

Abstract
Using a high-throughput, cell-based HCV luciferase reporter assay to screen a diverse small-molecule compound collection (~300 000 compounds), we identified a benzofuran compound class of HCV inhibitors. The optimization of the benzofuran scaffold led to the identification of several exemplars with potent inhibition (EC50 < 100 nM) of HCV, low cytotoxicity (CC50 > 25 µM), and excellent selectivity (selective index = CC50/EC50, > 371-fold). The structure–activity studies culminated in the design and synthesis of a 45-compound library to comprehensively explore the anti-HCV activity. The identification, design, synthesis, and biological characterization for this benzofuran series is discussed.
Keywords: hepatitis C, HCV inhibitor, benzofuran, antiviral, HCV replication
Graphical abstract
INTRODUCTION
The hepatitis C virus (HCV) is the causative agent of hepatitis C and represents the major underlying cause of hepatocellular carcinoma and chronic liver disease. Infection with HCV leads to chronic hepatitis in 85% of cases. HCV remains a significant public health burden and is a leading cause of liver transplantations and is responsible for at least 10 000 deaths annually in the US.1 Globally, an estimated 180 million people are chronically infected by HCV, with an additional 3–4 million people being newly infected annually.2 There is currently no vaccine to prevent HCV infection.3 Effective broad-spectrum therapy against different HCV genotypes is currently unavailable.4 The previous standard of care, peginterferon (pegIFN) and ribavirin (RBV) for 24–48 weeks, depending on HCV genotype, showed low sustained virological response (SVR) rates for patients infected with genotype 1, the most prevalent genotype in the US. This treatment is associated with severe adverse effects, including depression, myalgia, and hemolytic anemia. Recent research efforts have been directed toward the development of interferon-sparing regimens that contain one or more direct-acting antivirals (DAAs) that target one or more discrete biological targets for HCV (Figure 1). Prior clinical trials with DAAs as monotherapy were associated with rapid development of resistance, thereby necessitating their use as combinations.5 Two combinations of first-generation DAA, pegIFN, and ribavirin approved in 2011 have launched the era of DAA combination therapy for HCV.4 Current efforts toward interferon-free regimens involve combination therapy of two or three DAAs. Several such regimens that are associated with high treatment response (~90% sustained virologic response) have been approved over the past 2 years.6 These regimens, while very effective and more tolerable, are associated with high costs. Despite therapeutic advances for orally active DAAs, the high cost, potential for drug-resistance emergence, and drug–drug interactions suggest that a significant unmet need for additional therapies that act on novel HCV targets remains.
Figure 1.

Structures of representative HCV direct-acting antivirals.
RESULTS AND DISCUSSION
Hit Compound Identification and Initial SAR Screening
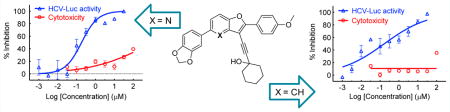
Hu and co-workers7 have developed a quantitative high-throughput screening (qHTS) assay platform representing the complete HCV replication cycle. The phenotypic assay incorporated two components: a recombinant infectious virus (HCVcc-Cre virus) and a stable host cell line (Huh7.5.1 LoxPGluc). This assay platform can identify inhibitors of all stages of the HCV replication cycle as well as compounds that interrupt host functions important for HCV infection and propagation.7 The NIH Molecular Libraries Small Molecule Repository (MLSMR) library of 339 561 compounds was screened with this assay platform, affording 11 624 preliminary hit compounds (PubChem AID 651820). Elimination of known reactive groups and focusing on hits with a minimum of 50% inhibition reduced the number to 655 promising hits. Concentration– response confirmatory assays (PubChem AID 720575) and cytotoxicity countersceening (PubChem AID 720576) further narrowed the field to a set of 206 validated compound hits. The general structure 1 was shared by a structural cluster of several hit compounds, including two benzofuran-based compounds (1a and 1b) displaying low-micromolar inhibitory activities in the HCV-Luc assay (recombinant HCV carrying the Renilla luciferase reporter, Figure 2). The hit compounds were originally synthesized and submitted to the MLSMR by Larock and co-workers8 as part of library development efforts within the KU Chemical Methodologies and Library Development Center (KU CMLD). Common functional groups between the two compounds include an acetylene-linked aliphatic alcohol system at the C3-position, aryl substitution at the C2-position, and an aryl/heteroaryl system at the C5-position of the benzofuran scaffold.
Figure 2.

Confirmed qHTS hit compounds from the benzofuran series.
We decided to further explore this chemotype because substituted benzofurans, such as nesbuvir (HCV-796, Figure 1), display potent HCV NS5B polymerase inhibitory activity and have advanced to clinical trials.9 Thus, the structure–activity relationship (SAR) around the benzofuran scaffold was explored through a two-pronged approach. The biological activity of existing benzofuran exemplars from the KU CMLD Center compound collection was surveyed first, providing a rapid survey of diverse chemical space. Next, a focused compound library was constructed on the basis of the results from the first step to systematically explore the SAR around the most promising chemical space. Figure 3 summarizes the analogs explored via this strategy; all three pendant groups on the benzofuran were investigated as well as nitrogen atom incorporation into the benzofuran centroid.
Figure 3.

Summary of explored modifications around 1a (PubChem compound CID 24747883).
