

Abstract
Background
Heart failure (HF) is associated with increased arrhythmia risk and triggered activity. Abnormal Ca2+ handling is thought to underlie triggered activity, and mitochondria participate in Ca2+ homeostasis.
Methods and Results
A model of nonischemic HF was induced in C57BL/6 mice by hypertension. Computer simulations were performed using a mouse ventricular myocyte model of HF. Isoproterenol‐induced premature ventricular contractions and ventricular fibrillation were more prevalent in nonischemic HF mice than sham controls. Isolated myopathic myocytes showed decreased cytoplasmic Ca2+ transients, increased mitochondrial Ca2+ transients, and increased action potential duration at 90% repolarization. The alteration of action potential duration at 90% repolarization was consistent with in vivo corrected QT prolongation and could be explained by augmented L‐type Ca2+ currents, increased Na+‐Ca2+ exchange currents, and decreased total K+ currents. Of myopathic ventricular myocytes, 66% showed early afterdepolarizations (EADs) compared with 17% of sham myocytes (P<0.05). Intracellular application of 1 μmol/L Ru360, a mitochondrial Ca2+ uniporter–specific antagonist, could reduce mitochondrial Ca2+ transients, decrease action potential duration at 90% repolarization, and ameliorate EADs. Furthermore, genetic knockdown of mitochondrial Ca2+ uniporters inhibited mitochondrial Ca2+ uptake, reduced Na+‐Ca2+ exchange currents, decreased action potential duration at 90% repolarization, suppressed EADs, and reduced ventricular fibrillation in nonischemic HF mice. Computer simulations showed that EADs promoted by HF remodeling could be abolished by blocking either the mitochondrial Ca2+ uniporter or the L‐type Ca2+ current, consistent with the experimental observations.
Conclusions
Mitochondrial Ca2+ handling plays an important role in EADs seen with nonischemic cardiomyopathy and may represent a therapeutic target to reduce arrhythmic risk in this condition.
Keywords: mitochondria, heart failure, arrhythmia, calcium
Subject Categories: Arrhythmias, Electrophysiology, Basic Science Research, Ion Channels/Membrane Transport, Remodeling
Clinical Perspective
What Is New?
Heart failure is associated with increased arrhythmia risk and triggered activity.
Abnormal Ca2+ handling is thought to underlie triggered activity, and mitochondria participate in Ca2+ homeostasis.
Nonischemic heart failure was accompanied by increased arrhythmic risk, and isolated myopathic myocytes showed increased mitochondrial Ca2+ transients, action potential duration, and triggered activity.
Arrhythmias and action potential prolongation could be improved by inhibiting mitochondrial Ca2+ entry through the mitochondrial Ca2+ uniporter.
What Are the Clinical Implications?
Mitochondrial Ca2+ handling may represent a therapeutic target to reduce arrhythmic risk in cardiomyopathy.
Introduction
The origin of arrhythmias in heart failure (HF) is unknown, but prolongation of the action potential (AP) accompanied by early afterdepolarizations (EADs) has been implicated as one possible cause.1, 2 EADs are secondary depolarizations during the AP plateau or repolarizing phases, which are thought to be caused by abnormal ion channel function and Ca2+ handling during the AP plateau.3
Cardiac mitochondria occupy as much as ≈33% to 40% of cell volume and are located beneath the plasma membrane and between myofibrils.4, 5 Mitochondria are known to be involved in Ca2+ handling, playing an important role in cytoplasmic Ca2+ homeostasis, especially when sarcoplasmic reticulum (SR) Ca2+ release is reduced in HF.6 Mitochondrial Ca2+ homeostasis is believed to involve Ca2+ influx into the matrix, mainly via the mitochondrial calcium uniporter (MCU), with the major efflux pathway being the mitochondrial Na+‐Ca2+ exchanger (NCX).7, 8 Mitochondrial membrane potential is the electromotive driving force for mitochondrial Ca2+ uptake.9
There are several possible mechanisms for an increase in mitochondrial Ca2+ flux in HF. Ca2+/calmodulin‐dependent protein kinase II activity is elevated in HF and positively correlates with enhanced mitochondria Ca2+ uptake.10 Mitochondrial numbers and area are increased in HF.11, 12 Reactive oxygen species are increased in HF, and oxidation of the MCU Cys‐97 residue can increase MCU activity.13 Given these changes, it is plausible that mitochondrial Ca2+ flux may increase in chronic mild or moderate HF models and thereby contribute to EADs in cardiomyopathy.
We tested whether mitochondrial Ca2+ influx contributed to EADs in HF. A computer model of mouse ventricular myocytes with or without changes observed in the experimental model was used to reveal mechanistic insights into the effect of MCU on EADs.
Methods
The data, analytic methods, and study materials have been made available to other researchers for purposes of reproducing the results or replicating the procedure. All supporting data are available within the article. Mice were randomly selected for the treatment or sham group. The observers were blinded to treatment groups.
Nonischemic HF Model
All animal protocols were in accordance with the guidelines of the Animal Care and Use Committee of the University of Minnesota (Minneapolis, MN) or of Lifespan Corporation and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Mice were anaesthetized using inhaled isoflurane (3% for induction and 1.5%–2% for maintenance). Nonischemic HF was induced by hypertension resulting from unilateral nephrectomy, deoxycorticosterone acetate (DOCA) treatment, and salt added to the drinking water, as described in the previous report.14 Nonischemic HF was induced by sustained hypertension in 17‐ to 18‐week‐old, male, C57BL/6 mice (22–25 g; Charles River, Wilmington, MA) by unilateral nephrectomy, SC implantation of a controlled‐release DOCA pellet (0.7 mg/d; Innovative Research of America, Sarasota, FL), and substituting drinking water with 1% saline for 6 weeks. Sham animals underwent a sham operation and received normal drinking water. CD1 wild‐type (WT) or MCU knockdown (MCU+/−) heterozygous mice (Texas A&M Institute for Genomic Medicine, College Station, TX; and Charles River) were used to test whether genetic inhibition of mitochondrial Ca2+ influx could decrease EADs. Mice were randomly selected for the nonischemic HF group.
Physiological Assessment
Blood pressure and heart rate were measured on acclimated conscious mice 6 weeks after surgery using tail‐cuff plethysmography (Columbus Instruments, Columbus, OH). Transthoracic echocardiography was performed using the Vevo 2100 system equipped with a RMV‐707B transducer (VisualSonics, Toronto, ON, Canada). During blood pressure and echocardiography measurements, mice were anesthetized with 1% isoflurane in oxygen and were closely monitored during the procedure. Images were obtained from the parasternal long axis view and parasternal short axis view at the midpapillary level. Wall thickness, chamber size, and ejection fraction were evaluated by 2‐dimensional and M‐mode echocardiography. Measurements were averaged from 3 consecutive beats.15
Telemetry Monitoring
Seven randomly selected nonischemic HF mice and 7 randomly selected C57BL/6 control mice, aged 17 weeks, were implanted with ETA‐F10 transmitters (Data Sciences International, St Paul, MN), as described before.6 Mice were anesthetized by IP injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). A skin incision was made in the right abdominal region, and a transmitter was inserted subcutaneously. The 2 electrocardiographic leads were tunneled and positioned under the skin to generate a lead II electrocardiographic configuration. One week after transmitter implantation, electrocardiographic signals were recorded for 24 hours. ECG recordings were performed at 6 weeks after the start of DOCA‐salt treatment. Heart rate calculations and cardiac rhythm analysis were performed by using Dataquest ART, version 4.1 (Data Sciences International) and LabChart 7 Pro, version 7.3.7 (ADinstruments). For arrhythmia provocations, 2 doses of isoproterenol were injected into the peritoneum (IP; 0.2 and 2.5 mg/kg).16, 17, 18, 19 After establishing the baseline rhythm for 24 hours, each mouse received low dose of isoproterenol. After 4 hours, high‐dose isoproterenol was applied, and the ECG was recorded for another 2 hours.
Cardiomyocyte and Mitoplast Isolation
Ventricular cardiomyocytes were isolated, as described before.14, 20 Randomly selected mice were anesthetized using inhaled isoflurane (3% for induction and 1.5%–2% for maintenance). After the anesthetized mouse was nonresponsive to toe pinch, a thoracotomy was performed. The rib cage was cut bilaterally and flipped over to expose the heart. The heart was then excised and placed immediately in a petri dish filled with ice‐cold Tyrode's solution. The heart was then perfused with buffer (in mmol/L: NaCl 113, KCl 4.7, Na2HPO4 0.6, KH2PO4 0.6, MgSO4 1.2, phenol red 0.032, NaHCO3 12, KHCO3 10, HEPES 10, taurine 30, and 2‐3‐butanedione monoxime 10) and digested with collagenase II (Worthington Biochemical Co, Lakewood, NJ). Cardiomyocytes were washed with control buffers (in mmol/L: NaCl 133.5, KCl 4, Na2HPO4 1.2, HEPES 10, and MgSO4 1.2) with serially increasing Ca2+ concentrations (0.2, 0.5, and 1 mmol/L). Then, myocytes were incubated in minimal essential medium (modified Eagle's medium with 1% insulin‐transferrin‐selenium, 0.1% bovine serum albumin, 1% l‐glutamine, and 1% penicillin/streptomycin) in a 95% O2/5% CO2 incubator at 37°C for 1 hour before being used for patch clamp recording and Ca2+ transient measurements.
Isolation of mitochondria was performed on ice. Freshly harvested mouse ventricle was washed 3 times with a buffer (in mmol/L: mannitol 225, sucrose 70, EGTA 1, HEPES 10, pH 7.4 with KOH). Then, the tissue was cut into small pieces with scissors. Tissue was digested by proteinase, bacterial (P8038‐50MG; Sigma; 5 mg/10 mL) at room temperature for 8 minutes. Bovine serum albumen (20%) was used to stop the digestion reaction. Tissue was homogenized. Nuclei and unbroken cells were pelleted by centrifugation at 1000g for 4 minutes twice. The mitochondrial and cytosolic fraction was obtained from the supernatant by centrifugation at 10 700g for 10 minutes twice. The pellet was resuspended in 500 μL of buffer without EGTA (pH 7.2 with KOH).10, 21
Electrophysiological Recordings
An Axopatch‐200B amplifier (Molecular Devices, Foster City, CA) was used to record APs or membrane currents by a perforated or ruptured patch technique.22 Measured currents included the L‐type Ca2+ current (ICa,L),23 K+ current,24 and NCX current.25 MCU current (IMCU) was recorded in the whole‐mitoplast voltage clamp configuration.10, 21
For AP recordings, pipettes were filled with (in mmol/L) the following: potassium gluconate 120, KCl 20, NaCl 5, HEPES 5, and MgATP 5 (pH 7.2). The extracellular bathing solution (Tyrode's solution) contained (in mmol/L) the following: NaCl 140, KCl 5.4, MgCl2 1, HEPES 10, CaCl2 1.8, and glucose 5.5 (pH 7.4). For perforated current‐clamp experiments, β‐escin (50 μmol/L) was added to pipette solution, and pipette resistance was ≈5 MΩ.22 For ruptured current‐clamp experiments, EGTA (20 μmol/L) was added to pipette solution, and pipette resistances were ≈2 MΩ. Electric stimuli (0.5 or 1 Hz) were used to induce 30 APs. Stimulus current was 1.2 times the threshold current. Records were low‐pass filtered at 10 kHz and digitized at 20 kHz. APs without EADs were used for AP analysis.
The ICa,L 23 was recorded at room temperature. The cells were depolarized every 10 seconds from a holding potential of −50 mV to test potentials between −40 and +60 mV (10‐mV steps) for 300 ms. Bath solution contained the following (mmol/L): TEA 135, MgCl2 0.53, CaCl2 1.8, CsCl 20, and HEPES 5, pH 7.4, with CsOH. Pipette solution contained the following (mmol/L): CsOH 110, aspartic acid 90, CsCl 20, tetraethylammonium chloride 10, HEPES 10, EGTA 10, Mg‐ATP 5, Na2 creatine phosphate 5, GTP (Tris) 0.4, and leupeptin 0.1, pH 7.2, with CsOH. After the cell membrane was broken by application of additional suction, cell capacitance and series resistance were electrically compensated. Current amplitudes were normalized to the cell capacitance and expressed as pA/pF.
