden cardiac death. J Electrocardiol. 2007;40:S118–S122. [DOI] [PubMed] [Google Scholar]
- 3. Antoons G, Sipido KR. Targeting calcium handling in arrhythmias. Europace. 2008;10:1364–1369. [DOI] [PubMed] [Google Scholar]
- 4. Heijman J, Voigt N, Nattel S, Dobrev D. Calcium handling and atrial fibrillation. Wien Med Wochenschr. 2012;162:287–291. [DOI] [PubMed] [Google Scholar]
- 5. Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–575. [DOI] [PubMed] [Google Scholar]
- 6. Gyorke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008;77:245–255. [DOI] [PubMed] [Google Scholar]
- 7. Brunello L, Slabaugh JL, Radwanski PB, Ho HT, Belevych AE, Lou Q, Chen H, Napolitano C, Lodola F, Priori SG, Fedorov VV, Volpe P, Fill M, Janssen PM, Gyorke S. Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc Natl Acad Sci USA. 2013;110:10312–10317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loaiza R, Benkusky NA, Powers PP, Hacker T, Noujaim S, Ackerman MJ, Jalife J, Valdivia HH. Heterogeneity of ryanodine receptor dysfunction in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2013;112:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belevych AE, Terentyev D, Terentyeva R, Ho HT, Gyorke I, Bonilla IM, Carnes CA, Billman GE, Gyorke S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ Res. 2012;110:569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Acsai K, Nagy N, Marton Z, Oravecz K, Varro A. Antiarrhythmic potential of drugs targeting the cardiac ryanodine receptor Ca2+ release channel: case study of dantrolene. Curr Pharm Des. 2015;21:1062–1072. [DOI] [PubMed] [Google Scholar]
- 11. Dulhunty AF, Casarotto MG, Beard NA. The ryanodine receptor: a pivotal ca2+ regulatory protein and potential therapeutic drug target. Curr Drug Targets. 2011;12:709–723. [DOI] [PubMed] [Google Scholar]
- 12. Betzenhauser MJ, Marks AR. Ryanodine receptor channelopathies. Pflugers Arch. 2010;460:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mackrill JJ. Ryanodine receptor calcium channels and their partners as drug targets. Biochem Pharmacol. 2010;79:1535–1543. [DOI] [PubMed] [Google Scholar]
- 14. Eisner DA, Kashimura T, Venetucci LA, Trafford AW. From the ryanodine receptor to cardiac arrhythmias. Circ J. 2009;73:1561–1567. [DOI] [PubMed] [Google Scholar]
- 15. Davis JP, Shettigar V, Tikunova SB, Little SC, Liu B, Siddiqui JK, Janssen PM, Ziolo MT, Walton SD. Designing proteins to combat disease: cardiac troponin C as an example. Arch Biochem Biophys. 2016;601:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sorensen AB, Sondergaard MT, Overgaard MT. Calmodulin in a heartbeat. FEBS J. 2013;280:5511–5532. [DOI] [PubMed] [Google Scholar]
- 17. Yang Y, Guo T, Oda T, Chakraborty A, Chen L, Uchinoumi H, Knowlton AA, Fruen BR, Cornea RL, Meissner G, Bers DM. Cardiac myocyte Z‐line calmodulin is mainly RyR2‐bound, and reduction is arrhythmogenic and occurs in heart failure. Circ Res. 2014;114:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balshaw DM, Xu L, Yamaguchi N, Pasek DA, Meissner G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J Biol Chem. 2001;276:20144–20153. [DOI] [PubMed] [Google Scholar]
- 19. Yamaguchi N, Xu L, Pasek DA, Evans KE, Meissner G. Molecular basis of calmodulin binding to cardiac muscle ca(2+) release channel (ryanodine receptor). J Biol Chem. 2003;278:23480–23486. [DOI] [PubMed] [Google Scholar]
