

Abstract
Background
Among the growing numbers of patients with heart failure, up to one half have heart failure with preserved ejection fraction (HFpEF). The lack of effective treatments for HFpEF is a substantial and escalating unmet clinical need—and the lack of HFpEF‐specific animal models represents a major preclinical barrier in advancing understanding of HFpEF. As established treatments for heart failure with reduced ejection fraction (HFrEF) have proven ineffective for HFpEF, the contention that the intrinsic cardiomyocyte phenotype is distinct in these 2 conditions requires consideration. Our goal was to validate and characterize a new rodent model of HFpEF, undertaking longitudinal investigations to delineate the associated cardiac and cardiomyocyte pathophysiology.
Methods and Results
The selectively inbred Hypertrophic Heart Rat (HHR) strain exhibits adult cardiac enlargement (without hypertension) and premature death (40% mortality at 50 weeks) compared to its control strain, the normal heart rat. Hypertrophy was characterized in vivo by maintained systolic parameters (ejection fraction at 85%–90% control) with marked diastolic dysfunction (increased E/E′). Surprisingly, HHR cardiomyocytes were hypercontractile, exhibiting high Ca2+ operational levels and markedly increased L‐type Ca2+ channel current. In HHR, prominent regions of reparative fibrosis in the left ventricle free wall adjacent to the interventricular septum were observed.
Conclusions
Thus, the cardiomyocyte remodeling process in the etiology of this HFpEF model contrasts dramatically with the suppressed Ca2+ cycling state that typifies heart failure with reduced ejection fraction. These findings may explain clinical observations, that treatments considered appropriate for heart failure with reduced ejection fraction are of little benefit for HFpEF—and suggest a basis for new therapeutic strategies.
Keywords: calcium handling, cardiac, cardiomyocyte, fibrosis, heart failure preserved ejection fraction, hypertrophy
Subject Categories: Basic Science Research, Animal Models of Human Disease, Arrhythmias, Hypertrophy, Heart Failure
Clinical Perspective
What Is New?
Heart failure with preserved ejection fraction (HFpEF) is a condition for which there is no specific treatment.
The lack of HFpEF‐specific animal models represents a major preclinical barrier in advancing understanding of HFpEF.
We validate and characterize a new rodent model of HFpEF, undertaking longitudinal investigations to delineate the associated cardiac and cardiomyocyte pathophysiology.
Surprisingly, HHR cardiomyocytes were hypercontractile, exhibiting high Ca2+ operational levels and markedly increased L‐type Ca2+ channel current.
What Are the Clinical Implications?
The cardiomyocyte remodeling process in the etiology of this HFpEF model contrasts dramatically with the suppressed Ca2+ cycling state that typifies heart failure with reduced ejection fraction.
These findings may explain clinical observations that treatments considered appropriate for heart failure with reduced ejection fraction are of little benefit for HFpEF—and suggest a basis for new therapeutic strategies.
Introduction
Among the growing numbers of patients with heart failure, as many as one half have heart failure with preserved ejection fraction (HFpEF), and clinical trials have yet to define an effective and specific treatment for this condition.1, 2 The pathophysiology of HFpEF is characterized by near normal systolic function coincident with diastolic dysfunction and inadequate ventricular filling at normal pressures. Surprisingly, while there has been considerable focus on refining differential diagnostic criteria for HFpEF relative to heart failure with reduced ejection fraction (HFrEF), an understanding of the cardiomyocyte functional phenotype that characterizes HFpEF is lacking. The lack of HFpEF‐specific animal models represents a major preclinical barrier in advancing understanding of HFpEF.3 Defining the cellular basis of HFpEF dysfunction is critical for guiding the development of new treatments for this failure condition.4
Although the HFpEF phenotype is not homogenous,2, 5, 6 cardiac hypertrophy is considered an important diagnostic element, and the extent of hypertrophy is a key prognostic indicator in HFpEF.7, 8 Hypertension (although often present as a comorbidity to a variable extent in different HFpEF populations) has not been identified as a baseline discriminator in determining the development of HFpEF in outcome trials.9 In addition to hypertrophy, heart failure guidelines also recognize other diagnostic features of HFpEF, including increased left ventricular filling pressures, normal left ventricular end‐diastolic volume, and fibrosis.7, 8, 10 Systemic inflammatory involvement and metabolic disturbances linked with comorbidities may also contribute.9
In HFrEF, the cellular “remodeling” processes generally understood to characterize the progression to failure include reduced cardiomyocyte contractile response, diminished systolic Ca2+ activator availability, and dysregulated sarcoplasmic reticulum (SR) Ca2+ store operation.11 This general etiology is inferred primarily from experimental observation of tissues and myocytes of various animal models of systolic dysfunction occurring as a result of abnormal volume or pressure loads and some studies of end‐stage failing human myocardium.12 Cell‐directed treatments for HFrEF have principally focused on interventions to augment availability or actions of cardiomyocyte activator Ca2+.13 These HFrEF therapies, which have largely relied on neurohumoral approaches involving angiotensin, aldosterone, and β‐adrenoceptor targets, have not been successful in the HFpEF context,3, 14 raising questions about the cardiomyocyte phenotypes in HFpEF.
There is a poor understanding of the cardiomyocyte pathophysiology of HFpEF, in large part because of the limited availability of appropriate HFpEF preclinical models. Indeed, this problematic lack of animal models has recently been highlighted as a major barrier in the progression of preclinical mechanistic work available to inform new therapeutic directions for HFpEF.3 Proposed models generally involve pressure overload interventions, with secondary development of diastolic dysfunction, and do not fully recapitulate clinical features.15 The cardiomyocyte HFpEF functional characteristics, wherein the intrinsic pathologic trigger is not a systolic loading challenge, could be expected to be very different from the HFrEF myocyte phenotype. There are no studies of intact HFpEF‐derived cardiomyocytes (human or animal) from disease settings involving primary cardiac pathology. Findings from membrane disrupted cell preparations derived from the myocardium of patients with diastolic heart failure have described sarcomeric structures exhibiting increased passive force response to stretch—but there has been no investigation of intact cardiomyocyte Ca2+ handling and contractile responsiveness in the HFpEF setting arising from underlying myocardial hypertrophic pathogenesis.
Here, we report and validate a novel rodent model of HFpEF—which exhibits cardiac hypertrophy and premature mortality independent of hemodynamic loading.16, 17 In the hypertrophic heart rat strain (HHR), we demonstrate that the in vivo progression to diastolic failure (with preservation of systolic performance) is accompanied by unexpected hyperfunction of cardiomyocytes with elevated systolic activator Ca2+ availability and contractile response. Molecular and electrophysiologic analyses identify arrhythmogenic cellular substrates. Thus, the cardiomyocyte remodeling process in the etiology of this HFpEF model contrasts dramatically with that seen in the HFrEF state. These findings may explain clinical observations that intervention strategies considered appropriate for HFrEF are of little benefit for HFpEF. New insights from our unique animal model of HFpEF provide opportunities to develop novel and targeted HFpEF therapeutic approaches.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure on reasonable request.
Detailed methods are available in Data S1.
Experimental Animals
A novel normotensive, hypertrophic rat strain generated through selection and cross breeding of Fischer 344 and spontaneously hypertensive rat strain progenitors has previously been described.16, 17 Briefly, initial spontaneously hypertensive rat genomic analysis had identified blood pressure–independent, hypertrophic genetic loci.18 Selection for large and small echocardiographic heart size and low blood pressure was used to derive simultaneously the hypertrophic heart rat (HHR) and its control, the normal heart rat (NHR), respectively,16 now beyond the F50 generation of inbreeding. The HHR constitutes a polygenic model of spontaneous cardiac hypertrophy with a natural disease history. Experiments were performed using male HHR and NHR at the failure and prefailure ages specified below. Normotension in HHR and NHR has been demonstrated previously16 and here equivalent diurnal mean arterial pressures were confirmed telemetrically (Figure S1A).