For K+ current recording,24 Tyrode's solution was used as a bath solution. The pipette solution was composed of the following (mmol/L): K+‐aspartate 110, KCl 20, NaCl 8, MgC12 1, CaC12 1, 1,2‐bis(o‐aminophenoxy)ethane‐N,N,N′,N′‐tetraacetic acid 10, K2ATP 4, and HEPES 10 (pH adjusted to 7.2 KOH). Series resistance in the whole‐cell mode was in the range of 4 to 8 MΩ; 80% to 90% series resistance compensation was always used. Voltage‐clamp currents were low‐pass filtered at 1 to 3 kHz and digitized at 4 to 10 kHz. Currents were elicited by a series of 5‐second test potentials at 10‐mV increments from −110 to +50 mV from a holding potential of −80 mV at a frequency rate of 0.1 Hz. Current amplitudes were normalized to the cell capacitance and expressed as pA/pF.
For NCX current,25 pipette solutions contained the following (mmol/L): CsCl 115, NaCl 20, MgCl2 0.2, EGTA 0.01, MgATP 3, and HEPES 10, adjusted to pH 7.2 with CsOH. Control external solutions contained the following (mmol/L): NaCl 140, MgCI2 1.0, CaCl2 2.7, dextrose 11.0, and HEPES 10, adjusted to pH 7.4 with NaOH. In the zero‐Na solution, Li salts were used to replace Na. The external solutions for the AP clamp experiments (both zero and normal Na) were slightly modified and contained 4.4 mmol/L KCl and either 140 mmol/L Na or Li. Ryanodine was used at a concentration of 10 μmol/L. Ten 100‐ms clamp pulses from −40 to +10 mV produced a tetanic contracture. After the last pulse, the cell was repolarized to −100 mV, and Na was rapidly applied. This activated a transient inward current and prompted relaxation of the cell.
IMCU was recorded in whole‐mitoplast voltage clamp configuration.10, 21 Mitoplasts were perfused with solution containing the following (mmol/L): Na gluconate 150, HEPES 10, and CaCl2 5 (pH 7.4, adjusted with NaOH). Pipettes, after filling with solution (in mmol/L) of Na gluconate, NaCl 5, sucrose 135, HEPES 10, and EGTA 1.5 (pH 7.2 with NaOH), had an access resistance of ≈25 MΩ. After formation of a GΩ seal between the patch‐clamp pipette and inner mitochondrial membrane, capacitance transients were completely compensated. Voltage of 0.2 or 1.3 V and 0.5 to 300 ms was then applied to rupture the membrane and obtain the whole‐mitoplast configuration, as monitored by reappearance of capacitance transients and an increase in baseline noise. After rupturing of mitoplast membranes, the capacitance was ≈1.8 pF. A ramp voltage command protocol from −160 to +80 mV, for 2 seconds, was applied from a holding potential of 0 mV to evoke currents. Records were low‐pass filtered at 5 kHz and digitized at 20 kHz.
Cytoplasmic Ca2+ and Mitochondrial Ca2+ Transients
The cells were loaded with fluo‐4 (Thermo Fisher Scientific, Carlsbad, CA) at room temperature (30 minutes in Tyrode's solution with 2.5 μmol/L fluo‐4 AM, followed by 15 minutes of deesterification) for cytosolic Ca2+ transient measurements.26, 27 Cells were transferred onto the stage of a real‐time florescence microscope (NIS Elements AR; Nikon, Japan). The images (256×256 pixels) were acquired at a rate of 62.5 Hz. Analysis of the signals was performed with the NIS Elements software (Nikon). Ca2+ transients are presented as background‐subtracted normalized fluorescence (F/F0). An average of 3 consecutive traces without EAD‐related peaks, unless specifically illustrated, were used for data analysis.
Mitochondrial Ca2+ transients were monitored by loading cells with Rhod‐2 AM (Thermo Fisher Scientific; 1 μmol/L; 1 hour, 37°C) combined with the ruptured current‐clamp technique with 20 μmol/L EGTA added in the pipette solution. Because of its positive charge, Rhod‐2 AM accumulated primarily in the mitochondrial matrix. Minor cytosolic traces of Rhod‐2 were eliminated by whole‐cell dialysis by the pipette solution.28, 29 Rhod‐2 intensity was sampled at a rate of 1 kHz by an IonOptix system (IonOptix LLC, Milton, MA). Mitochondrial Ca2+ transients were presented as F/F0. Three consecutive traces without EAD‐related peaks were averaged for data analysis.
Mitochondrial Imaging
To determine mitochondrial morphological features, cardiomyocytes were infected with a modified baculovirus vector (BacMam 2.0 encoding mitochondrial‐targeted green fluorescent protein; CellLight mitochondrial green fluorescent protein; Thermo Fisher Scientific; 5 μL/mL). Experiments were performed according to company's protocol. In brief, cells were fixed by 4% formaldehyde solution in PBS for 10 to 30 minutes at room temperature. Then, cells were permeabilized with 0.2% (100 μL/50 mL) Triton X‐100 solution in PBS for 5 minutes at room temperature. The CellLight reagent was mixed several times by inversion to ensure a homogeneous solution. The CellLight reagent was added directly to the cells and mixed gently. Cells were put into the culture incubator overnight (≥16 hours). Images were taken with excitation/emission at 485/520 nm.30 The average fluorescence density (mitochondrial green fluorescent protein fluorescence normalized by area) was used to analyze data.
Western Blotting and Phosphorylated MCU Protein Analysis
For MCU and mitochondrial NCX Western blotting, protein samples prepared from ventricular tissues were homogenized in a buffer containing 20 mmol/L Tris HCl (pH 7.4), 150 mmol/L NaCl, 2.5 mmol/L EDTA, 1% Triton X‐100, 0.5% deoxycholate, 0.1% SDS, 10% glycerol, 50 mmol/L NaF, 1 mmol/L Na3VO4, and 10 μL/mL protein inhibitor cocktail (Pierce, Rockford, IL). Equal amounts (30 μg) of protein were separated onto 4% to 10% mini‐PROTEAN TGX gels (Biorad, Hercules, CA) and transferred to nitrocellulose membranes. The membranes were blotted with a 1:250 dilution of MCU (CCDC109A[Q‐14]; Santa Cruz Biotechnology, Santa Cruz, CA) or anti‐SLC24A6 antibodies (ab83551; Abcam, Cambridge, MA). An anti‐GAPDH antibody (1:1000 dilution; Santa Cruz Biotechnology) was used as a loading control for all studies. Donkey anti‐goat horseradish peroxidase antibody (Santa Cruz Biotechnology) was used at a dilution 1:5000. Protein expression levels were detected using Clarity Western ECL Blotting Substrate (Biorad). Band intensities were detected by a ChemiDoc MP Imaging System and analyzed by Image Lab software (Biorad).
For phosphorylated MCU protein analysis, protein samples prepared from ventricular tissues were homogenized in a buffer containing 20 mmol/L Tris HCl (pH 7.4), 150 mmol/L NaCl, 2.5 mmol/L EDTA, 1% Triton X‐100, 0.5% deoxycholate, 50 mmol/L NaF, 1 mmol/L Na3VO4, and 10 μL/mL protein inhibitor cocktail (Pierce). Cell debris was removed by centrifugation at 13 000g for 10 minutes. MCU proteins were immunoprecipitated with anti‐MCU antibody (CCDC109A[Q‐14]; Santa Cruz Biotechnology) using Pierce Classic Magnetic IP/Co‐IP Kit (Thermo Fisher Scientific, Waltham, MA), according to the manufacturer's instructions. In brief, cells were harvested with ice‐cold immunoprecipitation lysis/wash buffer. Equal amounts of protein (1000 μg) lysates were mixed with 5 μg of anti‐MCU antibody and left overnight at 4°C in a tube rotator to form immune complexes. Pierce protein A/G magnetic beads were then washed with immunoprecipitation lysis/wash buffer and added to the antigen sample‐antibody mixture. After mixing for 1 hour at room temperature, beads were collected with a magnetic stand and washed 3 times with immunoprecipitation lysis/wash buffer and once with ultrapure water. Complexes were eluted from the beads with elution buffer, followed by neutralizing with neutralization buffer. Then, the supernatant was separated onto 4% to 10% mini‐PROTEAN TGX gels (Biorad) and transferred to nitrocellulose membranes. The membranes were blotted with a 1:1000 dilution of anti‐phosphotyrosine antibody (EMD Millipore, Temecula, CA) to detect phosphorylated MCU. Sheep anti‐mouse horseradish peroxidase antibody (Santa Cruz Biotechnology) was used at a dilution 1:5000. Protein expression levels were detected using Clarity Western ECL Blotting Substrate (Biorad). Band intensities were detected by ChemiDoc MP Imaging System and analyzed by Image Lab software (Biorad). MCU proteins pulled down were immuoblotted with anti‐MCU antibody and were used as a loading control.
Real‐Time Polymerase Chain Reaction Quantification
Total RNA was isolated from mouse ventricular tissue using the RNeasy Mini Kit (Qiagen, Valencia, CA), according to the manufacturer's instructions. Total RNA was reverse transcribed into cDNA using SuperScript VILO Master Mix (Thermo Fisher Scientific, Waltham, MA). Quantitative real‐time reverse transcription–polymerase chain reaction was performed using gene‐specific primers (Mcu sense: 5′‐ GGGATAGACCTCCTGCTCCT‐3′; Mcu anti‐sense: 5′‐TCGCTGCATCTTCATGGCT‐3′), with the Fast SYBR Green Master Mix and 7500 Fast Real‐Time PCR System. The quantitative real‐time reverse transcription–polymerase chain reaction was activated with an initial denaturation step at 95°C for 20 seconds, followed by cycles of denaturation at 95°C for 3 seconds and annealing and extension at 60°C for 30 seconds. Samples were run in triplicate and averaged. Gene expression levels were normalized to the level of β‐actin (sense primer: 5′‐GGCTGTATTCCCCTCCATCG ‐3′; anti‐sense primer: 5′‐ CCTCGTCACCCACATAGGAG‐3′).
Mitochondrial Membrane Potential Measurement
Ventricular myocytes were incubated with tetramethylrhodamine methyl ester (50 nmol/L) for 30 minutes in Tyrode's solution at 22°C. Cells were transferred onto the stage of a real‐time florescence microscope (Olympus IX81; Japan). The images (2048×2048 pixels) were acquired at room temperature with an interval of 2 minutes. Analysis of the signals was performed with the MetaMorph software, version 7.8.11.0 (Nashville, TN). Tetramethylrhodamine methyl ester was excited at 540 nm, and fluorescence was recorded at 605 nm. Carbonyl cyanide 4‐(trifluoromethoxy) phenylhydrazone (20 μmol/L) was added to the bath solution after 10 minutes of recording.
Mouse Ventricular Cell Computer Model and Simulation
On the basis of our previous Ca2+ cycling31 and metabolism32 models, we developed a mouse excitation‐contraction‐metabolism coupling model and used it to investigate the effects of MCU on EADs (Figure 1A). The cell contained 32768 (64×32×16) Ca2+ release units (CRUs) and 8192 mitochondria. The CRU/mitochondrion ratio was 1:1 in the longitudinal direction and 2:1 in the transverse direction. Mitochondrial membrane voltage was modeled following Yang et al,32 with MCU and the mitochondrial NCX being taken from Cortassa et al.33 In this model, mitochondrial free Ca2+ has a ≈10% diastolic‐to‐systolic variation in controls, in good agreement with a recent experimental study.34 The charge of the mitochondrial membrane potential was ≈−182 mV.
Figure 1.