- 20. Guo T, Fruen BR, Nitu FR, Nguyen TD, Yang Y, Cornea RL, Bers DM. Fret detection of calmodulin binding to the cardiac RyR2 calcium release channel. Biophys J. 2011;101:2170–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kornyeyev D, Petrosky AD, Zepeda B, Ferreiro M, Knollmann B, Escobar AL. Calsequestrin 2 deletion shortens the refractoriness of Ca(2)(+) release and reduces rate‐dependent Ca(2)(+)‐alternans in intact mouse hearts. J Mol Cell Cardiol. 2012;52:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B, Bicciato S, Arcelli D, Spedito A, Scelsi M, Villani L, Esposito G, Boncompagni S, Protasi F, Volpe P, Priori SG. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ Res. 2008;103:298–306. [DOI] [PubMed] [Google Scholar]
- 23. Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini‐Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shettigar V, Zhang B, Little SC, Salhi HE, Hansen BJ, Li N, Zhang J, Roof SR, Ho HT, Brunello L, Lerch JK, Weisleder N, Fedorov VV, Accornero F, Rafael‐Fortney JA, Gyorke S, Janssen PM, Biesiadecki BJ, Ziolo MT, Davis JP. Rationally engineered troponin c modulates in vivo cardiac function and performance in health and disease. Nat Commun. 2016;7:10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N. Recombinant adeno‐associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. [DOI] [PubMed] [Google Scholar]
- 26. Black DJ, Tikunova SB, Johnson JD, Davis JP. Acid pairs increase the n‐terminal Ca2+ affinity of cam by increasing the rate of Ca2+ association. Biochemistry. 2000;39:13831–13837. [DOI] [PubMed] [Google Scholar]
- 27. Liu B, Ho HT, Brunello L, Unudurthi SD, Lou Q, Belevych AE, Qian L, Kim do H, Cho C, Janssen PM, Hund TJ, Knollmann BC, Kranias EG, Gyorke S. Ablation of hrc alleviates cardiac arrhythmia and improves abnormal ca handling in casq2 knockout mice prone to cpvt. Cardiovasc Res. 2015;108:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gyorke S, Lukyanenko V, Gyorke I. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol. 1997;500(Pt 2):297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu B, Ho HT, Velez‐Cortes F, Lou Q, Valdivia CR, Knollmann BC, Valdivia HH, Gyorke S. Genetic ablation of ryanodine receptor 2 phosphorylation at Ser‐2808 aggravates Ca(2+)‐dependent cardiomyopathy by exacerbating diastolic Ca2+ release. J Physiol. 2014;592:1957–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Belevych AE, Ho HT, Bonilla IM, Terentyeva R, Schober KE, Terentyev D, Carnes CA, Gyorke S. The role of spatial organization of Ca2+ release sites in the generation of arrhythmogenic diastolic Ca2+ release in myocytes from failing hearts. Basic Res Cardiol. 2017;112:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hwang HS, Nitu FR, Yang Y, Walweel K, Pereira L, Johnson CN, Faggioni M, Chazin WJ, Laver D, George AL Jr, Cornea RL, Bers DM, Knollmann BC. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res. 2014;114:1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Makita N, Yagihara N, Crotti L, Johnson CN, Beckmann BM, Roh MS, Shigemizu D, Lichtner P, Ishikawa T, Aiba T, Homfray T, Behr ER, Klug D, Denjoy I, Mastantuono E, Theisen D, Tsunoda T, Satake W, Toda T, Nakagawa H, Tsuji Y, Tsuchiya T, Yamamoto H, Miyamoto Y, Endo N, Kimura A, Ozaki K, Motomura H, Suda K, Tanaka T, Schwartz PJ, Meitinger T, Kaab S, Guicheney P, Shimizu W, Bhuiyan ZA, Watanabe H, Chazin WJ, George AL Jr. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circulation. 2014;7:466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nyegaard M, Overgaard MT, Sondergaard MT, Vranas M, Behr ER, Hildebrandt LL, Lund J, Hedley PL, Camm AJ, Wettrell G, Fosdal I, Christiansen M, Borglum AD. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kubalova Z, Gyorke I, Terentyeva R, Viatchenko‐Karpinski S, Terentyev D, Williams SC, Gyorke S. Modulation of cytosolic and intra‐sarcoplasmic reticulum calcium waves by calsequestrin in rat cardiac myocytes. J Physiol. 2004;561:515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Terentyev D, Nori A, Santoro M, Viatchenko‐Karpinski S, Kubalova Z, Gyorke I, Terentyeva R, Vedamoorthyrao S, Blom NA, Valle G, Napolitano C, Williams SC, Volpe P, Priori SG, Gyorke S. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise‐induced sudden cardiac death. Circ Res. 2006;98:1151–1158. [DOI] [PubMed] [Google Scholar]
