

Abstract
Background
ECG global electrical heterogeneity (GEH) is associated with sudden cardiac death. We hypothesized that a genome‐wide association study would identify genetic loci related to GEH.
Methods and Results
We tested genotyped and imputed variants in black (N=3057) and white (N=10 769) participants in the ARIC (Atherosclerosis Risk in Communities) study and CHS (Cardiovascular Health Study). GEH (QRS‐T angle, sum absolute QRST integral, spatial ventricular gradient magnitude, elevation, azimuth) was measured on 12‐lead ECGs. Linear regression models were constructed with each GEH variable as an outcome, adjusted for age, sex, height, body mass index, study site, and principal components to account for ancestry. GWAS identified 10 loci that showed genome‐wide significant association with GEH in whites or joint ancestry. The strongest signal (rs7301677, near TBX3) was associated with QRS‐T angle (white standardized β+0.16 [95% CI 0.13–0.19]; P=1.5×10−26), spatial ventricular gradient elevation (+0.11 [0.08–0.14]; P=2.1×10−12), and spatial ventricular gradient magnitude (−0.12 [95% CI −0.15 to −0.09]; P=5.9×10−15). Altogether, GEH‐SNPs explained 1.1% to 1.6% of GEH variance. Loci on chromosomes 4 (near HMCN2), 5 (IGF1R), 11 (11p11.2 region cluster), and 7 (near ACTB) are novel ECG phenotype‐associated loci. Several loci significantly associated with gene expression in the left ventricle (HMCN2 locus—with HMCN2;IGF1R locus—with IGF1R), and atria (RP11‐481J2.2 locus—with expression of a long non‐coding RNA and NDRG4).
Conclusions
We identified 10 genetic loci associated with ECG GEH. Replication of GEH GWAS findings in independent cohorts is warranted. Further studies of GEH‐loci may uncover mechanisms of arrhythmogenic remodeling in response to cardiovascular risk factors.
Keywords: ECG, electrocardiography, genome wide association study, global electrical heterogeneity, spatial ventricular gradient, sum absolute QRST integral
Subject Categories: Electrophysiology; Electrocardiology (ECG); Genetic, Association Studies
Clinical Perspective
What Is New?
Ten genetic loci have a genome‐wide significant associations with electrocardiographic global electrical heterogeneity (GEH) and reflect the multifactorial nature of GEH.
Loci on chromosomes 4 (near HMCN2), 5 (IGF1R), 11 (11p11.2 region cluster), and 7 (near ACTB) have not been previously reported to be associated with any elctrocardiogram phenotype.
What Are the Clinical Implications?
Functional characterization of GEH‐associated genetic loci may uncover pathophysiology of previously unrecognized conditions manifested by abnormal GEH and increased risk of sudden cardiac death.
Functional characterization of GEH‐associated loci may guide the discovery of targets for new therapies which could modulate electrical heterogeneity and potentially prevent sudden cardiac death.
Future pharmacogenomic studies of GEH may identify individuals who are most likely to benefit from (or be hurt by) specific pharmacological therapies.
Introduction
The human heart generates electricity with every heartbeat, and the surface ECG records differences in electrical potentials produced by the heart as an electrical generator on the body surface. The cornerstone assumption of electrocardiology is that the entire heart can be represented by a single electromagnetic dipole vector, which rotates throughout the cardiac cycle. Rotation of this vector throughout the cardiac cycle forms closed loops, corresponding to atrial depolarization (P‐loop), ventricular depolarization (QRS‐loop), and ventricular repolarization (T‐loop). The surface ECG, therefore, characterizes global electrophysiological properties of the entire heart throughout both depolarization and repolarization phases of the cardiac cycle. It can be demonstrated that the vector sum of the depolarization (QRS) and repolarization (T) vectors defines a vector along which non‐uniformity in excitation and repolarization is most prominent.1, 2 This vector, called the “spatial ventricular gradient” (SVG), captures the magnitude and direction of the steepest gradient between the areas of the heart with the longest and the shortest total recovery time.1, 3, 4 In 1988, Mark Josephson's group demonstrated5 that susceptibility to ventricular arrhythmias is characterized by heterogeneity in total recovery time (which is a combination of both dispersion of endocardial activation and dispersion of refractoriness). A global measure of the dispersion of total recovery time is a surrogate for an underlying arrhythmogenic substrate, encompassing dispersion of endocardial activation (eg, electrophysiological substrate of post‐infarction ventricular arrhythmias), as well as dispersion of refractoriness (eg, electrophysiological substrate of inherited or iatrogenic long QT syndromes).
Experimental and theoretical studies have demonstrated that the SVG is related to global heterogeneity of both action potential duration and morphology6, 7 and that it is theoretically independent of the specific ventricular activation sequence.8 SVG is a vector sum of spatial QRS and T vectors, reflecting implication of the spatial QRS‐T angle.9 Spatial QRS‐T angle is a well‐known marker9 of the risk of ventricular tachyarrhythmias10 and sudden cardiac death (SCD)11, 12 in the general population, heart failure (HF) patients, and other populations.13, 14
The sum absolute QRS‐T integral (SAI QRST) is a scalar analog of the SVG15, 16, 17, 18 which is associated with ventricular tachyarrhythmias in HF patients.15, 16, 19 SAI QRST is associated with electrical dyssynchrony,20 the mechanical response to cardiac resynchronization therapy (CRT),21 and mortality in patients with implanted CRT devices.22 Patient‐specific time‐varying associations between SAI QRST and high sensitivity troponin I suggest that SAI QRST also reflects subclinical myocardial injury.23
Altogether, features of the SVG vector (vector magnitude, direction, a scalar value, and QRS‐T angle) characterize global electrical heterogeneity (GEH). Abnormal GEH is associated with a risk for ventricular arrhythmias10, 15, 16, 19 and SCD, independent of time‐updated CVD, its risk factors, left ventricular ejection fraction, and traditional ECG measurements (heart rate, corrected QT interval (QTc), QRS duration, ventricular conduction abnormalities, and left ventricular hypertrophy).12 To understand the biology underlying GEH, we performed a genome‐wide meta‐analysis using data from 2 genome‐wide association studies (GWAS) of GEH.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure.24 Study‐specific genotype and phenotype data are available through the respective study cohort coordinating centers. GWAS results will be available through the CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology) Consortium Summary Results from Genomic Studies (dbGaP Study Accession: phs000930.v5.p1).
Participating Studies: ARIC and CHS
The ARIC (Atherosclerosis Risk in Communities) study is an ongoing, prospective cohort study assessing risk factors, progression, and outcomes of CVD in 15 792 community participants aged 45 to 64 years (45% male, 74% white).25 The CHS (Cardiovascular Health Study) is an ongoing, prospective cohort study assessing risk factors, progression, and outcomes of CVD in 5888 community participants aged >65 years (42% male, 85% white) recruited from 4 US communities.26 We excluded participants with reported race other than white or black, uninterpretable ECGs,12 arrhythmias (second or third degree atrioventricular block, atrial fibrillation [AF]), Wolff–Parkinson–White ECG phenotype, implanted pacemaker, QRS duration ≥120 ms, using class I and III antiarrhythmic medications or digoxin, history of coronary heart disease (CHD) or heart failure (HF) as previously defined,12 participants with extreme phenotype (> ±4 SD), and participants missing genotype data. The final ARIC study population included 2465 black participants and 8038 white participants. The final CHS population included 592 blacks and 2731 whites. Together the 2 cohorts included 13 826 adults (3057 blacks and 10 769 whites). Both studies were approved by the institutional review boards of all participating institutions, and all participants gave informed consent.
The Phenotype of Global Electrical Heterogeneity
GEH ECG measurements were calculated as previously described12 (Figure 1). SAI QRST was calculated as the arithmetic sum of areas under the QRS‐T curve.15, 17 Spatial mean QRS‐T angle was measured as the 3‐dimensional angle between the mean QRS‐vector and the mean T‐vector.9 The magnitude, azimuth, and elevation of the SVG vector were measured as previously defined.12
Figure 1.