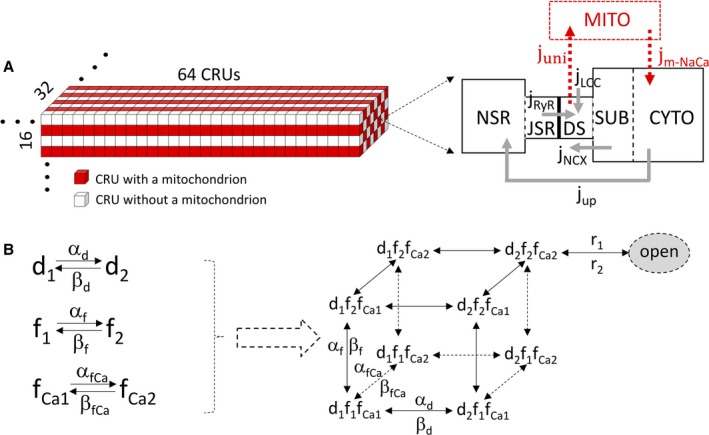
Computer model of mouse ventricular cell. A, Schematic diagrams of the 3‐dimensional structure of the cell model (left) and the Ca2+ release unit (CRU)–mitochondrial Ca2+ cycling model (right). B, The modified L‐type Ca2+ current model. Left: The Hodgkin‐Huxley (HH) scheme. Right: The equivalent Markov scheme of the HH scheme. To simulate a much lower channel open probability (≈5%–10%) observed in experiments, we added a new state (the final open state), with the opening rate from the d2f2fCa2 state being r1 and the closing rate being r2. CYTO indicates cytosolic space; DS, dyadic space; jm‐NaCa, mitochondrial Na+‐Ca2+ exchanger Ca2+ release; JSR, junctional sarcoplasmic reticulum; juni, mitochondrial Ca2+ uniporter uptake; jup, sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA) uptake; LCC, L‐type Ca2+ channel; MITO, mitochondrial space; NSR, network sarcoplasmic reticulum; RyR, ryanodine receptor; SUB, submembrane space.
Spatial Structure of Ventricular Myocyte Model
The ventricular myocyte model used in this study is a 3‐dimensional object containing 32768 CRUs (Figure 1A), with CRU spacing being 1.84 μm in the longitudinal direction and 0.9 μm in the transverse direction, corresponding to a dimension of 118×29×15 μm. The arrangement of mitochondria in heart muscle cells is shown as a “crystal‐like” pattern in the experiment by Vendelin et al,35 in which neighboring mitochondria are aligned along a rectangle, with the distance between the centers equal to 1.97±0.43 and 1.43±0.43 μm in the longitudinal and transverse directions, respectively. According to this measurement, the mitochondria in our model are designed to attach to every CRU in the longitudinal direction and every other CRU in the transverse direction. This arrangement results in 8192 (64×16×8) mitochondria in our cell model, which agrees with the physiological range of 7000 to 10 000 mitochondria in a cardiac myocyte.
The CRUs are coupled via Ca2+ diffusion in the cytosolic space and SR. The model is modified from the one developed by Restrepo et al.36 The details of the model are described in the later sections. Briefly, each CRU contains 5 subvolumes with defined volume ratios: network SR (NSR), junctional SR (JSR), dyadic space, submembrane space, and cytosolic space. Ca2+ from the extracellular space enters into dyadic space via L‐type Ca2+ channels (LCCs) and is released from the JSR to the dyadic space via ryanodine receptors (RyRs). Each CRU has a cluster of 100 RyR channels associated with a cluster of 10 LCCs, both simulated using random Markov transitions. Ca2+ is extruded from the submembrane space via NCX and taken up into the NSR from cytosolic space via sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA) pump. Ca2+ diffuses freely between the SR subvolumes and between the cytosolic subvolumes. CRUs are coupled via Ca2+ diffusion between neighboring NSR spaces, submembrane spaces, and cytosolic spaces, respectively. No Ca2+ diffusion exists directly between neighboring JSR spaces or between neighboring dyadic spaces. For CRUs attaching to mitochondria, we follow Cortassa et al33 in that Ca2+ enters into the mitochondrial space through the MCU and is extruded out of the mitochondria via the mitochondrial NCX.
Membrane Voltage and Ionic Currents
The ionic currents of mouse ventricular myocytes were originally formulated by Morotti et al,37 except for ICa,L, which is modeled by Hodgkin‐Huxley formalism. The total ionic current is as follows:
LCC model
The opening of individual LCCs is simulated by a stochastic 9‐state Markov model (Figure 1B). Each CRU is assumed to have NL LCCs under normal conditions. The Ca2+ flux into the proximal space of a CRU is given by the following:
where m, n, and k are the indexes of CRUs in the 3‐dimensional grid, L is the number of open LCCs in the (m, n, k)th CRU, and iCa,L is the single Ca2+ channel current given by the following:
cp(m,n,k) is the Ca2+ concentration in the corresponding proximal space of the CRU. Therefore, the whole‐cell ICa,L is summation of the Ca2+ currents of CRUs in the cell, ie,
The transition rates between different states of the LCC model are as follows:
where
where
where
r 1 and r 2 are constants. The parameters are listed in Table 1.
Table 1.
L‐Type Ca2+ Current Parameters
| Parameter | Description | Value | |
|---|---|---|---|
| PCa | L‐type channel permeability | 13.745 μmol/C ms | |
| γi, γo | Activity coefficient of Ca2+ | 0.341 | |
| NL | Number of LCCs in each CRU | 10 | |
|
|
Threshold of Ca2+‐dependent inactivation | 6 μ(mol−L)−1 | |
| τfCa | Time constant of Ca2+‐dependent inactivation | 10 ms | |
| r1 | Transition rate from d2f2fCa2 to the open state | 0.375/ms | |
| r2 | Transition rate from the open state to d2f2fCa2 | 6/ms |
CRU indicates Ca2+ release unit; LCC, L‐type Ca2+ channel.
The details of the other ionic currents are described in Morotti et al.37 The modified parameters are listed in Table 2.
Table 2.
Modified Ionic Current Parameters
| Parameter | Values | Units |
|---|---|---|
| gnal | 0.182 | mS/μF |
| gnak | 2.5 | mS/μF |
gnal, the conductance of late Na+ channel; gnak, the conductance of Na+‐K+ pump.
Mitochondrial Membrane Voltage and Ionic Currents
The mitochondrial membrane potential is given by the following:
where
Ca2+ uniporter
The mitochondrial Ca2+ uniporter was originally modeled by Magnus and Keizer.38 Herein, we replace the cytosolic Ca2+ concentration with the proximal Ca2+ concentration because the Ca2+ uniporter may directly face the dyad.39 The formula is as follows:
Na+/Ca2+ exchanger
The NCX was inherited from Cortassa et al33 The formula is as follows:
The corresponding parameters are listed in Table 3.
Table 3.
Mitochondrial Membrane Potential Parameters
| Parameter | Values | Units | |
|---|---|---|---|
| kΨ,U | 0.0192 | ms−1 | |
| Fc | 0.552 | mV·μ(mol L−1)−1 | |
|
|
0.0625 | μ(mol L−1)·ms−1 | |
| Ktrans | 19 | μmol L−1 | |
| Kact | 0.38 | μmol L−1 | |
|
|
0.208 | μmol L−1·ms−1 | |
| KNa | 9.4 | mmol/L | |
| KCa | 0.375 | μmol/L |
Intracellular and Mitochondrial Ca2+ Cycling
The dynamics of Ca2+ cycling in the individual CRUs are described by the following equations:
hmito=1, if a mitochondrion connects the CRU, otherwise hmito=0. The volumes of different compartments and other parameters are listed in Table 4. The diffusion flux and buffers of the Ca2+ cycling remain the same as in Song et al31
Table 4.
Intracellular Ca2+ Cycling Parameters
| Parameter | Values | Units |
|---|---|---|
| vi | 0.5 | μm3 |
| vs | 0.025 | μm3 |
| vp | 0.00126 | μm3 |
| vJSR | 0.02 | μm3 |
| vNSR | 0.025 | μm3 |
| vm | 0.125 | μm3 |
| βm | 0.0003 | |
| Ku | 0.000266 | μ(mol L−1)−2 ms−1 |
| Kb | 0.000035 | μ(mol L−1)−2 ms−1 |
| vup | 1.134 | μ(mol L−1)ms−1 |
| Ki | 0.1 | μmol L−1 |
| vnaca | 3.22 | μ(mol L−1)ms−1 |
JSR indicates junctional sarcoplasmic reticulum; NSR, network sarcoplasmic reticulum.
Parameter Changes in the HF Condition
In the condition of HF, the values of NCX and LCC conductance are increased by 30% and 20%, respectively, which agrees well with our experimental measurement. The close‐to‐open rates of RyRs are increased by 30%. The maximum SERCA activity is reduced by 67%. IKr, IKslow1, IKslow2, IK1, Ito,f, and Iss are reduced by 50%.
Numerical Analysis
The spatial cell model was coded in CUDA C. The ordinary differential equations were integrated numerically with the Euler method. Stochastic openings of LCCs and RyRs were numerically solved by the Gillespie algorithm. The gating variables were integrated using the method by Rush and Larsen. The time step was 0.01 ms. For all the simulations, we paced the cell at a pacing cycle length of 2 seconds for 50 beats. EADs were examined at the last 2 beats.
HF remodeling alterations were simulated on the basis of our previous study40 and experimental information from the current study: (1) RyR leakiness was increased by increasing the transition rate from the closed state to the open state by 30%; (2) the maximum SERCA activity was reduced by 67%, and the Kd was reduced from 0.6 to 0.3 μmol/L to simulate increased phospholamban phosphorylation; (3) the NCX, INCX, was increased by 30%, and the ICa,L was increased by 20%; (4) potassium currents, IKr, Ito,f, Ikslow1, Ikslow2, and IK1, were reduced by 50%; and (5) MCU activity was increased by 3‐fold from normal control. The cell was paced by a current pulse of 2 ms with an amplitude of −50 pA/pF (current‐clamp mode) at a pacing cycle length of 2 seconds for 50 beats to reach steady state. AP duration (APD) was defined by the duration during which the membrane voltage was >−80 mV.
Statistical Analysis
Data were shown as the mean±SEM. As noted in the text, Mann‐Whitney and Fisher's exact tests were used for statistical analysis. Bonferroni correction was used for multiple comparisons. P<0.05 was considered statistically significant. SigmaPlot, version 11.0 (Systat Software, Inc, San Jose, CA) was used for statistical analysis.
Results
Sustained Hypertension Caused Nonischemic Cardiomyopathy
At 6 weeks after surgery, nonischemic HF mice developed hypertension with a significant augmentation of both systolic and diastolic blood pressure, as measured by tail‐cuff plethysmography (Table 5). Echocardiography showed an impairment of systolic function. Ejection fraction decreased from 49±4% in control mice to 36±2% in nonischemic HF C57BL/6 mice (P<0.05). The left ventricular chamber was enlarged at both end systole and end diastole (Table 5). Ejection fraction decreased from 63±2% in control mice (n=6) to 51±3% in nonischemic HF CD1 mice (n=5, P<0.01).
Table 5.
Cardiac Dysfunction and High Blood Pressure Were Observed in Nonischemic HF Mice
| Variable | Sham Mice | Nonischemic HF Mice | ||
|---|---|---|---|---|
| Value | n | Value | n | |
| LVEDV, μL | 87±4 | 13 | 104±3* | 13 |
| LVESV, μL | 41±2 | 13 | 64±3** | 13 |
| EF, % | 51±2 | 13 | 38±2* | 13 |
| SBP, mm Hg | 99±7 | 5 | 118±3* | 4 |
| DBP, mm Hg | 74±5 | 5 | 90±4* | 4 |
Values are mean±SEM. DBP indicates diastolic artery blood pressure; EF, ejection fraction; HF, heart failure; LVEDV, left ventricular end‐diastolic volume; LVESV, left ventricular end‐systolic volume; SBP, systolic artery blood pressure.
*P<0.05, **P<0.01 compared with that in the sham group.
Nonischemic HF Mice Showed Induced Arrhythmias
ECG recordings were performed 6 weeks after the start of DOCA‐salt treatment. The heart rate remained constant, but the corrected QT (QTc) interval increased from 42±1 ms in sham mice to 52±2 ms in nonischemic HF mice (P<0.05, Figure 2B). With low‐dose isoproterenol (IP 0.2 mg/kg), 100% of nonischemic HF mice showed induced premature ventricular contractions versus only 29% of the sham mice (P<0.05 by Fisher's exact test). With high‐dose isoproterenol (2.5 mg/kg), 3 of 7 nonischemic HF mice and no control mice had ventricular fibrillation or tachycardia (Figure 2A). All nonischemic HF mice and control mice had premature ventricular contractions after high‐dose isoproterenol. These results implied that nonischemic HF mice were more prone to arrhythmia when compared with sham control mice.
Figure 2.
