

Abstract
Background
The joint effect of atherosclerosis and CRP (C‐reactive protein) on risk of ischemic stroke (IS) and myocardial infarction (MI) has been sparsely studied. The aim of this study was to explore whether CRP mediates the risk of events in subjects with prevalent carotid plaque, examine synergism, and test whether CRP and carotid plaque add to risk prediction beyond traditional risk factors.
Methods and Results
CRP and carotid total plaque area (TPA) were measured in 10 109 participants in the Tromsø Study from 1994 to 2008. Incident IS (n=671) and MI (n=1079) were registered until December 31, 2013. We calculated hazard ratios (HRs) of MI and IS according to categories of CRP (<1, 1–3, and >3 mg/L) and plaque status (no plaque and TPA below and above median) in Cox proportional hazard models with time‐varying covariates. Multivariable‐adjusted CRP >3 versus <1 mg/L was associated with risk of IS (HR, 1.84; 95% confidence interval, 1.49–2.26) and MI (HR, 1.46; 95% confidence interval, 1.23–1.73). TPA above median versus no plaque was associated with risk for IS (HR, 1.65; 95% confidence interval, 1.36–2.01) and MI (HR, 1.64; 95% confidence interval, 1.41–1.92). In participants with plaque, adjustment for CRP minimally attenuated the risk estimates. The highest incidence rates for MI and IS were seen in the group with both CRP >3 mg/L and TPA is above the median. TPA and CRP combined added to risk prediction beyond traditional risk factors.
Conclusions
The simultaneous presence of subclinical atherosclerosis and elevated CRP was associated with increased risk of IS and MI. The combined assessment of subclinical atherosclerosis and inflammatory biomarkers may improve cardiovascular disease risk stratification.
Keywords: atherosclerosis, carotid ultrasound, C‐reactive protein, ischemic stroke, myocardial infarction
Subject Categories: Epidemiology, Atherosclerosis, Ischemic Stroke, Ultrasound, Myocardial Infarction
Clinical Perspective
What Is New?
Repeated measures of carotid total plaque area and CRP (C‐reactive protein) were individually associated with increased risk of ischemic stroke and myocardial infarction.
CRP only minimally attenuated the risks in subjects with prevalent carotid plaque, contradictory to what would be expected if CRP and plaques represent the same underlying risk factor (ie, unstable plaques).
The highest incidence rates of ischemic stroke and myocardial infarction were found in subjects with both total plaque area above the median and CRP >3 mg/L.
Inclusion of total plaque area and CRP combined added to risk prediction models beyond traditional risk factors.
What Are the Clinical Implications?
The simultaneous presence of subclinical atherosclerosis and elevated CRP was associated with increased risk of ischemic stroke and myocardial infarction, indicating that the combined assessment of subclinical atherosclerosis and inflammatory biomarkers may improve cardiovascular disease risk stratification.
Introduction
Approximately one third of individuals who experience a first‐time cardiovascular event are misclassified as being at low risk on the basis of traditional risk factors (TRFs).1 Novel biomarkers that can improve cardiovascular disease (CVD) risk prediction are long awaited. Serum levels of CRP (C‐reactive protein)3 and subclinical atherosclerosis assessed by carotid ultrasound,2 have both repeatedly been found to predict future CVD independent of TRFs in large population‐based studies. However, the clinical utility of these factors in determination of cardiovascular risk is not established.2, 4
The development and manifestation of CVD is a complex process that encompasses several components, including atherosclerotic plaque development, plaque rupture, and thromboembolic events. Inflammation is recognized to play a pivotal role in initiation and progression of atherosclerosis. CRP is evidently linked to CVD risk, yet the underlying mechanisms behind these associations are not fully understood. Previous studies do not uniformly support CRP as a causal agent in plaque formation and progression.5, 6, 7 It is suggested that inflammatory active and rupture‐prone plaques may themselves be a source of CRP.8 In this setting, CRP would be expected to mediate the relationship between carotid atherosclerosis and CVD risk because CRP and atherosclerotic plaques would represent the same underlying risk factor (ie, unstable plaques). In addition, experimental studies have indicated that CRP may initiate mechanisms involved in plaque rupture and thrombus formation.9, 10, 11 Thus, an interaction between higher serum levels of CRP and inflammatory active plaques may increase the risk of plaque rupture and explain the attributable risk of CRP in CVD. In this scenario, we would expect the simultaneous presence of elevated CRP and subclinical atherosclerosis to have a synergistic effect on CVD risk.
Only a few studies have explored whether imaging measures of atherosclerosis and markers of inflammation interact with each other in determination of cardiovascular risk, and results are diverging.12, 13 In the Tromsø Study, carotid total plaque area (TPA) and CRP have been repeatedly measured in a general, middle‐aged, white population. By taking repeated measurements within individuals into account, we used Cox proportional hazard models with time‐varying covariates, to investigate the associations between CRP and carotid atherosclerosis, alone and in combination, with incident ischemic stroke (IS) and myocardial infarction (MI). We also examined whether CRP mediated the risk of MI and IS in subjects with carotid atherosclerosis. Finally, we compared the predictive performance of models including only TRFs with models that included TPA, CRP, and TPA+CRP by calculating net reclassification improvement (NRI) indexes.
Methods
The data, analytical methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Study Population
Participants were recruited from the fourth, fifth, and sixth surveys of the Tromsø Study (conducted in 1994–1995, 2001–2002, and 2007–2008, respectively).14 The Tromsø Study is a population‐based prospective study with repeated health surveys of the inhabitants in the municipality of Tromsø, Norway. Overall participation rates were high, ranging from 77% in the fourth survey to 66% in the sixth survey.14 Total birth cohorts and samples from other age groups were invited to the carotid ultrasound examination,14, 15 and 6727, 5454, and 7084 participants completed the fourth, fifth, and sixth surveys, respectively. Participants who attended ≥1 carotid ultrasound examinations were eligible for the present study. Participants without valid written consent (n=71), participants with known prebaseline history of IS (n=121) and MI (n=527), and participants who did not have information on CRP, ultrasound measurements, and relevant covariates in at least 1 of the completed surveys (n=467) were excluded. Our population thus consisted of 10 109 unique individuals, of whom 4932 completed 1, 2505 completed 2, and 2672 completed 3 surveys (Figure 1). Informed written consent was obtained from all participants; the study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Regional Committee for Medical and Health Research Ethics.
Figure 1.
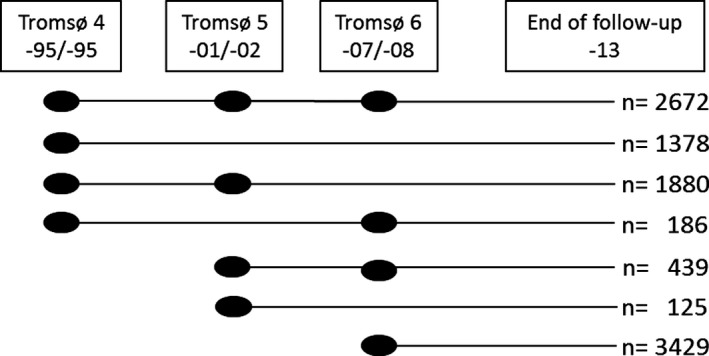
Overview of study inclusion. Dots indicate participation at the survey, and lines indicate observation periods. A total of 10 109 unique individuals were included in the study, of whom 4932 completed 1, 2505 completed 2, and 2672 completed 3 surveys.
