

Abstract
Background
Surgical palliation or repair of complex congenital heart disease in early infancy can produce right ventricular (RV) pressure overload, often leading to acute hemodynamic decompensation. The mechanisms causing this acute RV dysfunction remain unclear. We tested the hypothesis that the immature right ventricle lacks the ability to modify substrate metabolism in order to meet increased energy demands induced by acute pressure overloading.
Methods and Results
Twenty‐two infant male mixed breed Yorkshire piglets were randomized to a sham operation (Control) or pulmonary artery banding yielding >2‐fold elevation over baseline RV systolic pressure. We used carbon 13 (13C)‐labeled substrates and proton nuclear magnetic resonance to assess RV energy metabolism. [Phosphocreatine]/[ATP] was significantly lower after pulmonary artery banding. [Phosphocreatine]/[ATP] inversely correlated with energy demand indexed by maximal sustained RV systolic pressure/left ventricular systolic pressure. Fractional contributions of fatty acids to citric acid cycle were significantly lower in the pulmonary artery banding group than in the Control group (medium‐chain fatty acids; 14.5±1.6 versus 8.2±1.0%, long‐chain fatty acids; 9.3±1.5 versus 5.1±1.1%). 13C‐flux analysis showed that flux via pyruvate decarboxylation did not increase during RV pressure overloading.
Conclusions
Acute RV pressure overload yielded a decrease in [phosphocreatine]/[ATP] ratio, implying that ATP production did not balance the increasing ATP requirement. Relative fatty acids oxidation decreased without a reciprocal increase in pyruvate decarboxylation. The data imply that RV inability to adjust substrate oxidation contributes to energy imbalance, and potentially to contractile failure. The data suggest that interventions directed at increasing RV pyruvate decarboxylation flux could ameliorate contractile dysfunction associated with acute pressure overloading.
Keywords: congenital heart disease, myocardial metabolism, right ventricular pressure overload
Subject Categories: Congenital Heart Disease, Metabolism
Clinical Perspective
What Is New?
The immature right ventricle shows limited ability to maintain contractile function during acute pressure loading.
Right ventricular dysfunction during acute pressure overloading may be caused by defects in substrate metabolism.
The right ventricle is unable to increase carbohydrate oxidation during acute pressure overloading, indicating a defect in pyruvate decarboxylase regulation.
What Are the Clinical Implications?
Pyruvate decarboxylase may represent a therapeutic target for the immature right ventricle exposed to acute pressure overloading, such as occurs after surgery for congenital heart disease.
Introduction
Right ventricular (RV) pressure overloading represents an important usually unavoidable hemodynamic disturbance encountered after surgical palliation or repair of complex congenital heart diseases in early infancy.1 These palliations often involve placing a fixed obstruction in the form of a pulmonary artery banding (PAB) in order to reduce left‐to‐right shunting with elevated blood flow to the lungs. PAB occurs commonly for shunt defects such as muscular ventricular septal defects or unbalanced atrioventricular septal defects, which are not amenable to repair in early infancy. For those infants undergoing complete repair of these shunt lesions, postoperative reactive pulmonary hypertension can lead to acute RV pressure overload.2, 3 Additionally, many surgeries reroute blood flow in order to establish the RV as the systemic pump in cases of left ventricular (LV) hypoplasia. Surgical palliation often includes reconstruction of the aortic arch to assure adequate blood flow from the RV to the aorta. These relatively complex surgical procedures impose several acute stresses on the RV in addition to continuing postoperative high pressure and volume demands. The RV frequently fails in the immediate postoperative period following exposure to these multiple stresses imposed on increased pressure and volume workload. More than 40% of infants undergoing these palliative procedures leaving the RV as the systemic ventricle exhibit clinical low cardiac output syndrome.4 These patients require substantial inotropic support and mechanical ventilation. In some series, up to 14% of these patients cannot sustain circulation and require mechanical circulatory support in the form of extracorporeal membrane oxygenation, with extremely high morbidity and mortality (near 50%).5 Accordingly, strategies are necessary to improve RV function during these assaults by high‐stress phenomena. The cause of RV low cardiac output immediately after cardiac surgery is probably multifactorial, but some studies6 suggest that metabolic support provided either before or immediately after injury ameliorates functional decline of the ventricles. In particular, promotion of flux through pyruvate dehydrogenase (PDH) improves LV function after acute ischemia–reperfusion.7, 8, 9 This observation may apply to the RV, although few experimental models emulate acute RV loading, such as occurs during first‐stage palliation of complex single ventricle type abnormalities.
Our previous work shows that the immature LV responds to alterations in workload conditions, as occur with extracorporeal membrane oxygenation10, 11 or stimulation by thyroid hormone12, 13, 14 with robust switches in substrate accessed for oxidation. In fact, we have shown that thyroid hormone increases flux through PDH, particularly after ischemia–reperfusion injury, and that this shift in substrate flux yields improvements in LV function. Yet, under similar conditions the RV shows less robust changes in substrate utilization.15 These prior data suggest that the RV lacks the same capacity as the LV to moderate substrate utilization in response to various types of stress. However, limited work has been performed, analyzing RV metabolic responses in vivo. Some studies16, 17 have shown limitations in free fatty acid (FA) uptake along with decrease in phosphorylation potential in mature RV subjected to acute pressure overload. The relevance of those studies to conditions prevalent after surgery in the human infant remains unclear. In order to develop metabolic strategies to support the RV under pressure overload, a clear understanding of RV substrate utilization is required.
We postulated that the RV responds to stress, in this case pressure overloading, with a unique metabolic response compared with LV. Thus, we studied the immature swine heart in vivo, under acute PAB to produce RV pressure overload. We have used complex metabolic mapping with both nuclear magnetic resonance spectroscopy (NMR) and gas chromatography–mass spectroscopy (GC‐MS) data in RV tissue obtained during acute RV pressure overloading.
Materials and Methods
The data, analytic methods, and study materials will not be made publically available to other researchers for purposes of reproducing the results or replicating the procedure. However, researchers may contact the corresponding author for any questions related to the article.
Animals and Experimental Design
All experimental procedures in this study were approved by Seattle Children's Institutional Animal Use and Care Committee. Twenty‐two juvenile male mixed breed Yorkshire piglets with a mean weight of 12.5±0.5 kg were used in this experiment. Acute RV pressure overload was induced by the main PAB. Piglets were randomized to 2 groups; a sham operation (Control group) or PAB (PAB group).
Perioperative Management and Operative Technique
Pigs were initially sedated with an intramuscular injection of ketamine (33 mg/kg) and xylazine (2 mg/kg), and were placed on a circulating warming blanket for maintaining a rectal temperature of 36°C to 37.5°C. After intubation through surgical tracheostomy, the pigs were mechanically ventilated with an oxygen (40%–50%) and isoflurane (1%–2%) mixture in a monitored setting of ECG and oxygen saturation. Angio catheters were inserted into the right femoral artery and central vein through the left external jugular vein for continuous recording of the pressure, fluid infusion, and blood sample. After median sternotomy, a flow probe (Transonic Systems Inc, Ithaca, NY) around the ascending aorta was attached, and both ventricular pressure catheters (Millar Instruments, Houston, TX) and pulmonary artery pressure catheter (24‐gauge angio catheter) were directly inserted.
The main pulmonary artery was carefully dissected from the ascending aorta, and then banding tape was placed around the main pulmonary artery using a cotton umbilical tape ≈3 mm width (Ethicon, Somerville, NJ). In the Control group, the banding tape was just looped around the pulmonary artery without constriction. We performed preliminary experiments to validate our model of RV pressure overload. When the RV pressure with PAB tightening exceeded 2.5‐fold greater than baseline RV systolic pressure, all pigs had unrecoverable hemodynamic instability with a severe decline of LV and systemic arterial blood pressure (Figure 1). Accordingly, for the current protocol, the banding tape was gradually constricted until RV and LV pressure tracings showed either plateau or minimal decline, and then was slightly loosened and maintained at this level for 2 hours after verifying RV and LV pressure stability for 30 minutes (Figure 1). The RV systolic pressure to LV systolic pressure ratio under these conditions (RVSP/LVSPmax) provided a parameter for defining maximum sustainable RV work.
Figure 1.