Measurement of GEH. A, Spatial QRS‐T angle represents the angle between the QRS‐vector and T‐vector in three‐dimensional space. B, Spatial ventricular gradient (SVG) is a vector defined as the vector sum of the QRS‐vector and the T‐vector. SVG magnitude is the length of the SVG‐vector. SVG azimuth is the angle of the SVG‐vector projected onto the XY (horizontal) plane, and SVG elevation is the angle of the SVG‐vector projected in the XZ (vertical) plane. C, SAI QRST represents the sum of the area under the QRS complex and T‐wave using the isoelectric line as the reference (shaded area). X‐, Y‐, and Z‐lead QRS‐T complexes and their calculated SAI QRST results are shown.
GWAS Genotyping and Imputation
Both ARIC and CHS data were imputed to the 1000 Genomes Project Phase 1 reference panel (March 2012 release).27 Detailed methods are provided in Table S1. In the ARIC cohort 22 487 821 SNPs were available for analysis in whites, and 26 434 100 in blacks. For CHS white data, SNPs with variance in allele dosage less than or equal to 0.01 were excluded; SNPs with an effective number of minor alleles (2*N*MAF*imputation quality) less than or equal to 5 were excluded in CHS blacks. Thus, in CHS 9 117 794 SNPs were available for analysis in whites and 13 695 499 in blacks.
Statistical Methods
Within each cohort, and stratified by race, trait variables were transformed to make their distributions more normal, specifically using a square‐root transformation for all variables except SAI QRST for which a log‐transformation was used. Following transformation, the variables were then standardized to have a mean of 0 and a standard deviation of 1 (Z‐score). Each GEH z‐score was linearly regressed on allele dosage, adjusting for age, sex, height, body mass index (BMI), study site, and principal components to account for ancestry.
Meta‐analysis, both stratified by race and combined across all races, was performed using inverse variance weights as implemented in METAL,28 with adjustment for each study's genomic control correction factor, calculated from all analyzed SNPs.29 Associations were considered as genome‐wide significant at P<1×10−8.
Bioinformatics Analyses of the Loci
We used several bioinformatics approaches (LDlink,30 RegulomeDB,31 Genome‐Wide Annotation of Variants [GWAVA],32 and Data‐driven Expression Prioritized Integration for Complex Traits [DEPICT]33) to search and annotate SNPs in the regions containing genome‐wide significant SNPs. Publicly available reference haplotypes from Phase 3 (Version 5) of the 1000 Genomes Project (1000G)34 were used to calculate population‐specific measures of linkage disequilibrium (LD)30 in whites. To evaluate the potential functional effect of the identified loci, we identified all proxy SNPs in moderate to high LD (R 2≥0.5) to the index SNP within ±500‐kb regions. We used dbSNP's predicted30 functional effect of variants and considered SNPs with RegulomeDB31 score <4. RegulomeDB31 integrates the RoadMap Epigenomics and ENCODE projects to identify variants which have potential or demonstrated regulatory function, and predicts potential mechanisms of functional involvement. We used a GWAVA32 score to identify loci with likely functional non‐coding variants. We used an unmatched GWAVA score threshold ≥0.35, GWAVA transcription start site (TSS) score threshold ≥0.45, and region GWAVA score threshold ≥0.55 to mark potentially functional loci. In addition, we searched the National Human Genome Research Institute GWAS catalogue35 for the association of GEH SNPs and SNPs in high LD (R 2≥0.8) with other complex traits or diseases identified in previous GWAS.
Evidence of Expression in Heart Tissue
Expression quantitative trait loci (eQTLs) in the left ventricle (LV), and atrial appendage have been evaluated using the Genotype‐Tissue Expression (GTEx) project36 portal (analysis release V7; updated 09/05/2017; accessed 11/07/2017). All cis eQTLs, which have been precomputed in a ±1 Mb cis window around the TSS were included, if considered significant, as described below. Beta distribution‐adjusted empirical P‐values from FastQTL were used by the GTEx investigators to calculate q‐values, and a false discovery rate (FDR) threshold of ≤0.05 was applied to identify genes with a significant eQTL (“eGenes”). To identify the list of all significant variant‐gene pairs associated with eGenes, a genome‐wide empirical P‐value threshold, pt, was defined as the empirical P‐value of the gene closest to the 0.05 FDR threshold. pt was then used to calculate a nominal P‐value threshold for each gene based on the beta distribution model (from FastQTL) of the minimum P‐value distribution f(pmin) obtained from the permutations for the gene. Specifically, the nominal threshold was calculated as F−1(pt), where F−1 is the inverse cumulative distribution. For each gene, variants with a nominal P‐value below the gene‐level threshold were considered significant and included in the final list of variant‐gene pairs. A threshold of at least 70 samples per tissue was used, as it was determined to provide sufficient statistical power for eQTL discovery.
Evidence of Expression in Failing Hearts
HF is a well‐recognized substrate for SCD, which is characterized by differential gene expression and “fetal gene activation”.37 We, therefore, performed a look‐up for cis eQTL in 177 failing hearts in the Myocardial Applied Genomics Network (MAGNet) consortium database (http://www.med.upenn.edu/magnet/).38, 39, 40 The significance level was Bonferroni—adjusted for the number of tests performed.
Results
Meta‐Analysis of Genome‐Wide Association Results
We identified 10 loci (Figure 2) that showed genome‐wide significant association with GEH (Table 1). Top loci were associated with phenotype consistently in both ARIC and CHS studies (Figure 3). Four loci were associated with at ≥2 GEH traits: QRS‐T angle, SVG azimuth, and SVG magnitude, and SAI QRST (Figures 2 and 4). The loci associated with GEH are detailed in Figure 5, with the index SNP labeled for each independent signal. Pairwise correlations between the 5 GEH ECG variables and traditional ECG measurements ranged from negligible (r=0.007) to weak for all but one pair: SAI QRST‐SVG magnitude had moderate strength (r=0.7) correlation (Table 2). Most identified SNPs remained statistically significantly associated with GEH phenotypes after additional adjustment for traditional ECG metrics (QRS and QT duration, RR′ and PR intervals) (Table S2). GEH genome‐wide significant associations were largely consistent between whites and blacks (Table 1 and Figure 4). The total variance explained by the GEH‐associated SNPs was small, as expected: 1.6% for SVG magnitude; 1.48% for SAI QRST; 1.25% for spatial QRS‐T angle; 1.1% for SVG elevation, and 1.1% for SVG azimuth.
Figure 2.
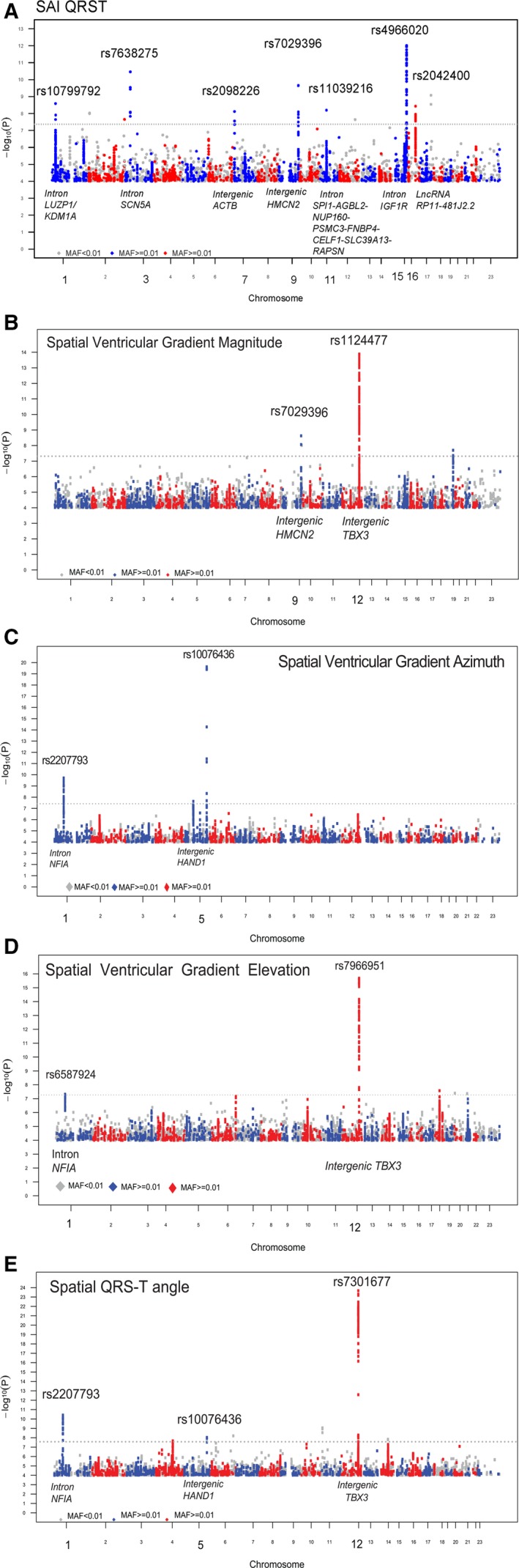
Manhattan plots of the GWAS meta‐analyses for (A) sum absolute QRS‐T integral (SAI QRST), (B) spatial ventricular gradient (SVG) magnitude, (C) SVG azimuth, (D) SVG elevation, and (E) spatial QRS‐T angle in individuals of both white and black ancestry. The dotted horizontal line represents the genome‐wide significance threshold.