All experimental animals were housed under a 12‐hour light/dark cycle with water and standard chow provided ad libitum. All investigations were undertaken in accordance with the National Health and Medical Research Council/Commonwealth Scientific and Industrial Research Organisation/ACC Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (1997), as approved (University of Melbourne Animal Ethics Committee).
Echocardiography, Electrocardiography, and Hemodynamic Measurements
Echo‐ and electrocardiography were performed under light anesthesia (inhalation of isoflurane at 1.5%). Cardiac structure and systolic and diastolic function were evaluated by transthoracic 2‐dimensional B‐ and M‐mode and blood flow and tissue Doppler echocardiography (GE Vivid 9; 15 mHz i13L linear array transducer). Acquisition and offline analysis was performed with GE EchoPac software. ECGs were obtained using external clip leads in standard configuration. Ambulatory 24‐hour blood pressure measurements were made using implanted devices to record from the intra‐abdominal aorta (TA11PA‐C40, DSI). To perform left ventricular catheterization, animals were anesthetized with sodium pentobarbital (60 mg/kg), intubated and ventilated, and a 2F miniaturized combined conductance catheter‐micromanometer (Model SPR‐838 Millar instruments, Houston, TX) was inserted into the right carotid and advanced into the left ventricle until stable pressure‐volume loops were obtained. Data were acquired under steady‐state conditions and during preload reduction.
Using the pressure conductance data, a range of functional parameters was then calculated including end‐diastolic pressure, maximum rate of pressure change in the ventricle (+dP/dt max), minimum rate of pressure change in the ventricle (−dP/dt min) and the slope of the end‐diastolic pressure volume relationship.
Histologic Analysis of Myocardial Collagen Content and Confocal Analysis of Cardiomyocyte Ultrastructure
To evaluate collagen histology, hearts were fixed in 10% formalin for histologic analysis using picrosirius red as previously described.19 Images were captured with brightfield microscopy using the Zeiss Imager D1, connected to a Zeiss AxioCam MRc5 color camera and using AxioVision 40 version 4.7.1.0 acquisition software (Zeiss, Germany) with 10 images per section, from 2 sections per heart. Image analysis was performed using Image Pro Plus (V4.5.1, Media Cybernetics, Bethesda, MD) in a “blinded” manner. For ultrastructural analysis to examine cardiomyocyte sarcomeric integrity, high‐resolution confocal microscopy was performed on fixed tissues labeled with the fluorescent marker wheat germ agglutinin to delineate cardiomyocyte T‐tubule geometry using a Zeiss LSM410 confocal microscope and ×63 NA 1.25 oil‐immersion objective. The t‐tubule images were converted to frequency space using fast Fourier transform, and the peak in the power spectrum corresponding to the sarcomere spacing was estimated by Gaussian fit (“T power”).
Cell Isolation and Cardiomyocyte Morphology
NHR and HHR single ventricular myocytes were isolated enzymatically using methods previously described.19, 20 Briefly, hearts were removed and perfused retrogradely on a Langendorff apparatus with Ca2+‐free bicarbonate‐buffered physiologic saline solution, maintained at 37°C, with the following composition (mmol/L): NaCl, 118; KCl, 4.8; KH2PO4, 1.2; MgSO4, 1.2; NaHCO3, 25, glucose, 11. Following 20 minutes of 0.45 mg/mL collagenase (Worthington, Type II) perfusion, hearts were removed from the perfusion apparatus, the left ventricle was isolated and placed in a conical flask, and myocytes were dispersed by gentle agitation. Fractions containing viable cells were resuspended at room temperature in 2 to 3 mL of HEPES‐buffered physiologic saline containing 1 mmol/L Ca2+ and were stored at room temperature. Cells were used within 8 hours.
Whole‐Cell Patch Clamp
Voltage‐gated Ca2+ currents (I‐Ca) and Na+/Ca2+ current (I‐NCX) were measured using a whole‐cell voltage clamp (Axopatch 200B, Digidata 1200B, Axon Instruments) with 1 to 3 MΩ pipette resistances (TW150F‐3, World Precision Instruments). For ICa, voltage step protocols with 3 holding potentials were applied: −115, −90, and −50 mV. For INCX, a combined voltage step and ramp protocol from holding potential −90 mV to −45 mV to 0 mV to +80 mV to −140 mV was implemented. Current analysis procedures were performed using Clampfit (pClamp8).
Cardiomyocyte Intracellular Ca2+ and Contractility Measurements
Intracellular Ca2+ and cell length were measured simultaneously by microfluorimetry and edge detection (IonOptix). Cells were fura‐2/AM‐loaded (2.5 μmol/L, 20 minutes, Molecular Probes), superfused with HEPES‐Krebs buffer and field stimulated. Cardiomyocyte performance was evaluated under basal paced conditions (37°C, 3 Hz, 1.5 mmol/L Ca2+), during a rest interval (37°C, 1.5 mmol/L Ca2+ 3 Hz for 5 minutes, stimulator off 30 seconds), with different inotropic challenges (4 mmol/L Ca2+, isoproterenol 10−8 mol/L and in response to rapid caffeine spritz to evaluate SR load (Ca2+ transient only).
Immunoblotting
Left ventricular tissue was homogenized and nonfractionated homogenates were reconstituted in sodium dodecylsulphate sample buffer. Equal volumes were loaded onto polyacrylamide gels for electrophoresis and immunoblot analysis. Antibody usage is tabulated in Figure S2A.
Statistical Analyses
In general (unless otherwise stated) data are presented as mean±SEM. Comparisons between two groups with normally distributed data were performed with Student unpaired t test. Data from experiments with two groups assessed at multiple points were evaluated by a one‐way ANOVA with repeated measures. Two‐way ANOVA was used for evaluation of data groups comprising 2 factors. Correlation analyses were performed to determine the Pearson coefficient within each strain and significance values evaluated. Data are shown depicting linear regression plots for each strain (SPSS c21.0; Graph Pad Prism V6). Data analysis was performed in a blinded manner. Survival (Kaplan–Meier) data were analyzed by log‐rank Mantel‐Cox test. Differences were considered significant at P<0.05 (SPSS v.21.0; Graph Pad Prism V6).
Results
Premature Mortality, Hypertrophy, and Diastolic Dysfunction With Preserved Systolic Function in the HHR
HHR and NHR cohorts were tracked longitudinally to characterize the functional and structural development and consequences of hypertrophy. HHRs died prematurely (primary end point) with a 40% mortality by 50 weeks of age compared with 0% NHR mortality (Figure 1A). Of those animals aged to end point or requiring euthanasia due to acute symptomatic deterioration, ≈40% exhibited pulmonary edema (lung weight index HHR 5.69±0.8 versus NHR 3.75±0.1, P<0.05). Other animals exhibited sudden premature death. Cardiac weight index at autopsy was substantially elevated (Figure 1B). In vivo function and structure was measured by echocardiography at 50 weeks (cohort surviving) and at 30 weeks (full cohort). Hemodynamic measures using cardiac catheterization were also obtained from a cohort of 50‐week animals (Table S1, Figure S1B and S1C). HHR ejection fraction was substantially preserved at 85% to 90% NHR level at 50 and 30 weeks, respectively (Figure 1C and 1D, Table S1). While these 10% to 15% differences could be resolved statistically, the systolic performance levels could be considered to be relatively preserved. In the clinical setting, the maintenance of ejection fraction at 85% to 90% of normal value would be identified as “preserved” and considered a diagnostic element in establishing HFpEF occurrence. Maintained systolic function in HHRs was evident in the absence of significant dilation (diastolic chamber dimension), while systolic chamber dimension changes matched ejection fraction maintenance at 85% to 90% NHR value (Figure 1E, Table S1). Diastolic dysfunction at 30 and 50 weeks was evident in HHRs with markedly elevated E/E′ (Figure 1F, Table S1) which was consistent with significantly increased end‐diastolic pressure‐volume relationship measurement from pressure‐volume loop analysis (Figure S1B and S1C). HHR tachycardia was apparent, consistent with progression to failure (Figure 1G, Table S1). Thus, HHRs exhibited premature mortality, preserved ejection fraction, and substantially degraded diastolic performance relative to controls (ie, HFpEF). Interestingly, evaluation of very young HHR cohorts showed the emergence of diastolic dysfunction early, with maximal systolic function (Figure S3).