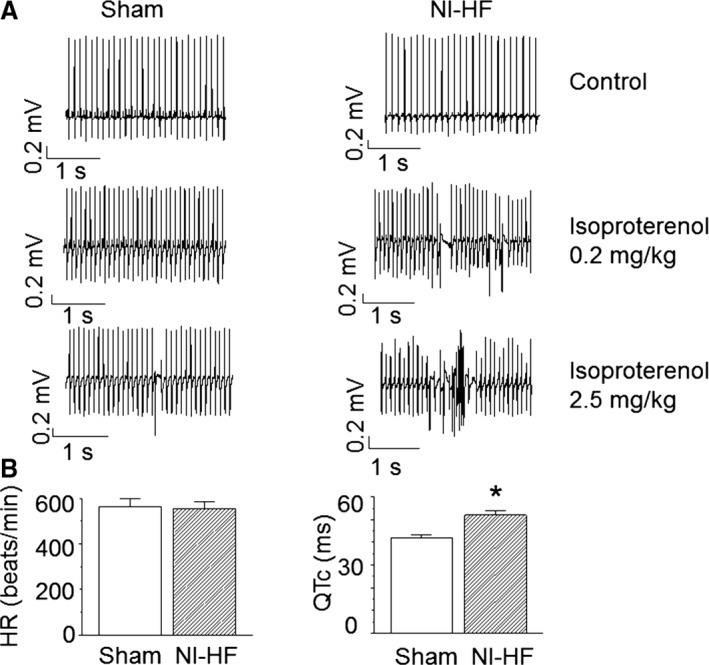
Telemetry of sham and nonischemic heart failure (NI‐HF) C57BL/6 mice. A, Examples of telemetric ECG recordings. ECG signals in the left and right panels were sampled from sham and NI‐HF mice, respectively. Waveforms were collected before (control) and after (0.2 and 2.5 mg/kg IP) isoproterenol injection. B, Heart rate (HR; beats per minute) and corrected QT (QTc) intervals (ms) were measured from lead II and plotted in left and right panels, respectively. Compared with the control mice, NI‐HF mice showed a longer QT interval. n=7 (mice) for each group. *P<0.05 compared with that in sham group.
Myopathic Mice Had Increased EADs
APs were recorded from acutely isolated mouse left ventricular cells by perforated current‐clamp. Compared with control APs, nonischemic HF cardiomyocytes showed an increase in the APD at 90% repolarization (APD90) from 83±21 to 242±45 ms (P<0.01, Table 6). APD90s were calculated from APs without EADs. The APD prolongation was qualitatively consistent, with the QTc prolongation seen in vivo (Figure 2B). The peak amplitudes of APs and the resting membrane potentials of ventricular cells were the same between nonischemic HF and sham mice (Table 6).
Table 6.
Characteristics of APs Recorded in Isolated Ventricular Cells From Sham and Myopathic Mice
| Group | APD90, ms | APA, mV | RMP, mV | EADn | n |
|---|---|---|---|---|---|
| Sham | 83±21 | 137±4 | −78±1 | 2 | 10 |
| Nonischemic HF | 242±45** | 135±5 | −74±1 | 8* | 12 |
| Nonischemic HF+Ru360 | 113±37† | 124±3 | −73±1 | 0† | 7 |
Cells in sham, nonischemic HF, and nonischemic HF+Ru360 (1 μmol/L) groups were from 4, 5, and 3 mice, respectively. Data are presented as mean±SEM. AP indicates action potential; APA, AP amplitude; APD90, AP duration at 90% repolarization (calculated from APs without EADs); EADn, number of cells in which early afterdepolarizations were observed; HF, heart failure; RMP, resting membrane potential.
*P<0.05, **P<0.01 compared with that in sham control group.
P<0.05 compared with that in nonischemic HF group.
Of nonischemic HF ventricular myocytes, 66% showed EADs compared with 17% of sham myocytes (Figure 3A, Table 6, P<0.05, by Fisher's exact test). Among 12 nonischemic HF ventricular myocytes, 3 cells had triggered extra systoles, whereas there were no triggered extra systoles in an equal number of control ventricular cells. EADs could be abolished by intracellular addition of 1 μmol/L Ru360, a mitochondrial calcium uniporter specific antagonist.41 Ru360 could substantially shorten APD90 (Figure 4, Table 6).
Figure 3.
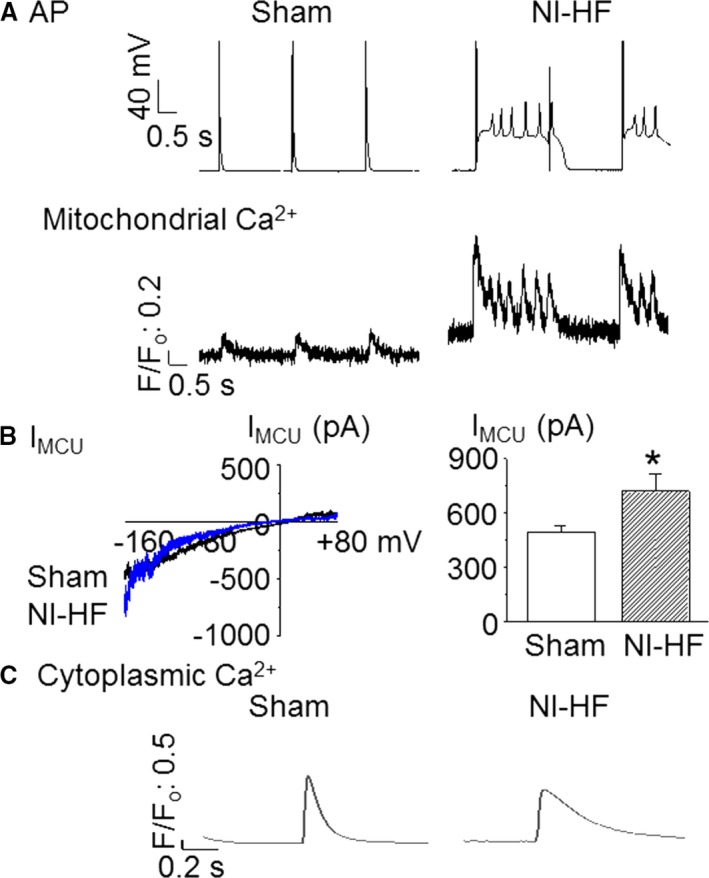
Representative action potentials (APs), mitochondrial Ca2+ transients, mitochondrial Ca2+ uniporter current (IMCU), and cytosol Ca2+ transients recorded from sham and nonischemic heart failure (NI‐HF) mouse ventricular myocytes. A, APs and mitochondrial Ca2+ transients simultaneously recorded from cardiomyopathic ventricular cells showing early afterdepolarizations (EADs). Stimulation (0.5 Hz) was used to evoke APs. The time scale bar is shown at 0 mV. Mitochondrial Ca2+ oscillations corresponded with EADs. B, Left panel: IMCU in mitoplasts isolated from both sham and NI‐HF ventricular cardiomyocytes. Right panel: The average IMCU at −160 mV (n=8 for sham, and n=7 for NI‐HF). C, Cytoplasmic Ca2+ transients without EADs. F/F0 indicates background‐subtracted normalized fluorescence.
Figure 4.
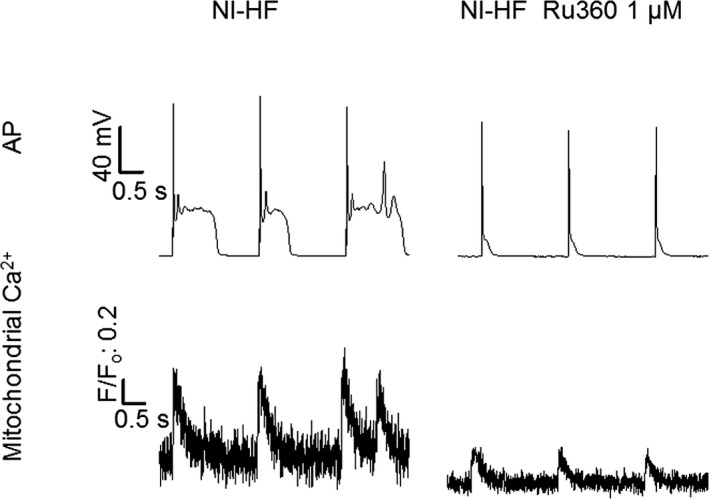
Typical action potential (AP) and mitochondrial Ca2+ traces recorded synchronously from nonischemic heart failure (NI‐HF) mice and NI‐HF mice treated with Ru360 (1 µmol/L). Top panel: APs. Bottom panel: Mitochondrial Ca2+ transients. Triggered activity was inhibited by intracellular application of 1 µmol/L Ru360 in NI‐HF cardiomyocytes. Stimulation (0.5 Hz) was used to evoke APs. The time scale bar is shown at 0 mV. F/F0 indicates background‐subtracted normalized fluorescence.
Electrophysiological Basis for EADs in Nonischemic HF Cardiomyocytes
Myopathic ventricular cells showed a reduction of the peak and steady‐state K+ currents (Figure 5A). In addition, the depolarizing ICa,L values were enhanced in nonischemic HF mice (Figure 5B). Finally, NCX currents were increased by 41% in nonischemic HF ventricular cells (1.11±0.09 versus 1.57±0.10 pA/pF, P<0.05, Figure 5C). The increase in depolarizing currents and the decrease in repolarizing currents could explain why APD90 increased in nonischemic HF mice.
Figure 5.
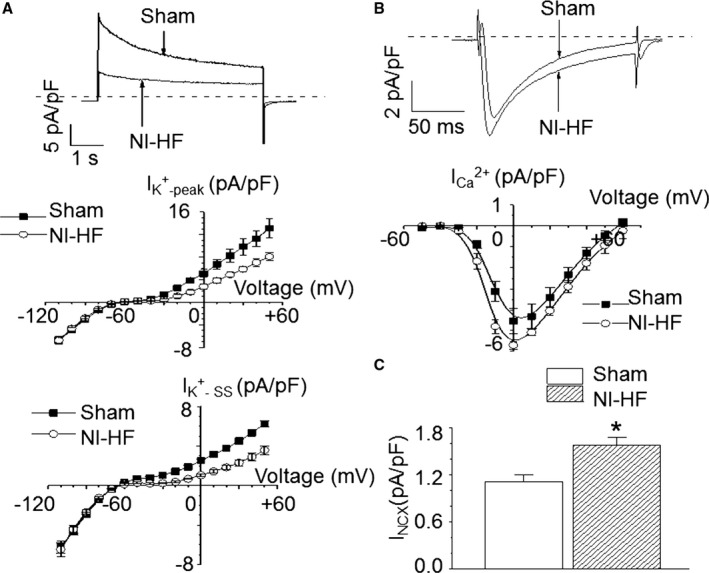
Comparison of membrane currents in sham and nonischemic heart failure (NI‐HF) cardiomyocytes. A, Top panel: K+ currents recorded at +50 mV from both sham and NI‐HF cardiomyocytes. Middle panel: The average peak amplitudes of total K+ currents (IK + ‐peak) are shown (n=4 cells from 2 sham mice, and n=3 cells from 1 NI‐HF mouse). The holding potential was −80 mV. Voltages were from −110 to +50 mV, with a step of +10 mV and a duration of 5 seconds. Bottom panel: The average steady‐state currents of total K+ currents (IK + ‐ SS) are drawn (n=4 cells from 2 sham mice, and n=3 cells from 1 NI‐HF mouse). B, Top panel: L‐type Ca2+ currents recorded at 0 mV from both sham and NI‐HF cardiomyocytes. Bottom panel: The average peak amplitudes of L‐type Ca2+ currents (n=4 cells from 1 sham mouse, and n=5 cells from 1 NI‐HF mouse). The holding potential was −50 mV. Voltages were from −40 to +60 mV, with a step of +10 mV and a duration of 300 ms. C, The average inward Na+‐Ca2+ exchanger (NCX) currents (n=7 cells from 2 mice in sham group, and n=9 cells from 2 mice in NI‐HF group). Cells were put in a solution containing no Na+ and 10 µmol/L ryanodine. The holding potential was −40 mV. Ten 100‐ms steps from −40 to +10 mV were followed by a step to −100 mV to repolarized cells. This step was accompanied by rapidly applied Na+ to record NCX currents. *P<0.05 compared with that in sham group.