Table 1.
Significant Loci at P<1×10−8 in GEH Phenotype Meta‐Analysis
| Locus | Chr | Closest Gene | SNP | Position | GEH Trait | E/O Allele | White Ancestry | Black Ancestry | Joint white/black Ethnicity | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EAF | β | se | P Value | EAF | β | se | P Value | EAF | β | se | P Value | |||||||
| 1 | 12 | TBX3 | rs7301677 | 115381147 | QRS‐T angle | T/C | 0.27 | +0.16 | 0.015 | 1.46E‐26a | 0.23 | +0.04 | 0.029 | 0.131 | 0.26 | +0.14 | 0.013 | 2.19e‐24a |
| IG + | 12 | TBX3 | rs7966951 | 115365211 | SVG elevation | A/G | 0.26 | +0.11 | 0.015 | 4.39E‐13a | 0.22 | +0.12 | 0.030 | 0.0001 | 0.26 | +0.11 | 0.014 | 2.07E‐16a |
| 12 | TBX3 | rs1124477 | 115349497 | SVG magnitude | T/C | 0.29 | −0.11 | 0.015 | 6.80e‐15a | 0.25 | −0.05 | 0.029 | 0.093 | 0.28 | −0.10 | 0.013 | 1.33E‐14a | |
| 2 IG | 5 | HAND1 | rs10076436 | 153871841 | SVG azimuth | C/G | 0.65 | +0.10 | 0.014 | 4.51e‐12a | 0.46 | +0.18 | 0.027 | 3.15e‐11a | 0.61 | +0.12 | 0.013 | 2.39e‐20a |
| ++ | 5 | HAND1 | rs13165478 | 153869040 | QRS‐T angle | A/G | 0.36 | −0.06 | 0.014 | 5.40e‐06a | 0.54 | −0.09 | 0.025 | 0.0003 | 0.40 | −0.07 | 0.012 | 9.36E‐09a |
| 3 | 1 | NFIA | rs2207793 | 61894653 | QRS‐T angle | T/C | 0.47 | −0.09 | 0.013 | 3.30E‐11a | 0.29 | −0.04 | 0.026 | 0.123 | 0.43 | −0.08 | 0.012 | 3.87E‐11a |
| IN | 1 | NFIA | rs2207793 | 61894653 | SVG azimuth | T/C | 0.47 | −0.08 | 0.014 | 1.77E‐08a | 0.29 | −0.08 | 0.028 | 0.003 | 0.43 | −0.08 | 0.012 | 1.94E‐10a |
| 4N | 9 | HMCN2 | rs7029396 | 133026250 | SAI QRST | C/G | 0.81 | −0.08 | 0.017 | 1.39e‐06 | 0.68 | −0.11 | 0.026 | 2.27e‐5 | 0.78 | −0.09 | 0.014 | 2.21E‐10a |
| IG; ++ | 9 | HMCN2 | rs7029396 | 133026250 | SVG magnitude | C/G | 0.81 | −0.08 | 0.018 | 6.61e‐06 | 0.68 | −0.12 | 0.029 | 5.17e‐5 | 0.78 | −0.09 | 0.015 | 2.40E‐09a |
| 5N, IN;++ | 15 | IGF1R | rs4966020 | 99284680 | SAI QRST | A/G | 0.64 | +0.08 | 0.013 | 1.29e‐10a | 0.38 | +0.08 | 0.025 | 0.002 | 0.58 | +0.08 | 0.011 | 9.69E‐13a |
| 6; IN | 3 | SCN5A | rs7638275 | 38665823 | SAI QRST | A/G | 0.02 | −0.30 | 0.047 | 8.71E‐11a | 0.006 | −0.25 | 0.178 | 0.169 | 0.02 | −0.30 | 0.045 | 3.49E‐11a |
| 7; IN; ++ | 1 | LUZP1‐KDM1A | rs10799792 | 23446441 | SAI QRST | T/G | 0.59 | +0.08 | 0.013 | 1.00E‐08a | 0.92 | +0.07 | 0.043 | 0.103 | 0.62 | +0.08 | 0.013 | 2.60E‐09a |
| 8; +; lncRNA | 16 | RP11‐481J2.2 | rs2042400 | 58464340 | SAI QRST | T/C | 0.39 | −0.07 | 0.013 | 7.32E‐09a | 0.45 | −0.04 | 0.023 | 0.083 | 0.40 | −0.07 | 0.011 | 3.70E‐09a |
| 9N; IN; ++ | 11 | SPI1‐AGBL2‐NUP160‐PSMC3‐FNBP4‐CELF1‐SLC39A13‐RAPSN | rs11039216 | 47406592 | SAI QRST | T/C | 0.49 | +0.06 | 0.013 | 1.71e‐06 | 0.46 | +0.08 | 0.024 | 0.0008 | 0.49 | +0.07 | 0.011 | 6.42E‐09a |
| 10N; IG; + | 7 | ACTB | rs2098226 | 5583045 | SAI QRST | A/T | 0.77 | +0.08 | 0.015 | 2.90e‐07 | 0.66 | +0.06 | 0.024 | 0.0071 | 0.74 | +0.07 | 0.013 | 7.67E‐09a |
+/++ indicates GWAVA score; Chr, chromosome; E/O allele, effect allele/other allele; EAF, effect allele frequency; GWAVA, Genome‐Wide Annotation of Variants; IG, intergenic variant; IN, intronic variant; lncRNA, long non‐coding RNA; N, novel loci; se, standard error of β; SAI QRST, sum absolute QRS‐T integral; SVG, spatial ventricular gradient; β, effect size.
Genome‐wide significant P‐values.
Figure 3.
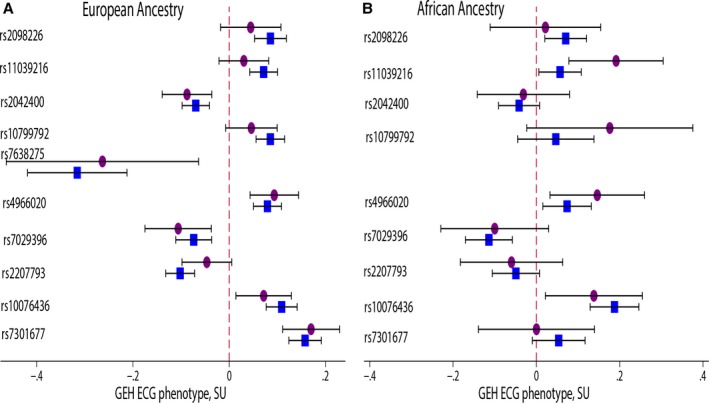
Comparison of the association of index SNPs markers with global electrical heterogeneity (GEH) ECG phenotypes in 10 loci in 2 cohorts: (A) ARIC and (B) CHS. Associations in ARIC are shown by a blue square; in CHS—by a purple circuit. SU, standardized units (per 1 standard deviation of ECG trait). ARIC indicates Atherosclerosis Risk in Communities Study; CHS, Cardiovascular Health Study.