Figure 1.
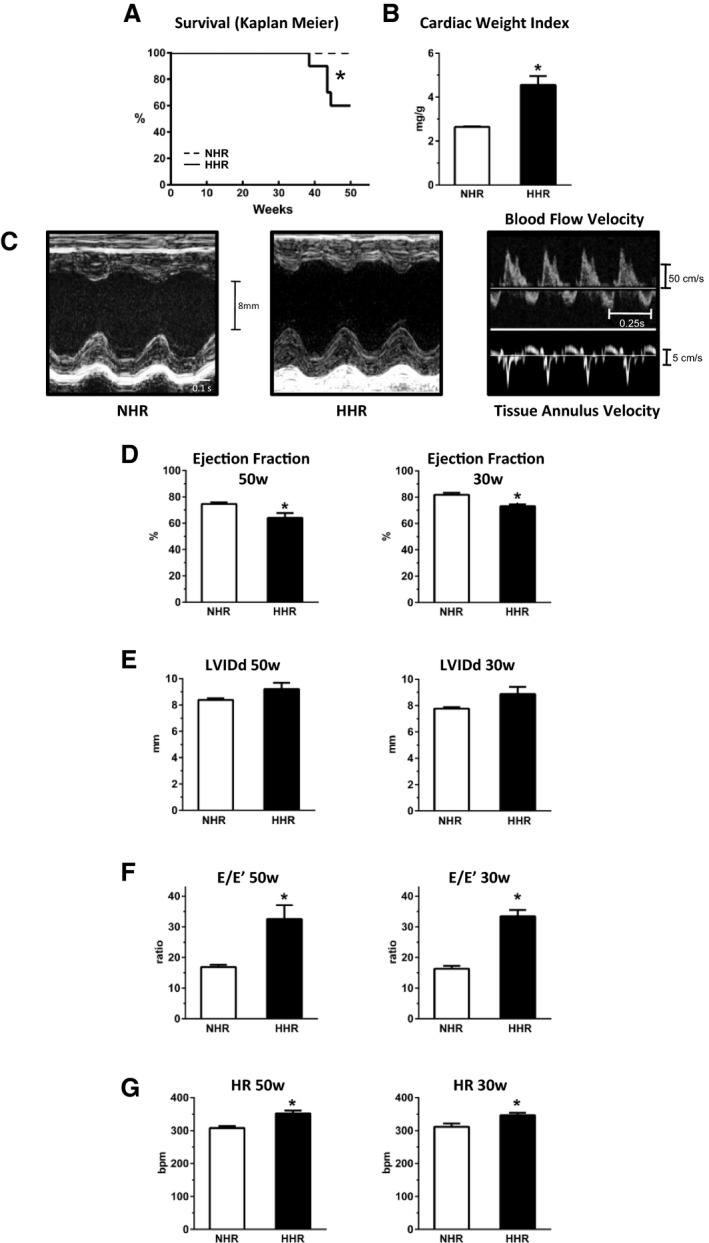
Premature mortality, hypertrophy, and diastolic dysfunction with preserved systolic function in the hypertrophic heart rat (end point age 50 weeks, prefailure 30 weeks). A, Survival, normal heart rat (NHR, dashed line) and hypertrophic heart rat (HHR, solid line). B, Postmortem cardiac weight index (mg/g). (50 weeks). C, Exemplar M‐mode echocardiographs HHR vs NHR hearts, and exemplar blood flow and tissue Doppler images (50 weeks). D, Preserved systolic function in HHR with minimal decrement of ejection fraction (EF). E, Left ventricular internal diameter (diastole) (LVIDd). F, Diastolic function—E/E′. G, Heart rate (HR, bpm). Graphs show mean ± SEM. For A, *P=0.0004 (log‐rank Mantel‐Cox test), for B and D through G, *P<0.05 (Student t test, n=6–10 hearts/group).
Histologic analyses in the 50‐week HHRs revealed the presence of dispersed focal fibrotic areas in the extracellular matrix that did not follow a typical perivascular localization. The regions of most prominent occurrence of fibrotic foci were mapped to the transverse midwall area of the left ventricle adjacent to the interventricular septum and the area of septal free wall confluence (Figure S4). Although the overall fibrotic load in HHR was significantly greater than NHR (Figure 2A through 2E), when focal areas were excluded, the interstitial matrix histology was similar in HHRs and NHRs. Thus, this failure‐associated histology comprised focal fibrotic regions indicative of localized reparative responses.
Figure 2.
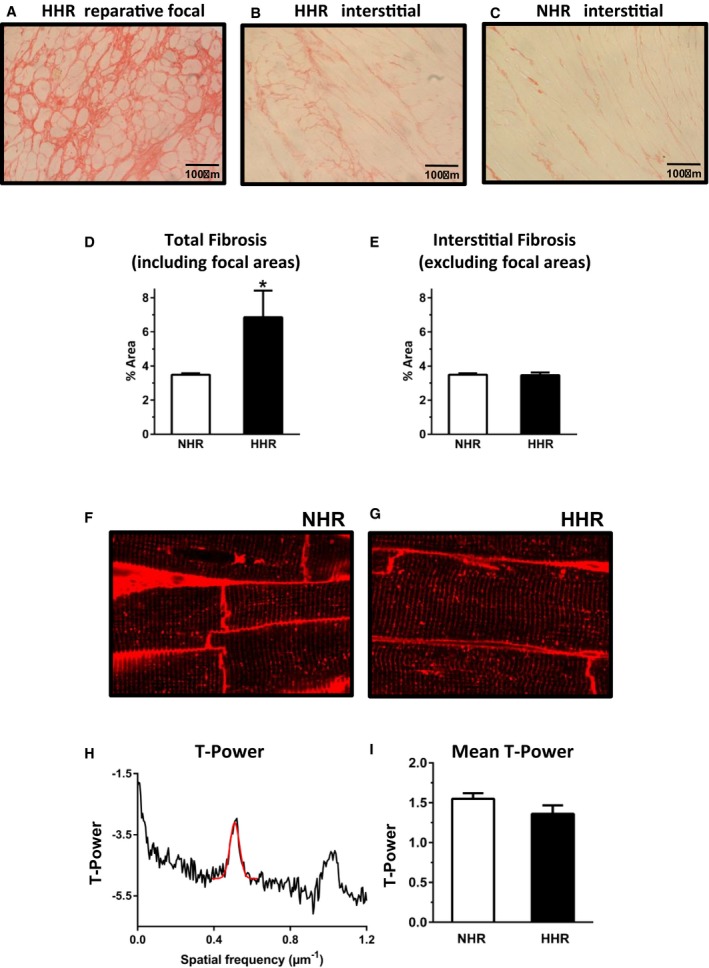
Extracellular matrix disrupted by fibrotic foci with progressing HFpEF, yet maintained cardiomyocyte ultrastructural integrity (50 weeks). A through C, Picrosirius red collagen histology. Fibrotic foci hypertrophic heart rat (HHR), interstitial fibrosis normal heart rat (NHR) and HHR. D and E, Fibrosis densitometry: total and interstitial excluding focal areas in HHR. F and G, T‐tubule labeling with wheat germ agglutinin Alex‐594 shows t‐system geometry regularity in sectioned myocardium of HHR and NHR. H and I, Depiction of T‐tubule power vs spatial frequency in longitudinal dimension for HHR and NHR evidenced high level of T‐tubule structural integrity in HHR and NHR. Graphs show mean ± SEM. For D and E, Student t test, n=20 images/N=5 hearts. For F and G, n=18/group; N=3 hearts/group.