Altered Cytoplasmic and Mitochondrial Ca2+ Transients in Myopathic Ventricular Cells
Compared with control cells, the peak amplitudes (F/F0) of cytoplasmic Ca2+ transients were reduced by 33% (F/F0: 3.82±0.35 versus 2.56±0.38; P<0.05) in nonischemic HF ventricular cells. There were no differences in the baseline fluorescence, the time to 90% peak, and the decay time constants of cytosolic Ca2+ transients among the sham and nonischemic HF groups (Figure 3C, Table 7). An average of 3 consecutive traces without EADs was used for data analysis. Cytoplasmic Ca2+ transients in the presence of EADs had peak amplitudes that were increased by 27% (F/F0: 3.82±0.35 versus 4.87±0.60; P>0.05, n=19) in nonischemic HF ventricular cells when compared with controls. Compared with control cells, failing myocytes had a faster mitochondrial Ca2+ influx rate (P<0.05) that could be slowed by adding 1 μmol/L Ru360 to the cytoplasm. On the other hand, the mitochondrial Ca2+ decay rate was unchanged in nonischemic HF ventricular cells, suggesting mitochondrial Ca2+ loading was the result of increased mitochondrial Ca2+ uptake (Table 8, Figure 3A and Figure 4). Meanwhile, there was a significant increase of IMCU in nonischemic HF mitoplasts (Figure 3B). The average IMCU at −160 mV was 496.6±33.9 pA in sham mitoplast versus 722.6±91.7 pA in nonischemic HF mitoplast (P<0.05). Single‐channel currents of IMCU recorded from mitoplasts isolated from sham and nonischemic HF mouse hearts (data not shown) indicate that MCU activity is increased in HF by ≈ 3‐fold.
Table 7.
Parameters of Cytoplasmic Ca2+ Transients in Isolated Ventricular Cells
| Group | Time to 90% Peak, ms | τdecay, ms | Peak Amplitude, F/F0 | Baseline, F/F0 | n |
|---|---|---|---|---|---|
| Sham | 46.7±2.5 | 254.4±17.3 | 3.82±0.35 | 1.9±0.1 | 19 |
| Nonischemic HF | 45.6±2.9 | 250.8±19.6 | 2.56±0.38a | 1.7±0.1 | 15 |
Time to 90% peak: the time between the beginnings of the transient to 90% of peak amplitude. Cells in sham and nonischemic HF group were from 3 and 4 mice, respectively. Data are presented as mean±SEM. τdecay indicates time constant of Ca2+ transient decay; F/F0, background‐subtracted normalized fluorescence; HF, heart failure.
P<0.05 compared with that in sham control group.
Table 8.
Characteristics of Mitochondrial Ca2+ Transients Recorded in Isolated Ventricular Cells From Sham and Nonischemic HF Mice
| Group | τrise, ms | τdecay, ms | Peak Amplitude, F/F0 | Baseline, F/F0 | n |
|---|---|---|---|---|---|
| Sham | 35±11 | 185±22 | 0.16±0.02 | 1.4±0.1 | 6 |
| Nonischemic HF | 11±3* | 213±34 | 0.66±0.17** | 1.8±0.2* | 8 |
| Nonischemic HF+Ru360 | 14±3* | 182±13 | 0.20±0.03 | 1.1±0.1 | 8 |
Cells in sham, nonischemic HF, and nonischemic HF+Ru360 (1 μmol/L) groups were from 4, 5, and 3 mice, respectively. Data are presented as mean±SEM. τdecay indicates time constant of Ca2+ transient decay; τrise, time constant of Ca2+ transient rise; F/F0, background‐subtracted normalized fluorescence; HF, heart failure.
*P<0.05, **P<0.01 compared with that in sham group.
Specific knockdown of MCU was undertaken, as shown in Figure 6A and 6B. MCU+/− nonischemic HF mice showed qualitatively similar alterations of APs and mitochondrial Ca2+ transients as internal application of 1 μmol/L Ru360 (Table 9, Table 10, and Figure 4, Figure 7). Although MCU+/− nonischemic HF mice displayed shorter APD90s (234±54 versus 577±92 ms; P<0.05) when compared with WT nonischemic HF mice, the MCU+/− mice had a significant lower basal mitochondrial Ca2+ (F/Fo: 1.2±0.1 versus 1.6±0.2; P<0.05) and a smaller mitochondrial Ca2+ (F/Fo: 0.18±0.04 versus 0.39±0.08; P<0.05) peak amplitude. The differences in APD90 between Tables 6 and 9 appeared to be mouse‐species dependent (C57BL/6 versus CD1).
Figure 6.
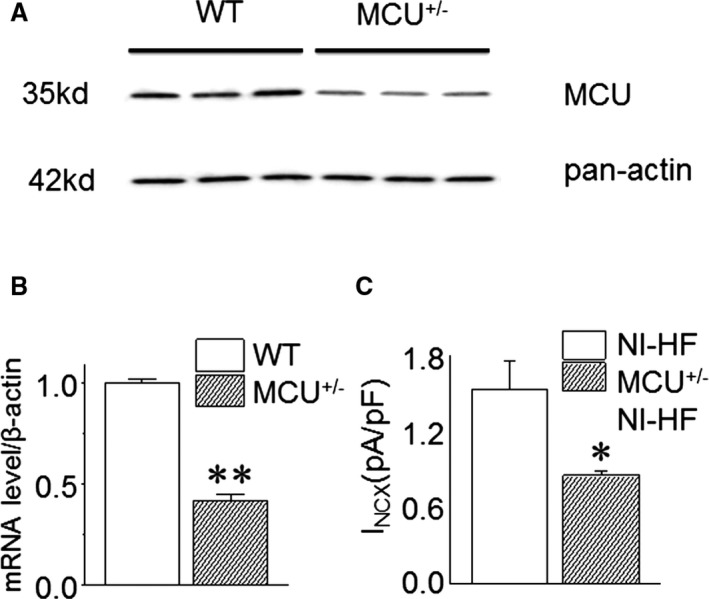
Mitochondrial Ca2+ uniporter (MCU) expression in wild‐type (WT) and MCU +/− mice and Na+‐Ca2+ exchanger (NCX) currents of WT nonischemic heart failure (NI‐HF) and MCU +/− NI‐HF mice. A, Representative Western blot results: Compared with WT mice, the MCU protein level is decreased in MCU +/− mice hearts. B, MCU mRNA expression obtained by quantitative real‐time reverse transcription–polymerase chain reaction is substantially reduced in MCU +/− mice hearts by 58% (n=6 mice in WT group, and n=8 mice in MCU +/− group). C, Compared with NI‐HF mice ventricular cardiomyocytes, the membrane NCX currents are significantly decreased in MCU +/− NI‐HF mice cardiomyocytes (n=7 cells from 2 mice in NI‐HF cardiomyocytes, and n=6 cells from 2 mice in MCU +/− NI‐HF cardiomyocytes). *P<0.05, **P<0.01 compared with that in NI‐HF group.
Table 9.
AP Parameters Obtained in Isolated Ventricular Cells From Nonischemic HF and MCU+/− Nonischemic HF Mice
| Group | APD90, ms | APA, mV | RMP, mV | EADn | n |
|---|---|---|---|---|---|
| Nonischemic HF | 577±92 | 107±30 | −76±7 | 3 | 3 |
| MCU+/− nonischemic HF | 234±54a | 102±4 | −74±1 | 0a | 8 |
| Nonischemic HF+CGP37157 | 261±53a | 112±9 | −75±2 | 0a | 6 |
Cells in nonischemic HF, MCU+/− nonischemic HF, and nonischemic HF+CGP37157 (1 μmol/L) groups were from 2, 2, and 3 mice, respectively. Data are presented as mean±SEM. AP indicates action potential; APA, AP amplitude; APD90, AP duration at 90% repolarization (calculated from APs without EADs); EADn, number of cells in which early afterdepolarizations were observed; HF, heart failure; MCU, mitochondrial Ca2+ uniporter; RMP, resting membrane potential.
P<0.05 compared with that in nonischemic HF group.
Table 10.
Parameters of Mitochondrial Ca2+ Transients Recorded in Isolated Ventricular Cells From Nonischemic HF and MCU+/− Nonischemic HF Mice
| Group | τrise, ms | τdecay, ms | Peak Amplitude, F/F0 | Baseline, F/F0 | n |
|---|---|---|---|---|---|
| Nonischemic HF | 11±2 | 211±50 | 0.39±0.08 | 1.6±0.2 | 5 |
| MCU+/− nonischemic HF | 9±1 | 173±9 | 0.18±0.04a | 1.2±0.1a | 9 |
| Nonischemic HF+CGP37157 | 138±29a | 276±31 | 0.39±0.07 | 1.6±0.1 | 6 |
Cells in nonischemic HF, MCU+/− nonischemic HF, and nonischemic HF+CGP37157 (1 μmol/L) groups were from 2, 2, and 3 mice, respectively. Data are presented as mean±SEM. τdecay indicates time constant of Ca2+ transient decay; τrise, time constant of Ca2+ transient rise; F/F0, background‐subtracted normalized fluorescence; HF, heart failure; MCU, mitochondrial Ca2+ uniporter.
P<0.05 compared with that in nonischemic HF group.
Figure 7.
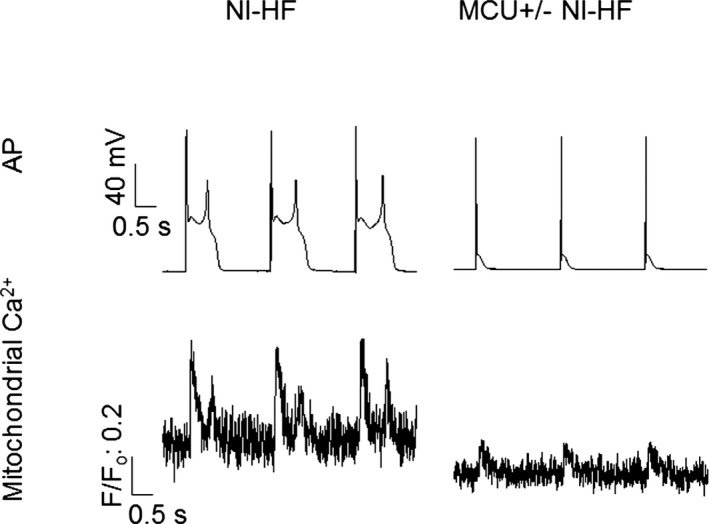
Action potentials (APs) and mitochondrial Ca2+ traces recorded simultaneously from nonischemic heart failure (NI‐HF) and mitochondrial Ca2+ uniporter (MCU) +/− NI‐HF mice. Top panel: APs. Early afterdepolarizations were eliminated in MCU +/− NI‐HF mouse cardiomyocytes. Bottom panel: Mitochondrial Ca2+ transients. Stimulation (0.5 Hz) was used to evoke APs. The time scale bar is shown at 0 mV. F/F0 indicates background‐subtracted normalized fluorescence.
CGP37157 (1 μmol/L) blocks mitochondrial NCX. Its effect on APs was similar to that seen in MCU+/− nonischemic HF mice. APD90 was shortened (261±53 ms) after CGP37157. With CGP37157, the mitochondrial Ca2+ transient increasing time was >10 times longer than those in the HF groups, with or without MCU knockdown. Cells from both MCU+/− nonischemic HF mice or WT nonischemic HF mice with CGP37157 application did not demonstrate EADs (Tables 9 and 10).
Inhibition of Mitochondrial Ca2+ Transients Reduced Arrhythmias
Compared with WT nonischemic HF mice, NCX current density was reduced in MCU+/− nonischemic HF mice (Figure 6C). Although all WT nonischemic HF mice had EADs, EADs were not observed in 8 MCU+/− nonischemic HF mice (Figure 7 and Table 9, P<0.05 by Fisher's exact test). Corresponding to the decrease of APD90 in MCU+/− nonischemic HF mice ventricular cells, QTc was significantly reduced in these myopathic mice (Figure 8B), and the ventricular tachycardia/fibrillation occurrence rate induced by 2.5 mg/kg isoproterenol decreased from 77.8% in 9 WT nonischemic HF mice to 16.7% in 6 MCU+/− nonischemic HF mice (P<0.01 by Fisher's exact test, Figure 8A).