Figure 4.

Forest plots for the association of index SNPs markers with 5 global electrical heterogeneity (GEH) ECG phenotypes in 10 loci. Associations for white (square), black (diamond) individuals, and joint ancestries (circuit) are shown. Red color indicates associations with SAI QRST; green color—with SVG magnitude; dark navy—with SCG azimuth; magenta color—with SVG elevation; blue color—with spatial QRS‐T angle. SU, standardized units (per 1 standard deviation of ECG trait). A, Locus 1. B, Locus 2. C, Locus 3. D, Locus 4. E, Locus 5. F, Locus 6. G, Locus 7. H, Locus 8. I, Locus 9. J, Locus 10.
Figure 5.
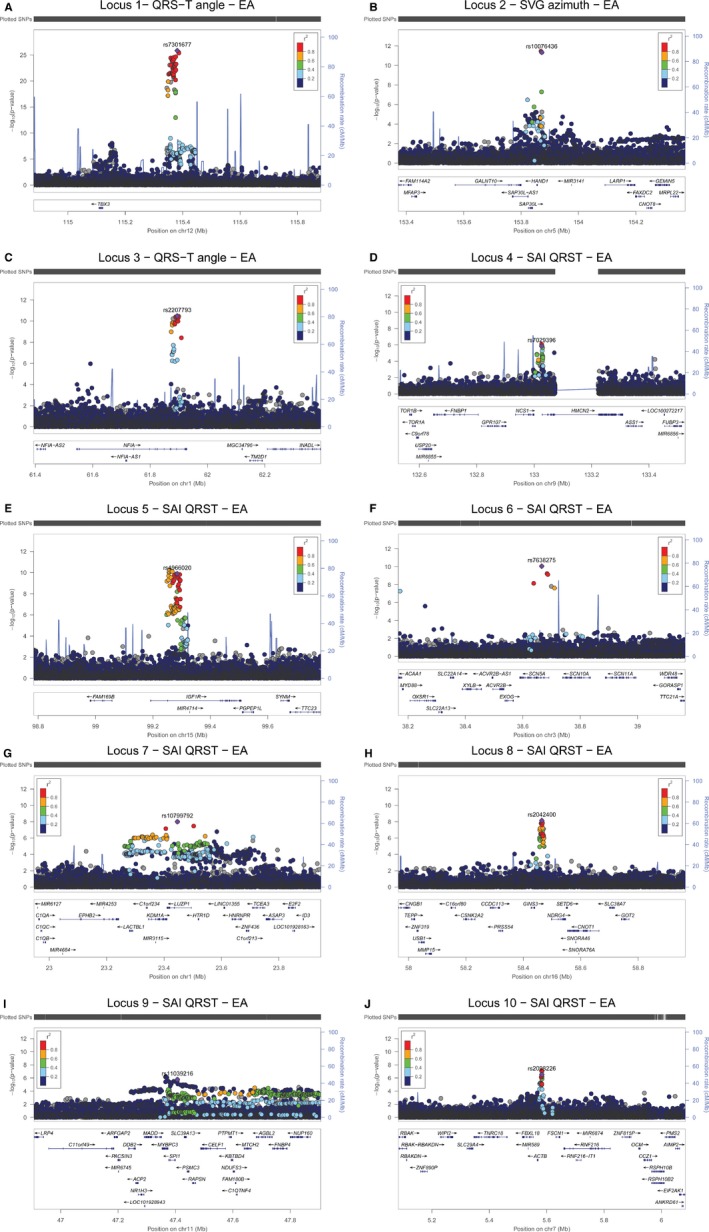
Regional association plots show association results at each significantly associated locus. Ten loci are displayed in the order listed in Table 1. Each SNP is plotted with respect to its chromosomal location ±500 kb (X‐axis) and its P‐value (Y‐axis on the left). Known gene transcripts are annotated at the bottom. The SNPs are colored according to their degree of linkage disequilibrium (r 2). The index SNP is shown as a purple diamond. The tall blue spikes indicate the recombination rate (Y‐axis on the right) at that region of the chromosome. A, Locus 1. B, Locus 2. C, Locus 3. D, Locus 4. E, Locus 5. F, Locus 6. G, Locus 7. H, Locus 8. I, Locus 9. J, Locus 10.
Table 2.
Pairwise Pearson Correlation Coefficients Between the Standardized GEH ECG Variables, and Standardized Traditional ECG Measurements
| QRS‐T Angle | SAI QRST | SVG Azimuth | SVG Elevation | SVG Magnitude | QT Duration | RR′ Interval | PR Interval | |
|---|---|---|---|---|---|---|---|---|
| SAI QRST | 0.243 | |||||||
| SVG azimuth | 0.405 | −0.016 | ||||||
| SVG elevation | 0.164 | −0.168 | −0.131 | |||||
| SVG magnitude | −0.249 | 0.696 | −0.281 | −0.205 | ||||
| QT duration | −0.059 | 0.227 | 0.048 | 0.0647 | 0.094 | |||
| RR′ interval | −0.040 | 0.320 | −0.013 | 0.042 | 0.155 | 0.855 | ||
| PR interval | 0.029 | 0.056 | −0.009 | 0.149 | 0.007 | 0.105 | 0.136 | |
| QRS duration | 0.258 | 0.394 | 0.179 | 0.0689 | 0.070 | 0.189 | 0.187 | 0.144 |
GEH indicates global electrical heterogeneity; SAI QRST, sum absolute QRS‐T integral; SVG, spatial ventricular gradient.
Potential Functional Impact of the GEH Variants
All detected SNPs were non‐coding. Therefore, we utilized eQTL databases and functional annotation tools that interpret non‐coding variants (Table S3). Several loci were significantly associated with gene expression in the heart. The fourth locus (near HMCN2) was associated with expression of HMCN2 (effect size 0.23; P‐value 1.7e‐5), and the fifth locus (IGF1R) was associated with expression of IGF1R (effect sie 0.24; P‐value 1.4e‐7) in LV. The IGF1R locus was also significantly associated with IGF1R gene expression in human failing hearts (adjusted P‐value 0.023). The eighth locus was associated with expression of a long non‐coding RNA RP11‐481J2.2 (effect size 0.24; P‐value 1.0e‐6) and NDRG4 (effect size 0.37; P‐value 1.1e‐9) in atrial appendage.
According to RegulomeDB score, all 10 loci demonstrated at least one SNP in LD >0.5 with an index SNP in the corresponding locus that had a functional potential (score <4). GWAVA annotation predicted functional variants in 8 out of 10 loci, as defined by either unmatched score (which has high accuracy with ROC AUC 0.97), or TSS score (ROC AUC 0.88). Five loci had both unmatched and TSS scores: second (near HAND1), fourth (near HMCN2), fifth (IGF1R), seventh (LUZP1‐KDM1A), and ninth (complex SPI1‐AGBL2‐NUP160‐PSMC3‐FNBP4‐CELF1‐SLC39A13‐RAPSN).
Several GEH loci have been previously reported to be associated with other phenotypes. The fifth locus (IGF1R) showed pleiotropy; it has been previously reported as having a genome‐wide significant association with fasting plasma glucose.41 Genetic variants in the first locus (near TBX3) have been previously shown to be associated with QRS duration42 and PR43 interval. Locus 2 (near HAND1) and locus 3 (NFIA) previously have been shown to be associated with QRS duration.42, 44, 45 Adjustment for QRS, PR, QT, and RR′ intervals (Table S2) strengthened association between the first locus (near TBX3) and GEH, suggesting that GEH is capturing an independent underlying electrophysiological phenomenon, as opposed to simply being a marker of QRS, PR, QT, or RR′ intervals. In contrast, the association between the second locus (near HAND1) and GEH was nearly fully explained by traditional ECG measurements. Attenuation of the association of GEH with the third locus (NFIA) after adjustment for QRS, PR, QT, and RR′ was negligible.