Progression to failure has been linked with loss of cardiomyocyte T‐tubule structural integrity, understood to compromise excitation‐contraction coupling efficacy. Despite the very marked cardiomyocyte size increase, ultrastructural analyses found no evidence of T‐tubule structural disturbance in HHR myocytes. Morphologically, there was no evidence of T‐tubule spatial dispersion. The wheat germ agglutinin–stained myocytes were similar for HHRs and NHRs, confirming the absence of significant changes in the T‐system morphology (Figure 2F through 2I).
Cardiomyocyte Hypercontractility Characterizes Progression to Failure With Preserved Systolic Function
Electromechanical performance of fura‐2 loaded isolated cardiomyocytes obtained from HHR and NHR hearts at age 20 to 30 weeks was evaluated, a prefailure time point selected as a disease progression stage before emergence of premature mortality in HHR. This was considered a time point at which the most consistent phenotypic information could be obtained about the status of electromechanical coupling priming the HHR for failure, and when cardiomyocyte isolation yield could be most reproducible.
HFpEF in the HHR was associated with significant cardiomyocyte enlargement (Figure 3A and 3B, Table S2) determined microscopically from single myocyte dimensions and electrophysiologically by membrane capacitance. Radial and longitudinal cell dimensions were increased similarly (>20%). Thus, hypertrophy at organ and cellular levels was established in the HHR. Functional analysis of shortening performance in isotonically contracting isolated intact cardiomyocytes revealed a dramatic HHR “hypercontractile” state, with almost doubled maximum shortening and shortening kinetic measures allowing overall maintenance of cell activation cycle timing (Figures 3C, 3E, and 4A through 4C). Interestingly, this in vitro hypercontractility was consistent with in vivo hemodynamic measurements obtained using cardiac catheterization (on 50‐week animals only), where the maximal rate of pressure development in the HHR was increased (Table S1). HHR myocyte responses to in vitro inotropic intervention (elevated Ca2+, β‐stimulation) were modestly potentiated (Figure 4E through 4H).
Figure 3.
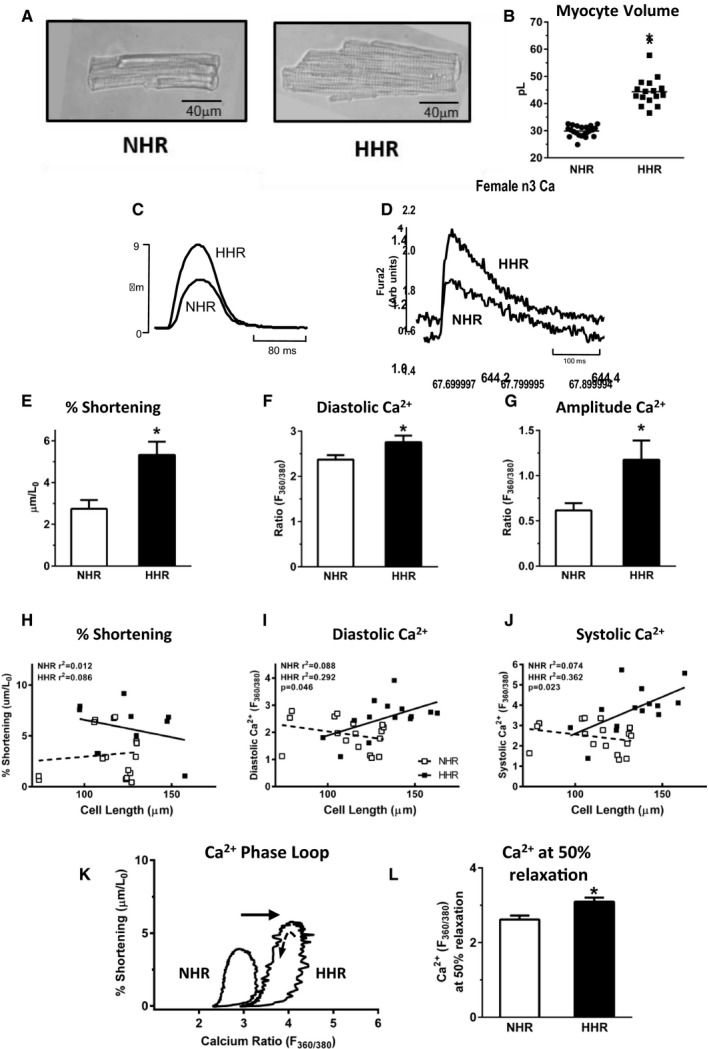
HHR cardiomyocyte hypercontractility characterizes progression to failure with preserved systolic function (20–30 weeks). A, Bright field normal heart rat (NHR) and hypertrophic heart rat (HHR) cardiomyocyte micrographs. B, Mean computed cardiomyocyte volume from 2‐dimensional isolated myocyte measurements (n=mean 50 cells for N=16–22 hearts). C and D, Fura‐2 loaded isolated myocyte exemplar records: shortening and Ca2+ transients. E through G, Cardiomyocyte mean maximum % shortening (normalized for myocyte length), and mean Ca2+ diastolic and twitch amplitude levels (n=16–17 cells for N=10 hearts/group). H through J, Regression plots: myocyte size (μm) vs % shortening, diastolic Ca2+ and peak systolic Ca2+ levels (n=16–17 cells from N=10 hearts/group). K and L, Exemplar “Phase‐loop” plots of cardiomyocyte length vs Ca2+ during activation cycle show HHR right‐shift, with significantly higher Ca2+ at 50% myocyte relaxation, indicating reduced sensitivity to Ca2+ levels (n=16–17 cells from N=10 hearts/group). Graphs show mean ± SEM, For B, E through G, and L, *P<0.05 (Student t test). For H through J, P value, Pearson's correlation.
Figure 4.
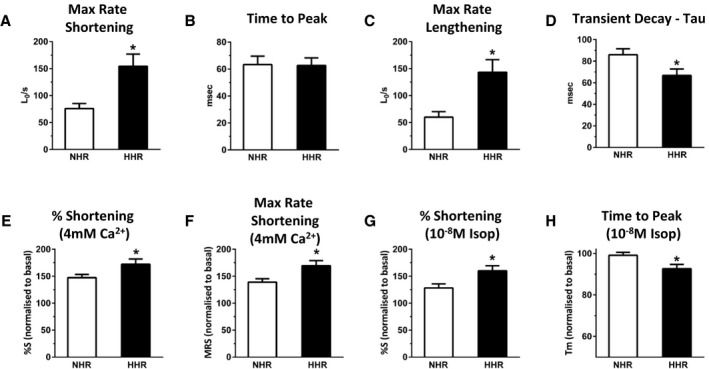
Isolated cardiomyocyte basal shortening parameters and responses to inotropic interventions (20–30 weeks). A through D, Mean maximum rate of shortening, time of peak shortening, maximum rate of lengthening and Ca2+ transient decay time constant (τ=1/e). E and F, Mean changes in shortening (%S) and maximum rate of shortening in response to an increase in superfusate [Ca2+] from 1.5 to 4 mmol/L for normal heart rat (NHR) and hypertrophic heart rat (HHR). G and H, Mean changes in shortening (%S) and time to peak shortening in response to isoproterenol treatment (10−8 mol/L) for NHR and HHR. Graphs show mean ± SEM. For each myocyte group, reference performance level under control pretreatment conditions designated 100%. (*P<0.05, Student t test, n=17 to 19 cells from N=4 to 6 hearts/group).