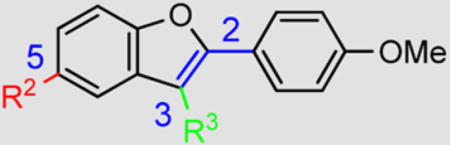
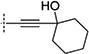
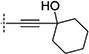
In the initial phase, 35 2,3,5-trisubstituted benzofuran compounds were selected from the KU CMLD Center compound collection and screened in the HCV-Luc assay in parallel with the ATPlite cytotoxicity assay.7 The results are summarized in Tables 1–3. The activity for our initial hit molecules 1a and 1b were independently validated (2{1} and 2{6}, respectively) with material directly obtained from the KU CMLD compound repository, showing potency and toxicity results that were comparable to the data from the qHTS (low-throughput assay data shown throughout the manuscript for consistency, qHTS data available online PubChem AIDs 651820 and 720575). Five analogs (2{2}–2{5} and 3{4}) displayed improved potency (EC50 ≤ 980 nM) compared with 2{1}. Gratifyingly, the five analogs that displayed improved potency also maintained a favorable selectivity index or, in some cases, possessed reduced cytotoxicity (CC50) against Huh7.5.1 cells (Table 1). All five analogs contain a propargyl alcohol moiety at the C3-position, suggesting that this group may be important for activity.
Table 1.
HCV Inhibition and Cytotoxicity of 2-(4-Methoxyphenyl)-Containing Benzofuran Analogs
| Cmpd | Structure | HCV-Luc EC50 (µM) |
ATPlite CC50 (µM) |
Selective Index (SI) |
|
|---|---|---|---|---|---|
| |||||
| R2 | R3 | ||||
| 2{1} |
|
|
0.98 (n=2) | 70.9 | 72 |
| 2{2} |
|
|
0.074 | 26.1 | 373 |
| 2{3} |
|
|
0.17 (n=3) | > 100 | > 588 |
| 2{4} |
|
|
0.52 | > 100 | > 192 |
| 2{5} |
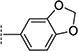
|
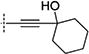
|
0.084 (n=3) | > 100 | > 1190 |
| 2{6} |
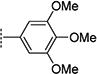
|
|
10.9 (n=3) | > 100 | > 9 |
| 2{7} |
|
|
> 31.6 | > 100 | N.A. |
| 2{8} |
|
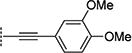
|
6.99 | > 100 | > 14 |
| 2{9} |
|
|
21.50 | > 100 | > 5 |
| 2{10} |
|
|
> 31.6 | > 100 | N.A. |
| 2{11} |
|
|
> 31.6 | > 100 | N.A. |
| 2{12} |
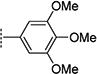
|
|
5.94 | > 100 | >17 |
| 2{13} |
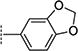
|
|
> 31.6 | > 100 | N.A. |
| 2{14} |
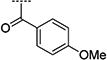
|
|
5.70 | 31.90 | 6 |
| 2{15} |
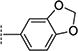
|
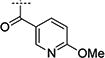
|
> 31.6 | > 100 | N.A. |
| 2{16} |
|
|
3.04 | 31.70 | 10 |
| 2{17} |
|
|
4.39 | 39.20 | 9 |
| 2{18} |
|
|
> 31.6 | > 100 | N.A. |
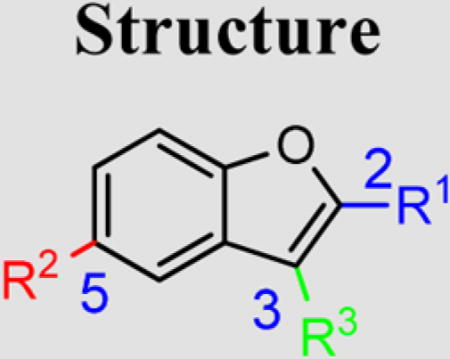
Table 3.
HCV Inhibition and Cytotoxicity of 2-Aryl-Containing Benzofuran Analogs
| Cmpd |
|
HCV-Luc EC50 (µM) |
ATPlite CC50 (µM) |
Selective Index (SI) |
||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | ||||
| 4{1} |
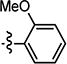
|
|
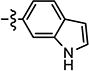
|
>31.6 | >100 | N.A. |
| 4{2} |
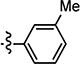
|
|
|
12.00 | >100 | >8 |
| 4{3} |
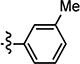
|
|
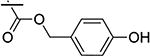
|
3.59 | 38.10 | 11 |
| 4{4} |
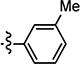
|
|
|
2.34 | >100 | >43 |
| 4{5} |
|
|
|
1.84 | >100 | >54 |
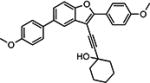
Compounds in Table 1 contain a 4-methoxyphenyl group at the C2-position of the benzofuran scaffold, and the substituents vary at its C3- and C5-positions. Compound 2{2}, which has a propargyl alcohol at the C3 position and morpholino substitution at C5, showed improved potency but relatively higher cytotoxicity. In 2{3}–2{5}, introduction of a cyclohexyl group at the 2′-position of the propargyl alcohol (resulting in 1-ethynylcyclohexanol) provided improved potency and greatly improved selectivity. Replacing the 4-methoxyphenyl group at the C5-position with the 2-methoxy-3-pyridyl ring (2{4}) reduced potency and cytotoxicity, resulting in a modest selectivity improvement. The best compound in Table 1, 2{5}, showed an EC50 of 84 nM and >1190-fold selectivity. Replacement of the alcohol with electron-rich aryl groups adjacent to the alkyne linker at the R3 position (2{6}–2{8}) led to reduced activity against HCV, although the low cytotoxicity (>100 µM) was preserved, indicating that the alcohol group is important for HCV inhibition. Even more dramatically, the lack of C3-alkynyl moiety (2{9}– 2{18}) led to either a significant or complete loss of HCV inhibitory activity.