Carotid Ultrasound Examination
The baseline and follow‐up measurements followed identical scanning and reading procedures. In the fourth and fifth survey, ultrasonography was performed with an Acuson Xp10 128 ART ultrasound scanner equipped with a 7.5‐MHz linear‐array transducer. In the sixth survey, a GE Vivid 7 scanner with a linear 12‐MHz transducer was used. The far wall and near wall of the right common carotid artery, the bifurcation (bulb), and the internal carotid artery (6 locations) were scanned for the presence of plaques. A plaque was defined as a localized thickening of the vessel wall of >50% compared with the adjacent intima‐media thickness (IMT). TPA was calculated as the sum of all plaque areas (mm2). To ensure equal and standardized examination techniques and measurement procedures, sonographers completed a 2‐month prestudy training protocol. Details about the interobserver and intraobserver reproducibility and interequipment variability have been published previously.16, 17, 18
Cardiovascular Risk Factors
Information on TRFs was collected by physical examination, nonfasting blood samples, and self‐administered questionnaires. Blood pressure was recorded with an automatic device (Dinamap Vital Signs Monitor 1846; Critikon Inc, Tampa, FL) by trained personnel. Participants rested for 2 minutes in a sitting position and then 3 readings were taken on the upper right arm at 1‐minute intervals. The average of the 2 last readings was used in the analyses. Hypertension was defined as systolic blood pressure >140 mm Hg and/or diastolic blood pressure >90 mm Hg and/or use of antihypertensive medication. Body mass index was calculated as weight in kilograms divided by the square of height in meters (kg/m2). Nonfasting blood samples were collected from an antecubital vein. Serum was prepared by centrifugation after 1 hour respite at room temperature and analyzed at the Department of Clinical Biochemistry, University Hospital of North Norway. Serum total cholesterol was analyzed by an enzymatic colorimetric method using a commercially available kit (CHOD‐PAP; Boehringer‐Mannheim, Mannheim, Germany). Serum high‐density lipoprotein cholesterol was measured after precipitation of lower‐density lipoproteins with heparin and manganese chloride. Determination of glycosylated hemoglobin in EDTA whole blood was based on an immunoturbidimetric assay (UNIMATES; F. Hoffmann‐La Roche AG). The glycosylated hemoglobin percentage value was calculated from the glycosylated hemoglobin/hemoglobin ratio. Information on former MI and stroke, prevalent diabetes mellitus, current smoking, and use of antihypertensive and lipid‐lowering medication was collected from self‐administered questionnaires. Diabetes mellitus was defined as self‐reported diabetes mellitus, daily use of oral diabetic medication or insulin, or glycosylated hemoglobin level >6.5%. CRP was analyzed in thawed aliquots after storage at −70°C (fourth survey) or −20°C (fifth and sixth surveys) with a particle‐enhanced immunoturbidimetric assay on a Modular P (fourth and sixth surveys) or Hitachi 917 (fifth survey) autoanalyzer (Roche Hitachi, Mannheim, Germany), with reagents from Roche Diagnostics (Mannheim, Germany). Samples from the fourth survey were analyzed after 12 years of storage, and samples from the fifth and sixth surveys were analyzed at the time of the surveys. The lower detection limit of the high‐sensitivity CRP assay was 0.03 mg/L, and measurements of CRP <0.03 mg/L were set at this value. The analytical coefficient of variation for CRP levels between 0.1 and 20 mg/L was <4%.
Outcome Assessment
On the basis of data from hospital and out‐of‐hospital records, autopsy records, and death certificates, an independent end point committee validated hospitalized and out‐of‐hospital events of incident IS and MI. The national unique 11‐digit identification number was linked to national and local diagnosis registries, including the National Causes of Death Registry, the Population Registry of Norway, and the discharge diagnosis registry (outpatient diagnoses included) at the University Hospital of North Norway, which is the only hospital in the municipality of Tromsø. Medical records, death certificates, autopsy reports, and information from additional sources, such as records from nursing homes, general practitioners, and ambulance services, were used for validation. IS was defined as rapidly developing clinical signs of focal or global disturbance of cerebral function, with symptoms lasting ≥24 hours or leading to death with no apparent cause other than vascular origin, when computed tomography, magnetic resonance imaging, or autopsy had ruled out intracerebral or subarachnoid hemorrhage.
Cases of incident MI were identified by linkage to the discharge diagnosis registry at University Hospital of North Norway with search for International Classification of Diseases, Ninth Revision (ICD‐9) codes 410 to 414 in the period from 1994 to 1998, and thereafter International Classification of Diseases, Tenth Revision (ICD‐10) codes I20 to I25. The hospital medical records were retrieved for case validation. Modified World Health Organization MONICA/MORGRAM19 criteria for MI were used and included clinical symptoms and signs, findings in ECG, values of cardiac biomarkers, and autopsy reports, if applicable. Furthermore, linkage to the national Causes of Death Registry at Statistics Norway allowed inclusion of fatal cases of MI that occurred out of hospital.
Statistical Analyses
We used the statistical software package SAS, 9.4 (SAS Institute, Cary, NC) for all data analyses. Differences in characteristics at time of study entrance between subjects with and without incident IS and MI were estimated separately for each outcome by analysis of covariance, adjusted for age and sex. When treated as continuous variables, CRP was log transformed and TPA was square root transformed to approximate normal distribution.
For each participant, person‐years of follow‐up were counted from the first date of enrollment in the fourth, fifth, or sixth survey to the date of event of interest (separately for incident IS or MI), emigration from Tromsø, death, or end of follow‐up (December 31, 2013), whichever came first. Follow‐up time and risk estimates were calculated separately for IS and MI.
In cohorts with long follow‐up, temporary fluctuations in exposure variables (CRP, TPA, and TRFs) over time may result in underestimation of the true association between exposure and outcome (regression dilution bias).20 An approach to minimize the impact of such bias is to perform analyses with time‐varying covariates, and this method was applied in the present study. Analysis with time‐varying covariates uses individual person data from repeated surveys and takes into account changes in exposure status during follow‐up, by assigning new observation periods with updated values of exposure variables at the time of subsequent study attendance. Thus, subjects who completed >1 survey contributed with 1 observation period per completed survey, and both exposure (CRP and TPA) and confounder (TRFs) data were updated at each completed survey. Because of differences in event censoring, the 10 109 participants contributed with 17 668 observation periods for IS and 17 454 observation periods for MI. If information on exposure or TRFs was missing, values from previous assessments were carried forward, when applicable.
To examine the association between CRP and TPA alone and in combination with risk of IS and MI, we used Cox proportional hazard models with time‐varying covariates and age as time scale.21 Sensitivity analyses were performed by regular Cox models with time‐fixed covariates, using values of exposure and confounder information at time of study entrance and each individual contributing data only once.
CRP was categorized into low‐risk (<1.0 mg/L), intermediate‐risk (1.0–3.0 mg/L), and high‐risk (>3.0 mg/L) groups in accordance with American Heart Association and the Centers for Disease Control and Prevention guidelines for cardiovascular risk.22 We calculated incidence rates and hazard ratios (HRs) with 95% confidence intervals (CIs) for IS and MI using the low‐risk group as reference, first in age‐adjusted models and second in models adjusted for TRFs. The TRFs included were current smoking status, total cholesterol, high‐density lipoprotein cholesterol, systolic blood pressure, diabetes mellitus, body mass index, and use of antihypertensive and lipid‐lowering medication, which were reliably assessed in the Tromsø Study, and have previously shown associations both with exposure (TPA and CRP)2, 3, 23 and outcome (IS and MI).
We defined 3 categories of plaque; categories of TPA were defined separately for men and women at each survey and divided below and above the median, whereas subjects with no plaque constituted the reference category. We estimated HRs with 95% CIs for IS and MI across plaque categories. To address the impact of CRP on the relationship between plaque and the outcomes, we performed age‐ and sex‐adjusted analyses (model 1) and analyses with additional adjustment for CRP (model 1+CRP) and calculated the percentage change in HR when CRP (log transformed) was added to the model. In the final model, we included the previously listed TRFs in addition to CRP (model 2).
Multiplicative interactions between CRP and TPA were assessed. To investigate synergistic effects of atherosclerosis and CRP on the risk of IS and MI, we calculated incidence rates and HRs for the other 8 constellations of atherosclerosis and CRP, and these were compared with the no plaque group with CRP <1 mg/L. Additive interaction and synergism was evaluated using the Rothman synergy index24 to determine whether the joint effects of CRP and atherosclerosis on the risk of IS and MI exceeded the sum of effects from each factor alone in age‐ and sex‐adjusted models. The synergy index, with corresponding 95% CIs, was calculated according to Andersson et al25 using an Excel sheet (epinet.se/res/xls/epinetcalculation.xls) comparing the following 4 constellations: no atherosclerosis and CRP <1 mg/L (reference), no atherosclerosis and CRP >3 mg/L, TPA>median and CRP <1 mg/L, TPA>median and CRP >3 mg/L. A synergy index >1.0 suggests that the effect of the joint exposures of 2 risk markers is greater than the sum of the separate effects.