HHR myocyte diastolic Ca2+ levels were elevated and transient amplitude values increased (Figure 3D, 3F, and 3G). Positive correlations between HHR myocyte size and Ca2+ levels (but not shortening responses) were detected (Figure 3H through 3J), suggesting a hypertrophy‐dependent phenomenon of Ca2+ load augmentation. For individual activation cycles, myocyte‐shortening Ca2+ “phase‐loop” plots were constructed. During the relaxation phase, the descending portion of the loop provides a dynamic index of myofilament Ca2+ sensitivity. A right shift in HHR myocyte loop relaxation phase provided evidence of decreased myofilament Ca2+ responsiveness in the hypertrophic myocytes, quantified by measurement of Ca2+ at 50% myocyte relaxation (Figure 3K and 3L).
No differences in HHR and NHR isoform type and total phosphorylation status of titin (an extensible protein with a role in determining sarcomeric stiffness) were identified (Figure S5, Table S3).
Elevated Myocyte Ca2+ Loading in Diastolic Dysfunction Linked With Increased I‐Ca2+ Current Density
I‐Ca measured using whole‐cell patch clamp methodology, were significantly elevated in prefailure HHR myocytes relative to NHR (holding potential −90 mV), with peak I‐Ca density (normalized for cell size) increased by ≈65% in HHRs (Figure 5A through 5C). Current measured from −90 mV substantially comprises the L‐type Ca2+ current. Consistent current density differences were also observed for current ensembles measured from holding potentials −50 and −115 mV. There was no evidence of HHR‐specific low‐voltage activated currents (absence of inflexion in current plots from −115 and −90 mV holding potentials) (Figure S6). Capacity for Ca2+ flux via the Na+/Ca2+ exchanger was examined electrophysiologically, as this transporter upregulation is implicated in arrhythmogenicity.21 In forward mode operation (ie, Ca2+ extrusion, inward current, from −90 mV) and reverse mode operation (to +40 mV), the I‐NCX densities (pA/pF) were not different for NHR and HHR (Figure 5D through 5F). Protein expression data indicate that NCX1 levels are also not different in the NHR and HHR (Figure S7).
Figure 5.
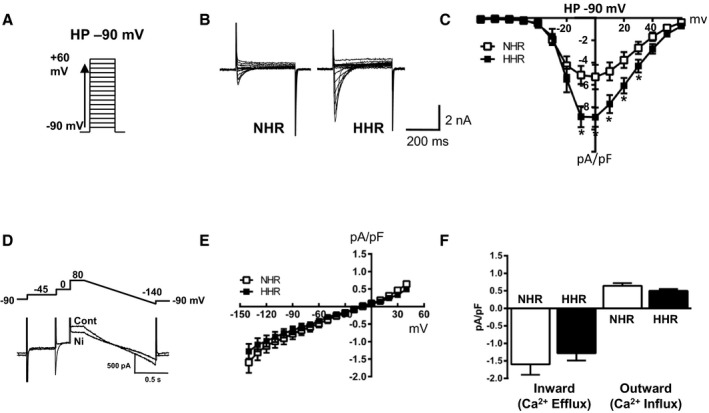
Elevated myocyte Ca2+ loading in diastolic dysfunction linked with increased I‐Ca current density (20–30 weeks). A, I‐Ca voltage clamp protocol, holding potential −90 mV. B, Representative I‐Ca current traces. C, Mean I‐Ca current densities (pA/pF). D, Na+/Ca2+ exchange current (I‐NCX) voltage clamp protocol and representative current traces, I‐NCX quantification by current subtraction ±5 mmol/L NiCl2. E, Mean normalized I‐NCX determined at ramp test potentials −140 to +40 mV. F, Mean maximum inward I‐NCX density (inward and outward mode operation). Graphs show mean ± SEM (*P<0.05, 1 way ANOVA with repeated measures, n=27–30 cells for N=9 hearts/group).
HHR In Vivo and In Vitro Dysrhythmic Substrates—Cardiomyocyte Ca2+ Handling Instability
In vivo occurrence of prominent arrhythmic episodes in HHR at 20 to 30 weeks and beyond was confirmed by external lead ECG—events not observed in NHR (Figure 6A). Significant prolongation of mean HHR QRS interval was quantified, consistent with ventricular abnormality (Figure S8). Electromechanical instability in isolated HHR cardiomyocytes was also quantifiable, with a 2‐fold increase in occurrence of spontaneous SR Ca2+ release events (Figure 6B) indicative of SR Ca2+ store overload susceptibility. Regulation of activator Ca2+ and SR Ca2+ load stability is crucially dependent on expression and activation levels of key proteins involved in SR Ca2+ uptake and release. In the HHR, total expression levels of the SR Ca2+ ATPase (SERCA2a), phospholamban (the SERCA2a negative regulator), and the SR Ca2+ release channel protein were significantly reduced. In contrast, the phosphorylation states of these proteins were markedly increased—effects that act to relieve SR pump inhibition and promote SR Ca2+ release channel trigger sensitivity and leak (Figure 6C and 6D). Expression‐level reduction and increased activation status of the upstream regulator Ca2+‐calmodulin dependent kinase II (CaMKII) was observed (Figure 6E), consistent with increased activation of the SR Ca2+ effector CaMKII target proteins. Evidence indicates that SERCA2 activity is increased on the basis of hyperphosphorylation of the SR pump regulator phospholamban (Figure 6C) and more rapid rate of transient decay in the HHR (Figure 4D). Pilot caffeine spritz experiments also indicate SR Ca load is elevated in the HHR (Figure S9).
Figure 6.
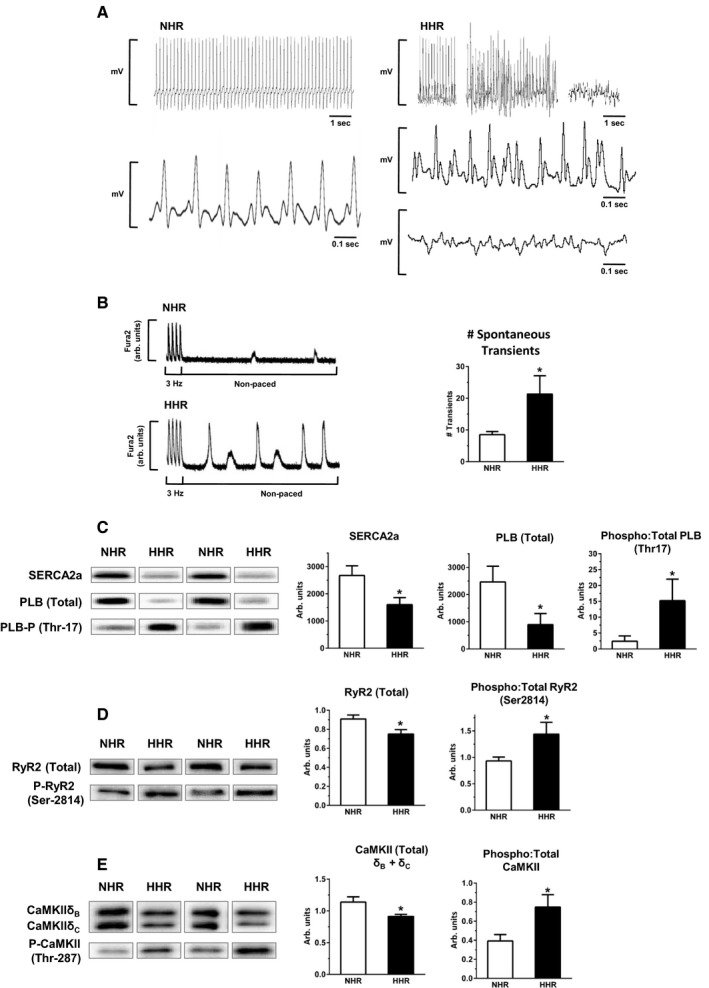
HHR in vivo and in vitro dysrhythmic substrates—cardiomyocyte Ca handling instability. A, Exemplar ECG records depicting stable sinus rhythm in normal heart rat (NHR) vs episodic hypertrophic heart rat (HHR) dysrhythmic periods. B, Spontaneous Ca2+ release events in nonpaced cardiomyocytes, quantified over 30 seconds. (*P<0.05, Student t test, n=21–23 cells/group). C through E, Immunoblot analyses of total protein expression and phosphorylation levels, left ventricular tissue homogenate, for SERCA2a, PLB (Ser‐17), RYR2 (Ser2814), CaMKII (combined δB & δC isoform, Thr287) (*P<0.05, Student t test, N=10 hearts/group). Graphs show mean ± SEM. For A, age 30 to 50 weeks. For B through E, age 20 to 30 weeks.