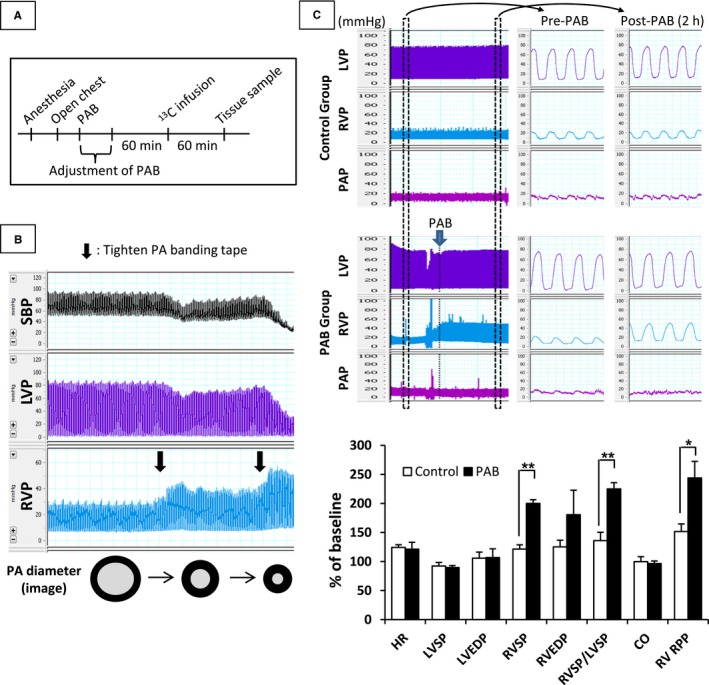
Diagram of experimental protocol (A) and typical representative pressure changes in Control and PAB groups (B and C). Target RV systolic pressure for PAB was greater than 2‐fold baseline RV systolic pressure obtained just after opening the chest. The banding tape was looped around the PA without constriction for pigs in the Control group. After stabilization of hemodynamic data for 60 minutes, 13C‐labeled substrates were infused through the right coronary artery for 60 minutes. Samples of RV tissue were collected at the end of the infusion. Pigs with severely tightened PAB exhibited unrecoverable hemodynamic instability with a severe decline of LV and systemic arterial blood pressure (SBP) (B). Values are means±SE; n=11 per group. *P<0.05; **P<0.01 vs Control. 13C indicates Carbon 13; CO, cardiac output; EDP, end‐diastolic pressure; HR, heart rate; LV, left ventricular; LVEDP, left ventricular end‐diastolic pressure; PAB, pulmonary artery banding; RPP, rate‐pressure product; RV, right ventricular; RVEDP, right ventricular end‐diastolic pressure; RVSP/LVSP, RV‐to‐LV systolic pressure ratio; SP, systolic blood pressure.
Metabolic Analysis
Blood analysis
Blood glucose was measured using a Bayer Contour point‐of‐care glucometer (Bayer HealthCare, Tarrytown, NY). Blood pH, pco 2, Po2, and hemoglobin were measured at regular intervals by a Radiometer ABL 800 (Radiometer America, Westlake, OH).
Infusion of carbon 13 (13C)‐labeled substrates
Two totally different 13C‐labeling strategies were used to provide robust analyses of RV substrate metabolism during acute pressure overload. [2‐13C]lactate and [2,4,6,8‐13C]octanoate, medium‐chain FA (MCFA), were obtained from Sigma–Aldrich (St. Louis, MO), and [U‐13C]mixed long‐chain FAs (LCFAs) and [U‐13C]glucose were obtained from Cambridge Isotope Laboratories (Andover, MA). In addition to lactate, both LCFA and MCFA were used in the first protocol (n=7 per group) because these FAs are major myocardial energy substrates typically supplied to postoperative infants, whereas they have different oxidation pathways; transporters and enzymes mediate LCFA to enter into mitochondria, while MCFA is directly oxidized into mitochondria.18, 19 Additionally, both LCFA and MCFA are used as nutritional supplements for human neonates after recover from cardiac surgery. LCFAs consist of palmitic acid (45%–55%), palmitoleic acid (10–15%), oleic acid (20–30%), and linoleic acid (10–15%), and these proportions are well matched with serum from healthy human subjects.20 Labeled substrates in all protocols were infused from the right coronary artery through a directly inserted 24‐gauge angio catheter for the final 60 minutes of the protocol. The intracoronary doses were adjusted to achieve 1.2 mmol/L [2‐13C]lactate, 0.1 mmol [2,4,6,8‐13C]octanoate, and 0.1 mmol/L [U‐13C]LCFAs elevations to closely match the physiological concentration in the right coronary artery and were based upon the mean coronary artery flow per body weight calculated in our preliminary pig experiments.9 Similarly in the second labeling protocol (n=4 per group) we infused [U‐13C]glucose to determine glucose contribution to oxidative substrates, as well as relative flux through other glycolytic pathways such as the pentose phosphate shunt. We delivered this uniformly labeled glucose at ≈2 mmol/L into the right coronary artery. This concentration maintains glucose levels within physiological range (3.5–5.5 mmol/L), but provided adequate signal for metabolic flux analysis of 13C‐glucose in this protocol. Freeze‐clamped sections of hearts in the region perfused by the right coronary artery were pulverized under liquid nitrogen for metabolic studies at the end of each protocol.
Nuclear magnetic resonance (NMR)
13C‐ and 1H‐NMR were performed on the RV extract tissue for measuring the fractional contribution (Fc) of each substrate to the acetyl coenzyme A (acetyl‐CoA) pool entering the citric acid cycle (CAC) and for measuring the concentration of myocardial energy metabolites, respectively, as previously described.10, 11, 12, 13, 14, 21, 22 13C‐NMR allows determination of fractional contribution of labeled substrates via analyses of the 13C‐glutamate spectrum. 1H‐NMR permits well‐validated high‐resolution and high‐sensitivity analyses for quantitation of a robust number of metabolites including high‐energy phosphates, such as phosphocreatine, adenosine triphosphate (ATP), nicotinamide adenine dinucleotide (NAD), and nicotinamide adenine dinucleotide phosphate (NADP).23
Freeze‐clamped hearts were ground into fine powder under liquid nitrogen and 0.5 mg was further homogenized in 2.5 mL of a methanol/ddH2O (1:0.25) mix. A 2:1 chloroform/ddH2O mix was added to the homogenate, vortexed, and placed on ice for 10 minutes. The samples were next centrifuged for 10 minutes at 2000g. The top layer was removed to a fresh tube and subjected to vacuum lyophilization. The resulting precipitate was dissolved in deuterium oxide (DLM11‐100, Cambridge Isotopes, Andover, MA) and Chenomx ISTD (IS‐1, Chenomx, Alberta Canada) at a 9:1 ratio, filtered through a 0.22‐μmol/L syringe filter into NMR sample tubes (WG‐1241‐8, Wilmad LabGlass, Vineland, NJ). 13C‐NMR data were acquired on a Varian Direct Drive (VNMRS) 600‐MHz spectrometer (Agilent Technologies) equipped with a Dell Precision T3500 Linux workstation running VNMRJ 4.0. The spectrometer system was outfitted with a Varian triple resonance salt‐tolerant cold probe with a cold carbon preamplifier. Fourier‐transformed spectra were fitted with commercial software (NUTS; Acorn NMR Inc, Livermore, CA), and then the data, which were determined from specific carbon glutamate labeling, were analyzed by the CAC analysis‐fitting algorithm TCAcalc (kindly provided by the Advanced Imaging Research Center at the University of Texas, Southwestern). A Varian standard 1‐dimensional proton noesy with presaturation (tnnoesy) was collected on each sample, using a nonselective 90‐degree excitation pulse (≈7 μs at 57 dB), a 100‐ms mixing time, acquisition time of 4 s, a presaturation delay of 1.5 s, spectral width of 12 parts‐per‐million (ppm), and temperature control set to 25°C. Collected spectra by 1H‐NMR were analyzed using Chenomx software (version 8.3, Chenomx) with quantifications based on spectral intensities relative to 0.5 mmol/L 2,2‐dimethyl‐2‐silapentane‐5‐sulfonate, which was added as a spike to each sample. Values were normalized relative to total creatine.