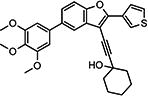
Compounds in Table 2 contain a 3′-thiophene group at the C2-position of the benzofuran scaffold and vary at the C3- and C5-positions. Compounds with a C5-morpholine group (3{1}–3{3} and 3{5}–3{7}) show reduction in HCV inhibitory activity compared with the analog 2{1}. C5-morpholine-containing compounds 3{2}, 3{3}, and 3{7} show increased cytotoxicity compared with 2{1}. The best compound in this series, 3{4} with 3-(1-ethynylcyclohexanol) and 5-(3′,4′,5′-trimethoxyphenyl) substitution on the benzofuran scaffold, showed an EC50 of 177 nM and >568-fold selectivity against Huh7.5.1 cells. Direct comparison between 3{3} and 3{4} in which the only aryl group at R2 is replaced with a morpholine moiety suggests that this replacement is not favorable for anti-HCV potency.
Table 2.
HCV Inhibition and Cytotoxicity of 2-Thiophene-Containing Benzofuran Analogs
| Cmpd |
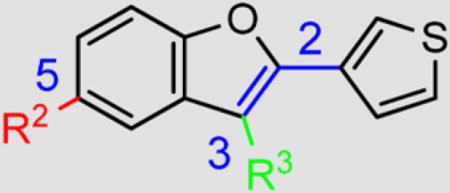
|
HCV-Luc EC50 (µM) |
ATPlite CC50 (µM) |
Selective Index (SI) |
|
|---|---|---|---|---|---|
| R2 | R3 | ||||
| 3{1} |
|
|
>31.6 | > 100 | N.A. |
| 3{2} |
|
|
> 31.6 | 34.20 | < 1 |
| 3{3} |
|
|
2.44 | 43.60 | 18 |
| 3{4} |
|
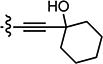
|
0.177 (n=3) | > 100 | > 568 |
| 3{5} |
|
|
11.7 | > 100 | > 9 |
| 3{6} |
|
|
8.56 | > 100 | > 12 |
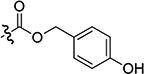
| 3{7} |
|
|
1.52 | 26.40 | 17 |
Analogs in Table 3 explored substituent changes at the C2-, C3-, and C5-positions simultaneously, often providing single forays into more drastic structural changes compared with the qHTS hit compounds. Compound 4{1} lacks the C5-substituent on the benzofuran scaffold and was found to be inactive in the HCV-Luc assay. Compounds 4{2}–4{5}, which contain a C5-morpholine moiety and vary at the C2- and C3-positions of the benzofuran scaffold, were found to lose activity against HCV. Thus, the screening results for compounds in Tables 1–3 indicate that HCV inhibitory activity is sensitive to substitutions at all three positions on the benzofuran scaffold. An aryl or heteroaryl group at the C5-position of the benzofuran scaffold is important for the inhibitory activity in the HCV-Luc assay. Substitution at the C2-position of the benzofuran is well-tolerated, with the introduction of various substituted aryl groups imparting nanomolar inhibitory potency. However, replacement of the propargyl alcohol moiety at the C3-position of the benzofuran with an aryl or heteroaryl group led to a complete loss of potency.
Library Design, Synthesis and Evaluation
On the basis of these encouraging results, we designed a library of 2,3,5-trisubstituted benzofuran compounds to systematically explore the SAR, utilizing the previously established synthetic sequence8 to readily access novel analogs of this chemotype (Figure 4). All target analogs were synthesized via the synthetic route outlined in Scheme 1. The synthesis of the 4-bromo-2-iodo-1-methoxybenzene intermediate 6 was achieved by regioselective iodination of commercially available 4-bromoanisole 5 using Selectfluor and iodine at room temperature.10 Sonogashira coupling of 6 with substituted aryl alkynes afforded 7, which was further diversified by Suzuki–Miyaura metal-catalyzed cross-coupling using various aryl boronic acid components to give 8. Iodocyclization of 8 using iodine monochloride provided the intermediate 9. The final diversification step was achieved in parallel using Sonogashira cross-coupling with various alkyne fragments on the Bohdan MiniBlock platform. The target compounds 10 were purified by reversed phase, preparative, mass-directed HPLC fractionation (MDF), and the purity was determined using analogous analytical HPLC conditions.
Figure 4.

Library design and building blocks.
Scheme 1.