Discussion
In this study, we present and characterize a new experimental model of HFpEF—and identify a cardiomyocyte pathology that contrasts conspicuously with that generally reported in HFrEF conditions. In vivo, the HHR displays marked diastolic dysfunction in the context of relatively preserved systolic function. The in vitro cardiomyocyte functional studies of Ca2+ handling and shortening performance surprisingly reveal a hypercontractile state underlying the in vivo diastolic dysfunction. Other notable features include those of focal and relatively discrete areas of interstitial fibrosis and the in vivo predisposition to dysrhythmia and sudden death. The cardiomyocyte phenotypes of higher incidence of spontaneous Ca2+ transients and molecular changes consistent with SR Ca2+ instability provide evidence of cellular arrhythmogenic substrate. Thus, preservation of EF is apparently achieved through hypercontractility, which compensates for cardiomyocyte deficit and structural slippage at fibrotic foci. These findings represent a potentially important advance in understanding the cellular etiology of HFpEF. We demonstrate that the cardiomyocyte Ca2+ handling and contractile abnormalities in HFpEF are fundamentally different to the cellular electromechanical features usually observed in association with HFrEF.
The HHR provides a model that closely reflects clinical HFpEF. We have previously identified early emergence of cardiac hypertrophy in adult HHR, linked with deficient cardiomyocyte endowment in the neonatal heart.16, 17 Now in this longitudinal study, we demonstrate that hypertrophic progression to failure occurs with animal aging and without additional intervention. At failure, diastolic dysfunction is marked, while ejection fraction and fractional shortening are largely maintained, with normal end‐diastolic volume index. We show that premature mortality in the HHR is associated with evidence of ventricular stiffness as indexed by elevated end‐diastolic pressure‐volume relationship (EDPVR) and impaired filling, focal reparative (interstitial) fibrosis, and prominent cardiomyocyte hypertrophy. Elevated brain natriuretic peptide has been shown previously.17 Together, these findings indicate the HHRs die prematurely from both fatal cardiac arrhythmias and chronic diastolic heart failure. This constellation of phenotypes is consistent with generally accepted clinical benchmarks for HFpEF diagnosis—notwithstanding the inherent syndrome heterogeneity.7, 8, 9
HHR cardiomyocytes exhibit hypercontractile status and high Ca2+ operational levels linked with arrhythmogenic vulnerability. These cardiomyocyte operational characteristics contrast dramatically with the canonical view of the hypertrophied and failing cardiomyocyte—a phenotype of HFrEF defined primarily in the setting of hemodynamic loading and typified by impaired contractility and reduced activator Ca2+ availability.11 Systolic function preservation in the HHR likely derives from the large increase in density of the L‐type Ca2+ channel, underpinning cardiomyocyte hypercontractility. The developmental mechano‐transduction link between increased myocyte dimension and hyperperformance is unknown but appears to be a characteristic specific for pathologic myocyte enlargement as expressed in the HHR. In HHRs there was a significant and positive correlation of cell dimension with systolic and with diastolic Ca2+ levels (ie, larger myocytes exhibited more elevation in Ca2+ operational levels). This relationship was absent in the NHR. This finding supports the speculation that a signaling process may operate to link hypertrophy induction and shift of Ca2+ operational levels in the evolving HFpEF context. Temporally and spatially averaged elevations in cardiomyocyte Ca2+ levels have been linked with induction of experimental hypertrophy.22 Elevated Ca2+ levels have been observed in ventricular biopsy specimens of patients diagnosed with hypertensive heart disease exhibiting HFpEF symptoms (in the absence of any disturbance of Na+ perturbation).23 Further work is required to evaluate the myocyte hypertrophy‐Ca2+ relationship and potential interventional opportunities specifically in the HFpEF setting. In the HHR, we also found an indirect indication of myofilament Ca2+ desensitization—this may suggest a protective adaptation in response to the high Ca2+ cycling environment. In the HHR, it will be etiologically informative to confirm that the elevated Ca2+ transient phenotype persists to point of failure, and further longitudinal studies are required.
It may be hypothesized that the cardiomyocyte deficit in HHR mediates compensatory hypertrophy of surviving myocytes through mechano‐transduced shifts in molecular management of Ca2+ influx. Increased voltage‐activated Ca2+ current was linked with both increased Ca2+ transient amplitude and peak myocyte shortening. Our data do not allow quantitative dissection of the Ca2+ channel/SERCA2 contributions to the transient but do support the proposition that the increased L‐type Ca2+ channel current provides the primary driver for elevated Ca2+ transient, and a consequentially activated SERCA2 allows for enhanced SR Ca2+ releasable load. Further experimental work with cardiomyocytes using pharmacologic agents is required to resolve these questions, including longitudinal studies that examine the early emergence of the Ca2+ channel phenotype.
Notably, no functional difference in Na+/Ca2+ current density was detected between HHR and NHR cardiomyocytes. This contrasts with previous experimental demonstration of Na+/Ca2+ exchanger upregulation as a key process in transition to failure in HFrEF.21 These findings also differ from observations made in a setting of diastolic dysfunction where the primary insult is acute renal intervention (nephrectomy) and cardiomyocyte systolic Ca2+ levels and contractility are unchanged.24 Protein expression data indicate that NCX1 levels are also not different in the two strains. Thus, an explanation of increased activation of the NCX in HHR seems likely, to ensure balance between Ca2+ influx/efflux. NCX activity is known to be allosterically regulated by intracellular Ca2+. There is also evidence of regulation of exchanger activity by protein posttranslation modification—either directly or by accessory protein (eg, phospholemman) modification. A comprehensive understanding of the manner in which posttranslational modifications regulate NCX has yet to be resolved.
The notion that early development myocardial stress events independent of systemic load may be involved in hypertrophy initiation is consistent with evidence that diastolic dysfunction in the spontaneously hypertensive rat is detectable before onset of hypertrophy and hypertension.25 Our finding that diastolic dysfunction is apparent in very young animals is also indicative of early compliance abnormality contributing to abnormal myocardial mechanical and structural modeling.