Diverse Biological Patterns of Global Electrical Heterogeneity
Nearly all the 10 identified loci had specific “electrical signatures”—a specific pattern of associations with the GEH ECG phenotype (Figure 4)—suggesting variable underlying biological and electrophysiological phenomena underlying each locus. Locus 1 (near TBX3) was characterized by the GEH phenotype of ventricular conduction abnormalities (widening QRS‐T angle, decreasing SVG magnitude, and rotation of the SVG vector backward and upward) as both SVG elevation and azimuth are increasing. Locus 2 (near HAND1) was characterized by rotation of SVG vector backward and downward (increasing SVG azimuth, but decreasing SVG elevation). Locus 3 (NFIA) was characterized by a “super‐normal” GEH pattern: narrowing of QRS‐T angle, rotation of SVG forward and downward, and increasing SVG magnitude. Locus 4 (near HMCN2) was characterized by rotation of SVG backward and decreasing SVG magnitude without QRS‐T angle widening. The fifth locus (IGF1R) was characterized by increased SVG magnitude despite QRS‐T angle widening, and rotation of SVG vector backward and downward. Patterns of the sixth (SCN5A) and eighth (lncRNA) loci were similar to each other (decreasing scalar values of SVG, without changes in SVG direction). However, the effect of adjustment for traditional ECG metrics was different for each locus (Table S2): adjustment strengthened the association with GEH in the eighth (lncRNA) locus but attenuated the association with GEH in the sixth (SCN5A) locus. Similar patterns were also demonstrated by locus 7 (LUZP1‐KDM1A), 9 (complex SPI1‐AGBL2‐NUP160‐PSMC3‐FNBP4‐CELF1‐SLC39A13‐ RAPSN), and 10 (near ACTB), which were manifest by increased scalar values of the SVG without significant changes in SVG direction. Adjustment for traditional ECG measurements strengthened the association between GEH and the ninth locus, but slightly attenuated associations in the seventh and tenth loci, suggesting differences in underlying biology.
Discussion
This meta‐analysis of GEH GWAS yielded 10 genetic loci that contained genome‐wide significant SNPs in white ancestry individuals, or in joint ancestry. Despite a smaller number of participants with black ancestry, consistent direction of the associations was also observed in individuals of black ancestry. The identified loci characterize different aspects of biology and electrophysiology and reflect the multifactorial nature of GEH.
The strongest association between GEH and locus 1 was mapped near the TBX3 gene, which plays a critical role in the development of the cardiac conduction system.46 TBX3 is expressed throughout the entire central cardiac conduction system (sinoatrial node, atrioventricular node, His bundle, and proximal bundle branches), in a pattern mutually exclusive to that of chamber‐specific genes (connexin 40, connexin 43, natriuretic peptide A gene).46 TBX3 represses chamber‐specific promoters which is crucial for normal development of the central conduction system. At late stages of fetal development connexin 40 expression is initiated in the His bundle and proximal bundle branches which are locations where TBX3 is expressed at relatively low levels. Reduced expression of TBX3 in proximal bundle branches is associated with greater expression of connexins 40 and 43, and accordingly, faster conduction,47 which is manifest by shorter QRS duration. TBX3 deficiency can also lead to insufficient development of AVN,46 which might manifest as prolonged PR interval. Indeed, supporting this connection between QRS and PR interval, the GWAS variant associated with increased QRS interval is also associated with decreased PR interval.42, 43 Importantly, in this locus, several variants were associated with GEH, but not with other ECG variables, suggesting different underlying biology, which should be studied further.
Locus 2 was mapped to the intronic region of HAND1. HAND1 is essential for cardiac development and postnatal structural remodeling. HAND1 affects the development of the ventricular outflow tract, and is responsible for cardiac left‐right asymmetry, which might be affected via epigenetic mechanisms. Local HAND1 expression level represents a switch between cardiomyocyte proliferation and differentiation. HAND1 also has a direct role in LV hypertrophy; it is a part of a metabolic pathway adapting the heart to hypoxia during development and adulthood, and is one of the “fetal” genes upregulated in failing cardiomyocytes. Re‐initiation of the fetal gene program instructs cell to increase their cytoplasmic mass, ie, permits the development of LV hypertrophy, which can lead to cardiac arrhythmias. Two putative HAND1 targets include T‐box transcription factor 5 (TBX5), participating in conduction system development, and the homebox factor IRX4, which has been implicated in the development of ventricular arrhythmias and SCD.
Locus 3 (NFIA) was previously reported to be associated with QRS duration in the Japan Pharmacogenomics Data Science Consortium (JPDSC) project, and the CHARGE QRS and eMERGE GWASs. Nuclear factor I‐A (NFIA) gene encodes a site‐specific DNA‐binding protein.
Locus 4 (near HMCN2) expresses HMCN2 (Hemicentin 2) in the LV. Hemicentin 2 belongs to the fibulin family of extracellular matrix proteins that play pivotal roles in fibroblast migration and development of interstitial fibrosis. HMCN2 is implicated in calcium ion binding and bicuspid aortic valve with concomitant ascending aortic dilation.
Locus 5 (insulin‐like growth factor 1 receptor, IGF1R) is a pleiotropic variant. IGF1R is a key step in growth hormone signaling and physiologic cardiomyocyte hypertrophy.48 Variation in expression of IGF1R in the LV could lead to variations in hypertrophy that in turn influences GEH.
Locus 6 (SCN5A) was mapped in the intron of SCN5A. SCN5A encodes a voltage‐gated sodium channel which is crucially important for cardiac action potential initiation and propagation. SCN5A mutations cause well‐known arrhythmic/SCD syndromes (Brugada and long QT‐3) manifested by life‐threatening arrhythmias. Locus 6 is independent (not in LD) from other previously reported variants of SCN5A and warrants further study.
Locus 7 (LUZP1‐KDM1A): Leucine zipper protein 1 LUZP1 encodes a protein that contains a leucine zipper motif. The exact function of the encoded protein is not known, but available data suggest that LUZP1 can be implicated in cardiac development, malformations, and cardiomyopathy development. Lysine‐specific demethylase‐1 (KDM1A) induces demethylation of histone H3 and, therefore, cause epigenetic modifications and alterations in gene transcription. Functional annotation of locus 7 (LUZP1‐KDM1A) suggests that it is an active histone modification site (active TSS, strong transcription in cardiac myocytes).
Functional annotation of locus 8 (lncRNA RP11‐481J2.2) and locus 9 (SPI1‐AGBL2‐NUP160‐PSMC3‐FNBP4‐CELF1‐SLC39A13‐RAPSN) suggest that both loci are likely functionally active. There are no previous reports of phenotype associations at GWAS significance for either loci. Locus 9 is a large cluster of genes at the 11p11.2 region of chromosome 11. The exact function of locus 9 is unknown. Neighboring common variations at the 11p11.2 region, encompassing MADD and SPI1, have been associated with fasting glucose and insulin in previous GWASs. Previously reported glycemic traits‐associated MADD loci are not in LD with locus 9 (R 2<0.2). Previously reported diastolic HF‐associated MADD loci are also not in LD with locus 9 (R 2<0.2).
Locus 8 is a long non‐coding RNA (lncRNA) RP11‐481J2.2. It is expressed in the human heart although its function is unknown. In addition, locus 8 contains eQTL for expression of NDRG4. While the exact function of NDRG4 in the heart is unknown, NDRG4 is responsible for sodium channel trafficking in the nervous system and has been associated with cardiomyopathy.
Genetic Determinants of Vectorcardiogram Morphology Phenotype
In this study, we discovered 4 novel ECG phenotype—associated genetic loci. Loci on chromosomes 4 (near HMCN2), 5 (IGF1R), 11 (11p11.2 region cluster), and 7 (near ACTB) have not been previously reported associated with any ECG phenotype. Future functional studies of these loci might discover novel molecular mechanisms and underlying biology of electrophysiological processes, affecting vectorcardiogram morphology.