General Synthetic Schemea
a(a) I2, Selectfluor, rt, 4 h (77% yield); (b) PdCl2(PPh3)2 (3 mol %), CuI (2 mol %), Et3N, 23 °C 18 h (75–93% isolated yield); (c) “various Suzuki-Miayura cross coupling conditions” (41–93% isolated yield); (d) ICl, DCM, rt, 4 h (30–89% yield); (e) PdCl2(PPh3)2 (3 mol %), CuI (2 mol %), Et2NH, DMF, 80 °C 15 h (library synthesis and MDF purification)8
Thiophene groups have been reported to present metabolic challenges;11–13 hence, we decided to explore more stable replacements for the thiophene group at the C2-position of analog 3{4} (R1 group in Figure 4), such as the 4-methoxyphenyl building block 11a (Figure 4), which was present in the C2-position of several promising compounds (2{1}–2{5}; Table 1) and the more sterically similar imidazole 11b (Figure 4). We also attempted to improve the aqueous solubility through replacement of the benzofuran scaffold with a furo[3,2-b]pyridine scaffold using the building block 11c (Figure 4). During physiochemical property assessment for KU CMLD Center compounds in Tables 1–3, it was observed that the potent compound 2{5} showed poor (<1 mg/mL) aqueous solubility (Table 6). Thus, it was also of interest to improve solubility as a secondary design element for compounds in the library. Introduction of the polar C2-imidazole moiety and the furo[3,2-b]pyridine moieties into the scaffold were anticipated to improve solubility for these analogs.
Table 6.
Physicochemical Properties of Selected Compounds
| Cmpd | Structure | HCV-Luc EC50 (µM) |
ATPlite CC50 (µM) |
Selective Index (SI) |
cLogPb | Microsomal Stabilityc t1/2 (min) |
Aqueous Solubility d (µg/ml) |
Permeability e (10−6 cm/s) |
|---|---|---|---|---|---|---|---|---|
| 3{4} |
|
0.177a | >100 | >565 | 6.47 | >30.0 | <1.0 | <5.6 |
| 2{3} |
|
0.171a | >100 | > 585 | 7.36 | >30.0 | <1.0 | N.D. |
| 2{5} |
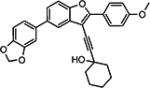
|
0.084a | >100 | > 1190 | 7.41 | >30.0 | <1.0 | < 13.9 |
| 10{11} |
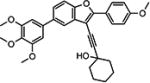
|
0.162a | >100 | >617 | 6.74 | >30.0 | <1.0 | N.D. |
| 10{13} |
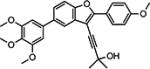
|
0.0975a | 86.1 | 883 | 5.43 | >30.0 | <1.0 | < 19.8 |
| 10{21} |
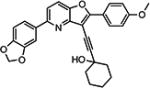
|
0.184 | >100 | >543 | 6.40 | >30.0 | <1.0 | < 15.4 |
| 10{32} |
|
0.777 | >100 | > 129 | 3.47 | >30.0 | <1.0 | <1.0 |
Sample was tested independently (n ≥ 2), and value is an average.
cLogP values calculated using CambridgeSoft Chemdraw.
CD 1 mouse liver microsomes, timepoints at 0, 5, 15, 30 and 45 min.
µSOL Evolution protocol (fully automated HTP platform, pION Inc.).
Double sink method (fully automated HTP platform, pION Inc.).
Substituents at the R2-position were incorporated from the most active compounds from Tables 1–3. Substitutions at the R3-position retain the propargyl alcohols (13a and 13b in Figure 4), which were found to be important in the preliminary SAR studies. We focused instead on exploring the effects of increased steric bulk (13c in Figure 4) or introduction of additional cyclic oxygen atoms (13d and 13e in Figure 4) on potency and cellular toxicity.
Results from the biological evaluation of the library compounds 10{1}–10{45} are shown in Table 4. Compounds 10{1}–10{5} (R1 and R2 = 4-methoxyphenyl) were found to be less potent compared with the leading compounds from previous SAR exploration (i.e., 2{2}, 2{3}, and 2{5}). The most active compound among 10{1}–10{5} was the propargylic alcohol 10{2}, possessing an EC50 of 210 nM and good selectivity. Steric bulk at the carbon atom adjacent to the alcohol moiety (10{3} and 10{1}) or introducing the oxygen-containing heterocycles oxetane (10{4}) or tetrahydropyran (10{5}) were all detrimental to potency. Among compounds 10{6}–10{10} (R1 = 4-methoxyphenyl; R2 = dioxolophenyl), the tetrahydropyran-containing analog (10{10}) was the most potent (130 nM), with compounds 10{7} and 10{9} only slightly less potent. Other modifications at the R3 position were less conducive to HCV inhibition. Among compounds 10{11}–10{15} (R1 = 4-methoxyphenyl, R2 = 3′,4′,5′-trimethoxyphenyl), compound 10{13} (R3 = 2,2-dimethylpropargyl alcohol) was the most potent compound (EC50 = 98 nM) and possessed excellent selectivity (SI = 879). In this subseries, HCV inhibition was reduced by 6-fold when the cyclohexyl ring on the propargyl alcohol at R3 (10{11}) was replaced with an oxetane ring (10{14}).
Table 4.
HCV Inhibition and Cytotoxicity of Benzofuran Library Analogs

|
Sample was tested independently (n ≥ 2), and value is an average.
Compounds 10{16}–10{30} built upon the pyrido[3,2-b]furan moiety generally showed HCV inhibitory potency better than the imidazole analogs 10{31}–10{45} (R1 = N1-methyl imidazole), although they were slightly less active when compared with the corresponding benzofuran compounds 10{1}–10{15}. The pyrido[3,2-b]furan analogs displayed a similar cytotoxicity level when compared with the corresponding benzofuran compounds 10{1}–10{15}. The best compound in this series was 10{21} (R1 = 4-methoxyphenyl; R2 = dioxolophenyl; R3 = 1-ethynylcyclohexanol) with an HCV EC50 of 180 nM and ~555-fold selectivity.