The added value by TPA and CRP in risk prediction was evaluated by comparing the discrimination power of a model based on the Framingham risk factors (FRFs) with models that additionally included TPA alone, CRP alone, and TPA+CRP together. Original Framingham risk score coefficients were not used because of possible issues of the applicability to different populations.26 For each outcome, a baseline Cox proportional hazard model with time‐fixed covariates was created, using values of FRFs (sex, age, systolic blood pressure, high‐density lipoprotein cholesterol, total cholesterol, smoking, and antihypertensive medication) at time of study entrance. The exposure variables (TPA and CRP), at time of study entrance, were then subsequently included alone and in combination to estimate individual 10 years’ risk for MI and IS. CRP and TPA were included both as continuous and categorical variables. We calculated Harrell's C‐index,27 which is an extension of the receiver operating characteristic curve for survival data. Finally, we computed the relative integrative discrimination improvement and NRI. We considered categories of predicted risk (0%–5%, 5%–10%, 10%–20%, and >20%) and applied SAS macros available in the article by Cook and Ridker.28, 29 Bootstrapping methods (n=500 replications), available at Cook's web page,30 were used to compute 95% CIs for Harrell's C‐index, integrative discrimination improvement, and NRI and test for difference between models by evaluation of P values estimated by the bootstrapping methods. We also considered improvements in discrimination indexes separately for the groups classified to be at intermediate risk (5%–20%) by the FRF‐based models.
For all Cox proportional hazard regression models, the proportional hazard assumption was verified by visual inspection of log‐log survival plots.
Results
Mean age at inclusion was 59.4±8.9 years (range, 25–84 years). Median observation time was 11.0 years (range, 0.01–19.3 years). The study population consisted of 5704 women and 4405 men with a total of 114 716 person‐years for IS and 112 817 person‐years for MI. Table 1 shows crude characteristics of the study population at each survey; sex‐stratified characteristics are presented in Table S1.
Table 1.
Study Population Characteristics in the Different Surveys: The Tromsø Study
| Variable | Fourth Survey (1994–1995) | Fifth Survey (2001–2002) | Sixth Survey (2007–2008) |
|---|---|---|---|
| No. of observations | 6116 | 5116 | 6726 |
| Men, % (n) | 47.8 (2922) | 42.4 (2168) | 42.0 (2824) |
| Age, mean (SD), y | 59.8 (10.3) | 65.5 (9.6) | 63.5 (9.2) |
| BMI, mean (SD), kg/m2 | 26.0 (3.9) | 26.8 (4.2) | 27.0 (4.2) |
| Carotid plaque, % (n) | 47.4 (2896) | 58.3 (2983) | 44.9 (3020) |
| TPA, median (IQR), mm2 a | 15.0 (8.7–26.2) | 20.3 (11.3–35.8) | 19.1 (11.0–31.5) |
| CRP, median (IQR), mg/L | 1.21 (0.61–2.54) | 1.55 (0.83–3.13) | 1.37 (0.77–2.55) |
| Systolic blood pressure, mean (SD), mm Hg | 145 (22) | 143 (22) | 141 (23) |
| Diastolic blood pressure, mean (SD), mm Hg | 83 (13) | 82 (13) | 78 (11) |
| Hypertension, % (n) | 56.3 (3446) | 60.6 (3100) | 58.4 (3931) |
| Antihypertensive medication, % (n) | 11.6 (711) | 23.4 (1197) | 27.3 (1836) |
| HDL cholesterol, mean (SD), mmol/L | 1.6 (0.4) | 1.5 (0.4) | 1.6 (0.4) |
| Total cholesterol, mean (SD), mmol/L | 6.7 (1.3) | 6.3 (1.2) | 5.8 (1.1) |
| Lipid‐lowering medication, % (n) | 1.7 (101) | 12.6 (643) | 16.6 (1115) |
| Diabetes mellitus, % (n) | 3.7 (227) | 8.3 (426) | 7.6 (514) |
| Smoking, % (n) | 31.1 (2025) | 25.0 (1276) | 17.8 (1200) |
BMI indicates body mass index; CRP, C‐reactive protein; HDL, high‐density lipoprotein; IQR, interquartile range; TPA, total plaque area.
In subjects with prevalent carotid plaque. In total, 10 109 participants were included in the study; of these, 4932 completed 1, 2505 completed 2, and 2672 completed 3 surveys.
Table 2 shows age‐ and sex‐adjusted population characteristics at time of study entrance, according to incident IS and MI. In general, levels of TRFs, CRP, plaque prevalence, and TPA were higher in subjects who experienced incident IS or MI during the study period. High‐density lipoprotein cholesterol was lower in subjects who experienced IS or MI.
Table 2.
Age‐ and Sex‐Adjusted Baseline Characteristics of the Study Population, According to First‐Ever IS and MI: The Tromsø Study
| Characteristics | Subjects Without IS | Subjects With Incident IS | P Value | Subjects Without MI | Subjects With Incident MI | P Value |
|---|---|---|---|---|---|---|
| No. of participants | 9438 | 671 | … | 9030 | 1079 | … |
| Men, % (n)a | 43.0 (4060) | 51.4 (345) | <0.0001 | 41.8 (3773) | 58.7 (633) | <0.0001 |
| Age, mean (95% CI), ya | 59.0 (58.8–59.2) | 64.9 (64.3–65.6) | <0.0001 | 58.9 (58.7–59.1) | 63.7 (63.2–64.2) | <0.0001 |
| BMI, mean (95% CI), kg/m2 | 26.3 (26.2–26.4) | 26.5 (26.2–26.8) | 0.438 | 26.3 (26.2–26.4) | 26.6 (26.4–26.9) | 0.034 |
| Carotid plaque, % (n) | 42.7 (4030) | 55.2 (370) | <0.0001 | 42.0 (3793) | 55.9 (603) | <0.0001 |
| TPA, mean (95% CI), mm2 b | 4.2 (4.2–4.3) | 4.4 (4.3–4.6) | 0.0028 | 4.2 (4.1–4.2) | 4.4 (4.3–4.5) | 0.0009 |
| CRP, mean (95% CI), mg/Lc | 1.34 (1.31–1.37) | 1.59 (1.46–1.72) | <0.0001 | 1.34 (1.31–1.37) | 1.53 (1.44–1.63) | <0.0001 |
| Systolic blood pressure, mean (95% CI), mm Hg | 141 (140–141) | 149 (148–151) | <0.0001 | 140 (140–141) | 148 (147–150) | <0.0001 |
| Diastolic blood pressure, mean (95% CI), mm Hg | 81.3 (81.0–81.5) | 85.4 (84.5–86.3) | <0.0001 | 81.1 (80.9–81.3) | 85.3 (84.5–86.0) | <0.0001 |
| Hypertension, % (n) | 51.0 (4817) | 63.8 (428) | <0.0001 | 50.4 (4559) | 63.7 (687) | <0.0001 |
| Antihypertensive medication, % (n) | 14.7 (1387) | 18.2 (122) | 0.062 | 14.5 (1309) | 17.9 (193) | 0.019 |
| Total cholesterol, mean (95% CI), mmol/L | 6.39 (6.36–6.41) | 6.62 (6.53–6.72) | <0.0001 | 6.35 (6.33–6.38) | 6.83 (6.76–6.91) | <0.0001 |
| HDL cholesterol, mean (95% CI), mmol/L | 1.56 (1.55–1.57) | 1.52 (1.49–1.55) | 0.0092 | 1.57 (1.56–1.58) | 1.47 (1.44–1.49) | <0.0001 |
| Lipid‐lowering medication, % (n) | 5.1 (481) | 3.3 (22) | 0.007 | 5.2 (470) | 3.2 (35) | 0.006 |
| Diabetes mellitus, % (n) | 4.3 (406) | 7.4 (50) | 0.002 | 4.2 (379) | 6.6 (71) | 0.003 |
| Smoking, % (n) | 28.1 (2652) | 33.6 (225) | 0.002 | 27.3 (2466) | 40.4 (436) | <0.0001 |
Each subject contributed with observations at time of study inclusion. P value for equality between subjects with incident events and subjects without events during follow‐up. BMI indicates body mass index; CI, confidence interval; CRP, C‐reactive protein; HDL, high‐density lipoprotein; IS, ischemic stroke; MI, myocardial infarction; TPA, total plaque area.
Unadjusted.
Square root–transformed TPA in subjects with prevalent carotid plaque.
Geometric means.
TPA showed a significant weak correlation to CRP, with Spearman correlation coefficient of 0.13 (P<0.001). CRP level was >3 mg/L in 22.4% of all observations. Risk estimates for IS and MI across CRP risk categories are shown in Table 3. CRP level >3 mg/L compared with <1 mg/L was associated with increased risk of IS (HR, 1.84; 95% CI, 1.49–2.26) and MI (HR, 1.46; 95% CI, 1.23–1.73) in multivariable‐adjusted models. Sex‐stratified analyses are displayed in Tables S2 and S3; however, there was no significant interaction with sex for either outcome.