Concentration of metabolites in RV tissue
In addition to analysis of metabolite concentration by 1H‐NMR, commercially available kits were used to measure glycogen (Cayman, Ann Arbor, MI), triglycerides (Cayman), and free FA (BioVision, Mountain View, CA) concentrations in RV. Furthermore, the absolute quantity of glutamate, glutamine, and CAC intermediates; citrate, α‐ketoglutarate (α‐KG), succinate, fumarate, and malate in the extracted RV tissue was measured by GC‐MS using an Agilent 6890N gas chromatograph equipped with a HP‐5 column coupled to a 5975N mass spectrometer (Agilent Technologies, Santa Clara, CA) as described elsewhere.10, 21, 22 GC‐MS data are also reported as the 13C‐molar percent enrichment (MPE) of each metabolite. Mass isotopomers of metabolites containing 1 to n 13C‐labeled atoms were identified as Mi, with i=1, 2, … n, and the total MPE of individual 13C‐labeled mass isotopomers (Mi) of a given metabolite was calculated as follows: total MPE=%∑AMi/(AM+∑AMi), where A and M represent the peak area of each fragmentogram and unlabeled mass isotopomer, respectively. The pathway from pyruvate entering into the CAC via acetyl‐CoA is called pyruvate decarboxylation (PDC). PDC flux rate relative to citrate synthase (CS) was determined using the following formula: PDC/CS=(total 13C‐MPE of citrate—total 13C‐MPE of malate)/M3 of intracellular pyruvate. We also measured CS activity, a biomarker of mitochondrial function in RV tissue, by a commercially available assay kit (Cayman).
Immunoblotting
The primary antibodies used in this study were 5′ adenosine monophosphate‐activated protein kinase α (AMPKα), phospho‐AMPKα‐Thr172, acetyl‐CoA carboxylase (ACC), phospho‐ACC‐Ser79, and PDH obtained from Cell Signaling Technology (Danvers, MA), phospho‐PDH‐Ser293 obtained from Millipore (Billerica, MA), glutamine‐fructose‐6‐phosphate transaminase 2 obtained from Santa Cruz Biotechnology) (Callas, TX), O‐linked N‐acetylglucosamine (O‐GlcNAc), and O‐GlcNAcase obtained from Abcam (Cambridge, MA) and O‐GlcNAc transferase obtained from Sigma‐Aldrich. Fifty micrograms of total protein extract from heart tissue were electrophoresed through 4.5% stacking and 10% running SDS‐polyacrylamide gels and electroblotted onto PVDF‐plus membranes. Equal protein loading of samples was determined by a protein assay (BioRad, Hercules, CA) and confirmed by reversible protein stain kit for PVDF membranes (Thermo Scientific, Rockford, IL) and probing with antibodies against α‐Tubulin (Santa Cruz Biotechnology). Membranes were probed overnight at 4°C with primary antibodies dissolved in PBS‐T containing 5% milk or bovine serum albumin. Blots were incubated at room temperature for 1 hour with the appropriate secondary antibody conjugated to horseradish peroxidase. The blots were visualized with enhanced chemiluminescence after exposure to X‐ray film. The densitometric intensities were determined using Image J analysis software (National Institutes of Health, Bethesda, MD). Western blots were repeated in triplicate to confirm the findings.
Statistical Analysis
Reported values are means±standard error (SE) in text, figures, and tables. Significant differences from the baseline value for each group in Tables 1 and 2 were estimated by a paired t test. Other data were compared and analyzed with the Mann‐Whitney U‐test. Pearson test was used for evaluation of correlations in Figures 2C and 3D. Criterion for significance was P<0.05 for all comparisons.
Table 1.
Hemodynamic Data
| Baseline | P Value | End | P Value | |||
|---|---|---|---|---|---|---|
| Control | PAB | Control | PAB | |||
| Temperature, °C | 37.3±0.3 | 37.3±0.3 | 0.95 | 36.8±0.3 | 36.4±0.2 | 0.32 |
| HR, bpm | 101±4 | 105±9 | 0.67 | 125±5 | 127±15 | 0.90 |
| LVSP, mm Hg | 79±4 | 83±4 | 0.46 | 72±3 | 74±3 | 0.66 |
| LVEDP, mm Hg | 6±1 | 6±1 | 0.95 | 6±1 | 7±1 | 0.86 |
| RVSP, mm Hg | 25±2 | 24±1 | 0.55 | 30±3 | 49±4 | 0.002 |
| RVEDP, mm Hg | 6±1 | 6±1 | 0.98 | 8±1 | 11±2 | 0.17 |
| RVSP/LVSPmax, % | 32±3 | 29±2 | 0.37 | 43±5 | 67±4 | 0.007 |
| CO, L/min | 1.09±0.09 | 1.08±0.06 | 0.97 | 1.06±0.09 | 1.05±0.09 | 0.94 |
| RV RPP | 32±3 | 29±2 | 0.37 | 43±5 | 66±4 | 0.007 |
Values are means±SE; n=11 per group. P value, Control vs PAB. bpm indicates beats per minute; CO, cardiac output; HR, heart rate; LVEDP, left ventricular end‐diastolic pressure; LVSP, left ventricular systolic blood pressure; RPP, rate‐pressure product; RVEDP, right ventricular end‐diastolic pressure; RVSP, right ventricular systolic blood pressure.
Table 2.
Blood and Tissue Concentrations
| Baseline | P Value | End | P Value | |||
|---|---|---|---|---|---|---|
| Control | PAB | Control | PAB | |||
| Blood or plasma | ||||||
| Hgb, g/dL | 9.9±0.6 | 9.9±0.3 | 0.88 | 10.2±0.5 | 10.7±0.4 | 0.44 |
| Glucose, mg/dL | 87.2±1.4 | 78.8±14.3 | 0.12 | 98.0±7 | 93.7±17.9 | 0.69 |
| RV tissue (normalized by total creatine) | ||||||
| Glucose | 0.134±0.027 | 0.128±0.018 | 0.94 | |||
| Glutamate | 0.293±0.037 | 0.289±0.013 | 0.94 | |||
| Glutamine | 0.540±0.032 | 0.468±0.033 | 0.13 | |||
| Aspartate | 0.063±0.012 | 0.080±0.020 | 0.79 | |||
| Carnitine | 0.034±0.003 | 0.034±0.005 | 0.94 | |||
| RV tissue (normalized by RV tissue protein weight) | ||||||
| Glycogen, mg/g | 50.4±6.6 | 62.1±3.9 | 0.14 | |||
| Free FA, mmol/g | 28.9±1.7 | 26.3±1.1 | 0.23 | |||
| TG, mg/g | 30.0±2.2 | 35.3±5.0 | 0.43 | |||
Values are means±SE; n=11 per group. P value, Control vs PAB. FA indicates fatty acid; Hgb, hemoglobin; PAB, pulmonary artery banding; RV, right ventricular; TG, triglycerides.
Figure 2.