Compounds 10{31}–10{45} (R1 = N1-methyl imidazole) showed reduced potency against HCV inhibition and reduced selectivity compared with the most active compounds from previous SAR exploration (i.e., 2{2}, 2{3}, and 2{5}), indicating that an imidazole ring is not well tolerated in this position. The most active compound in this series was 10{43} (R2 = 3′,4′,5′-trimethoxyphenyl; R3 = 2,2-dimethylpropargyl alcohol) with an HCV EC50 of 320 nM and ~100-fold selectivity. Overall, the systematic SAR investigation revealed the activity of individual analogs to be intricately dependent on the substitution at all positions combined. This is illustrated across the substitution at R3 for each series, for which, in some cases, the unsubstituted propargylic alcohol moiety (13b) was found to be most potent, but in other cases, the more substituted moieties (i.e., 1-ethynylcyclohexanol (13a), 2-methylbut-3-yn-2-ol (13c), or 4-ethynyltetrahydro-2H-pyran-4-ol (13e)) were found to be preferred.
Anti-HCV Characterization
From the SAR campaign, promising compounds were selected on the basis of their antiviral activity, selectivity, and potential improvement in physicochemical properties. The activity of the promising compounds was confirmed for wild-type HCV (HCVcc, genotype 2a) infection in Huh7.5.1 cells. Both extracellular and intracellular viral levels were dramatically inhibited by the test compounds, among which 10{13} and 10{21} showed the most promising activity (Figure 5). It is worth noting that more reduction was observed in the intracellular viral levels with most compounds, suggesting this chemotype might be targeting HCV early stage infection. One advantage of this series of antivirals is their low cytotoxicity toward human hepatocytes. For further confirmation, the cytotoxicity of advanced compounds was determined in HepG2 cells and primary human hepatocytes. As shown in Table 5, all test compounds exhibited low cytotoxicity toward both types of cells (CC50 > 31.6 µM).
Figure 5.

Inhibition of wild-type HCV infection in human hepatocytes by selected analogs. Huh7.5.1 cells were seeded in 12-well plates (105 cells/well) and cultured overnight. HCVcc was used to infect the cells with the treatment of compounds at 10 µM. Virus-containing medium was removed after 4 h incubation, and compound treatment was added back, followed by incubation for an additional 48 h. Intracellular and extracellular viral RNA levels were evaluated by quantitative real-time PCR. The results shown are the means of three replicates ± SE. Cyclosporin A at 10 µM was used as a positive control.
Table 5.
Selectivity and HCV Life Cycle Target for Selected Compounds
| Cmpd | Structure | HCV-Luc EC50 (µM) |
ATPlite CC50 (µM) |
HCVsc DMSO (%)b |
HCVpp DMSO (%)b |
VSVpp DMSO (%)b |
HCV Replicon DMSO (%)b |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||||
| Huh7.5.1 | HepG2 | Primary Human Hepatocytes |
GT la |
GT lb |
GT lb |
GT 2a |
Transient la |
|||||
| 3{4} |
|
0.177a | >100 | >100 | >100 | 27.3 | 80.3 | 88.4 | 119 | 103 | 74.7 | 76.6 |
| 2{3} |
|
0.17la | >100 | >100 | > 100 | 33.7 | 86.1 | 76.5 | 104 | 109 | 89.0 | 137 |
| 2{5} |
|
0.084a | >100 | >100 | > 100 | 24.9 | 85.5 | 83.8 | 106 | 109 | 100 | 108 |
| 10{11} |
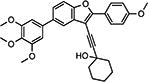
|
0.162a | >100 | > 31.6 | > 31.6 | 33.2 | 69.3 | 77.0 | 83.4 | 106 | 112 | 86.4 |
| 10{13} |
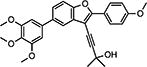
|
0.098a | 86.1 | > 31.6 | > 31.6 | 26.1 | 83.7 | 79.1 | 116 | 78.7 | 118 | 106 |
| 10{21} |
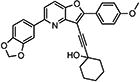
|
0.184 | > 100 | 92.3 | N.D. | 20.0 | 41.2 | 78.1 | 121 | 82.1 | 121 | 194 |
| 10{32} |
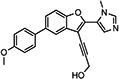
|
0.777 | >100 | > 100 | N.D. | 9.30 | 23.7 | 41.3 | 81.3 | 106 | 99.3 | 136 |
Sample was tested independently (n ≥ 2), and value is an average.
Sample was tested at 10 µM.