Figure 8.
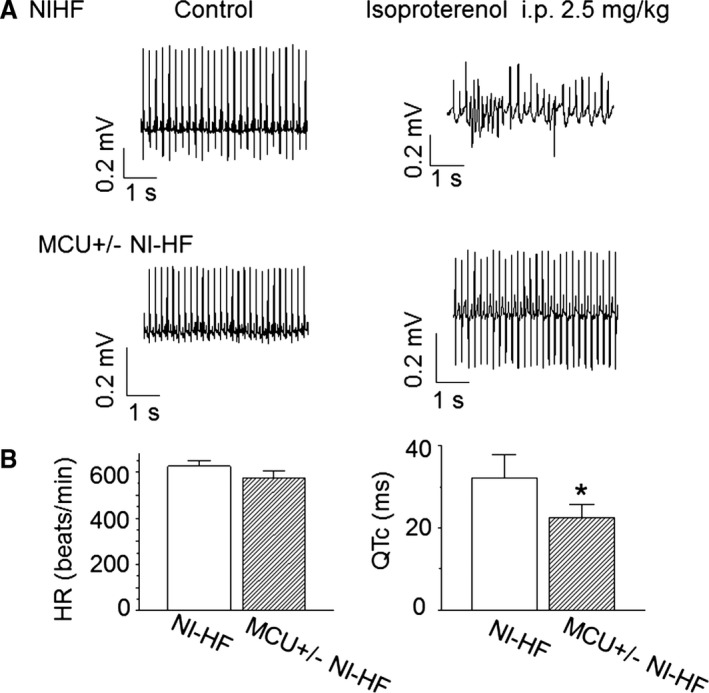
Telemetry of nonischemic heart failure (NI‐HF) and mitochondrial Ca2+ uniporter (MCU)+/− NI‐HF CD1 mice. A, Examples of telemetric ECG recordings. ECG signals in top and bottom panels were sampled from NI‐HF (n=5) and MCU +/− NI‐HF (n=6) mice, respectively. Waveforms were collected before (control) and after (IP 2.5 mg/kg) isoproterenol injection. B, Heart rate (HR) and corrected QT (QTc) were measured from lead II and plotted in left and right panels, respectively. Compared with the NI‐HF mice, MCU +/− NI‐HF mice showed a reduced QT interval (n=4 mice for the NI‐HF group, and n=5 mice for the MCU +/− NI‐HF group). *P<0.05 compared with that in NI‐HF group.
Possible Mechanism of Increased Mitochondrial Ca2+ Transients in Nonischemic HF Cardiomyocytes
Fluorescence staining results revealed that the overall mitochondrial mass increased in the nonischemic HF group, consistent with previous reports (Figure 9A).11, 12 Moreover, phosphorylation of MCU protein was significantly enhanced from 0.30±0.04 to 1.45±0.46 with nonischemic HF. There was no significant difference of MCU or mitochondrial NCX expressions between sham and nonischemic HF hearts (Figure 9C and 9D). Together, a larger mass of mitochondrial and the presence of activated phosphorylated MCU could explain the larger Ca2+ transients in nonischemic HF cardiomyocytes. Change in mitochondrial membrane potential in nonischemic HF ventricular cells was slightly depolarized and collapsed when applying 20 μmol/L carbonyl cyanide 4‐(trifluoromethoxy) phenylhydrazone (Figure 9B), suggesting that changes in mitochondrial membrane polarization were unlikely to be a cause of the increased transients.
Figure 9.
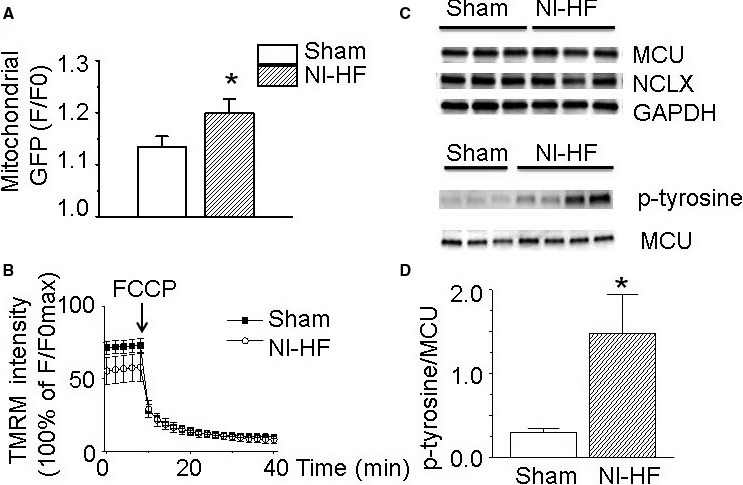
The alterations of mitochondrial properties in nonischemic heart failure (NI‐HF) mice. A, Fluorescence staining results revealed that the overall mitochondrial mass increased from background‐subtracted normalized fluorescence (F/Fo) of 1.13±0.02 in sham ventricular cardiomyocytes (n=16) to 1.20±0.03 in NI‐HF cardiomyocytes (n=19). B, Compared with sham cardiomyocytes (n=12), the mitochondrial membrane potential was slightly depolarized in NI‐HF cardiomyocytes (n=10). Mitochondrial membrane potential was collapsed by 20 µmol/L carbonyl cyanide 4‐(trifluoromethoxy) phenylhydrazone (FCCP) in both sham and NI‐HF cardiomyocytes. C, Western blots of mitochondrial Ca2+ uniporter (MCU), mitochondrial Na+‐Ca2+ exchange (NCLX), and phosphorylated MCU. D, The expression of phosphorylated MCU was upregulated from 0.30±0.04 in sham group to 1.45±0.46 in NI‐HF group. GFP indicates green fluorescent protein; p‐tyrosine, phosphorylated tyrosine; TMRM, tetramethylrhodamine methyl ester. *P<0.05 compared with that in sham group.
Computer Simulations
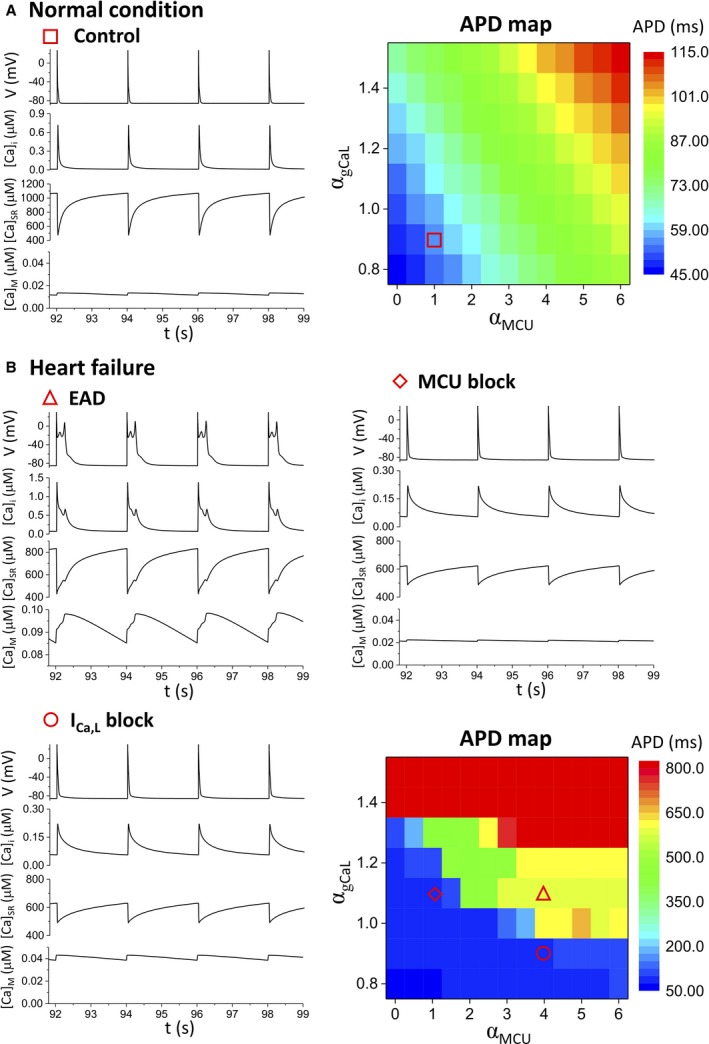
Under normal control conditions (left panel in Figure 10A), the APD is ≈35 ms, and the amplitude of the cytosolic Ca2+ transient is ≈0.2 μmol/L with a pacing cycle length of 2 seconds. The SR Ca2+ load and nadir are ≈700 and ≈500 μmol/L, respectively. The mitochondria free Ca2+ concentration is ≈20 nmol/L and has an ≈10% diastolic‐to‐systolic variation. To investigate the effects of MCU activity and LCC conductance on AP dynamics under the normal condition (right panel in Figure 10A), we performed simulations scanning the MCU activity (multiplying the original MCU activity by a factor αMCU) and the LCC conductance (multiplying the original LCC conductance by a factor αgCaL), and measured the corresponding APD for each set of parameters. Increasing MCU up to 6‐fold or LCC conductance by 50% resulted in an ≈1‐fold increase in APD, but no EADs were observed in these simulations.
Figure 10.
Computer simulation results. A, Action potential duration (APD) and Ca2+ cycling dynamics under the normal condition. Left panel: Time traces of membrane voltage, cytosolic Ca2+ concentration, sarcoplasmic reticulum Ca2+ concentration, and mitochondrial free Ca2+ concentration under the normal control condition. Right panel: Dependence of the APD on mitochondrial Ca2+ uniporter (MCU) activity (αMCU) and L‐type Ca2+ channel (LCC) conductance (αgCaL). B, APD and Ca2+ cycling dynamics under the heart failure condition. The layout of time traces for each case is the same as in A. Top left: Heart failure controls with early afterdepolarizations (EADs). Top right: MCU activity was reduced by 3‐fold (αMCU changed from 4 to 1). Bottom left: LCC conductance was reduced by 20% (αgCaL changed from 1.1 to 0.9). Bottom right: Dependence of the APD on MCU activity and LCC conductance. In this map, the APD jumps suddenly from ≈50 to >300 ms when EADs occur. The 3 specific cases shown above were marked on the map with different symbols. ICa,L indicates L‐type Ca2+ current.
Under the HF condition (Figure 10B), EADs occurred at the same pacing cycle length of 2 seconds (triangle). The APD increased to ≈600 ms, which agrees with the experimental data (Table 9). Under this condition, the cytosolic Ca2+ concentration, SR Ca2+ load, and mitochondrial free Ca2+ concentration were substantially higher than those under the normal control conditions during the diastolic period. Despite increased mitochondrial mass and, presumably, mitochondrial buffering potential, diastolic Ca2+ was increased in nonischemic HF. The EADs were suppressed by blocking MCU activity by 3‐fold (diamond), agreeing with the experimental observations that blocking MCU with Ru360 (Figure 4) or MCU knockout (Figure 7) suppressed EADs. After MCU blockade, the APD, cytosolic Ca2+ concentration, SR Ca2+ load, and mitochondrial free Ca2+ concentration all recovered to approximately the same levels as in the normal control conditions. The EADs could also be suppressed by reducing LCC conductance (circle), but in this case, the mitochondrial free Ca2+ concentration was twice that of normal controls, whereas the APD, cytosolic Ca2+ concentration, and SR load became normal. Similar to the normal condition, we also scanned the MCU activity and LCC conductance (the APD map in Figure 10B) to systematically investigate the AP dynamics under HF conditions. The APD map shows that the efficacy of MCU block in suppressing EADs depended on LCC conductance, with a larger LCC conductance requiring a stronger MCU blockage.
To understand the underlying mechanisms and reveal the role of mitochondrial Ca2+ handling in the genesis of EADs, further simulations were performed, which showed that there was a positive feedback between the membrane potential and intracellular Ca2+ cycling, resulting in a bistable behavior of the AP. Bistability means that a system exhibits 2 stable states under the same conditions, and the system could stay at either of the 2 states, depending on the initial conditions. In our system, the 2 stable states are a long APD state with EADs and with high Ca2+ (high state) and a short APD state without EADs and with low Ca2+ (low state). The bistable behavior depends on the strength of MCU. Figure 11A through 11C plots the maximum mitochondrial free Ca2+ concentration, peak cytosolic Ca2+ concentration, and SR Ca2+ load versus αMCU, respectively. When αMCU<2.4, the Ca2+ concentrations in the 3 compartments stayed at the low state (without EADs), and when αMCU>30, the Ca2+ concentrations stayed at the high state (with EADs). But when 2.4<αMCU<30, the system could stay at either the low or the high state, depending on the initial conditions. In other words, if the initial condition is close to the low state, it will approach the low state, and vice versa.