Table 3.
Crude IRs and HRs With 95% CIs of First‐Ever IS and MI From Time‐Varying Cox Models Across Risk Categories of CRP: The Tromsø Study (1994–2013)
| CRP, mg/L | na | Events | IR (95% CI)b | HR (95% CI) | |
|---|---|---|---|---|---|
| Model 1c | Model 2d | ||||
| IS | |||||
| <1 | 6690 | 161 | 3.7 (3.2–4.3) | Reference | Reference |
| 1–3 | 7024 | 261 | 5.7 (5.0–6.4) | 1.27 (1.04–1.55) | 1.15 (0.94–1.41) |
| >3 | 3954 | 249 | 10.0 (8.8–11.3) | 2.18 (1.79–2.67) | 1.84 (1.49–2.26) |
| MI | |||||
| <1 | 6605 | 262 | 6.1 (5.4–6.9) | Reference | Reference |
| 1–3 | 6938 | 467 | 10.4 (9.5–11.3) | 1.45 (1.25–1.69) | 1.25 (1.07–1.46) |
| >3 | 3911 | 350 | 14.2 (12.8–15.7) | 1.95 (1.66–2.29) | 1.46 (1.23–1.73) |
CI indicates confidence interval; CRP, C‐reactive protein; HR, hazard ratio; IR, incidence rate; IS, ischemic stroke; MI, myocardial infarction.
Observations.
Crude IRs per 1000 person‐years.
Age as time scale, adjusted for sex.
Age as time scale, adjusted for sex, total cholesterol, high‐density lipoprotein cholesterol, diabetes mellitus, systolic blood pressure, smoking, body mass index, lipid‐lowering medication, and antihypertensive medication.
HRs for IS and MI across predefined plaque categories are shown in Table 4. In age‐ and sex‐adjusted models, both TPA values below and above the median were associated with higher risk of IS and MI compared with no plaque. Adding CRP to these models led to minimal attenuation of the risk estimates, with absolute attenuation varying from 1.7% to 8.6%. Additional adjustment for TRFs (model 2) led to further attenuation of the risk estimates, but plaque still remained a significant predictor of IS, with HRs (95% CIs) of 1.33 (1.08–1.65) and 1.65 (1.36– 2.01), referring to TPA below and above median, respectively. For MI, the corresponding HRs (95% CIs) were 1.31 (1.11–1.55) and 1.64 (1.41–1.92). Sex‐specific estimates are presented in Tables S4 and S5. For MI but not for IS, there was a significant interaction between plaque category and sex (P=0.02). A stronger association between TPA and risk of MI in women than in men was suggested (Table S5).
Table 4.
Crude IRs and HRs With 95% CIs of First‐Ever IS and MI From Time‐Varying Cox Models Across Categories of TPA Before and After Adjustment for CRP: The Tromsø Study (1994–2013)
| TPA | na | Events | IR (95% CI)b | Model 1 HR (95% CI)c | Model 1 +CRP HR (95% CI)c | Absolute Attenuation of HR After Inclusion of CRP in the Modeld | Attenuation of HR After Inclusion of CRP in the Model, % | Model 2 HR (95% CI)e |
|---|---|---|---|---|---|---|---|---|
| IS | ||||||||
| No plaque | 8945 | 177 | 3.1 (2.6–3.5) | Reference | Reference | … | … | Reference |
| TPA below median | 4362 | 177 | 6.2 (5.3–7.2) | 1.37 (1.11–1.69) | 1.36 (1.10–1.68) | 0.01 | 1.7 | 1.33 (1.08–1.65) |
| TPA above median | 4361 | 317 | 11.2 (10.1–12.6) | 1.93 (1.31–1.77) | 1.85 (1.53–2.24) | 0.08 | 8.6 | 1.65 (1.36–2.01) |
| MI | ||||||||
| No plaque | 8881 | 300 | 5.2 (4.7–5.9) | Reference | Reference | … | … | Reference |
| TPA below median | 4285 | 291 | 10.4 (9.3–11.7) | 1.47 (1.25–1.73) | 1.46 (1.24–1.72) | 0.01 | 2.1 | 1.31 (1.11–1.55) |
| TPA above median | 4288 | 488 | 17.7 (16.2–19.4) | 2.14 (1.84–2.49) | 2.07 (1.78–2.41) | 0.07 | 6.1 | 1.64 (1.41–1.92) |
CI indicates confidence interval; CRP, C‐reactive protein; HR, hazard ratio; IR, incidence rate; IS, ischemic stroke; MI, myocardial infarction; TPA, total plaque area.
Observations.
Crude IRs per 1000 person‐years.
Age as time scale, adjusted for sex.
Change in HR from model 1 to model 1+CRP. CRP was log transformed.
Age as time scale, adjusted for sex, total cholesterol, high‐density lipoprotein cholesterol, diabetes mellitus, systolic blood pressure, smoking, body mass index, lipid‐lowering medication, antihypertensive medication, and CRP (log transformed).
Age‐ and sex‐adjusted HRs of IS and MI across the different constellations of CRP and plaque categories are displayed in Figure 2. Incidence rates and HRs for IS and MI across these categories are listed in Table S6. Subjects with the joint presence of CRP >3 mg/L and TPA above median had the highest incidence rates for both outcomes. For IS, there was a significant excess additive risk when both TPA was above median and CRP was >3 mg/L, with a synergy index of 1.72 (95% CI, 1.06–2.81). The attributable proportion because of interaction was 31.6%. However, there was no indication of synergistic effects between TPA and CRP on risk of MI (Table S7).
Figure 2.
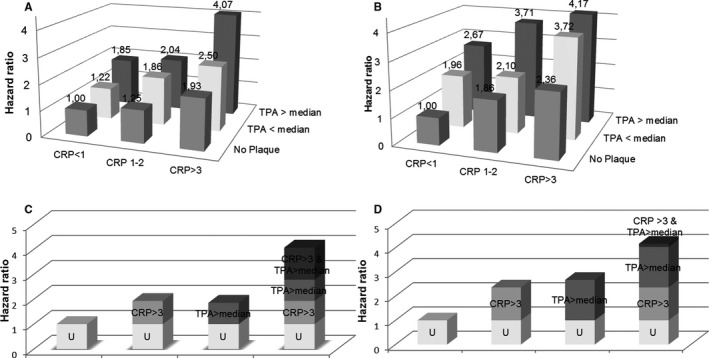
Age‐ and sex‐adjusted hazard ratios of ischemic stroke (A) and myocardial infarction (B) across different constellations of C‐reactive protein (CRP) and total plaque area (TPA). Bottom panels show contributions from different exposure categories on risk for ischemic stroke (C) and myocardial infarction (D). U indicates common reference category for each outcome.
There were no significant multiplicative interactions between CRP and TPA category for either outcome. However, there was a nonsignificant trend of increasing magnitude of CRP risk estimates for IS by increasing TPA (Table S8).
In sensitivity analyses, considering values of exposure (CRP and TPA) and TRFs at time of study entrance in regular time‐fixed Cox models (Tables S9 through S13), the risk estimates were slightly weaker for both outcomes compared with time‐varying analyses, but significance remained unchanged.
When TPA was added as a continuous variable to FRF‐based models, the C‐index improved for prediction of both IS (P=0.040) and MI (P=0.013) (Table S14 and S15). When considering NRI across risk categories (<5%, 5%–10%, 10%–20%, and >20%), the overall NRI for IS was 2.8% (P=0.226), and net improvement was 2.3% for cases and 0.6% for noncases. Overall NRI for MI when adding TPA to FRFs was 3.8% (P=0.030), with a net improvement of 2.5% for MI cases and 1.2% for noncases. The estimate of relative integrative discrimination improvement was 0.16 (P=0.0023) for IS and 0.07 (P<0.0001) for MI. When considering only the intermediate‐risk group, overall NRI was 14.4% (P<0.001) for IS and 10.5% (P=0.035) for MI (Table S16 and S17).
There were no significant differences in C‐index, integrative discrimination improvement, or overall categorical NRI for either outcome after addition of CRP as a continuous variable to FRFs in the whole population. Categorical NRI was 3.6% for IS and 3.7% for MI when CRP was included as a categorical variable. In the intermediate‐risk group, NRI was 12.6% for IS and 8.2% for MI after addition of CRP. For IS, the highest categorical NRIs were seen when including both variables (CRP+TPA) as continuous variables, 6.6% (P=0.007) for the population and 21.6% (P<0.001) for the intermediate‐risk group. For MI, the highest NRIs of 5.0% (P=0.01) for the population and 12.0% (P=0.02) for the intermediate‐risk group were seen when both variables were included as categorical variables.