To explore the biological target of these compounds in the HCV replication cycle, HCV replication cycle assays were performed by infecting Huh7.5.1 cells with different types of modified viral particles. In the HCV single-cycle assay, single-round infectious HCV (HCVsc) was used to detect inhibition in the HCV replication steps prior to assembly. All the test compounds showed an inhibitory effect in the HCVsc assay (<35% of DMSO control results), suggesting a target in early stage HCV infection (Table 5). This is consistent with what was observed with the HCVcc infection assay. HCV pseudoparticle (HCVpp) and subgenomic replicon assays were then employed to detect whether HCVpp entry and HCV replication, respectively, were targeted by these compounds. The HCVpp entry was not significantly inhibited by the test compounds (≥69% of DMSO treatment), except for 10{32} and, to a lesser extent, 10{21} (Table 5). Even less inhibition was observed in either HCV replicon cell lines of genotype 1b or 2a as well as in the transient genotype 1a replicon assay (all ≥74% of DMSO treatment, Table 5). Together, these replicon experiments indicate that replication is highly unlikely to be the target of these compounds. Overall, this series of compounds inhibited infection at early stage and targeted neither HCVpp viral entry or HCV RNA replication. One possible mechanism of action could be through inhibition of viral late entry or trafficking in the infected cells. We previously reported a similar inhibitory pattern in the HCV replication cycle assays in a series of promising antiviral chlorcyclizine analogs.14
Physicochemical Properties
To assess the ADME (absorption, distribution, metabolism, and excretion) of the test compounds, their physicochemical properties were evaluated (Table 6). Half-lives (microsomal stability) greater than 30 min were observed with all selected analogs, suggesting a potentially useful therapeutic half-life in vivo. The aqueous solubility was relatively low for all the compounds in Table 6. This solubility may be improved by further structural modifications, through formulation, or by preparing salt forms of the compounds (only applicable to compounds 10{21} and 10{32}). Low experimental solubility for all compounds was observed, despite improvements in the calculated cLogP values (e.g., 10{13} and 10{32}). Compounds 10{6}, 10{13}, and 10{21} showed moderate permeability, and compounds 3{4} and 10{32} exhibited relatively lower permeability (Table 6).
CONCLUSION
In conclusion, we discovered a new chemical series with antiviral activity against HCV and explored the SAR around this chemotype utilizing our compound collection and the synthesis of a 45-member library of 2,3,5-trisubstituted compounds. In general, these compounds showed good potency (submicromolar to low-nanomolar) in the HCV infection assay and showed good selectivity compared to the initial qHTS hit. Representative compounds appear to inhibit an early stage of the viral lifecycle, possibly viral entry or trafficking. Further characterization of the precise biological target of these compounds is currently underway.
EXPERIMENTAL PROCEDURES
General Biological Assay Procedures
Human hepatoma cell lines Huh7.5.1 and HepG2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Life technologies, Grand Island, NY, USA) with 10% fetal bovine serum (FBS) (Life Technologies, Grand Island, NY, USA) and antibiotics in 5% CO2 at 37 °C. The primary human hepatocytes (obtained from S. Strom at the University of Pittsburgh through the NIH Liver Tissue and Cell Distribution System) were maintained in Williams E medium containing cell maintenance supplement reagents (Life Technologies, Grand Island, NY, USA). The primary screen and related bioinformatics were carried out as reported.7 Modified HCV particles for infection assays were produced and stocked as reported before.7,15,16 In all HCV infection assays, Huh7.5.1 cells were plated in 96-well plates at 104 cells/well and incubated overnight unless noted otherwise. HCV viral particles were used to infect the cells in the presence of the compounds of interest at a fixed concentration (10 µM) or in concentration–response. After 48 h of incubation, the viral level was measured by luminescence unless noted otherwise.
HCV-Luc Infection and ATPlite Assays
HCV-Luc infection was carried out as described in the general procedure. Renilla luciferase signal was measured for viral load using the Renilla Luciferase assay system (Promega, Madison, WI, USA). Cytotoxicity was tested with the ATPlite assay kit (PerkinElmer, Waltham, MA, USA) in parallel. The compound concentration causing 50% effect (antiviral and cytotoxicity) was calculated as EC50 and CC50 values using the nonlinear regression equation in GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA, USA).
HCV Replication Cycle Assays
In the HCVcc assay, Huh7.5.1 cells were cultured in 12-well plates at 105 cells/well overnight before infection in the presence of compounds. After infection overnight, the virus-containing medium was removed, and the compound was added back, followed by 48 h of incubation. Quantitative real-time PCR was carried out to measure the viral RNA levels. The HCV single-cycle assay was performed as described in the general procedure. In the HCVpp assay, HCVpp was added to the cells in the presence of compound for 4 h and washed away. Cells were incubated for an additional 48 h, followed by a luciferase assay to detect inhibition on HCVpp entry. In the HCV subgenomic replicon assay, HCV replicon cells were plated as described in the general procedure and treated with test compounds for 48 h before luciferase readout. In the transient replicon assay, Huh7.5.1 cells were transiently transfected with subgenomic replicon RNA before 48 h of incubation with compound treatment. Results were obtained as described in the general procedure. Cyclosporin A at 10 µM (in HCVsc and replicon assays) and bafilomycin A1 at 10 nM (in HCVpp assay) were tested as positive controls.
Physicochemical Properties
The cLogP values were calculated using CambridgeSoft Chemdraw software. For the measurement of in vitro microsomal stability, compounds were incubated with human, mouse, or rat microsomes at 37 °C with cofactor NADPH. At 0, 5,15, 30 and 45 min, the concentration of compounds was measured by LC–MS/MS. Half-life (t1/2) was calculated as described before.17 The fully automated HTP platform (pION Inc., Billerica, MA, USA) was used to measure the solubility and permeability of compounds following the protocol of µSOL Evolution and the double-sink PAMPA (parallel artificial memebrane permeability assay), respectively.