We also identified several loci in proximity to previously reported variants, but not in LD with them, thus representing independent signals. Locus on chromosome 16 (lncRNA RP11‐481J2.2) is located between two previously reported loci, associated with QT interval and ST‐T wave amplitudes. Locus on chromosome 1 (LUZP1‐KDM1A) is located near loci, previously reported associated with QT interval, height, longevity, and salt‐sensitive hypertension. Locus on chromosome 3 (SCN5A) is located on a well‐known sodium channel gene. Further functional studies will help to shed important light on the biology of ECG.
Finally, 3 GEH‐associated loci (near TBX3, HAND1, and NFIA) are known, previously reported loci, associated with QRS duration and PR interval. Nevertheless, our findings helped to interpret previously puzzling discordant associations of these loci with QRS and PR intervals.
Impact on Research, Clinical Care, and Prevention
Improving our understanding of genetic variants related to variations in GEH may have important clinical implications. First, it improves our understanding of the mechanisms underlying GEH (eg, ventricular conduction abnormalities, left‐to‐right ventricular ion channels distribution asymmetry). Functional characterization of GEH‐associated genetic loci may uncover underlying pathophysiology of previously clinically unrecognized conditions which are manifest by significantly abnormal GEH and increased risk of SCD. Second, functional characterization of GEH‐associated loci may aid the discovery of targets for new drugs which could modulate electrical heterogeneity and, thus, potentially prevent ventricular tachyarrhythmias and SCD. The recent success of PCSK9‐inhibitors is an inspirational example of this novel therapeutic approach guided by genomic discovery. Development of safe and effective therapeutics to prevent SCD is urgently needed. Third, future pharmacogenomic studies of GEH may identify individuals who are most likely to benefit from (or be hurt by) specific pharmacological treatments. Limitations of QT interval as an assay for an assessment of pro‐arrhythmic compounds is well recognized. Many pharmaceutical compounds prolong QT interval without pro‐arrhythmic risk, while others manifest by an increased risk of ventricular tachyarrhythmia without significant QT prolongation. GEH is a measure of dispersion of depolarization and repolarization and therefore integrates underlying electrophysiological substrate more than traditional ECG measurements. Future studies of the effect of medications on GEH are needed. Finally, in the future, GEH‐associated loci may become clinically important for prediction of SCD and ventricular arrhythmia risk. As expected for common genetic variants, the effect size of the identified variants is small. However, combining several SNPs into a genetic risk score might potentially allow early identification of individuals at risk for ventricular conduction abnormalities or ventricular arrhythmias.
Limitations
Important limitations of the study should be noted. Replication of the GEH GWAS in independent cohorts is needed. A GWAS with larger sample size is also needed as other studies become available in the future, especially for stratified analysis in different ancestries and ethnicities, and for comparison of males and females. While our study included both white and black ancestries, our results may not be generalizable to other ancestries. It is also important to emphasize that we have identified only markers of genetic regions associated with GEH ECG phenotype. The causal genes remain unknown. Targeted mapping of identified loci and functional analyses are needed to elucidate the underlying biology.
In summary, this genome‐wide meta‐analysis of >10 000 white and 3000 black individuals with GEH ECG phenotypes elucidated plausible biological mechanisms behind GEH, which was recently shown to be associated with SCD in the general population.12 Further studies of the underlying biology of GEH‐associated loci will help to uncover mechanisms of arrhythmogenic remodeling in the heart in response to traditional cardiovascular risk factors.
Sources of Funding
The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute (NHLBI), National Institutes of Health (NIH), Department of Health and Human Services, under Contract nos. (HHSN268201700001I, HHSN268201700003I, HHSN268201700005I, HHSN268201700004I, HHSN2682017000021); National Human Genome Research Institute contract U01HG004402; and NIH contract HHSN268200625226C. Infrastructure was partly supported by UL1RR025005, a component of the NIH and NIH Roadmap for Medical Research. This CHS research was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, HHSN268200960009C; and NHLBI grants U01HL080295, R01HL087652, R01HL105756, R01HL103612, R01HL120393, R01HL130114, and R01HL085251 with additional contribution from the National Institute of Neurological Disorders and Stroke. Additional support was provided through R01AG023629 from the National Institute on Aging. A full list of principal CHS investigators and institutions can be found at CHS‐NHLBI.org. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR000124, and the National Institute of Diabetes and Digestive and Kidney Diseases Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The Genotype‐Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health. This work was supported by 1R01HL118277 (Tereshchenko), R01 HL111089 (Sotoodehnia), R01 HL116747 (Sotoodehnia and Arking) and the Laughlin Family.
Disclosures
None.
Supporting information
Table S1. Genotyping and Imputation of the Genetic Data
Table S2. Adjustment for QRS, PR, QT, and RR′ Intervals
Table S3. Functional Characteristics of GEH‐Associated SNPs
Acknowledgments
The authors thank the staff and participants of the ARIC study and CHS for their important contributions.
(J Am Heart Assoc. 2018;7:e008160 DOI: 10.1161/JAHA.117.008160.)29622589
Contributor Information
Larisa G. Tereshchenko, Email: tereshch@ohsu.edu.
Dan E. Arking, Email: arking@jhmi.edu.
References
- 1. Wilson FN, Macleod AG, Barker PS, Johnston FD. The determination and the significance of the areas of the ventricular deflections of the electrocardiogram. Am Heart J. 1934;10:46–61. [Google Scholar]
- 2. Burch GE, Winsor T. A Primer of Electrocardiography. Philadelphia: Lea & Febiger; 1945. [Google Scholar]
- 3. Burger HC. A theoretical elucidation of the notion ventricular gradient. Am Heart J. 1957;53:240–246. [DOI] [PubMed] [Google Scholar]
- 4. Wilson FN, MacLeod AG, Barker PS. The T deflection of the electrocardiogram. Trans Assoc Am Physicians. 1931;46:29–38. [Google Scholar]
- 5. Vassallo JA, Cassidy DM, Kindwall KE, Marchlinski FE, Josephson ME. Nonuniform recovery of excitability in the left ventricle. Circulation. 1988;78:1365–1372. [DOI] [PubMed] [Google Scholar]
- 6. Plonsey R. A contemporary view of the ventricular gradient of Wilson. J Electrocardiol. 1979;12:337–341. [DOI] [PubMed] [Google Scholar]
- 7. Geselowitz DB. The ventricular gradient revisited: relation to the area under the action potential. IEEE Trans Biomed Eng. 1983;30:76–77. [DOI] [PubMed] [Google Scholar]
- 8. Abildskov JA, Burgess MJ, Millar K, Roland W. New data and concepts concerning the ventricular gradient. Chest. 1970;58:244–248. [DOI] [PubMed] [Google Scholar]