Discussion
Serum CRP levels and carotid atherosclerosis were individually associated with increased risk of IS and MI, independent of TRFs. Risk estimates for subjects with atherosclerosis were only slightly attenuated after adjustment for CRP. For both outcomes, the joint presence of TPA above median and CRP >3 mg/L was associated with the highest incidence rates. However, a synergistic effect was evident for IS only. TPA alone and the combination of CRP and TPA achieved small, but significant, improvements in risk prediction beyond FRFs, with most prominent effects in the group classified to be at intermediate risk by FRFs.
TRFs have well‐known limitations for accurate assessment of individual cardiovascular risk.1, 31 It is crucial to identify biomarkers that may improve the identification of subjects at risk and guide preventive treatment. Carotid ultrasound is noninvasive and easily accessible, and it can provide direct evidence for the presence and extent of subclinical atherosclerosis with the potential for a more accurate personalized risk assessment and treatment approach.31 Ultrasound assessed measures of subclinical atherosclerosis in carotid arteries; plaque presence,32 plaque echogenicity,33, 34 plaque area,35, 36 and IMT37 are reliable predictors of CVD, even after adjustment for TRFs. The European Guidelines on CVD prevention suggest that imaging methods for atherosclerotic burden are relevant, especially in individuals at intermediate risk based on TRFs, to improve cardiovascular risk stratification and preventive strategy.38, 39 Methodological issues about measurement of carotid IMT on the individual level have been raised, and in the most recent guidelines,40, 41 IMT screening is not recommended. On the other hand, carotid artery plaque assessment, including thickness and TPA, has been proposed as risk modifiers in CVD risk prediction, but formal reclassification analyses have not yet been fully evaluated.40 NRI added by plaque measures in CVD risk prediction has previously been reported by the ARIC (Atherosclerosis Risk in Communities) Study and Three City Study, with overall categorical NRI ranging from 7.7% to 13.1%.42, 43 Differences in plaque assessment, outcome of interest, definition of plaque categories, and incidence rates exist. This may explain discrepancies in results and complicates comparison between studies. For plaque area, the risk estimates in our study were stronger in women than in men, suggesting that assessment of carotid plaque may be a more important tool in risk classification of women than in men. It is suggested that <10% of the population who test positive for atherosclerosis will experience a near‐term event.1 Identification of reliable imaging and serological markers of disease activity is therefore essential to improve the selection of vulnerable patients and cost‐effectiveness of screening with carotid ultrasound in the primary prevention setting.
Inflammation plays a pivotal role in the initiation, progression, and complications of atherosclerosis. Hence, the prognostic value of circulating inflammatory markers in CVD prediction has been assessed in numerous epidemiologic studies. CRP is the marker of inflammation that has been most extensively studied in relation to CVD.3 Most epidemiological studies have reported a moderate dose‐responsive relationship between CRP and clinically relevant CVD outcomes after adjusting for TRFs. Increase in relative risk estimates for CVD ranges from 1.45‐ to ≈2‐fold when comparing the highest with the lowest CRP tertile.44 This is comparable to the effect of TRFs, such as blood cholesterol and blood pressure.44 A meta‐analysis comprising individual participant records from 54 long‐term prospective studies3 reported 1.37 (95% CI, 1.27–1.48) relative risk increase for coronary heart disease and 1.27 (95% CI, 1.15– 1.40) relative risk increase for IS per SD increase in log‐transformed CRP after adjustment for TRFs. These results concur with our risk estimate for IS, but the risk estimate for MI was weaker in our study (1.13; 95% CI, 1.06–1.20) (Table S8). Our results are concordant with the meta‐analysis by Shah et al, which concludes that CRP does not perform better than the FRFs for discrimination in coronary heart disease.4
Despite the evident association between CRP and CVD, the pathogenic role of CRP in CVD remains unclear. Large population‐based cohort studies failed to demonstrate an independent association between CRP and early stages and progression of atherosclerosis measured by carotid IMT.45 These findings are supported by recent results of genomic,6, 46 epidemiological,7, 45 and experimental studies on CRP, which have not proved a causal role of CRP in the formation and progression of atherosclerosis.5, 47 In addition, some controversy about the prognostic value of CRP in CVD prediction still remains,48 and few studies have explored whether CRP's ability to predict CVD is dependent on the presence of atherosclerosis.12, 13 Cao and colleagues concluded that CRP >3 mg/L was a particularly useful predictor in the presence of subclinical atherosclerosis, with a 72% increase in risk for CVD and a 52% increase in total mortality.12 However, CRP did not add predictive power in the absence of carotid atherosclerosis, and an additive interaction for composite CVD and all‐cause mortality was suggested.12 Contradictory, CRP was associated with CVD events with a similar magnitude in the presence and absence of atherosclerosis in the ARIC Study population, but additive interaction of these measures was not assessed.13
In our study, adjustment for CRP led to only minimal attenuation of the risk estimates in participants with plaque. This questions the theory that inflammatory active rupture‐prone plaques secrete CRP.8 In this scenario, CRP and carotid plaques should represent the same underlying risk factor (ie, unstable plaques), and a more substantial attenuation of the risk estimates would be expected on adjustment for CRP. In line with these findings, it is not firmly established that CRP correlates to vulnerable plaque characteristics.49, 50, 51
Our study suggests synergistic effects of CRP and plaque in determination of IS risk. Elevated CRP may be related to mechanisms involved in plaque rupture in acute CVD syndromes, such as production of proteolytic metalloproteinases (matrix metalloproteinases 2 and 9).9 In addition, CRP is closely correlated to obesity, diabetes mellitus, hypercholesterolemia, and cigarette smoking.1, 3, 8 These are all conditions that lead to a prothrombotic state.1 CRP has been shown to induce tissue factor expression by vascular endothelial cells and smooth muscle cells and increase plasminogen activator inhibitor‐1 activity with concomitant reduction in tissue type plasminogen activator activity, resulting in overall impaired fibrinolysis.10 The role of the coagulation system in the outcome of plaque complications is essential. An interaction between CRP and inflammatory active plaques may thus increase risk of plaque rupture and thrombus formation.11 Because mendelian randomization studies and animal studies have not supported a causal role of CRP in CVD, it may be more likely that CRP as a nonspecific marker of inflammation increases secondarily to upstream processes, which are more directly linked to the pathogenesis of CVD.5 However, one limitation of mendelian randomization studies is that the power to detect meaningful gene‐environment interaction is low.52 To our knowledge, it has not been tested whether gene polymorphisms associated with increased serum levels of CRP may have different effects in determining CVD events in the presence and absence of atherosclerosis. Although carotid atherosclerosis may be considered a direct part taker in IS, it is more indirectly correlated with coronary disease, and this may partly explain the lack of synergistic effects on risk of MI in the present study. Assessment of atherosclerosis in coronary arteries may provide evidence of synergistic effects in regard to MI. Unfortunately, coronary computed tomographic scans were not performed in the Tromsø Study.
The strengths of this study are the population‐based design, the large sample of repeated individual data, standardized diagnostic criteria, rigorous validation of cases, and high attendance rate. The unavailability for follow‐up is negligible because of use of the unique personal identity number to search official health registries. One single hospital provides all hospital care in the region, which facilitates the completeness of our outcome registries. However, case identification was retrospective, and some nonhospitalized nonfatal cases may not have been identified. Although we used a standardized protocol for TPA assessment, these measurements are prone to measurement error. The use of different ultrasonography equipment in the fourth and the sixth survey and nonstandardized uptake angles are likely to have increased the measurement error between surveys. We aimed to diminish the effect of measurement errors by defining TPA medians separately at each survey. A limitation of our study is that our ultrasound protocol included examination of only the right carotid artery, and plaques in the left artery were not acknowledged. Our classification of atherosclerosis was designated to study the interaction of carotid atherosclerosis and CRP, and this limits the comparability with other studies. If the stability of CRP is affected by freezing, thawing, or storage, bias may be introduced by the use of frozen blood samples. In the present study, CRP was analyzed in thawed serum aliquots after 12 years (fourth survey) or consecutively during the course of the study (fifth and sixth surveys). CRP stability in frozen samples was previously reported to be acceptable, with high correlations between CRP values obtained before and after storage.53
The use of updated exposure variables on subsequent surveys may have diminished regression dilution effects and survival bias related to subsequent study attendance. Response bias may have distorted the validity of covariates, such as self‐reported smoking, diabetes mellitus, and medication use. Selection bias may have affected the estimates, because attendance rates were lower in elderly people, who are at higher risk of CVD. We did not perform competing risk analyses, meaning that the occurrence of the event of interest (IS and MI) could have been impeded by competing events. Our study population consisted of middle‐aged whites and our results may not be generalizable to populations of other racial and age compositions.