General Synthetic Procedures
All chemicals were used as received from commercial sources. Commercial anhydrous organic solvents (EtOAc, CHCl3, MeOH, EtOH, MeCN, DMF, hexane, toluene, etc.) were used for all reactions. The parallel chemistry reactions were carried out in a Mettler Toledo Miniblock or sealed microwave vials. Stirring was achieved with oven-dried, magnetic stir bars. Analytical thin layer chromatography (TLC) was performed using commercially prepared polyester-backed silica gel plates (200 µm), and visualization was effected with short wavelength UV light (254 nm). Flash column chromatography was carried out using Teledyne Isco CombiFlash Rf employing normal phase disposable columns. 1H and 13C NMR spectra were recorded on a Bruker Avance spectrometer (400 MHz/500 MHz 1H and 101/126 MHz 13C). Chemical shifts are reported in parts per million (ppm), and referenced to the solvent: CDCl3 with TMS as internal reference (0.0 ppm for 1H and 0.0 ppm for 13C) and DMSO-d6. Coupling constants (J) are reported in Hertz (Hz). Purification via preparative HPLC was achieved utilizing a Waters X-Bridge C18 column (19 × 150 mm, 5 µm, with 19 × 10 mm guard column) at a flow rate of 20 mL/min. Samples were diluted in DMSO and purified using an elution mixture of water and MeCN, running a concentration gradient that increased by 20% MeCN over a 4 min period. Analytical analysis after preparative chromatography utilized a Waters Acquity system with UV detection and mass detection (Waters LCT Premier). The analytical method conditions included a Waters Aquity BEH C18 column (2.1 × 50 mm, 1.7 µm) and elution with a linear gradient of 5% MeCN in water to 100% MeCN at 0.6 mL/min flow rate. The purity of each sample was determined using the UV peak area detected at 214 nm wavelength. High-resolution mass spectra for the diversified products were recorded using a time-of-flight mass spectrometer.
All synthesized compounds tested were determined to be >95% purity by HPLC/HRMS determination, with the following exceptions: 10{13} (92.8%), 10{21} (93.9%), 10{34} (90.2%), 10{41} (92.9%), 10{42} (94.0%), 10{43} (94.4%), and 10{44} (94.6%). A table of the purity for all synthesized final analogs is provided in the Supporting Information.
Representative synthetic procedure, synthesis of 10{21} (experimental details for all synthetic intermediates and the preparation of an additional 19 library compounds is provided in the Supporting Information):
6-Chloro-2-iodo-3-methoxypyridine (6b)
In an oven-dried round-bottomed flask, 6-chloro-2-iodopyridin-3-ol (2.5 g, 9.8 mmol) was dissolved in THF (20 mL), and then a 1 M solution of potassium tert-butoxide in THF (15 mL, 15 mmol) was added slowly to the reaction mixture at room temperature. The resulting reaction mixture was stirred for 15 min, and then iodomethane (0.914 mL, 15 mmol) was added slowly to the mixture, upon which the colorless solution turned into a white suspension. The reaction was quenched after 8 h with 20 mL of water and extracted with EtOAc (2 × 40 mL). The organic layers were pooled, washed with brine, dried over Na2SO4, and concentrated in vacuo. Silica gel column chromatography with EtOAc/hexane provided the desired compound as an off-white solid (1.5 g, 5.6 mmol, 57% yield). mp = 64–71 °C (uncorrected). 1H NMR (500 MHz, chloroform-d) δ 7.16 (d, J = 8.5 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 3.84 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 155.04, 141.38, 123.65, 119.26, 108.97, 56.83. HRMS (ESI) m/z calcd for C6H6ClINO+ [M + H]+, 269.9177; found, 269.9171.
6-Chloro-3-methoxy-2-((4-methoxyphenyl)ethynyl)-pyridine (7c)
A solution of 6-chloro-2-iodo-3-methoxypyridine 6b (1.43 g, 5.31 mmol), PdCl2(PPh3)2 (112 mg, 0.16 mmol, 3 mol %) and CuI (61 mg, 0.31 mmol,6 mol %) in Et3N (12 mL) was stirred briefly at 0 °C, and then 1-ethynyl-4-methoxybenzene 11a (1.0 mL, 8 mmol) was added to the reaction mixture. The reaction mixture was warmed to rt and allowed to proceed under vigorous stirring for ~18 h under an argon atmosphere. The resulting mixture was diluted with EtOAc (2 × 50 mL). The separated organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel using EtOAc/hexane as the eluent to afford the corresponding product 7c as a yellow, amorphous solid (1.35g, 4.93 mmol, 93% yield). 1H NMR (400 MHz, chloroform-d) δ 7.64–7.54 (m, 2H), 7.27–7.18 (m, 2H), 6.95–6.86 (m, 2H), 3.96 (s, 3H), 3.86 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 160.31, 155.90, 141.65, 133.76, 133.43, 123.67, 120.73, 114.23, 114.02, 95.60, 83.33, 56.39, 55.34. HRMS (ESI) m/z calcd for C15H13ClNO2+ [M + H]+, 274.0629; found, 274.0602.