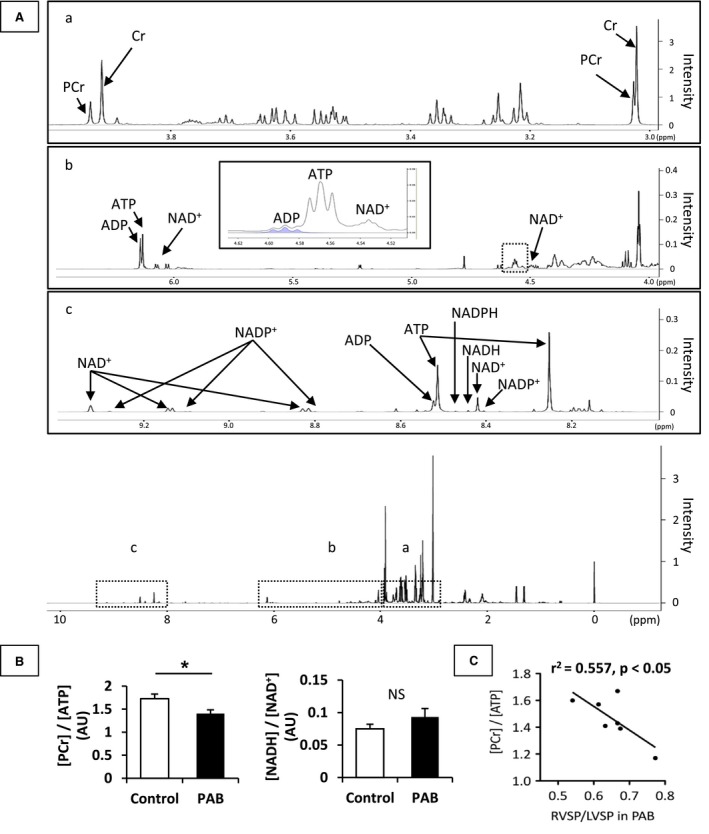
Myocardial energy metabolite ratios. These data were analyzed by 1H‐NMR spectra from RV samples obtained by freeze clamping at the end of the protocol. Representative 1H‐NMR spectra are shown in (A); special regions (a) 3.0 to 4.0 ppm, (b) 4.0 to 6.5 ppm and (c) 8.0 to 9.4 ppm. Analyzed data (B) showed that PAB group demonstrated significantly lower [PCr]/[ATP] than the Control group. [NADH]/[NAD +] was not statistically different between the 2 groups. [PCr]/[ATP] in PAB group inversely correlated with RVSP/LVSP (C). Values are presented as means±SE; n=7 per group. *P<0.05 vs Control. ADP indicates adenosine diphosphate; ATP, adenosine triphosphate; AU, arbitrary units; Cr, creatine; PCr, phosphocreatine; NAD+, the oxidized form of nicotinamide adenine dinucleotide; NADH, the reduced form of nicotinamide adenine dinucleotide; NADP+, the oxidized form of nicotinamide adenine dinucleotide phosphate; NADPH, the reduced form of nicotinamide adenine dinucleotide phosphate; NMR, nuclear magnetic resonance spectroscopy; NS, non‐significant; ppm, parts per million; PAB, pulmonary artery banding; RVSP/LVSP, right ventricular to left ventricular systolic pressure ratio; SP, systolic pressure.
Figure 3.
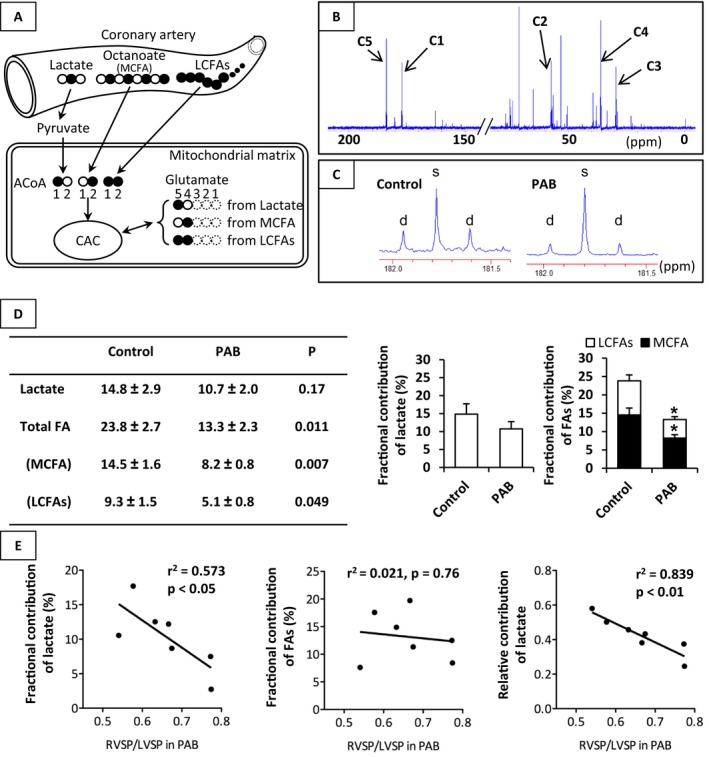
Fractional contribution (Fc) of each substrate to the acetyl‐CoA entering the CAC. Schematic demonstrating of our 13C‐labeled substrates and typical 13C‐NMR full spectrum (A and B), representative spectra for glutamates carbon 5 (C), analyzed data (D) in both groups, and correlation between Fc of substrates and RVSP/LVSP in PAB group (E). Chemical shifts in parts per million (ppm) were as follows: C3, 27.5; C4, 34.2; C2, 55.2; C1, 175.0; C5 of glutamate, 182.1. C5 of glutamate showed lower doublets spike area (d) in the PAB group than that in the Control group, when being measured with reference to singlet spike area (s). PAB group demonstrated significantly lower Fc of MCFA and LCFAs than the Control group. On the other hand, there was a negative linear relation between RVSP/LVSP and lactate oxidation in the PAB group. Values are presented as means±SE; n=7 per group for (D) and n=7 in PAB group for (E). *P<0.05 vs Control. 13C indicates Carbon 13; ACoA, acetyl‐CoA; CAC, citric acid cycle; Fc, fractional contribution; LCFAs, long‐chain fatty acids; MCFA, medium‐chain fatty acid; NMR, nuclear magnetic resonance spectroscopy; RVSP/LVSP, systolic blood pressure ratio between right ventricle and left ventricle.
Results
The mean age and preoperative body weight were similar between 2 groups (Control; 42±5 days, 12.6±0.7 kg, PAB; 42±4 days, 12.2±0.9 kg). There were no operative or technical complications in any piglets. Blood pH, pco 2 and po 2 in all animals were maintained within expected normal range (a pH of 7.35–7.45, an arterial pco 2 of 35–45 mm Hg and an arterial po 2 of >100 mm Hg) during protocol.
Hemodynamics
Table 1 shows that parameters of cardiac function measured at a baseline after opening chest were similar between the 2 groups. PAB (49±4 mm Hg) successfully increased RV systolic pressure 2‐fold over baseline (24±1 mm Hg) (Figure 1C). RVSP/LVSPmax after PAB was ≈67%; it was markedly higher than in the Control group (43±5%, P=0.007). Rate pressure product calculated by heart rate*systolic blood pressure/1000 in RV was also higher in the PAB group than in the Control group. LV pressure did not decline with PAB under these conditions.
Metabolic Studies
Energy metabolites in RV tissue were measured by 1H‐NMR. A representative 1H‐NMR spectrum is shown in Figure 2A. Chemical shifts of 1H‐NMR resonance peaks on energy metabolites were creatine (Cr, 3.022 and 3.915 ppm); phosphocreatine (3.028 and 3.935 ppm); ADP (4.204, 4.247, 4.370, 4.592, 6.135, 8.251, 8.524 ppm); ATP (4.230, 4.275, 4.400, 4.566, 6.136, 8.254, 8.514 ppm); the reduced form of NAD (NADH, 4.696, 5.976, 6.932, 8.443 ppm); the oxidized form of NAD (NAD+, 4.535, 6.030, 6.073, 8.160, 8.184, 8.420, 8.822, 9.139, 9.325 ppm); the reduced form of NADP (NADPH, 4.972, 5.965, 6.195, 6.616, 6.956, 8.235, 8.472 ppm); the oxidized form of NADP (NADP+,4.975, 6.042, 8.132, 8.180, 8.406, 8.808, 9.098, 9.280 ppm) relative to the 2,2‐dimethyl‐2‐silapentane‐5‐sulfonate peak. 1H‐NMR showed that the [phosphocreatine]/[ATP] ratio was lower in the PAB group than in the Control group (Figure 2B). However, the overall workload on the RV varied substantially according to PAB tolerability as defined earlier. We then performed regression analyses to determine whether an association existed between our (RVSP/LVSPmax) and [phosphocreatine]/[ATP]. The RVSP/LVSPmax inversely correlated with [phosphocreatine]/[ATP] (R 2=0.557, P<0.05) (Figure 2C). We analyzed substrate contribution to CAC under acute RV pressure overload by PAB by 13C‐NMR (Figure 3A through 3D). These data were determined from specific carbon glutamate labeling. We confirmed that a similar RV glutamate concentration existed between 2 groups by GC‐MS (Control, 7.5±1.1 nmol/mg; PAB, 5.8±0.4; P=0.18) and 1H‐NMR (Table 2). Acute RV pressure overloading caused a significant decrease in relative contributions of both labeled MCFA and LCFAs to the CAC (MCFA; 14.5±1.6 versus 8.2±1.0%, P<0.01, LCFAs; 9.3±1.5 versus 5.1±1.1%, P<0.05, Control versus PAB group) (Figure 3D). These decreases in labeled substrate contribution for free FAs were not accompanied by any change in the lactate Fc to CAC. However, because the hemodynamic response to PAB was highly variable, we performed correlation studies to determine the relationship between RV compensation and substrate utilization. Our hemodynamic indicator RVSP/LVSPmax following PAB inversely correlated to lactate Fc (r 2=0.573, P<0.05), but not FAs oxidation, in the PAB group (Figure 3E). We also analyzed the relative contribution to CAC among the 13C‐labeled substrates. Relative lactate contribution also correlated RVSP/LVSPmax (r 2=0.839, P<0.01). [Lactate]/[pyruvate] by 1H‐NMR, absolute quantity of CAC intermediates by GC‐MS, and CS activity by assay kit in RV tissue were not statistically different between the 2 groups (Figure 4).