- 9. Oehler A, Feldman T, Henrikson CA, Tereshchenko LG. QRS‐T angle: a review. Ann Noninvasive Electrocardiol. 2014;19:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borleffs CJ, Scherptong RW, Man SC, van Welsenes GH, Bax JJ, van Erven L, Swenne CA, Schalij MJ. Predicting ventricular arrhythmias in patients with ischemic heart disease: clinical application of the ECG‐derived QRS‐T angle. Circ Arrhythm Electrophysiol. 2009;2:548–554. [DOI] [PubMed] [Google Scholar]
- 11. Kors JA, Kardys I, van der Meer IM, van Herpen G, Hofman A, van der Kuip DA, Witteman JC. Spatial QRS‐T angle as a risk indicator of cardiac death in an elderly population. J Electrocardiol. 2003;36(suppl):113–114. [DOI] [PubMed] [Google Scholar]
- 12. Waks JW, Sitlani CM, Soliman EZ, Kabir M, Ghafoori E, Biggs ML, Henrikson CA, Sotoodehnia N, Biering‐Sorensen T, Agarwal SK, Siscovick DS, Post WS, Solomon SD, Buxton AE, Josephson ME, Tereshchenko LG. Global electric heterogeneity risk score for prediction of sudden cardiac death in the general population: the Atherosclerosis Risk in Communities (ARIC) and Cardiovascular Health (CHS) Studies. Circulation. 2016;133:2222–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Bie MK, Koopman MG, Gaasbeek A, Dekker FW, Maan AC, Swenne CA, Scherptong RW, van Dessel PF, Wilde AA, Schalij MJ, Rabelink TJ, Jukema JW. Incremental prognostic value of an abnormal baseline spatial QRS‐T angle in chronic dialysis patients. Europace. 2013;15:290–296. [DOI] [PubMed] [Google Scholar]
- 14. Tereshchenko LG, Kim ED, Oehler A, Meoni LA, Ghafoori E, Rami T, Maly M, Kabir M, Hawkins L, Tomaselli GF, Lima JA, Jaar BG, Sozio SM, Estrella M, Kao WH, Parekh RS. Electrophysiologic substrate and risk of mortality in incident hemodialysis. J Am Soc Nephrol. 2016;27:3413–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tereshchenko LG, Cheng A, Fetics BJ, Butcher B, Marine JE, Spragg DD, Sinha S, Dalal D, Calkins H, Tomaselli GF, Berger RD. A new electrocardiogram marker to identify patients at low risk for ventricular tachyarrhythmias: sum magnitude of the absolute QRST integral. J Electrocardiol. 2011;44:208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tereshchenko LG, McNitt S, Han L, Berger RD, Zareba W. ECG marker of adverse electrical remodeling post‐myocardial infarction predicts outcomes in MADIT II study. PLoS One. 2012;7:e51812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sur S, Han L, Tereshchenko LG. Comparison of sum absolute QRST integral, and temporal variability in depolarization and repolarization, measured by dynamic vectorcardiography approach, in healthy men and women. PLoS One. 2013;8:e57175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kozmann G, Tuboly G, Szathmáry V, Švehlíková J, Tyšler M. Computer modelling of beat‐to‐beat repolarization heterogeneity in human cardiac ventricles. Biomed Signal Process Control. 2014;14:285–290. [Google Scholar]
- 19. Tereshchenko LG, Cheng A, Fetics BJ, Marine JE, Spragg DD, Sinha S, Calkins H, Tomaselli GF, Berger RD. Ventricular arrhythmia is predicted by sum absolute QRST integralbut not by QRS width. J Electrocardiol. 2010;43:548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tereshchenko LG, Ghafoori E, Kabir MM, Kowalsky M. Electrical dyssynchrony on noninvasive electrocardiographic mapping correlates with SAI QRST on surface ECG. Comput Cardiol. 2015;42:69–72. [PMC free article] [PubMed] [Google Scholar]
- 21. Tereshchenko LG, Cheng A, Park J, Wold N, Meyer TE, Gold MR, Mittal S, Singh J, Stein KM, Ellenbogen KA; Investigators S‐AT . Novel measure of electrical dyssynchrony predicts response in cardiac resynchronization therapy: results from the SMART‐AV Trial. Heart Rhythm. 2015;12:2402–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jacobsson J, Borgquist R, Reitan C, Ghafoori E, Chatterjee NA, Kabir M, Platonov PG, Carlson J, Singh JP, Tereshchenko LG. Usefulness of the sum absolute QRST integral to predict outcomes in patients receiving cardiac resynchronization therapy. Am J Cardiol. 2016;118:389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tereshchenko LG, Feeny A, Shelton E, Metkus T, Stolbach A, Mavunga E, Putman S, Korley FK. Dynamic changes in high‐sensitivity cardiac troponin I are associated with dynamic changes in sum absolute QRST integral on surface electrocardiogram in acute decompensated heart failure. Ann Noninvasive Electrocardiol. 2017;22:e12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology) . Consortium Summary Results from Genomic Studies. dbGaP study accession: Phs000930.V5.P1. dbGaP. 2018;5.
- 25. The ARIC Investigators . The Atherosclerosis Risk in Community (ARIC) study: design and objectives. Am J Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 26. Mittelmark MB, Psaty BM, Rautaharju PM, Fried LP, Borhani NO, Tracy RP, Gardin JM, O'Leary DH. Prevalence of cardiovascular diseases among older adults. The Cardiovascular Health Study. Am J Epidemiol. 1993;137:311–317. [DOI] [PubMed] [Google Scholar]
- 27. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Willer CJ, Li Y, Abecasis GR. Metal: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Devlin B, Roeder K, Wasserman L. Genomic control, a new approach to genetic‐based association studies. Theor Popul Biol. 2001;60:155–166. [DOI] [PubMed] [Google Scholar]
- 30. Machiela MJ, Chanock SJ. LDlink: a web‐based application for exploring population‐specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ritchie GR, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11:294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pers TH, Karjalainen JM, Chan Y, Westra HJ, Wood AR, Yang J, Lui JC, Vedantam S, Gustafsson S, Esko T, Frayling T, Speliotes EK; Genetic Investigation of ATC , Boehnke M, Raychaudhuri S, Fehrmann RS, Hirschhorn JN, Franke L. Biological interpretation of genome‐wide association studies using predicted gene functions. Nat Commun. 2015;6:5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP‐trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H, Syron J, Fleming J, Siminoff L, Traino H, Mosavel M, Barker L, Jewell S, Rohrer D, Maxim D, Filkins D, Harbach P, Cortadillo E, Berghuis B, Turner L, Hudson E, Feenstra K, Sobin L, Robb J, Branton P, Korzeniewski G, Shive C, Tabor D, Qi L, Groch K, Nampally S, Buia S, Zimmerman A, Smith A, Burges R, Robinson K, Valentino K, Bradbury D, Cosentino M, Diaz‐Mayoral N, Kennedy M, Engel T, Williams P, Erickson K, Ardlie K, Winckler W, Getz G, DeLuca D, MacArthur D, Kellis M, Thomson A, Young T, Gelfand E, Donovan M, Meng Y, Grant G, Mash D, Marcus Y, Basile M, Liu J, Zhu J, Tu Z, Cox NJ, Nicolae DL, Gamazon ER, Im HK, Konkashbaev A, Pritchard J, Stevens M, Flutre T, Wen X, Dermitzakis ET, Lappalainen T, Guigo R, Monlong J, Sammeth M, Koller D, Battle A, Mostafavi S, McCarthy M, Rivas M, Maller J, Rusyn I, Nobel A, Wright F, Shabalin A, Feolo M, Sharopova N, Sturcke A, Paschal J, Anderson JM, Wilder EL, Derr LK, Green ED, Struewing JP, Temple G, Volpi S, Boyer JT, Thomson EJ, Guyer MS, Ng C, Abdallah A, Colantuoni D, Insel TR, Koester SE, Little AR, Bender PK, Lehner T, Yao Y, Compton CC, Vaught JB, Sawyer S, Lockhart NC, Demchok J, Moore HF. The genotype‐tissue expression (GTEx) project. Nat Genet. 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cappola TP. Molecular remodeling in human heart failure. J Am Coll Cardiol. 2008;51:137–138. [DOI] [PubMed] [Google Scholar]