In conclusion, we found that repeated measurements of CRP and plaque burden, assessed by TPA in carotid arteries, individually were predictors of IS and MI, independent of TRFs. The joint presence of elevated CRP and carotid atherosclerosis was associated with the highest incidence rates of IS and MI. Our results extend previous findings and indicate that these measures may have synergistic effects in the determination of CVD risk. CRP has been linked to mechanisms involved in plaque rupture and thrombus formation, which may explain synergism. Future research should focus on whether addition of emerging biomarkers, particularly indicative of unstable plaque features, improves individualized risk assessment and should evaluate cost‐effectiveness of measuring these biomarkers in primary and secondary CVD prevention.
Sources of Funding
The Tromsø Study has been supported by the Research Council of Norway, the Norwegian Council on Cardiovascular Disease, the Northern Norway Regional Health Authority, UiT The Arctic University of Norway, the Norwegian Foundation for Health and Rehabilitation, the Odd Berg Research Foundation, and the Simon Fougner Hartmann's Family Fund. Eltoft receives a research grant from the University Hospital of North Norway (Tromsø, Norway). The publication charges for this article have been funded by a grant from the publication fund of UiT The Arctic University of Norway.
Disclosures
None.
Supporting information
Table S1. Distribution of Risk Factors in the Different Surveys Stratified By Sex. The Tromsø Study
Table S2. Sex Stratified Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke From Time‐Varying Cox Models Across Risk Categories of C‐Reactive Protein (CRP). The Tromsø Study
Table S3. Sex Stratified Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Myocardial Infarction From Time‐Varying Cox Models Across Risk Categories of C‐Reactive Protein (CRP). The Tromsø Study
Table S4. Sex Stratified Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke From Time‐Varying Cox Models Across Categories of Total Plaque Area (TPA) Before and After Adjustment for C‐Reactive Protein (CRP). The Tromsø Study
Table S5. Sex Stratified Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Myocardial Infarction From Time‐Varying Cox Models Across Categories of Total Plaque Area (TPA) Before and After Adjustment for C‐Reactive Protein (CRP). The Tromsø Study
Table S6. Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke (IS) and Myocardial Infarction (MI) From Time‐Varying Cox Models Across Constellations of C‐Reactive Protein (CRP) and Categories of Total Plaque Area (TPA). The Tromsø Study
Table S7. Additive Interaction of C‐Reactive Protein (CRP) and Categories of Total Plaque Area (TPA) on Risk of First‐Ever Ischemic Stroke and Myocardial Infarction. The Tromsø Study
Table S8. Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) Per Standard Deviation Increase in C‐Reactive Protein (CRP)* Across Categories of Total Plaque Area (TPA). Assessment of Multiplicative Interaction. The Tromsø Study
Table S9. Crude Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke From Time‐Fixed* Cox Models Across Risk Categories of C‐Reactive Protein (CRP). The Tromsø Study
Table 10. Crude Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Myocardial Infarction From Time‐Fixed* Cox Models Across Risk Categories of C‐Reactive Protein (CRP). The Tromsø Study
Table S11. Crude Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke From Time‐Fixed* Cox Models Across Categories of Total Plaque Area (TPA), Before and After Adjustment for C‐Reactive Protein (CRP). The Tromsø Study
Table S12. Crude Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Myocardial Infarction From Time‐Fixed* Cox Models Across Categories of Total Plaque Area (TPA), Before and After Adjustment for C‐Reactive Protein (CRP). The Tromsø Study
Table S13. Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke (IS) and Myocardial Infarction (MI) From Time‐Fixed* Cox Models Across Constellations of C‐Reactive Protein (CRP) and Categories of Total Plaque Area (TPA). The Tromsø Study
Table S14. Performance of Time‐Fixed* Cox Regression Models for Ischemic Stroke (IS) With Addition of Total Plaque Area (TPA), C‐Reactive Protein (CRP) and Both (TPA+CRP) Compared to Framingham Risk Factor Based Model. The Tromsø Study
Table S15. Performance of Time‐Fixed* Cox Regression Models for Myocardial Infarction (MI) With Addition Total Plaque Area (TPA), C‐Reactive Protein (CRP) and Both (TPA+CRP) Compared to Framingham Risk Factor‐Based Model. The Tromsø Study
Table S16. Performance of Time‐Fixed* Cox Regression Models for Ischemic Stroke (IS) in Subjects At Intermediate Risk (5–20%, n=2994) With Addition of Total Plaque Area (TPA), C‐Reactive Protein (CRP) and Both (TPA+CRP) Compared to Framingham Risk Factor‐Based Model. The Tromsø Study
Table S17. Performance of Time‐Fixed* Cox Regression Models for Myocardial Infarction (MI) in Subjects At Intermediate Risk (5–20%, n=5250) With Addition of Total Plaque Area (TPA), C‐Reactive Protein (CRP) and Both (TPA+CRP) Compared to Framingham Risk Factor‐Based Model. The Tromsø Study
(J Am Heart Assoc. 2018;7:e008951 DOI: 10.1161/JAHA.118.008951.)29773576
References
- 1. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P, Pasterkamp G, Fayad Z, Stone PH, Waxman S, Raggi P, Madjid M, Zarrabi A, Burke A, Yuan C, Fitzgerald PJ, Siscovick DS, de Korte CL, Aikawa M, Airaksinen KE, Assmann G, Becker CR, Chesebro JH, Farb A, Galis ZS, Jackson C, Jang IK, Koenig W, Lodder RA, March K, Demirovic J, Navab M, Priori SG, Rekhter MD, Bahr R, Grundy SM, Mehran R, Colombo A, Boerwinkle E, Ballantyne C, Insull W Jr, Schwartz RS, Vogel R, Serruys PW, Hansson GK, Faxon DP, Kaul S, Drexler H, Greenland P, Muller JE, Virmani R, Ridker PM, Zipes DP, Shah PK, Willerson JT. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part II. Circulation. 2003;108:1772–1778. [DOI] [PubMed] [Google Scholar]
- 2. Naqvi TZ, Lee MS. Carotid intima‐media thickness and plaque in cardiovascular risk assessment. JACC Cardiovasc Imaging. 2014;7:1025–1038. [DOI] [PubMed] [Google Scholar]
- 3. Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J. C‐reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta‐analysis. Lancet. 2010;375:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shah T, Casas JP, Cooper JA, Tzoulaki I, Sofat R, McCormack V, Smeeth L, Deanfield JE, Lowe GD, Rumley A, Fowkes FG, Humphries SE, Hingorani AD. Critical appraisal of CRP measurement for the prediction of coronary heart disease events: new data and systematic review of 31 prospective cohorts. Int J Epidemiol. 2009;38:217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ridker PM. From C‐reactive protein to interleukin‐6 to interleukin‐1: moving upstream to identify novel targets for atheroprotection. Circ Res. 2016;118:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zacho J, Tybjaerg‐Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C‐reactive protein and ischemic vascular disease. N Engl J Med. 2008;359:1897–1908. [DOI] [PubMed] [Google Scholar]
- 7. Eltoft A, Arntzen KA, Hansen JB, Wilsgaard T, Mathiesen EB, Johnsen SH. C‐reactive protein in atherosclerosis—a risk marker but not a causal factor? A 13‐year population‐based longitudinal study: the Tromso study. Atherosclerosis. 2017;263:293–300. [DOI] [PubMed] [Google Scholar]
- 8. Ben‐Yehuda O. High‐sensitivity C‐reactive protein in every chart? The use of biomarkers in individual patients. J Am Coll Cardiol. 2007;49:2139–2141. [DOI] [PubMed] [Google Scholar]
- 9. Cimmino G, Ragni M, Cirillo P, Petrillo G, Loffredo F, Chiariello M, Gresele P, Falcinelli E, Golino P. C‐reactive protein induces expression of matrix metalloproteinase‐9: a possible link between inflammation and plaque rupture. Int J Cardiol. 2013;168:981–986. [DOI] [PubMed] [Google Scholar]