Figure 11.
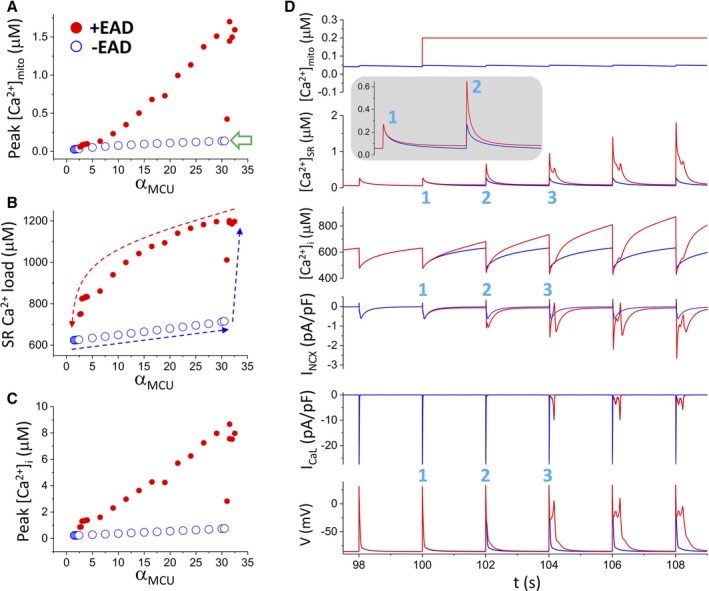
Mechanistic insights from the computer model into the roles of mitochondrial Ca2+ handling in the genesis of early afterdepolarizations (EADs). A through C, Ca2+ concentrations in different compartments of the cell model vs the strength of mitochondrial Ca2+ uniporter (MCU), showing bistability. The simulations were performed as follows. We first started the simulations from the normal control MCU activity (αMCU=1) and increased αMCU gradually to 35, as indicated by the blue arrows (B). The system switches from no EADs (also low Ca2+ concentration states) to EADs (also high Ca2+ concentration states) at αMCU≈30, at which the mitochondrial free Ca2+ reaches 136 nmol/L (open arrow). For each αMCU, 50 beats were simulated for the cell to reach steady state before changing to a larger αMCU. We then started the simulations in the same way but from a high MCU activity (αMCU=35) to the normal control value (αMCU=1), as indicated by the red arrow (B). The system switches from EADs to no EADs at αMCU≈2.4. D. [Ca2+]mito, [Ca2+]i, [Ca2+]SR, INCX, IC a,L, and V vs time from 2 simulations for αMCU=5. In the first simulation (blue traces), the mitochondrial Ca2+ is free running (unclamped), and the cell is in the low Ca2+ state without EADs. In the second simulation (red traces), [Ca2+]mito is suddenly elevated to 200 nmol/L and held constant (clamped) for the rest of the simulation. 1, 2, and 3 mark the first 3 beats during the clamped phase. [Ca2+]mito indicates mitochondrial Ca2+ concentration; [Ca2+]i, intracellular Ca2+ concentration; [Ca2+]SR, SR Ca2+ concentration; INCX, NCX current; IcaL, L‐type Ca2+ current; V, membrane potential.
To understand the positive feedback loop responsible for the bistable behavior and the role of mitochondrial Ca2+ handling, we performed simulations by holding the mitochondrial Ca2+ concentration at different constant levels. Figure 11D shows an example (αMCU=5) when the mitochondrial Ca2+ concentration was suddenly elevated to 200 nmol/L from the low state (≈50 nmol/L) at time=100 seconds. EADs were induced from the third beat after the elevation. As seen in this simulation, the elevation of the mitochondrial Ca2+ concentration caused an elevated diastolic intracellular Ca2+ concentration ([Ca2+]i) but not the peak [Ca2+]i in the first beat (inset). Elevation of diastolic [Ca2+]i caused an elevated SR Ca2+ load, which caused a larger SR Ca2+ release and thus a higher [Ca2+]i in the second beat. A higher [Ca2+]i resulted in a larger NCX current, which lengthened the APD. The SR Ca2+ load became even higher in the second beat, which caused an even larger [Ca2+]i in the third beat. At this beat, the elevated [Ca2+]i was large enough to result in an NCX current that caused EADs. When EADs occurred, reactivation of LCCs brought extra Ca2+ into the cell to maintain the high Ca2+ state of the cell. In this specific example, EADs could be induced when we elevated mitochondrial Ca2+ to >120 nmol/L (open arrow in Figure 11A for the [Ca2+] mitochondrial space threshold), but it took many more beats to load the SR Ca2+ to the level for EADs to occur. Therefore, the occurrence of the EADs is facilitated by the following positive feedback loop: an elevated [Ca2+]mito causes an elevation in diastolic [Ca2+]i, which results in a higher SR Ca2+ load; a higher SR load causes a larger Ca2+ release and thus a higher [Ca2+]i; and a higher [Ca2+]i results in an increased INCX, which lengths the APD and triggers reactivation of the LCC to cause EADs. Reactivation of the LCC brings in extra Ca2+ to elevate Ca2+ concentrations of the cell. Mechanistic insights into the positive feedback loops and the role of mitochondrial handling are detailed in the Discussion.
Discussion
Cardiomyopathy is associated with increased arrhythmic risk, at least in part as a result of increased EADs. After depolarizations are thought to involve alterations in Ca2+ handling.6, 12 Mitochondria are known to sequester Ca2+ and are altered in cardiomyopathy.12 Therefore, we set out to determine if alterations in mitochondrial Ca2+ flux could contribute to EADs in cardiomyopathy.
We used sustained hypertension to induce nonischemic cardiomyopathy in mice. Consistent with previous reports in other models of cardiomyopathy, these DOCA‐treated nonischemic HF mice showed reduced ejection fraction and reduced systolic cytoplasmic Ca2+ transients.14, 15, 42, 43, 44 Similar to other myopathic models, these nonischemic HF mice showed electrical remodeling with prolonged QTc intervals and APDs.1 The enhancement of the NCX inward current in nonischemic HF mice likely contributed to the increased APD.45 These changes were accompanied by increased spontaneous and induced arrhythmias that could be suppressed by inhibition of mitochondria Ca2+ handling.
Ru360 is a fast, potent (inhibitory concentration of 50%=0.2 nmol/L), and specific (even at a high concentration of 10 μmol/L) antagonist of MCU.41 As expected, Ru360 reduced mitochondrial Ca2+ uptake. The mechanism of reduced EADs with Ru360 appeared to be reduced sarcolemmal NCX current, a key depolarizing current known to be involved in EADs.2 This effect is most easily explained by Ru360 reducing mitochondrial Ca2+ loading, reducing mitochondrial Ca2+ efflux, and decreasing sarcolemmal NCX. The involvement of MCU in the EAD production was supported by the data from MCU+/− mice, which had nearly identical reductions in mitochondrial Ca2+ uptake and EADs as those induced by Ru360. Because a global knockout of MCU is subject to extensive remodeling and has a limited life expectancy,46 the MCU+/− mice were selected in these experiments.
In nonischemic HF animals, the mitochondria appeared to take up more Ca2+ compared with WT animals. This increased mitochondrial uptake and sarcolemmal NCX inward current occurred concomitantly with a reduction in SR Ca2+ release. Ru360 was able to reduce mitochondrial Ca2+ uptake and improve SR Ca2+ release, suggesting that mitochondrial Ca2+ uptake may contribute to the reduced cytoplasmic Ca2+ transient and to contractile dysfunction in cardiomyopathy.
In the systolic period, nonischemic HF mice had an increased mitochondrial Ca2+ influx and a reduced SR Ca2+ release. However, the baseline of cytosolic Ca2+ and the SR Ca2+ uptake rates were not changed in diastolic period. Therefore, EADs, which occur in this period, were not evoked by reduced SR Ca2+ activity. On the other hand, increased mitochondrial Ca2+ uptake could explain the elevated mitochondrial Ca2+ release in the diastolic period, and the increased mitochondrial Ca2+ release likely was the driving force for activating membrane NCX and initiating EADs. Consistent with the idea that mitochondrial Ca2+ flux contributes to EADs, inhibiting Ca2+ entry or efflux from mitochondria has similar antiarrhythmic effects.
We investigated possible reasons for the increase in mitochondrial Ca2+ flux with HF. Consistent with previous reports, we found an increase in total mass of mitochondria in nonischemic HF. Moreover, MCU is known to be activated by phosphorylation.47 In nonischemic HF, we found that MCU was significantly more phosphorylated than in WT mice and IMCU was significantly increased in nonischemic HF group. Finally, mitochondrial reactive oxygen species overproduction is observed in DOCA‐salt mice myocytes.15 Reactive oxygen species can oxidize Cys‐97 of MCU and increase MCU activity.13 Consistent with the latter 2 possibilities, we observed increased whole‐mitoplast MCU current. Our result differs from that of Michels et al, who reported a decrease in mitochondrial Ca2+ uptake in a heterogeneous group of human failing hearts.48 The reason for this difference is not clear, but our results clearly show that mitochondrial Ca2+ handling can contribute to EADs and arrhythmia in chronic and moderate HF.
Computer simulations demonstrated that EADs caused by HF remodeling could be abolished by blocking the MCU, consistent with the experimental observations. These simulations revealed that intracellular Ca2+ cycling (including both SR and mitochondria) and the occurrence of EADs formed a positive feedback loop to result in an all‐or‐none behavior or bistability. Figure 12 illustrates the interactions between the APD (EADs) and intracellular Ca2+ cycling on the basis of our simulations. Specifically, increasing MCU activity causes an elevated [Ca2+]mito, which causes a higher diastolic [Ca2+]i because of the slow Ca2+ extrusion via mitochondrial NCX. The higher [Ca2+]i causes more Ca2+ uptake via the SERCA pump of the SR. A higher SR Ca2+ load results in a larger Ca2+ release via RyRs. This, in turn, causes a higher NCX current to lengthen APD. When NCX is strong enough to maintain the voltage at the window range of ICa,L, LCCs are reactivated to cause depolarizations in the AP plateau to result in EADs. Reactivation of LCCs brings extra Ca2+ to load the cell with more Ca2+, which causes the SR and mitochondria to take up more Ca2+ via SERCA and MCU, respectively.
Figure 12.
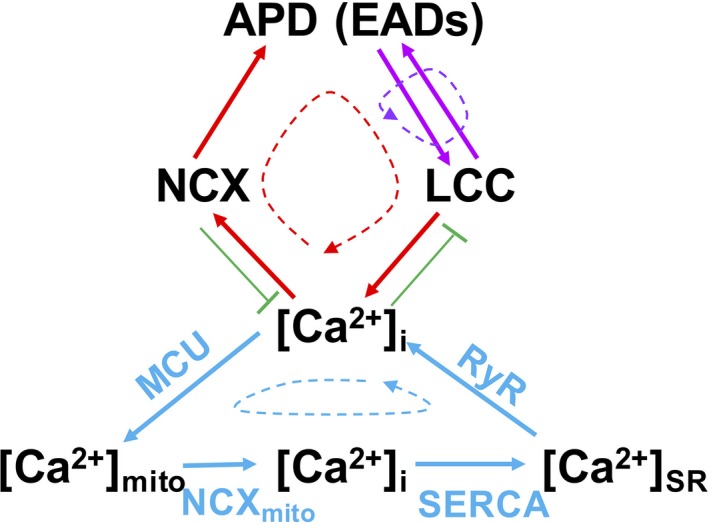
Schematic plot of the feedback loops involved in the action potential (AP) and Ca2+ cycling dynamics. APD indicates AP duration; [Ca2+]mito, mitochondrial Ca2+ concentration; [Ca2+]i, intracellular Ca2+ concentration; [Ca2+]SR, SR Ca2+ concentration;EAD, early afterdepolarization; LCC, L‐type Ca2+ channel; MCU, mitochondrial Ca2+ uniporter; NCX, Na+‐Ca2+ exchange; RyR, ryanodine receptor; SERCA, sarco/endoplasmic reticulum Ca2+‐ATPase.