Figure 4.
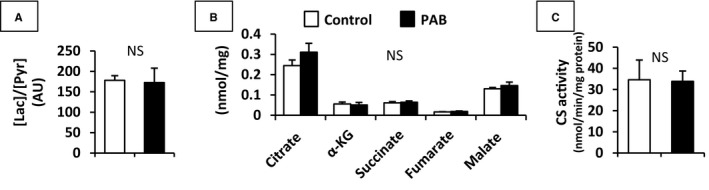
[Lactate]/[pyruvate] by 1H‐NMR (A), absolute quantity of CAC intermediates by GC‐MS (B), and citrate synthase (CS) activity (C) in RV. In addition to [lactate]/[pyruvate], neither these absolute concentrations nor citrate synthase activity in RV tissue were not statistically different between 2 groups. Values are presented as means±SE; n=7 per group. AU indicates arbitrary units; CAC, citric acid cycle; GC‐MS, gas chromatography–mass spectrometry; NMR, nuclear magnetic resonance spectroscopy; NS, non‐significant; α‐KG, α‐ketoglutarate; Lac, lactate; Pyr, pyruvate; RV, right ventricle.
We also estimated relative activity for enzymes regulating metabolic pathways during control and pressure overloaded states (Figure 5). We previously showed that changes in activity during this acute protocol occur through rapid changes in phosphorylation at select enzyme locations.24 AMPK‐ACC signaling axis controls myocardial FA oxidation. Phospho‐AMPKα activates the β‐oxidation of FAs in the mitochondria through the phosphorylation of ACC. The phosphorylated to total protein ratios on both AMPKα and ACC were similar between the 2 groups. Phospho‐PDH (inactive form of PDH), which inhibits PDC, showed no difference between the 2 groups.
Figure 5.
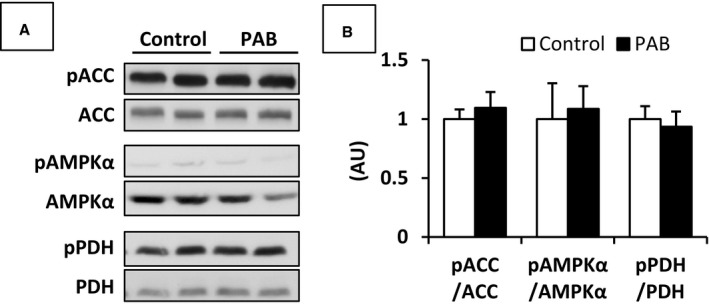
Representative immunoblot data (A) and pooled data (B) for proteins involved in the regulation of FA and carbohydrate metabolism. Phosphorylated ACC, AMPKα, and PDH were at Ser79, Thr172, and Ser293, respectively. These data were not significantly different between the 2 groups. Values are presented as mean±SE; n=7 per group. ACC indicates acetyl‐CoA carboxylase; AMPKα, 5′adenosine monophosphate activated protein kinase; AU, arbitrary unit; FA, fatty acid; PAB, pulmonary artery banding; PDH, pyruvate dehydrogenase.
We evaluated whether acute PAB caused depletion of available substrate within the RV myocardium in order to potentially explain the decrease in free FA oxidation. However, the data showed that there was no difference in tissue levels of free FA, glycogen, or triglycerides between the control and PAB hearts (Table 2). Furthermore, there were no differences in tissue glucose or in absolute levels of multiple amino acids, which could be detected by 1H‐NMR (Table 2).
We also performed separate experiments in Control and PAB piglets using the [U‐13C]glucose infusions as described above in order to evaluate glycolytic pathways during these acute pressure overloading conditions. 13C‐labeled glucose generally contributes relatively low Fc to the CAC in vivo.25 Accordingly, we submitted tissue obtained after this protocol to highly sensitive GC‐MS analyses, rather than 13C‐NMR. The [U‐13C]glucose studies showed that PDC flux supplied ≈40% of acetyl‐CoA metabolized by the CS to form citrate from oxaloacetate in the CAC (Figure 6). As lactate oxidation in the 13C‐NMR experiments supplied 10% to 15% of acetyl‐CoA, we estimate that glucose and other sources of pyruvate such as glycogen supply the remaining ≈20%. However, relative flux through PDH did not differ between PAB and Control or account for differences between these 2 groups in contribution through the unlabeled fraction found in the 13C‐NMR studies. Overall, both the 13C‐NMR and GC‐MS studies show that the RV does not increase flux through PDH during acute pressure overloading (Figures 3D and 6).
Figure 6.
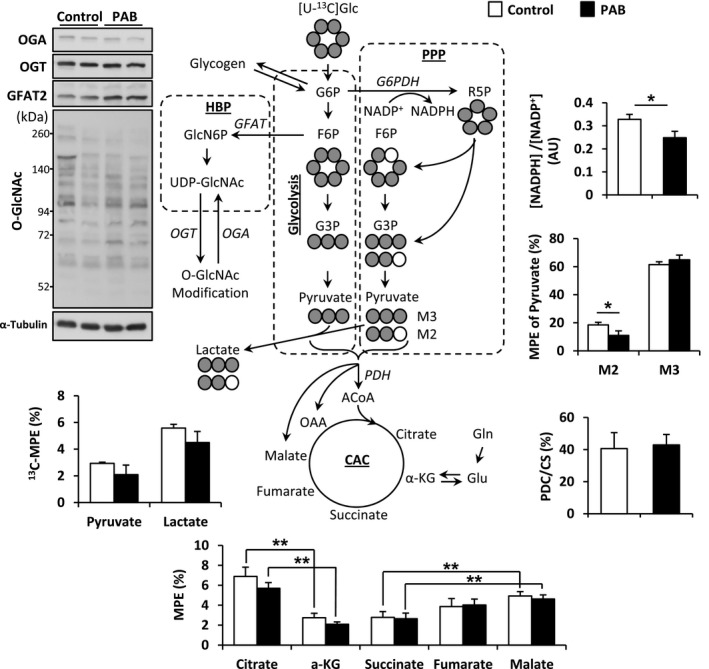
Schematic demonstrating of [U‐13C]glucose to CAC and PPP, and protein expression levels in HBP. NADPH and M2 pyruvate, produced by PPP, were significantly low in PAB compared with Control. Values are presented as means±SE; n=4 per group. *P<0.05, **P<0.01 vs Control. ACoA indicates acetyl‐CoA; AU, arbitrary units; CAC, citric acid cycle; CS, citrate synthase; GFAT, glutamine‐fructose‐6‐phosphate transaminase; Glc, glucose; Gln, glutamine; Glu, glutamate; HBP, hexosamine biosynthetic pathway; α‐KG, α‐ketoglutarate; MPE, molar percent enrichment; NADP+, the oxidized form of nicotinamide adenine dinucleotide phosphate; NADPH, the reduced form of nicotinamide adenine dinucleotide phosphate; OAA, oxaloacetate; OGA, O‐GlcNAcase; O‐GlcNAc, O‐linked N‐acetylglucosamine; OGT, O‐GlcNAc transferase; PAB, pulmonary artery banding; PDC, pyruvate decarboxylation; PDH, pyruvate dehydrogenase; PPP, pentose phosphate pathway; R5P, ribose‐5‐phosphate.
Additionally, we found that a substantial portion of pyruvate originating from glucose labels lactate. 13C‐MPE of lactate (Figure 6) far exceeded 13C‐MPE of pyruvate, indicating substantially different kinetics for lactate dehydrogenase than documented in previous studies of LV metabolism using similar methodology.22, 26 These data imply that the aerobic RV, unlike the LV, depends substantially on anaerobic glycolysis to lactate for ATP generation. However, RV pressure overloading does not change these relationships.