- 10. Bisoendial RJ, Boekholdt SM, Vergeer M, Stroes ES, Kastelein JJ. C‐reactive protein is a mediator of cardiovascular disease. Eur Heart J. 2010;31:2087–2091. [DOI] [PubMed] [Google Scholar]
- 11. Montero I, Orbe J, Varo N, Beloqui O, Monreal JI, Rodriguez JA, Diez J, Libby P, Paramo JA. C‐reactive protein induces matrix metalloproteinase‐1 and ‐10 in human endothelial cells: implications for clinical and subclinical atherosclerosis. J Am Coll Cardiol. 2006;47:1369–1378. [DOI] [PubMed] [Google Scholar]
- 12. Cao JJ, Arnold AM, Manolio TA, Polak JF, Psaty BM, Hirsch CH, Kuller LH, Cushman M. Association of carotid artery intima‐media thickness, plaques, and C‐reactive protein with future cardiovascular disease and all‐cause mortality: the Cardiovascular Health Study. Circulation. 2007;116:32–38. [DOI] [PubMed] [Google Scholar]
- 13. Agarwala A, Virani S, Couper D, Chambless L, Boerwinkle E, Astor BC, Hoogeveen RC, Coresh J, Sharrett AR, Folsom AR, Mosley T, Ballantyne CM, Nambi V. Biomarkers and degree of atherosclerosis are independently associated with incident atherosclerotic cardiovascular disease in a primary prevention cohort: the ARIC study. Atherosclerosis. 2016;253:156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jacobsen BK, Eggen AE, Mathiesen EB, Wilsgaard T, Njolstad I. Cohort profile: the Tromso Study. Int J Epidemiol. 2012;41:961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eggen AE, Mathiesen EB, Wilsgaard T, Jacobsen BK, Njolstad I. The sixth survey of the Tromso Study (Tromso 6) in 2007–08: collaborative research in the interface between clinical medicine and epidemiology: study objectives, design, data collection procedures, and attendance in a multipurpose population‐based health survey. Scand J Public Health. 2013;41:65–80. [DOI] [PubMed] [Google Scholar]
- 16. Joakimsen O, Bonaa KH, Stensland‐Bugge E. Reproducibility of ultrasound assessment of carotid plaque occurrence, thickness, and morphology: the Tromso Study. Stroke. 1997;28:2201–2207. [DOI] [PubMed] [Google Scholar]
- 17. Fosse E, Johnsen SH, Stensland‐Bugge E, Joakimsen O, Mathiesen EB, Arnesen E, Njolstad I. Repeated visual and computer‐assisted carotid plaque characterization in a longitudinal population‐based ultrasound study: the Tromso study. Ultrasound Med Biol. 2006;32:3–11. [DOI] [PubMed] [Google Scholar]
- 18. Herder M, Johnsen SH, Arntzen KA, Mathiesen EB. Risk factors for progression of carotid intima‐media thickness and total plaque area: a 13‐year follow‐up study: the Tromso Study. Stroke. 2012;43:1818–1823. [DOI] [PubMed] [Google Scholar]
- 19. MORGAM project . MORGAM manual. 2001. http://www.thl.fi/publications/morgam/manual/contents.htm. Accessed November 15, 2017.
- 20. Hutcheon JA, Chiolero A, Hanley JA. Random measurement error and regression dilution bias. BMJ. 2010;340:c2289. [DOI] [PubMed] [Google Scholar]
- 21. Thiebaut AC, Benichou J. Choice of time‐scale in Cox's model analysis of epidemiologic cohort data: a simulation study. Stat Med. 2004;23:3803–3820. [DOI] [PubMed] [Google Scholar]
- 22. Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO III, Criqui M, Fadl YY, Fortmann SP, Hong Y, Myers GL, Rifai N, Smith SC Jr, Taubert K, Tracy RP, Vinicor F. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. [DOI] [PubMed] [Google Scholar]
- 23. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 24. Rothman KJ. The estimation of synergy or antagonism. Am J Epidemiol. 1976;103:506–511. [DOI] [PubMed] [Google Scholar]
- 25. Andersson T, Alfredsson L, Kallberg H, Zdravkovic S, Ahlbom A. Calculating measures of biological interaction. Eur J Epidemiol. 2005;20:575–579. [DOI] [PubMed] [Google Scholar]
- 26. Polak JF, Szklo M, Kronmal RA, Burke GL, Shea S, Zavodni AEH, O'Leary DH. The value of carotid artery plaque and intima‐media thickness for incident cardiovascular disease: the multi‐ethnic study of atherosclerosis. J Am Heart Assoc. 2013;2:e000087 DOI: 10.1161/JAHA.113.000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harrell FE Jr, Lee KL, Mark DB. Multivariable prognostic models: issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med. 1996;15:361–387. [DOI] [PubMed] [Google Scholar]
- 28. Cook NR, Ridker PM. The use and magnitude of reclassification measures for individual predictors of global cardiovascular risk. Ann Intern Med. 2009;150:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pencina MJ, D'Agostino RB Sr, D'Agostino RB Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172; discussion 207–212 [DOI] [PubMed] [Google Scholar]
- 30. Cook N. Risk prediction modeling ‐ SAS Macros. 2014. http://ncook.bwh.harvard.edu/sas-macros.html. Accessed September 24, 2017.
- 31. Weber LA, Cheezum MK, Reese JM, Lane AB, Haley RD, Lutz MW, Villines TC. Cardiovascular imaging for the primary prevention of atherosclerotic cardiovascular disease events. Curr Cardiovasc Imaging Rep. 2015;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Inaba Y, Chen JA, Bergmann SR. Carotid plaque, compared with carotid intima‐media thickness, more accurately predicts coronary artery disease events: a meta‐analysis. Atherosclerosis. 2012;220:128–133. [DOI] [PubMed] [Google Scholar]
- 33. Mathiesen EB, Bonaa KH, Joakimsen O. Echolucent plaques are associated with high risk of ischemic cerebrovascular events in carotid stenosis: the Tromso study. Circulation. 2001;103:2171–2175. [DOI] [PubMed] [Google Scholar]
- 34. Park TH. Evaluation of carotid plaque using ultrasound imaging. J Cardiovasc Ultrasound. 2016;24:91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spence JD, Eliasziw M, DiCicco M, Hackam DG, Galil R, Lohmann T. Carotid plaque area: a tool for targeting and evaluating vascular preventive therapy. Stroke. 2002;33:2916–2922. [DOI] [PubMed] [Google Scholar]
- 36. Johnsen SH, Mathiesen EB. Carotid plaque compared with intima‐media thickness as a predictor of coronary and cerebrovascular disease. Curr Cardiol Rep. 2009;11:21–27. [DOI] [PubMed] [Google Scholar]
- 37. Lorenz MW, Markus HS, Bots ML, Rosvall M, Sitzer M. Prediction of clinical cardiovascular events with carotid intima‐media thickness: a systematic review and meta‐analysis. Circulation. 2007;115:459–467. [DOI] [PubMed] [Google Scholar]
- 38. Perk J, De Backer G, Gohlke H, Graham I, Reiner Z, Verschuren M, Albus C, Benlian P, Boysen G, Cifkova R, Deaton C, Ebrahim S, Fisher M, Germano G, Hobbs R, Hoes A, Karadeniz S, Mezzani A, Prescott E, Ryden L, Scherer M, Syvanne M, Scholte op Reimer WJ, Vrints C, Wood D, Zamorano JL, Zannad F. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): the Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J. 2012;33:1635–1701 [DOI] [PubMed] [Google Scholar]
- 39. Peters SA, den Ruijter HM, Bots ML, Moons KG. Improvements in risk stratification for the occurrence of cardiovascular disease by imaging subclinical atherosclerosis: a systematic review. Heart. 2012;98:177–184. [DOI] [PubMed] [Google Scholar]
- 40. Piepoli MF, Hoes AW, Agewall S, Albus C, Brotons C, Catapano AL, Cooney MT, Corra U, Cosyns B, Deaton C, Graham I, Hall MS, Hobbs FD, Lochen ML, Lollgen H, Marques‐Vidal P, Perk J, Prescott E, Redon J, Richter DJ, Sattar N, Smulders Y, Tiberi M, van der Worp HB, van Dis I, Verschuren WM, De Backer G, Roffi M, Aboyans V, Bachl N, Bueno H, Carerj S, Cho L, Cox J, De Sutter J, Egidi G, Fisher M, Fitzsimons D, Franco OH, Guenoun M, Jennings C, Jug B, Kirchhof P, Kotseva K, Lip GY, Mach F, Mancia G, Bermudo FM, Mezzani A, Niessner A, Ponikowski P, Rauch B, Ryden L, Stauder A, Turc G, Wiklund O, Windecker S, Zamorano JL. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: the Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts): developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur J Prev Cardiol. 2016;23:Np1–Np96. [DOI] [PubMed] [Google Scholar]