- 36. Gyorke S, Belevych AE, Liu B, Kubasov IV, Carnes CA, Radwanski PB. The role of luminal ca regulation in ca signaling refractoriness and cardiac arrhythmogenesis. J Gen Physiol. 2017;149:877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Walton SD, Ho HT, Elizaga NM, Siddiqui JK, O'Neil AJ, Neilson NA, Belevych A, Liu B, Radwanski P, Gyorke S, Davis JP. Engineering an anti‐arrhythmic calmodulin. Biophys J. 2016;110:217a. [Google Scholar]
- 38. Her C, McCaffrey JE, Thomas DD, Karim CB. Calcium‐dependent structural dynamics of a spin‐labeled RyR peptide bound to calmodulin. Biophys J. 2016;111:2387–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Lierop JE, Wilson DP, Davis JP, Tikunova S, Sutherland C, Walsh MP, Johnson JD. Activation of smooth muscle myosin light chain kinase by calmodulin. Role of LYS(30) and GLY(40). J Biol Chem. 2002;277:6550–6558. [DOI] [PubMed] [Google Scholar]
- 40. Walton SD, Chakravarthy H, Shettigar V, O'Neil AJ, Siddiqui JK, Jones BR, Tikunova SB, Davis JP. Divergent soybean calmodulins respond similarly to calcium transients: insight into differential target regulation. Front Plant Sci. 2017;8:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sondergaard MT, Tian X, Liu Y, Wang R, Chazin WJ, Chen SR, Overgaard MT. Arrhythmogenic calmodulin mutations affect the activation and termination of cardiac ryanodine receptor‐mediated Ca2+ release. J Biol Chem. 2015;290:26151–26162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tikunova SB, Liu B, Swindle N, Little SC, Gomes AV, Swartz DR, Davis JP. Effect of calcium‐sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry. 2010;49:1975–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tikunova SB, Rall JA, Davis JP. Effect of hydrophobic residue substitutions with glutamine on Ca(2+) binding and exchange with the N‐domain of troponin C. Biochemistry. 2002;41:6697–6705. [DOI] [PubMed] [Google Scholar]
- 44. di Barletta MR, Viatchenko‐Karpinski S, Nori A, Memmi M, Terentyev D, Turcato F, Valle G, Rizzi N, Napolitano C, Gyorke S, Volpe P, Priori SG. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006;114:1012–1019. [DOI] [PubMed] [Google Scholar]
- 45. Hino A, Yano M, Kato T, Fukuda M, Suetomi T, Ono M, Murakami W, Susa T, Okuda S, Doi M, Kobayashi S, Yamamoto T, Koseki N, Kyushiki H, Ikemoto N, Matsuzaki M. Enhanced binding of calmodulin to the ryanodine receptor corrects contractile dysfunction in failing hearts. Cardiovasc Res. 2012;96:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kato T, Yamamoto T, Nakamura Y, Nanno T, Fukui G, Sufu Y, Hamada Y, Maeda T, Nishimura S, Ishiguchi H, Murakami W, Fukuda M, Xu X, Hino A, Ono M, Oda T, Okuda S, Kobayashi S, Koseki N, Kyushiki H, Yano M. Correction of impaired calmodulin binding to RyR2 as a novel therapy for lethal arrhythmia in the pressure‐overloaded heart failure. Heart Rhythm. 2017;14:120–127. [DOI] [PubMed] [Google Scholar]
- 47. Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol. 1996;270:C148–C159. [DOI] [PubMed] [Google Scholar]