The 13C‐labeling detected by GC‐MS shows distinct RV patterns. Panchal et al26 reported that LV labeled by [U‐13C]pyruvate and/or [U‐13C]lactate did not produce M1 or M2 lactate or pyruvate. However, our experiments, which derived both [U‐13C]pyruvate and/or [U‐13C]lactate from [U‐13C]glucose, did produce M1 and M2 labeling, implying that either the pentose phosphate pathway (PPP) operates or cataplerotic production of pyruvate from malate occurs in the RV. If we assume that M2 is produced only by PPP, then this pathway accounts for pyruvate production from glucose in the RV compared with a negligible amount reported in prior LV studies. M2 pyruvate in PAB was lower than that in Control (Figure 6). NADPH is a product of the PPP,27 and works as a reductant in various synthetic (anabolic) pathways, including FA synthesis. 1H‐NMR showed low [NADPH]/[NADP+] in the PAB group (Figure 6) consistent with lower flux through the PPP.
The hexose biosynthetic pathway provides another alternative route for glucose metabolism. Several studies have shown that O‐GlcNAcylation of proteins resulting from this pathway provides cardiac protection during oxidative stress, calcium overload, endoplasmic reticulum stress, and ischemia–reperfusion.28, 29, 30, 31 However, using standard immunoblot methodology, we found no increase in RV O‐GlcNAc during acute increases in pressure overloading (Figure 6). The implications for this apparent deficiency in stress signaling in this immature RV model require elucidation.
13C‐MPE for CAC intermediates are displayed in Figure 6. Although our experiments showed no difference in these parameters between experimental groups, we again detected unique RV labeling patterns. Unlike similar experiments performed in the LV, we detected substantial changes in 13C‐MPE for intermediates when proceeding through the CAC. First, we detected a sharp decline in 13C‐MPE of α‐KG compared with 13C‐MPE of citrate, implying dilution of the 13C‐labeled α‐KG pool by unlabeled α‐KG, likely derived from glutamate. Second, we also detected a marked increase in 13C‐MPE proceeding around the CAC from succinate to malate and fumarate. This finding suggests anaplerotic entry of 13C3‐labeled substrate, presumably pyruvate, through malic enzyme to form malate, which exchanges with fumarate.
Discussion
We performed these experiments using a translational animal model that emulates many aspects of the infants undergoing palliative or corrective surgery for complex congenital heart defects. Surgery in these infants results in acute or continuing RV loading to near systemic or greater pressure levels. This model emulates the neonatal response to acute RV overloading as occurs with PAB instituted to protect the lungs from volume and pressure overloading. The model does not necessarily reflect the RV that has undergone hypertrophy after more prolonged banding. Human neonates exhibit variable ability to acutely tolerate RV pressure overloading after surgery. The physiological basis for this intolerance and the variable hemodynamic compensation remain undefined, particularly in the immature heart. Prior studies have shown that the immature LV compared with the mature LV displays a more limited ability to maintain balance between ATP utilization and consumption during acute increases in cardiac work.32, 33 This imbalance is caused in part by immature mitochondrial structure and respiratory control. However, a limited capacity to increase production of reducing equivalent delivery to the mitochondrial respiratory chain might also contribute. Our prior studies have suggested that the immature RV capacity to modify substrate oxidation in response to changes in workload is even more limited than apparent in the LV.15 Our current data indicate that the immature RV demonstrates an inability to maintain energy balance, manifested by a decrease in [phosphocreatine]/[ATP], a surrogate measure for energy supply/demand, during increases in pressure overload commensurate with acute PAB in infants. Furthermore, we demonstrate an inverse correlation between the degree of RV pressure overload in an individual animal and the [phosphocreatine]/[ATP]. Similar acute decreases in [phosphocreatine]/[ATP] are noted in the LV in intact mature animals only after ischemia–reperfusion or at maximal workloads driven by cardiac pacing.34, 35 Both of these conditions create deficits in ATP production compared with utilization. Thus, our data support a hypothesis that the ability of the immature RV to acutely adapt to increase in RV pressure work directly relates to the capacity of the mitochondria to maintain balance between ATP supply and demand.
The mature LV in vivo responds to abrupt elevations in work and ATP utilization by modifying substrate preference in order to increase production of reducing equivalents presented for oxidative phosphorylation. Under these inotropic conditions, the LV overrides the preference for oxidation of FAs and switches to substrates that metabolize to pyruvate. Some studies36, 37 performed in vivo suggest that promotion of FA oxidation increases myocardial oxygen consumption without impacting contractile function. Thus, FA oxidation induces mechanical inefficiency, although the mechanisms for this action remain controversial. In response to stress, the LV in vivo then increases the relative contribution of acetyl‐CoA to the CAC through PDH while decreasing FA oxidation.38 In porcine LV in vivo, lactate represents the principal substrate providing pyruvate for decarboxylation to acetyl‐CoA, followed preferentially by glucose. Our studies show that acute pressure loading does not induce an alteration in steady‐state exchange between lactate and pyruvate as determined by 13C‐labeling (Figure 6). We have previously shown that the RV demonstrates less robust ability than the LV to shift substrate utilization during volume and pressure unloading by mechanical circulatory support.15 Similarly, both our current 13C‐NMR and GC‐MS studies show no increase in substrate flux through PDH when the RV encounters acute pressure overload and relative free FA contribution to the CAC is reduced. The data suggest that the RV under acute pressure overloading conditions somewhat lacks the ability to promote flux through PDH or adequately increase substrate flux to this enzyme complex. The typical enzyme phosphorylation cascades that respond rapidly during other types of stress, such as ischemia–reperfusion in the LV,24 did not appear to react to pressure overloading in the RV. This finding suggests that direct pharmacological manipulation of enzymes such as PDH might serve as a therapeutic modality to enhance RV function. The RV inability to increase flux through PDH may also relate to the higher‐than‐anticipated pyruvate shunting to the CAC via carboxylation to malate and/or oxaloacetate. This phenomenon became apparent through the increase in 13C‐MPE for malate relative to succinate (Figure 6), indicating entry of 13C‐pyruvate along the distal portion of the CAC. This 13C‐labeling pattern has not occurred in our prior studies performed in pig LV in vivo,10, 12, 19, 22, 39 suggesting that the RV demonstrates a higher rate of anaplerosis. Similarly, we demonstrated a decrease in13C‐MPE for α‐KG relative to citrate, implying the existence of a high rate of unlabeled glutamate transamination. Thus the RV appears to rely heavily on anaplerotic pathways in order to maintain concentration and cycling of the CAC intermediates.
Following phosphorylation by hexokinase, glucose can be shunted into the oxidative PPP through glucose‐6‐phosphate dehydrogenase rather than undergoing conversion to fructose‐6‐phosphate and entering glycolysis (Figure 6). The glucose‐6‐phosphate dehydrogenase reaction is the main supplier of cytosolic NADPH, which at least acutely maintains redox homeostasis and protects cardiomyocytes from oxidative stress.40, 41, 42, 43 Surprisingly, validation of flux through PPP in the heart has occurred only indirectly through assessment metabolite concentrations during glucose‐6‐phosphate dehydrogenase inhibition.44, 45 We did not specifically design our protocol to evaluate the PPP. Unexpectedly, we noted appearance of M2 pyruvate, which we assume must occur through [U‐13C]glucose metabolism in the PPP. We estimate that PPP provides in the range of 10% to 15% of labeled pyruvate production. The M2 pyruvate component decreases with RV pressure overloading in association with an expected decrease in [NADPH]/[NADP+]. Thus, these data imply that acute RV pressure overloading decreases glucose shunting to the PPP, resulting in increased oxidative stress. As we have not used this glucose labeling pattern previously, we do not know if this phenomenon is restricted to the RV.