As illustrated in Figure 12, there are different feedback loops involved in this process. The first such loop is the positive feedback between voltage and ICa,L, which is minimally required for EADs to occur.49, 50 The second loop is the one between intracellular Ca2+ cycling and EADs, in which reactivation of LCCs during EADs causes Ca2+ elevation, which results in a larger NCX current to potentiate EADs. This is the positive feedback loop that maintains the bistability between no EADs and EADs in the APs shown in Figure 11. Mitochondrial Ca2+ handling and SR Ca2+ involves another such loop (ie, a higher SR Ca2+ load results in a larger Ca2+ transient, which causes a higher mitochondrial uptake via MCU). A key feature of mitochondrial Ca2+ handling is that Ca2+ is taken up quickly into the mitochondria via MCU but releases slowly via mitochondrial NCX, causing an elevation of the diastolic [Ca2+]i. Although it was not possible to resolve experimentally whether the mitochondrial Ca2+ transient preceded the sarcolemmal voltage changes related to an EAD, EADs were eliminated when inhibiting mitochondrial Ca2+ uptake, suggesting mitochondrial Ca2+ flux is involved in EAD generation.
In summary, cardiomyopathic mice showed increased arrhythmic risk with prolonged QTc, increased APDs, and EADs. These changes were accompanied by reduced cytoplasmic Ca2+ transients and enhanced mitochondrial Ca2+ transients. An MCU blocker was able to inhibit mitochondrial Ca2+ uptake and reduce EADs. Consistent with an effect on mitochondrial Ca2+ flux, knockdown of MCU prevented the increased Ca2+ flux in mitochondria and reduced arrhythmic risk. These findings with nonischemic cardiomyopathy are summarized in Figure 10, and they suggest that changes in mitochondrial Ca2+ handling are important in the arrhythmic risk seen in cardiomyopathy.
Sources of Funding
This work was supported by the National Institutes of Health (R01 HL104025 and R01 HL106592 to Dudley and R01 HL133294 to Qu and Xie) and Veterans Affairs MERIT grant (BX000859 to Dudley).
Disclosures
None.
(J Am Heart Assoc. 2018;7:e007805 DOI: 10.1161/JAHA.117.007805.)29627768
References
- 1. Nuss HB, Kaab S, Kass DA, Tomaselli GF, Marban E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol. 1999;277:H80–H91. [DOI] [PubMed] [Google Scholar]
- 2. Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhao Z, Xie Y, Wen H, Xiao D, Allen C, Fefelova N, Dun W, Boyden PA, Qu Z, Xie LH. Role of the transient outward potassium current in the genesis of early afterdepolarizations in cardiac cells. Cardiovasc Res. 2012;95:308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruiz‐Meana M, Fernandez‐Sanz C, Garcia‐Dorado D. The SR‐mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc Res. 2010;88:30–39. [DOI] [PubMed] [Google Scholar]
- 5. Porter GA Jr, Hom J, Hoffman D, Quintanilla R, de Mesy Bentley K, Sheu SS. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog Pediatr Cardiol. 2011;31:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choudhary G, Dudley SC Jr. Heart failure, oxidative stress, and ion channel modulation. Congest Heart Fail. 2002;8:148–155. [DOI] [PubMed] [Google Scholar]
- 7. Campanella M, Pinton P, Rizzuto R. Mitochondrial Ca2+ homeostasis in health and disease. Biol Res. 2004;37:653–660. [DOI] [PubMed] [Google Scholar]
- 8. Brown DA, O'Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc Res. 2010;88:241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aon MA, Cortassa S, Marban E, O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278:44735–44744. [DOI] [PubMed] [Google Scholar]
- 10. Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 2009;84:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hohendanner F, Ljubojevic S, Macquaide N, Sacherer M, Sedej S, Biesmans L, Wakula P, Platzer D, Sokolow S, Herchuelz A, Antoons G, Sipido K, Pieske B, Heinzel FR. Intracellular dyssynchrony of diastolic cytosolic [Ca2+] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ Res. 2013;113:527–538. [DOI] [PubMed] [Google Scholar]
- 13. Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P, Riitano MF, Worth AM, Seelam A, Carvalho E, Subbiah R, Jana F, Soboloff J, Peng Y, Cheung JY, Joseph SK, Caplan J, Rajan S, Stathopulos PB, Madesh M. Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell. 2017;65:1014–1028.e1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Silberman GA, Fan TH, Liu H, Jiao Z, Xiao HD, Lovelock JD, Boulden BM, Widder J, Fredd S, Bernstein KE, Wolska BM, Dikalov S, Harrison DG, Dudley SC Jr. Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation. 2010;121:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu M, Gu L, Sulkin MS, Liu H, Jeong EM, Greener I, Xie A, Efimov IR, Dudley SC Jr. Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol. 2013;54:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gellen B, Fernandez‐Velasco M, Briec F, Vinet L, LeQuang K, Rouet‐Benzineb P, Benitah JP, Pezet M, Palais G, Pellegrin N, Zhang A, Perrier R, Escoubet B, Marniquet X, Richard S, Jaisser F, Gomez AM, Charpentier F, Mercadier JJ. Conditional FKBP12.6 overexpression in mouse cardiac myocytes prevents triggered ventricular tachycardia through specific alterations in excitation‐contraction coupling. Circulation. 2008;117:1778–1786. [DOI] [PubMed] [Google Scholar]
- 17. Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, De BM, Wehrens XH, Pautler RG, Roden DM, Taffet GE, Dirksen RT, Anderson ME, Hamilton SL. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine‐induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:12179–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Knollmann BC, Blatt SA, Horton K, de Freitas F, Miller T, Bell M, Housmans PR, Weissman NJ, Morad M, Potter JD. Inotropic stimulation induces cardiac dysfunction in transgenic mice expressing a troponin T (I79N) mutation linked to familial hypertrophic cardiomyopathy. J Biol Chem. 2001;276:10039–10048. [DOI] [PubMed] [Google Scholar]
- 19. Vranjkovic O, Hang S, Baker DA, Mantsch JR. β‐Adrenergic receptor mediation of stress‐induced reinstatement of extinguished cocaine‐induced conditioned place preference in mice: roles for β1 and β2 adrenergic receptors. J Pharmacol Exp Ther. 2012;342:541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. O'Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–296. [DOI] [PubMed] [Google Scholar]
- 21. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. [DOI] [PubMed] [Google Scholar]
- 22. Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and Na+‐Ca2+ exchanger: molecular partners in pacemaker regulation. Circ Res. 2001;88:1254–1258. [DOI] [PubMed] [Google Scholar]
- 23. Wang Y, Tandan S, Cheng J, Yang C, Nguyen L, Sugianto J, Johnstone JL, Sun Y, Hill JA. Ca2+/calmodulin‐dependent protein kinase II‐dependent remodeling of Ca2+ current in pressure overload heart failure. J Biol Chem. 2008;283:25524–25532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. El GG, Fiset C. 4‐Hydroxytamoxifen inhibits K+ currents in mouse ventricular myocytes. Eur J Pharmacol. 2010;629:96–103. [DOI] [PubMed] [Google Scholar]
- 25. Bridge JH, Smolley J, Spitzer KW, Chin TK. Voltage dependence of sodium‐calcium exchange and the control of calcium extrusion in the heart. Ann N Y Acad Sci. 1991;639:34–47. [DOI] [PubMed] [Google Scholar]
- 26. Kapur N, Banach K. Inositol‐1,4,5‐trisphosphate‐mediated spontaneous activity in mouse embryonic stem cell‐derived cardiomyocytes. J Physiol. 2007;581:1113–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. MacKenzie L, Bootman MD, Laine M, Berridge MJ, Thuring J, Holmes A, Li WH, Lipp P. The role of inositol 1,4,5‐trisphosphate receptors in Ca2+ signalling and the generation of arrhythmias in rat atrial myocytes. J Physiol. 2002;541:395–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O'Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation‐contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL. Dynamin‐related protein 1 (Drp1)‐mediated diastolic dysfunction in myocardial ischemia‐reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014;28:316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song Z, Ko CY, Nivala M, Weiss James N, Qu Z. Calcium‐voltage coupling in the genesis of early and delayed afterdepolarizations in cardiac myocytes. Biophys J. 2015;108:1908–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang L, Korge P, Weiss JN, Qu Z. Mitochondrial oscillations and waves in cardiac myocytes: insights from computational models. Biophys J. 2010;98:1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cortassa S, Aon MA, O'Rourke B, Jacques R, Tseng H‐J, Marbán E, Winslow RL. A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys J. 2006;91:1564–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu X, Ginsburg KS, Kettlewell S, Bossuyt J, Smith GL, Bers DM. Measuring local gradients of intramitochondrial [Ca2+] in cardiac myocytes during sarcoplasmic reticulum Ca2+ release. Circ Res. 2013;112:424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vendelin M, Beraud N, Guerrero K, Andrienko T, Kuznetsov AV, Olivares J, Kay L, Saks VA. Mitochondrial regular arrangement in muscle cells: a “crystal‐like” pattern. Am J Physiol Cell Physiol. 2005;288:C757–C767. [DOI] [PubMed] [Google Scholar]
- 36. Restrepo JG, Weiss JN, Karma A. Calsequestrin‐mediated mechanism for cellular calcium transient alternans. Biophys J. 2008;95:3767–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morotti S, Edwards AG, McCulloch AD, Bers DM, Grandi E. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J Physiol. 2014;592:1181–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Magnus G, Keizer J. Minimal model of beta‐cell mitochondrial Ca2+ handling. Am J Physiol. 1997;273:C717–C733. [DOI] [PubMed] [Google Scholar]
- 39. O'Rourke B, Blatter LA. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol. 2009;46:767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nivala M, Song Z, Weiss JN, Qu Z. T‐tubule disruption promotes calcium alternans in failing ventricular myocytes: mechanistic insights from computational modeling. J Mol Cell Cardiol. 2015;79:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause‐Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, Bers DM. Oxygen‐bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem. 1998;273:10223–10231. [DOI] [PubMed] [Google Scholar]
- 42. Jeong EM, Monasky MM, Gu L, Taglieri DM, Patel BG, Liu H, Wang Q, Greener I, Dudley SC Jr, Solaro RJ. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J Mol Cell Cardiol. 2013;56:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lovelock JD, Monasky MM, Jeong EM, Lardin HA, Liu H, Patel BG, Taglieri DM, Gu L, Kumar P, Pokhrel N, Zeng D, Belardinelli L, Sorescu D, Solaro RJ, Dudley SC Jr. Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ Res. 2012;110:841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yemane H, Busauskas M, Burris SK, Knuepfer MM. Neurohumoral mechanisms in deoxycorticosterone acetate (DOCA)‐salt hypertension in rats. Exp Physiol. 2010;95:51–55. [DOI] [PubMed] [Google Scholar]
- 45. Ferreiro M, Petrosky AD, Escobar AL. Intracellular Ca2+ release underlies the development of phase 2 in mouse ventricular action potentials. Am J Physiol Heart Circ Physiol. 2012;302:H1160–H1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Uchi J, Jhun BS, Xu S, Hurst S, Raffaello A, Liu X, Yi B, Zhang H, Gross P, Mishra J, Ainbinder A, Kettlewell S, Smith GL, Dirksen RT, Wang W, Rizzuto R, Sheu SS. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2‐dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid Redox Signal. 2014;21:863–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Michels G, Khan IF, Endres‐Becker J, Rottlaender D, Herzig S, Ruhparwar A, Wahlers T, Hoppe UC. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage‐gated Ca2+ channels. Circulation. 2009;119:2435–2443. [DOI] [PubMed] [Google Scholar]
- 49. Tran DX, Sato D, Yochelis A, Weiss JN, Garfinkel A, Qu Z. Bifurcation and chaos in a model of cardiac early afterdepolarizations. Phys Rev Lett. 2009;102:258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Qu Z, Xie L‐H, Olcese R, Karagueuzian HS, Chen P‐S, Garfinkel A, Weiss JN. Early afterdepolarizations in cardiac myocytes: beyond reduced repolarization reserve. Cardiovasc Res. 2013;99:6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]