Diastolic dysfunction can arise from altered internal cardiomyocyte conditions including high interbeat diastolic Ca2+ levels that limit relaxation and/or increase sarcomere structural stiffness.9, 26 In the HHR, the elevated resting Ca2+ levels likely contribute to diastolic dysfunction, and the data suggest that this may be attributed to increased SR Ca2+ “leakiness” mediated via CaMKII activation‐induced ryanodine receptor sensitization through hyperphosphorylation. Direct CaMKII hyperphosphorylation of titin has also been associated with sarcomere stiffness.27 In this study, it was not possible to detect a net change in total titin phosphorylation state, suggesting that any CaMKII phosphorylation actions are offset by other kinase signaling events. Titin isoform expression was not different in HHR—consistent with observations in other rodent models of diastolic dysfunction.27
Arrhythmogenic vulnerability in various settings has been associated with ectopic diastolic events of spontaneous SR Ca2+ release. In the HHR the preserved SR “reloading” capacity (through phospho‐phospholamban‐mediated SERCa2 disinhibition), in association with ryanodine receptor 2 sensitization (a combination of ryanodine receptor 2 hyperphosphorylation and SR load elevation), provides a substrate for spontaneous SR Ca2+ release activity. Our findings show that neither altered Na+/Ca2+ exchanger functional capacity or disrupted T‐tubule geometry are preconditions for occurrence of SR spontaneous Ca2+ release triggered arrhythmogenic events—that high Ca2+ operational levels independently confer instability. Data from in vitro (intact cardiomyocytes) and in vivo (ECG) experiments produced direct evidence of HHR arrhythmogenic propensity, consistent with sudden cardiac death–related HHR mortality during failure progression. This arrhythmogenic vulnerability evaluated in vitro may be even more accentuated in vivo given that the HHR neurohumoral milieu involves factors that differentially elevate heart rate and may predispose to arrhythmia. In considering the demonstrated in vitro differences in HHR and NHR cardiomyocyte performance, it is important to appreciate that the in vivo disparity in heart rate is an additional major consideration.
Myocardial fibrosis/extracellular matrix remodeling also plays a role in diastolic dysfunction.13 In the HHR, regions of focal fibrosis were evident, particularly prominent near the interventricular septum, which were not present in the NHR. Our histologic findings in HHR ventricular tissue might reflect regional vulnerability to localized perimysial layer shear and rupture28 more extensive in the septum confluence, producing areas of reparative fibrosis associated with, and possibly contributing to, the development of global ventricular stiffness. Involvement of cardiomyocyte‐specific inflammatory response may also be involved—we have recently demonstrated activation of lipocalin‐2 mediated inflammatory pathways in the HHR.29 A role for cardiomyocyte hypercontractility in predisposing for mechanical shear damage is feasible. Indeed, fibrosis and slippage might change the mechanics of the cardiac syncytium such that myocyte hypercontractility is not translated to increased systolic function in vivo. In overview, the etiologic processes that culminate in HHR HFpEF likely have a long‐term latent origin. We have previously reported that hypertrophy in the HHR is linked with reduced cardiomyocyte population at an early growth stage.16, 17 This myocyte deficit apparently mediates compensatory hypertrophy and hypercontractility of diminished myocyte endowment via a mechano‐transduction response (yet to be characterized). Hypercontractility is associated with regions of tissue mechanical vulnerability—myocyte rupture and fibrotic replacement. Thus, with progression toward failure, even while the surviving cardiomyocyte population may be hypercontractile, the remodeled tissue geometry and accumulating focal fibrosis undermines integrity. Overall, ejection fraction is relatively conserved while function (primarily diastolic) is compromised. Clinically, a range of earlier life cardiac stresses and genetic factors could be expected to contribute to HFpEF predisposition and ultimate disease emergence.
In summary, here we report the characterization of a new model of HFpEF and provide the first demonstration of cardiomyocyte cellular pathophysiology underlying this increasingly prevalent form of heart failure. Therapies that have been largely successful in treatment of HFrEF have been targeted at suppressing neurohumoral signaling axes involving the renin‐angiotensin‐aldosterone system and β‐adrenoceptors.3, 14 Recently, evaluation of phosphodiesterase‐5 inhibition also reported lack of clinical improvement in HFpEF.30 While these therapies may reduce morbidity in some HFpEF cohorts, mortality reduction has not be achieved in trials to date.31 Now this investigation provides important mechanistic insight into the contrasting cellular etiologies of HFpEF and HFrEF, and represents a conceptual advance to be exploited for targeted development of HFpEF‐specific therapeutic intervention. Based on this study's novel findings, further studies of the signaling pathways involved and the effects of systemic secondary insults in shaping HFpEF failure development may now be pursued. As a new preclinical platform, the HHR provides a model for building multisystem pathology states through additional interventions that capture the spectrum of HFpEF phenotypes and comorbidities observed clinically. With this new understanding of the cardiopathology mechanism, there is potential opportunity for preclinical innovation in responding to the HFpEF challenge.
Sources of Funding
Career Fellowship support provided through the National Heart Foundation of Australia (Bell, Porrello), the National Health and Medical Research Council (NHMRC) of Australia (Porrello) and the University of Melbourne R. Douglas Wright Faculty Trust (Bell), Research support provided through NHMRC (Delbridge, Harrap, Bell, Erickson, Kalman).
Disclosures
None.
Supporting information
Data S1. Supplemental methods.
Table S1. Echocardiographic and Hemodynamic (Cardiac Catheterization) Analysis of 50‐ and 30‐Week HHR and NHR Hearts
Table S2. HHR and NHR Cardiac and Cardiomyocyte Morphology During Progression to Failure (30 Weeks)
Table S3. Gel Staining Summary Information
Figure S1. Telemetry blood pressure (20–30 weeks) and LV hemodynamic measurements (50 weeks).
Figure S2. Immunoblot primary antibody summary information and Coomassie stained membranes.
Figure S3. Longitudinal systolic and diastolic NHR and HHR functional measurements.
Figure S4. Locational fibrotic mapping of HHR cardiac transverse sections (age 50 weeks).
Figure S5. Titin‐isoform composition and titin phosphorylation are unchanged in HHR (age 50 weeks).
Figure S6. Enhanced sarcolemmal Ca2+ current density in HHR vs NHR.
Figure S7. NCX1 protein expression is unchanged in HHR (age 20–30 weeks).
Figure S8. NHR and HHR electrocardiogram parameters (age 50 weeks).
Figure S9. Increased cardiomyocyte SR Ca2+ load in HHR.
(J Am Heart Assoc. 2018;7:e007451 DOI: 10.1161/JAHA.117.007451.)29858360
These data were presented at the Symposium of the Basic Cardiovascular Sciences Council (BCVS) of the American Heart Association, July 10 to 13, 2017, in Portland, OR.