- 41. Goff DC Jr, Lloyd‐Jones DM, Bennett G, Coady S, D'Agostino RB, Gibbons R, Greenland P, Lackland DT, Levy D, O'Donnell CJ, Robinson JG, Schwartz JS, Shero ST, Smith SC Jr, Sorlie P, Stone NJ, Wilson PW, Jordan HS, Nevo L, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Smith SC Jr, Tomaselli GF. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S49–S73. [DOI] [PubMed] [Google Scholar]
- 42. Nambi V, Chambless L, Folsom AR, He M, Hu Y, Mosley T, Volcik K, Boerwinkle E, Ballantyne CM. Carotid intima‐media thickness and presence or absence of plaque improves prediction of coronary heart disease risk: the ARIC (Atherosclerosis Risk In Communities) study. J Am Coll Cardiol. 2010;55:1600–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Plichart M, Celermajer DS, Zureik M, Helmer C, Jouven X, Ritchie K, Tzourio C, Ducimetiere P, Empana JP. Carotid intima‐media thickness in plaque‐free site, carotid plaques and coronary heart disease risk prediction in older adults: the Three‐City Study. Atherosclerosis. 2011;219:917–924. [DOI] [PubMed] [Google Scholar]
- 44. Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, Lowe GD, Pepys MB, Gudnason V. C‐reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–1397. [DOI] [PubMed] [Google Scholar]
- 45. Willeit P, Thompson SG, Agewall S, Bergstrom G, Bickel H, Catapano AL, Chien KL, de Groot E, Empana JP, Etgen T, Franco OH, Iglseder B, Johnsen SH, Kavousi M, Lind L, Liu J, Mathiesen EB, Norata GD, Olsen MH, Papagianni A, Poppert H, Price JF, Sacco RL, Yanez DN, Zhao D, Schminke U, Bulbul A, Polak JF, Sitzer M, Hofman A, Grigore L, Dorr M, Su TC, Ducimetiere P, Xie W, Ronkainen K, Kiechl S, Rundek T, Robertson C, Fagerberg B, Bokemark L, Steinmetz H, Ikram MA, Volzke H, Lin HJ, Plichart M, Tuomainen TP, Desvarieux M, McLachlan S, Schmidt C, Kauhanen J, Willeit J, Lorenz MW, Sander D; PROG‐IMT study group . Inflammatory markers and extent and progression of early atherosclerosis: meta‐analysis of individual‐participant‐data from 20 prospective studies of the PROG‐IMT collaboration. Eur J Prev Cardiol. 2016;23:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, Shah T, Engert JC, Clarke R, Davey‐Smith G, Nordestgaard BG, Saleheen D, Samani NJ, Sandhu M, Anand S, Pepys MB, Smeeth L, Whittaker J, Casas JP, Thompson SG, Hingorani AD, Danesh J. Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ. 2011;342:d548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Strang F, Schunkert H. C‐reactive protein and coronary heart disease: all said—is not it? Mediators Inflamm. 2014;2014:757123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Blaha MJ, Budoff MJ, DeFilippis AP, Blankstein R, Rivera JJ, Agatston A, O'Leary DH, Lima J, Blumenthal RS, Nasir K. Associations between C‐reactive protein, coronary artery calcium, and cardiovascular events: implications for the JUPITER population from MESA, a population‐based cohort study. Lancet. 2011;378:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gronholdt ML, Sillesen H, Wiebe BM, Laursen H, Nordestgaard BG. Increased acute phase reactants are associated with levels of lipoproteins and increased carotid plaque volume. Eur J Vasc Endovasc Surg. 2001;21:227–234. [DOI] [PubMed] [Google Scholar]
- 50. Halvorsen DS, Johnsen SH, Mathiesen EB, Njolstad I. The association between inflammatory markers and carotid atherosclerosis is sex dependent: the Tromso Study. Cerebrovasc Dis. 2009;27:392–397. [DOI] [PubMed] [Google Scholar]
- 51. Noh TS, Moon SH, Cho YS, Hong SP, Lee EJ, Choi JY, Kim BT, Lee KH. Relation of carotid artery 18F‐FDG uptake to C‐reactive protein and Framingham risk score in a large cohort of asymptomatic adults. J Nucl Med. 2013;54:2070–2076. [DOI] [PubMed] [Google Scholar]
- 52. Smith GD, Ebrahim S. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. [DOI] [PubMed] [Google Scholar]
- 53. Lewis MR, Callas PW, Jenny NS, Tracy RP. Longitudinal stability of coagulation, fibrinolysis, and inflammation factors in stored plasma samples. Thromb Haemost. 2001;86:1495–1500. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Distribution of Risk Factors in the Different Surveys Stratified By Sex. The Tromsø Study
Table S2. Sex Stratified Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke From Time‐Varying Cox Models Across Risk Categories of C‐Reactive Protein (CRP). The Tromsø Study
Table S3. Sex Stratified Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Myocardial Infarction From Time‐Varying Cox Models Across Risk Categories of C‐Reactive Protein (CRP). The Tromsø Study
Table S4. Sex Stratified Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke From Time‐Varying Cox Models Across Categories of Total Plaque Area (TPA) Before and After Adjustment for C‐Reactive Protein (CRP). The Tromsø Study
Table S5. Sex Stratified Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Myocardial Infarction From Time‐Varying Cox Models Across Categories of Total Plaque Area (TPA) Before and After Adjustment for C‐Reactive Protein (CRP). The Tromsø Study
Table S6. Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke (IS) and Myocardial Infarction (MI) From Time‐Varying Cox Models Across Constellations of C‐Reactive Protein (CRP) and Categories of Total Plaque Area (TPA). The Tromsø Study
Table S7. Additive Interaction of C‐Reactive Protein (CRP) and Categories of Total Plaque Area (TPA) on Risk of First‐Ever Ischemic Stroke and Myocardial Infarction. The Tromsø Study
Table S8. Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) Per Standard Deviation Increase in C‐Reactive Protein (CRP)* Across Categories of Total Plaque Area (TPA). Assessment of Multiplicative Interaction. The Tromsø Study
Table S9. Crude Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke From Time‐Fixed* Cox Models Across Risk Categories of C‐Reactive Protein (CRP). The Tromsø Study
Table 10. Crude Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Myocardial Infarction From Time‐Fixed* Cox Models Across Risk Categories of C‐Reactive Protein (CRP). The Tromsø Study
Table S11. Crude Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke From Time‐Fixed* Cox Models Across Categories of Total Plaque Area (TPA), Before and After Adjustment for C‐Reactive Protein (CRP). The Tromsø Study
Table S12. Crude Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Myocardial Infarction From Time‐Fixed* Cox Models Across Categories of Total Plaque Area (TPA), Before and After Adjustment for C‐Reactive Protein (CRP). The Tromsø Study
Table S13. Incidence Rates (IRs) and Hazard Ratios (HRs) With 95% Confidence Intervals (CIs) of First‐Ever Ischemic Stroke (IS) and Myocardial Infarction (MI) From Time‐Fixed* Cox Models Across Constellations of C‐Reactive Protein (CRP) and Categories of Total Plaque Area (TPA). The Tromsø Study
Table S14. Performance of Time‐Fixed* Cox Regression Models for Ischemic Stroke (IS) With Addition of Total Plaque Area (TPA), C‐Reactive Protein (CRP) and Both (TPA+CRP) Compared to Framingham Risk Factor Based Model. The Tromsø Study
Table S15. Performance of Time‐Fixed* Cox Regression Models for Myocardial Infarction (MI) With Addition Total Plaque Area (TPA), C‐Reactive Protein (CRP) and Both (TPA+CRP) Compared to Framingham Risk Factor‐Based Model. The Tromsø Study
Table S16. Performance of Time‐Fixed* Cox Regression Models for Ischemic Stroke (IS) in Subjects At Intermediate Risk (5–20%, n=2994) With Addition of Total Plaque Area (TPA), C‐Reactive Protein (CRP) and Both (TPA+CRP) Compared to Framingham Risk Factor‐Based Model. The Tromsø Study
Table S17. Performance of Time‐Fixed* Cox Regression Models for Myocardial Infarction (MI) in Subjects At Intermediate Risk (5–20%, n=5250) With Addition of Total Plaque Area (TPA), C‐Reactive Protein (CRP) and Both (TPA+CRP) Compared to Framingham Risk Factor‐Based Model. The Tromsø Study