- 48. Terentyev D, Viatchenko‐Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc Natl Acad Sci USA. 2003;100:11759–11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramay HR, Liu OZ, Sobie EA. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc Res. 2011;91:598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. DelPrincipe F, Egger M, Niggli E. Calcium signalling in cardiac muscle: refractoriness revealed by coherent activation. Nat Cell Biol. 1999;1:323–329. [DOI] [PubMed] [Google Scholar]
- 51. Polakova E, Illaste A, Niggli E, Sobie EA. Maximal acceleration of Ca2+ release refractoriness by beta‐adrenergic stimulation requires dual activation of kinases PKA and CaMKII in mouse ventricular myocytes. J Physiol. 2015;593:1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Szentesi P, Pignier C, Egger M, Kranias EG, Niggli E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+‐induced Ca2+ release refractoriness in heart muscle. Circ Res. 2004;95:807–813. [DOI] [PubMed] [Google Scholar]
- 53. Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Gyorke S. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. 2011;90:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sondergaard MT, Sorensen AB, Skov LL, Kjaer‐Sorensen K, Bauer MC, Nyegaard M, Linse S, Oxvig C, Overgaard MT. Calmodulin mutations causing catecholaminergic polymorphic ventricular tachycardia confer opposing functional and biophysical molecular changes. FEBS J. 2015;282:803–816. [DOI] [PubMed] [Google Scholar]
- 55. Denegri M, Avelino‐Cruz JE, Boncompagni S, De Simone SA, Auricchio A, Villani L, Volpe P, Protasi F, Napolitano C, Priori SG. Viral gene transfer rescues arrhythmogenic phenotype and ultrastructural abnormalities in adult calsequestrin‐null mice with inherited arrhythmias. Circ Res. 2012;110:663–668. [DOI] [PubMed] [Google Scholar]
- 56. Denegri M, Bongianino R, Lodola F, Boncompagni S, De Giusti VC, Avelino‐Cruz JE, Liu N, Persampieri S, Curcio A, Esposito F, Pietrangelo L, Marty I, Villani L, Moyaho A, Baiardi P, Auricchio A, Protasi F, Napolitano C, Priori SG. Single delivery of an adeno‐associated viral construct to transfer the CASQ2 gene to knock‐in mice affected by catecholaminergic polymorphic ventricular tachycardia is able to cure the disease from birth to advanced age. Circulation. 2014;129:2673–2681. [DOI] [PubMed] [Google Scholar]
- 57. Belevych AE, Radwanski PB, Carnes CA, Gyorke S. ‘Ryanopathy’: causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc Res. 2013;98:240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CPTV CaM N541 increased Ca waves frequency and shortened RyR2 refractoriness. A, Representative line‐scan images of SCWs in permeabilized WT myocytes exposed to cAMP. B, Average frequency of SCWs (n=46–51 cells) and refractoriness (n=21–22 cells), *P<0.05 vs wtCaM. Ca indicates calcium; CaM, calmodulin; cAMP, cyclic adenosine monophosphate; CPTV, catecholaminergic polymorphic ventricular tachycardia; RyR2, Ry00anodine receptor 2; SCW, spontaneous Ca waves; WT, wild type.
Figure S2. TCaM did not alter the extent of myocyte shortening in R33Q myocytes. A, Representative shortening traces of R33Q myocytes infected with control (mCherry) or TCaM virus. B, Average percentage of shortening (n=45–47 cells), *P<0.05 vs mCherry. TCaM indicates therapeutic calmodulin.
Figure S3. EC‐coupling in R33Q myocytes noninfected (control) and infected with TCaM. A, Voltage‐dependence of Ca currents (ICa) and corresponding Ca transients recorded in control R33Q myocytes (n=6) and in R33Q myocyte expressing TCaM (n=4). B, Representative traces of Ca transients and ICa evoked by depolarizing steps from −50 to −20, 0, and 20 mV in control and TCaM myocytes. *P<0.05 vs control. Ca indicates calcium; EC, excitation contraction; ICa, Calcium currents; TCaM, therapeutic calmodulin.