References
- 1. Greenberg B. Heart failure preserved ejection fraction with coronary artery disease: time for a new classification? J Am Coll Cardiol. 2014;63:2828–2830. [DOI] [PubMed] [Google Scholar]
- 2. Borlaug BA. The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2014;11:507–515. [DOI] [PubMed] [Google Scholar]
- 3. Roh J, Houstis N, Rosenzweig A. Why don't we have proven treatments for HFpEF? Circ Res. 2017;120:1243–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Senni M, Paulus WJ, Gavazzi A, Fraser AG, Diez J, Solomon SD, Smiseth OA, Guazzi M, Lam CS, Maggioni AP, Tschope C, Metra M, Hummel SL, Edelmann F, Ambrosio G, Stewart Coats AJ, Filippatos GS, Gheorghiade M, Anker SD, Levy D, Pfeffer MA, Stough WG, Pieske BM. New strategies for heart failure with preserved ejection fraction: the importance of targeted therapies for heart failure phenotypes. Eur Heart J. 2014;35:2797–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Simone G, Gottdiener JS, Chinali M, Maurer MS. Left ventricular mass predicts heart failure not related to previous myocardial infarction: the Cardiovascular Health Study. Eur Heart J. 2008;29:741–747. [DOI] [PubMed] [Google Scholar]
- 6. Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, Paulus WJ. Phenotype‐specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation. 2016;134:73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shah SJ, Katz DH, Selveraj S, Burke MA, Yancy CW, Gheorghiade M, Bonow RO, Huang CC, Deo RC. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation. 2015;131:269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kelly JP, Mentz RJ, Mebazaa A, Voors AA, Butler J, Roessig L, Fiuzat M, Zannad F, Pitt B, O'Connor CM, Lam CS. Patient selection in heart failure with preserved ejection fraction clinical trials. J Am Coll Cardiol. 2015;65:1668–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 10. Ponikowski P; Task Force Members . 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 11. Luo M, Anderson ME. Mechanisms of altered Ca2+ handling in heart failure. Circ Res. 2013;113:690–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, Koch WJ; American Heart Association Council on Basic Cardiovascular Sciences CoCC, Council on Functional Genomics and Translational Biology . Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res. 2012;111:131–150. [DOI] [PubMed] [Google Scholar]
- 13. Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2014;385:812–824. [DOI] [PubMed] [Google Scholar]
- 14. Desai AS, Jhund PS. After TOPCAT: what to do now in heart failure with preserved ejection fraction. Eur Heart J. 2016;37:3135–3140. [DOI] [PubMed] [Google Scholar]
- 15. Conceição G, Heinonen I, Lourenço AP, Duncker DJ, Falcão‐Pires I. Animal models of heart failure with preserved ejection fraction. Neth Heart J. 2016;24:275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harrap SB, Danes VR, Ellis JA, Griffiths CD, Jones EF, Delbridge LM. The hypertrophic heart rat: a new normotensive model of genetic cardiac and cardiomyocyte hypertrophy. Physiol Genomics. 2002;9:43–48. [DOI] [PubMed] [Google Scholar]
- 17. Porrello ER, Bell JR, Schertzer JD, Curl CL, McMullen JR, Mellor KM, Ritchie RH, Lynch GS, Harrap SB, Thomas WG, Delbridge LM. Heritable pathologic cardiac hypertrophy in adulthood is preceded by neonatal cardiac growth restriction. Am J Physiol Regul Integr Comp Physiol. 2009;296:R672–R680. [DOI] [PubMed] [Google Scholar]
- 18. Innes BA, McLauglin MG, Kapuscinski MK, Jacob HJ, Harrap SB. Independent genetic susceptibility to cardiac hypertrophy in inherited hypertension. Hypertension. 1998;31:741–746. [DOI] [PubMed] [Google Scholar]
- 19. Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose‐fed mice. J Mol Cell Cardiol. 2011;50:1035–1043. [DOI] [PubMed] [Google Scholar]
- 20. Domenighetti AA, Danes VR, Curl CL, Favaloro JM, Proietto J, Delbridge LM. Targeted GLUT‐4 deficiency in the heart induces cardiomyocyte hypertrophy and impaired contractility linked with Ca2+ and proton flux dysregulation. J Mol Cell Cardiol. 2010;48:663–672. [DOI] [PubMed] [Google Scholar]
- 21. Rodriguez JS, Velez Rueda JO, Salas M, Becerra R, Di Carlo MN, Said M, Vittone L, Rinaldi G, Portiansky EL, Mundiña‐Weilenmann C, Palomeque J, Mattiazzi A. Increased Na⁺/Ca²⁺ exchanger expression/activity constitutes a point of inflection in the progression to heart failure of hypertensive rats. PLoS One. 2014;9:e96400. doi: 10.1371/journal.pone.0096400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3‐dependent perinuclear Ca2+ signaling in cardiac myocyte excitation‐transcription coupling. J Clin Invest. 2006;116:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Runte KE, Bell SP, Selby DE, Häußler TN, Ashikaga T, LeWinter MM, Palmer BM, Meyer M. Relaxation and the role of calcium in isolated contracting myocardium from patients with hypertensive heart disease and heart failure with preserved ejection fraction. Circ Heart Fail. 2017;10:e004311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Primessnig U, Schönleitner P, Höll A, Pfeiffer S, Bracic T, Rau T, Kapl M, Stojakovic T, Glasnov T, Leineweber K, Wakula P, Antoons G, Pieske B, Heinzel FR. Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur J Heart Fail. 2016;18:987–997. [DOI] [PubMed] [Google Scholar]
- 25. Dupont S, Maizel J, Mentaverri R, Chillon JM, Six I, Giummelly P, Brazier M, Choukroun G, Tribouilloy C, Massy ZA, Slama M. The onset of left ventricular diastolic dysfunction in SHR rats is not related to hypertrophy or hypertension. Am J Physiol Heart Circ Physiol. 2012;302:H1524–H1532. [DOI] [PubMed] [Google Scholar]
- 26. van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–1973. [DOI] [PubMed] [Google Scholar]
- 27. Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Krüger M, Backs J, Linke WA. Crucial role for Ca2(+)/calmodulin‐dependent protein kinase‐II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res. 2013;112:664–674. [DOI] [PubMed] [Google Scholar]
- 28. Pope AJ, Sands GB, Smaill BH, LeGrice IJ. Three‐dimensional transmural organization of perimysial collagen in the heart. Am J Physiol. 2008;295:H1243–H1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marques FZ, Prestes PR, Byars SG, Ritchie SC, Würtz P, Patel SK, Booth SA, Rana I, Minoda Y, Berzins SP, Curl CL, Bell JR, Wai B, Srivastava PM, Kangas AJ, Soininen P, Ruohonen S, Kähönen M, Lehtimäki T, Raitoharju E, Havulinna A, Perola M, Raitakari O, Salomaa V, Ala‐Korpela M, Kettunen J, McGlynn M, Kelly J, Wlodek ME, Lewandowski PA, Delbridge LM, Burrell LM, Inouye M, Harrap SB, Charchar FJ. Experimental and human evidence for lipocalin‐2 (neutrophil gelatinase‐associated lipocalin [NGAL]) in the development of cardiac hypertrophy and heart failure. J Am Heart Assoc. 2017;6:e005971. doi: 10.1161/JAHA.117.005971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E; RELAX Trial . Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kosmala W, Rojek A, Przewlocka‐Kosmala M, Wright L, Mysiak A, Marwick TH. Effect of aldosterone antagonism on exercise tolerance in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2016;68:1823–1834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Table S1. Echocardiographic and Hemodynamic (Cardiac Catheterization) Analysis of 50‐ and 30‐Week HHR and NHR Hearts
Table S2. HHR and NHR Cardiac and Cardiomyocyte Morphology During Progression to Failure (30 Weeks)
Table S3. Gel Staining Summary Information
Figure S1. Telemetry blood pressure (20–30 weeks) and LV hemodynamic measurements (50 weeks).
Figure S2. Immunoblot primary antibody summary information and Coomassie stained membranes.
Figure S3. Longitudinal systolic and diastolic NHR and HHR functional measurements.
Figure S4. Locational fibrotic mapping of HHR cardiac transverse sections (age 50 weeks).
Figure S5. Titin‐isoform composition and titin phosphorylation are unchanged in HHR (age 50 weeks).
Figure S6. Enhanced sarcolemmal Ca2+ current density in HHR vs NHR.
Figure S7. NCX1 protein expression is unchanged in HHR (age 20–30 weeks).
Figure S8. NHR and HHR electrocardiogram parameters (age 50 weeks).
Figure S9. Increased cardiomyocyte SR Ca2+ load in HHR.