Limitations
Our studies operated under limitations, which are unavoidable and inherent in study of the heart in vivo and have been reviewed in detail in a recent American Heart Association scientific statement.46 We demonstrated that the decrease in [phosphocreatine]/[ATP] was proportional to the pressure work demand indexed by RVSP/LVSPmax. However, we could not definitively link total acute RV failure to further decreases in [phosphocreatine]/[ATP], as hemodynamic decompensation with demise occurred with pressure overloading beyond a certain limit. Nevertheless, we feel that it is logical to assume that accelerated energy depletion leads to the abrupt RV failure. Unlike metabolic studies using isolated perfused hearts, unlabeled circulating substrates contribute a fraction of the acetyl‐CoA to the CAC. These unlabeled substrates could impact our Fc calculations derived from 13C‐NMR data, particularly if there are major differences in substrate concentrations occurring between groups or during our protocol. This problem is obviated somewhat by delivery of 13C‐labeled substrates directly into the coronary artery, thereby diluting the influence of these unlabeled circulating substrates. In the PAB group, we did note an insignificant increase in circulating free FA. This increase in circulating unlabeled free FA might have contributed to the decrease in Fc of free FAs with PAB, although RV tissue free FA concentrations were not affected. The GC‐MS calculations showed no difference in CAC intermediate concentrations per group, supporting that the dilution effect from these substrates was not substantial. However, the circulating unlabeled substrates, as well as the substrates in the liver, would dilute the very small pool of labeled substrates exiting heart and entering the systemic circulation. Accordingly, potential metabolism and scrambling of 13C within the liver or other organs would have negligible impact on our data. Additionally, the PDC/CS algorithms accommodate for unlabeled substrate.47 The PDC/CS data showed no differences between groups and confirm our premise that relative PDH flux does not increase with RV pressure loading. Finally, the RV coronary anatomy and venous drainage into the coronary sinus precludes accurate assessment of RV oxygen consumption. TCAcalc analyses to determine total CAC flux, as can be performed in isolated perfused hearts, are not applicable to the heart in vivo. Thus, substrate flux in these studies must be reported as fractional or relative to other substrates. In our previous studies in immature piglets, we cannulated the coronary sinus in order to measure coronary flow and oxygen content, thereby providing LV oxygen consumption. However, the majority of the veins draining the RV empty directly into the right atrium. Additionally, cannulation of the right coronary artery in these experiments precludes placement of flow probes, which could further distort the vessel. Thus, we were unable to measure RV oxygen consumption, a limitation of these studies.48 Additionally, differences in RVSP/LVSP and [phosphocreatine]/[ATP] could remotely relate to changes in perfusion with pressure loading that we were not able to measure. Capillary rarefaction with perfusion abnormalities occur with RV hypertrophy and failure.49 However, perfusion deficits are less likely to occur in our experimental conditions without RV hypertrophy, particularly in the immature heart with relatively high capillary density.50
Conclusions
The immature and nonhypertrophied RV in vivo exposed to acute pressure overload, as often occurs in the infant undergoing complex congenital heart disease, demonstrates limited ability to modulate substrate metabolism and increase ATP production. The data suggest that the RV inability to sustain balance between ATP production and utilization contributes to contractile dysfunction and even hemodynamic decompensation. PDH flux does not accelerate in response to increased pressure work and decreases in FA oxidation as expected from studies in the LV.38 Thus, the PDH pathway may serve as an important target for intervention in order to bolster RV function during abrupt increases in workload. We also noted a novel potential reduction in the PPP, which could increase oxidative stress and injury, and thereby promote RV dysfunction during acute increases in pressure.
Sources of Funding
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number R01HL60666. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. A portion of the research was performed using Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory.
Disclosures
None.
(J Am Heart Assoc. 2018;7:e008570 DOI: 10.1161/JAHA.118.008570.)29848498
References
- 1. Kohler D, Arnold R, Loukanov T, Gorenflo M. Right ventricular failure and pathobiology in patients with congenital heart disease—implications for long‐term follow‐up. Front Pediatr. 2013;1:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brunner N, de Jesus Perez VA, Richter A, Haddad F, Denault A, Rojas V, Yuan K, Orcholski M, Liao X. Perioperative pharmacological management of pulmonary hypertensive crisis during congenital heart surgery. Pulm Circ. 2014;4:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lindberg L, Olsson AK, Jogi P, Jonmarker C. How common is severe pulmonary hypertension after pediatric cardiac surgery? J Thorac Cardiovasc Surg. 2002;123:1155–1163. [DOI] [PubMed] [Google Scholar]
- 4. Helbing WA, Hansen B, Ottenkamp J, Rohmer J, Chin JG, Brom AG, Quaegebeur JM. Long‐term results of atrial correction for transposition of the great arteries. Comparison of mustard and senning operations. J Thorac Cardiovasc Surg. 1994;108:363–372. [PubMed] [Google Scholar]
- 5. Alsoufi B, McCracken C, Shashidharan S, Kogon B, Border W, Kanter K. Palliation outcomes of neonates born with single‐ventricle anomalies associated with aortic arch obstruction. Ann Thorac Surg. 2017;103:637–644. [DOI] [PubMed] [Google Scholar]
- 6. Pascual F, Coleman RA. Fuel availability and fate in cardiac metabolism: a tale of two substrates. Biochim Biophys Acta. 2016;1861:1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T, Wagg CS, Jaswal JS, Harris RA, Clanachan AS, Dyck JR, Lopaschuk GD. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res. 2012;94:359–369. [DOI] [PubMed] [Google Scholar]
- 8. Kudej RK, White LT, Kudej AB, Vatner SF, Lewandowski ED. Brief increase in carbohydrate oxidation after reperfusion reverses myocardial stunning in conscious pigs. Circulation. 2002;106:2836–2841. [DOI] [PubMed] [Google Scholar]
- 9. Olson AK, Hyyti OM, Cohen GA, Ning XH, Sadilek M, Isern N, Portman MA. Superior cardiac function via anaplerotic pyruvate in the immature swine heart after cardiopulmonary bypass and reperfusion. Am J Physiol Heart Circ Physiol. 2008;295:H2315–H2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kajimoto M, O'Kelly Priddy CM, Ledee DR, Xu C, Isern N, Olson AK, Des Rosiers C, Portman MA. Myocardial reloading after extracorporeal membrane oxygenation alters substrate metabolism while promoting protein synthesis. J Am Heart Assoc. 2013;2:e000106 DOI: 10.1161/jaha.113.000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kajimoto M, O'Kelly Priddy CM, Ledee DR, Xu C, Isern N, Olson AK, Portman MA. Extracorporeal membrane oxygenation promotes long chain fatty acid oxidation in the immature swine heart in vivo. J Mol Cell Cardiol. 2013;62:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Files MD, Kajimoto M, O'Kelly Priddy CM, Ledee DR, Xu C, Des Rosiers C, Isern N, Portman MA. Triiodothyronine facilitates weaning from extracorporeal membrane oxygenation by improved mitochondrial substrate utilization. J Am Heart Assoc. 2014;3:e000680 DOI: 10.1161/jaha.113.000680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kajimoto M, Ledee DR, Xu C, Kajimoto H, Isern NG, Portman MA. Triiodothyronine activates lactate oxidation without impairing fatty acid oxidation and improves weaning from extracorporeal membrane oxygenation. Circ J. 2014;78:2867–2875. [PMC free article] [PubMed] [Google Scholar]