- 38. Liu Y, Morley M, Brandimarto J, Hannenhalli S, Hu Y, Ashley EA, Tang WH, Moravec CS, Margulies KB, Cappola TP, Li M. RNA‐Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 2015;105:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith JG, Felix JF, Morrison AC, Kalogeropoulos A, Trompet S, Wilk JB, Gidlöf O, Wang X, Morley M, Mendelson M, Joehanes R, Ligthart S, Shan X, Bis JC, Wang YA, Sjögren M, Ngwa J, Brandimarto J, Stott DJ, Aguilar D, Rice KM, Sesso HD, Demissie S, Buckley BM, Taylor KD, Ford I, Yao C, Liu C; Consortium C‐S, EchoGen C, consortium Q‐I, Consortium C‐Q , Sotoodehnia N, van der Harst P, Stricker BHC, Kritchevsky SB, Liu Y, Gaziano JM, Hofman A, Moravec CS, Uitterlinden AG, Kellis M, van Meurs JB, Margulies KB, Dehghan A, Levy D, Olde B, Psaty BM, Cupples LA, Jukema JW, Djousse L, Franco OH, Boerwinkle E, Boyer LA, Newton‐Cheh C, Butler J, Vasan RS, Cappola TP, Smith NL. Discovery of genetic variation on chromosome 5q22 associated with mortality in heart failure. PLoS Genet. 2016;12:e1006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wild PS, Felix JF, Schillert A, Teumer A, Chen MH, Leening MJG, Volker U, Grossmann V, Brody JA, Irvin MR, Shah SJ, Pramana S, Lieb W, Schmidt R, Stanton AV, Malzahn D, Smith AV, Sundstrom J, Minelli C, Ruggiero D, Lyytikainen LP, Tiller D, Smith JG, Monnereau C, Di Tullio MR, Musani SK, Morrison AC, Pers TH, Morley M, Kleber ME, Aragam J, Benjamin EJ, Bis JC, Bisping E, Broeckel U, Cheng S, Deckers JW, Del Greco MF, Edelmann F, Fornage M, Franke L, Friedrich N, Harris TB, Hofer E, Hofman A, Huang J, Hughes AD, Kahonen M; Investigators K , Kruppa J, Lackner KJ, Lannfelt L, Laskowski R, Launer LJ, Leosdottir M, Lin H, Lindgren CM, Loley C, MacRae CA, Mascalzoni D, Mayet J, Medenwald D, Morris AP, Muller C, Muller‐Nurasyid M, Nappo S, Nilsson PM, Nuding S, Nutile T, Peters A, Pfeufer A, Pietzner D, Pramstaller PP, Raitakari OT, Rice KM, Rivadeneira F, Rotter JI, Ruohonen ST, Sacco RL, Samdarshi TE, Schmidt H, Sharp ASP, Shields DC, Sorice R, Sotoodehnia N, Stricker BH, Surendran P, Thom S, Toglhofer AM, Uitterlinden AG, Wachter R, Volzke H, Ziegler A, Munzel T, Marz W, Cappola TP, Hirschhorn JN, Mitchell GF, Smith NL, Fox ER, Dueker ND, Jaddoe VWV, Melander O, Russ M, Lehtimaki T, Ciullo M, Hicks AA, Lind L, Gudnason V, Pieske B, Barron AJ, Zweiker R, Schunkert H, Ingelsson E, Liu K, Arnett DK, Psaty BM, Blankenberg S, Larson MG, Felix SB, Franco OH, Zeller T, Vasan RS, Dorr M. Large‐scale genome‐wide analysis identifies genetic variants associated with cardiac structure and function. J Clin Invest. 2017;127:1798–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hwang J‐Y, Sim X, Wu Y, Liang J, Tabara Y, Hu C, Hara K, Tam CHT, Cai Q, Zhao Q, Jee S, Takeuchi F, Go MJ, Ong RTH, Ohkubo T, Kim YJ, Zhang R, Yamauchi T, So WY, Long J, Gu D, Lee NR, Kim S, Katsuya T, Oh JH, Liu J, Umemura S, Kim Y‐J, Jiang F, Maeda S, Chan JCN, Lu W, Hixson JE, Adair LS, Jung KJ, Nabika T, Bae J‐B, Lee MH, Seielstad M, Young TL, Teo YY, Kita Y, Takashima N, Osawa H, Lee S‐H, Shin M‐H, Shin DH, Choi BY, Shi J, Gao Y‐T, Xiang Y‐B, Zheng W, Kato N, Yoon M, He J, Shu XO, Ma RCW, Kadowaki T, Jia W, Miki T, Qi L, Tai ES, Mohlke KL, Han B‐G, Cho YS, Kim B‐J. Genome‐wide association meta‐analysis identifies novel variants associated with fasting plasma glucose in East Asians. Diabetes. 2015;64:291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sotoodehnia N, Isaacs A, de Bakker PI, Dorr M, Newton‐Cheh C, Nolte IM, van der Harst P, Muller M, Eijgelsheim M, Alonso A, Hicks AA, Padmanabhan S, Hayward C, Smith AV, Polasek O, Giovannone S, Fu J, Magnani JW, Marciante KD, Pfeufer A, Gharib SA, Teumer A, Li M, Bis JC, Rivadeneira F, Aspelund T, Kottgen A, Johnson T, Rice K, Sie MP, Wang YA, Klopp N, Fuchsberger C, Wild SH, Mateo Leach I, Estrada K, Volker U, Wright AF, Asselbergs FW, Qu J, Chakravarti A, Sinner MF, Kors JA, Petersmann A, Harris TB, Soliman EZ, Munroe PB, Psaty BM, Oostra BA, Cupples LA, Perz S, de Boer RA, Uitterlinden AG, Volzke H, Spector TD, Liu FY, Boerwinkle E, Dominiczak AF, Rotter JI, van Herpen G, Levy D, Wichmann HE, van Gilst WH, Witteman JC, Kroemer HK, Kao WH, Heckbert SR, Meitinger T, Hofman A, Campbell H, Folsom AR, van Veldhuisen DJ, Schwienbacher C, O'Donnell CJ, Volpato CB, Caulfield MJ, Connell JM, Launer L, Lu X, Franke L, Fehrmann RS, te Meerman MG, Groen HJ, Weersma RK, van den Berg LH, Wijmenga C, Ophoff RA, Navis G, Rudan I, Snieder H, Wilson JF, Pramstaller PP, Siscovick DS, Wang TJ, Gudnason V, van Duijn CM, Felix SB, Fishman GI, Jamshidi Y, Stricker BH, Samani NJ, Kaab S, Arking DE. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Muller M, Sotoodehnia N, Sinner MF, Verwoert GC, Li M, Kao WH, Kottgen A, Coresh J, Bis JC, Psaty BM, Rice K, Rotter JI, Rivadeneira F, Hofman A, Kors JA, Stricker BH, Uitterlinden AG, van Duijn CM, Beckmann BM, Sauter W, Gieger C, Lubitz SA, Newton‐Cheh C, Wang TJ, Magnani JW, Schnabel RB, Chung MK, Barnard J, Smith JD, Van Wagoner DR, Vasan RS, Aspelund T, Eiriksdottir G, Harris TB, Launer LJ, Najjar SS, Lakatta E, Schlessinger D, Uda M, Abecasis GR, Muller‐Myhsok B, Ehret GB, Boerwinkle E, Chakravarti A, Soliman EZ, Lunetta KL, Perz S, Wichmann HE, Meitinger T, Levy D, Gudnason V, Ellinor PT, Sanna S, Kaab S, Witteman JC, Alonso A, Benjamin EJ, Heckbert SR. Genome‐wide association study of PR interval. Nat Genet. 2010;42:153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ritchie MD, Denny JC, Zuvich RL, Crawford DC, Schildcrout JS, Bastarache L, Ramirez AH, Mosley JD, Pulley JM, Basford MA, Bradford Y, Rasmussen LV, Pathak J, Chute CG, Kullo IJ, McCarty CA, Chisholm RL, Kho AN, Carlson CS, Larson EB, Jarvik GP, Sotoodehnia N, Manolio TA, Li R, Masys DR, Haines JL, Roden DM. Genome‐ and phenome‐wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127:1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sano M, Kamitsuji S, Kamatani N, Hong KW, Han BG, Kim Y, Kim JW, Aizawa Y, Fukuda K; Japan Pharmacogenomics Data Science C . Genome‐wide association study of electrocardiographic parameters identifies a new association for PR interval and confirms previously reported associations. Hum Mol Genet. 2014;23:6668–6676. [DOI] [PubMed] [Google Scholar]
- 46. Hoogaars WMH, Tessari A, Moorman AFM, de Boer PAJ, Hagoort J, Soufan AT, Campione M, Christoffels VM. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc Res. 2004;62:489–499. [DOI] [PubMed] [Google Scholar]
- 47. Rentschler S, Vaidya DM, Tamaddon H, Degenhardt K, Sassoon D, Morley GE, Jalife J, Fishman GI. Visualization and functional characterization of the developing murine cardiac conduction system. Development. 2001;128:1785–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weeks KL, Bernardo BC, Ooi JYY, Patterson NL, McMullen JR. The IGF1‐PI3K‐Akt signaling pathway in mediating exercise‐induced cardiac hypertrophy and protection. Adv Exp Med Biol. 2017;1000:187–210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genotyping and Imputation of the Genetic Data
Table S2. Adjustment for QRS, PR, QT, and RR′ Intervals
Table S3. Functional Characteristics of GEH‐Associated SNPs