- 14. Kajimoto M, O'Kelly Priddy CM, Ledee DR, Xu C, Isern N, Olson AK, Portman MA. Effects of continuous triiodothyronine infusion on the tricarboxylic acid cycle in the normal immature swine heart under extracorporeal membrane oxygenation in vivo. Am J Physiol Heart Circ Physiol. 2014;306:H1164–H1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kajimoto M, Ledee DR, Isern NG, Portman MA. Right ventricular metabolism during venoarterial extracorporeal membrane oxygenation in immature swine heart in vivo. Am J Physiol Heart Circ Physiol. 2017;312:H721–H727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schwartz GG, Greyson CR, Wisneski JA, Garcia J, Steinman S. Relation among regional O2 consumption, high‐energy phosphates, and substrate uptake in porcine right ventricle. Am J Physiol. 1994;266:H521–H530. [DOI] [PubMed] [Google Scholar]
- 17. Schwartz GG, Greyson C, Wisneski JA, Garcia J. Inhibition of fatty acid metabolism alters myocardial high‐energy phosphates in vivo. Am J Physiol. 1994;267:H224–H231. [DOI] [PubMed] [Google Scholar]
- 18. Labarthe F, Gelinas R, Des Rosiers C. Medium‐chain fatty acids as metabolic therapy in cardiac disease. Cardiovasc Drugs Ther. 2008;22:97–106. [DOI] [PubMed] [Google Scholar]
- 19. Kajimoto M, Ledee DR, Olson AK, Isern NG, Des Rosiers C, Portman MA. Differential effects of octanoate and heptanoate on myocardial metabolism during extracorporeal membrane oxygenation in an infant swine model. Am J Physiol Heart Circ Physiol. 2015;309:H1157–H1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Puttmann M, Krug H, von Ochsenstein E, Kattermann R. Fast HPLC determination of serum free fatty acids in the picomole range. Clin Chem. 1993;39:825–832. [PubMed] [Google Scholar]
- 21. Des Rosiers C, Lloyd S, Comte B, Chatham JC. A critical perspective of the use of (13)C‐isotopomer analysis by GCMS and NMR as applied to cardiac metabolism. Metab Eng. 2004;6:44–58. [DOI] [PubMed] [Google Scholar]
- 22. Priddy CM, Kajimoto M, Ledee DR, Bouchard B, Isern N, Olson AK, Des Rosiers C, Portman MA. Myocardial oxidative metabolism and protein synthesis during mechanical circulatory support by extracorporeal membrane oxygenation. Am J Physiol Heart Circ Physiol. 2013;304:H406–H414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen JH, Wu YV, DeCarolis P, O'Connor R, Somberg CJ, Singer S. Resolution of creatine and phosphocreatine 1H signals in isolated human skeletal muscle using HR‐MAS 1H NMR. Magn Reson Med. 2008;59:1221–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ledee D, Kang MA, Kajimoto M, Purvine S, Brewer H, Pasa‐Tolic L, Portman MA. Quantitative cardiac phosphoproteomics profiling during ischemia‐reperfusion in an immature swine model. Am J Physiol Heart Circ Physiol. 2017;313:H125–H137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lloyd S, Brocks C, Chatham JC. Differential modulation of glucose, lactate, and pyruvate oxidation by insulin and dichloroacetate in the rat heart. Am J Physiol Heart Circ Physiol. 2003;285:H163–H172. [DOI] [PubMed] [Google Scholar]
- 26. Panchal AR, Comte B, Huang H, Kerwin T, Darvish A, Des Rosiers C, Brunengraber H, Stanley WC. Partitioning of pyruvate between oxidation and anaplerosis in swine hearts. Am J Physiol Heart Circ Physiol. 2000;279:H2390–H2398. [DOI] [PubMed] [Google Scholar]
- 27. Hecker PA, Leopold JA, Gupte SA, Recchia FA, Stanley WC. Impact of glucose‐6‐phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013;304:H491–H500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laczy B, Fulop N, Onay‐Besikci A, Des Rosiers C, Chatham JC. Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein o‐glcnacylation. PLoS One. 2011;6:e18417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Darley‐Usmar VM, Ball LE, Chatham JC. Protein O‐linked beta‐N‐acetylglucosamine: a novel effector of cardiomyocyte metabolism and function. J Mol Cell Cardiol. 2012;52:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hardiville S, Hart GW. Nutrient regulation of signaling, transcription, and cell physiology by O‐glcnacylation. Cell Metab. 2014;20:208–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martinez MR, Dias TB, Natov PS, Zachara NE. Stress‐induced o‐glcnacylation: an adaptive process of injured cells. Biochem Soc Trans. 2017;45:237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Portman MA, Heineman FW, Balaban RS. Developmental changes in the relation between phosphate metabolites and oxygen consumption in the sheep heart in vivo. J Clin Invest. 1989;83:456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ingwall JS, Kramer MF, Woodman D, Friedman WF. Maturation of energy metabolism in the lamb: changes in myosin ATPase and creatine kinase activities. Pediatr Res. 1981;15:1128–1133. [DOI] [PubMed] [Google Scholar]
- 34. Kay L, Saks VA, Rossi A. Early alteration of the control of mitochondrial function in myocardial ischemia. J Mol Cell Cardiol. 1997;29:3399–3411. [DOI] [PubMed] [Google Scholar]
- 35. Hou M, Hu Q, Chen Y, Zhao L, Zhang J, Bache RJ. Acute effects of febuxostat, a nonpurine selective inhibitor of xanthine oxidase, in pacing induced heart failure. J Cardiovasc Pharmacol. 2006;48:255–263. [DOI] [PubMed] [Google Scholar]
- 36. Korvald C, Elvenes OP, Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol. 2000;278:H1345–H1351. [DOI] [PubMed] [Google Scholar]
- 37. Korvald C, Elvenes OP, Aghajani E, Myhre ES, Myrmel T. Postischemic mechanoenergetic inefficiency is related to contractile dysfunction and not altered metabolism. Am J Physiol Heart Circ Physiol. 2001;281:H2645–H2653. [DOI] [PubMed] [Google Scholar]
- 38. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. [DOI] [PubMed] [Google Scholar]
- 39. Olson AK, Bouchard B, Ning XH, Isern N, Des Rosiers C, Portman MA. Triiodothyronine increases myocardial function and pyruvate entry into the citric acid cycle after reperfusion in a model of infant cardiopulmonary bypass. Am J Physiol Heart Circ Physiol. 2012;302:H1086–H1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vimercati C, Qanud K, Mitacchione G, Sosnowska D, Ungvari Z, Sarnari R, Mania D, Patel N, Hintze TH, Gupte SA, Stanley WC, Recchia FA. Beneficial effects of acute inhibition of the oxidative pentose phosphate pathway in the failing heart. Am J Physiol Heart Circ Physiol. 2014;306:H709–H717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jain M, Cui L, Brenner DA, Wang B, Handy DE, Leopold JA, Loscalzo J, Apstein CS, Liao R. Increased myocardial dysfunction after ischemia‐reperfusion in mice lacking glucose‐6‐phosphate dehydrogenase. Circulation. 2004;109:898–903. [DOI] [PubMed] [Google Scholar]
- 42. Hecker PA, Lionetti V, Ribeiro RF Jr, Rastogi S, Brown BH, O'Connell KA, Cox JW, Shekar KC, Gamble DM, Sabbah HN, Leopold JA, Gupte SA, Recchia FA, Stanley WC. Glucose 6‐phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circ Heart Fail. 2013;6:118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, Kita T, Kimura T, Shioi T. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. 2010;3:420–430. [DOI] [PubMed] [Google Scholar]
- 44. Goodwin GW, Cohen DM, Taegtmeyer H. [5‐3H]glucose overestimates glycolytic flux in isolated working rat heart: role of the pentose phosphate pathway. Am J Physiol Endocrinol Metab. 2001;280:E502–E508. [DOI] [PubMed] [Google Scholar]
- 45. Zuurbier CJ, Eerbeek O, Goedhart PT, Struys EA, Verhoeven NM, Jakobs C, Ince C. Inhibition of the pentose phosphate pathway decreases ischemia‐reperfusion‐induced creatine kinase release in the heart. Cardiovasc Res. 2004;62:145–153. [DOI] [PubMed] [Google Scholar]
- 46. Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley‐Usmar V, Des Rosiers C, Gerszten R, Glatz JF, Griffin JL, Gropler RJ, Holzhuetter HG, Kizer JR, Lewandowski ED, Malloy CR, Neubauer S, Peterson LR, Portman MA, Recchia FA, Van Eyk JE, Wang TJ; American Heart Association Council on Basic Cardiovascular S . Assessing cardiac metabolism: a scientific statement from the American Heart Association. Circ Res. 2016;118:1659–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Khairallah M, Labarthe F, Bouchard B, Danialou G, Petrof BJ, Des Rosiers C. Profiling substrate fluxes in the isolated working mouse heart using 13C‐labeled substrates: focusing on the origin and fate of pyruvate and citrate carbons. Am J Physiol Heart Circ Physiol. 2004;286:H1461–H1470. [DOI] [PubMed] [Google Scholar]
- 48. Crick SJ, Sheppard MN, Ho SY, Gebstein L, Anderson RH. Anatomy of the pig heart: comparisons with normal human cardiac structure. J Anat. 1998;193:105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smolich JJ, Walker AM, Campbell GR, Adamson TM. Left and right ventricular myocardial morphometry in fetal, neonatal, and adult sheep. Am J Physiol. 1989;257:H1–H9. [DOI] [PubMed] [Google Scholar]