6-(Benzo[d][1,3]dioxol-5-yl)-3-methoxy-2-((4-methoxyphenyl)ethynyl)pyridine (8g)
To a solution of chloroalkyne 7c (370 mg, 1.35 mmol), X-PHOS (129 mg, 0.27 mmol, 20 mol %) and Pd(OAc)2 (30 mg, 0.135 mmol, 10 mol %) in a dioxane (10 mL)/water (2 mL) mixture, and K3PO4 (860 mg, 4.0 mmol) were added under an argon atmosphere to a 20 mL microwave vial. To the resulting mixture was added boronic acid 12b (450 mg, 2.7 mmol), and the mixture was vacuum-purged three times. The reaction mixture was heated at 100 °C for 120 min under microwave irradiation. Upon cooling to room temperature, the reaction mixture was extracted with EtOAc (2 × 40 mL). The combined organic extracts were dried over Na2SO4, concentrated, and purified by silica gel flash column chromatography using EtOAc/hexane as the eluent to afford the corresponding product 8g as a yellow solid (335 mg, 0.932 mmol, 69% yield). mp = 188–192 °C (uncorrected). 1H NMR (400 MHz, chloroform-d) δ 7.55–7.50 (m, 2H), 7.47 (d, J = 8.7 Hz, 1H), 7.43 (d, J = 1.7 Hz, 1H), 7.38 (dd, J = 8.1, 1.8 Hz, 1H), 7.18 (d, J = 7.5 Hz, 1H), 6.83 (s, 1H), 6.83–6.76 (m, 2H), 5.93 (s, 2H), 3.88 (s, 3H), 3.77 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 160.03, 155.63, 149.59, 148.16, 147.92, 133.71, 133.26, 133.20, 120.48, 119.86, 118.50, 114.81, 113.93, 108.35, 107.30, 101.22, 93.93, 84.57, 56.06, 55.32. HRMS (ESI) m/z calcd for C22H18NO4+ [M + H]+, 360.1230; found, 360.1199.
5-(Benzo[d][1,3]dioxol-5-yl)-3-iodo-2-(4-methoxyphenyl)-furo[3,2-b]pyridine (9i)
To a solution of the alkyne 8g (359 mg, 1.0 mmol) in CH2Cl2 (15 mL) was added gradually iodine monochloride (244 mg, 1.5 mmol) dissolved in CH2Cl2 (10 mL). The reaction mixture was allowed to stir at rt for 2 h and was monitored by TLC to establish completion. The excess iodine monochloride was removed by washing with saturated aqueous Na2S2O3 solution (2 × 30 mL). The organic layers were dried over anhydrous Na2SO4 and concentrated under vacuum to afford the crude product, which was purified by flash chromatography on silica gel using EtOAc/hexanes as the eluent to provide the benzofuran product 9i as an off-white solid (350 mg, 0.743 mmol, 74% yield). mp = 178–182 °C (uncorrected). 1H NMR (500 MHz, chloroform-d) δ 8.23–8.21 (m, 2H), 7.73–7.68 (m, 2H), 7.63–7.58 (m, 2H), 7.08–7.01 (m, 2H), 6.96–6.88 (m, 1H), 6.04 (d, J = 2.7 Hz, 2H), 3.90 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 160.89, 156.78, 154.10, 149.28, 148.25, 146.00, 133.90, 129.25, 122.22, 121.18, 118.43, 116.94, 114.06, 110.14, 108.42, 107.85, 101.26, 62.92, 55.44. HRMS (ESI) m/z calcd for C21H15INO4+ [M + H]+, 472.0040; found, 472.0038.
1-((5-(Benzo[d][1,3]dioxol-5-yl)-2-(4-methoxyphenyl)furo-[3,2-b]pyridin-3-yl)ethynyl)cyclohexanol (10{21})
To a 4 dram vial was added successively the 3-iodobenzofuran 9i (42 mg, 0.09 mmol), the alkyne 13a (22 mg, 0.18 mmol), PdCl2(PPh3)2 (35 mg, 0.009 mmol, 5 mol %), CuI (2 mg, 0.009 mmol, 5 mol %), DMF (1 mL), and Et2NH (0.5 mL). The solution was stirred and flushed with argon and then heated to 80 °C for 15 h. The TLC revealed complete conversion of the starting material. The solution was allowed to cool and diluted with EtOAc (3 mL). The combined organic layers were concentrated in vacuo and purified by mass-directed, preparative HPLC to afford the trisubstituted product 10{21} (19 mg, 0.04 mmol, 44% yield). 1H NMR (500 MHz, chloroform-d) δ 8.32–8.21 (m, 2H), 7.78 (d, J = 8.6 Hz, 1H), 7.58–7.49 (m, 3H), 7.05–6.98 (m, 2H), 6.92 (d, J = 8.1 Hz, 1H), 6.04 (s, 2H), 3.91 (s, 3H), 2.19 (d, J = 12.3 Hz, 2H), 1.88–1.73 (m, 6H), 1.71–1.61 (m, 2H), 1.45–1.28 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 161.20, 160.31, 153.61, 148.58, 148.19, 147.78, 145.98, 128.05, 122.01, 121.89, 119.28, 117.07, 114.19, 108.45, 108.06, 102.73, 101.40, 97.93, 74.40, 69.19, 55.48, 40.98, 39.94, 25.38, 23.55. HRMS (ESI) m/z calcd for C29H26NO5+ [M + H]+, 468.1806; found, 468.1843.
Supplementary Material
Acknowledgments
We thank Patrick Porubsky for compound management services. This work was supported by grants from the Molecular Libraries Initiative 5U54HG005031 (J.A.) and the National Institute for General Medical Sciences KU-CMLD PO50-GM069663 (J.A.). Support for the NMR instrumentation was provided by NIH Shared Instrumentation Grant No. S10RR024664 and NSF Major Research Instrumentation Grant No. 0320648. This work was partially supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, and the National Center for Advancing Translational Sciences, National Institutes of Health.
Footnotes
ASSOCIATED CONTENT
- Experimental details and characterization for all new synthetic intermediates and the preparation of an additional 19 library compounds (PDF)
The authors declare no competing financial interest.
References
- 1.Belousova V, Abd-Rabou AA, Mousa SA. Recent advances and future directions in the management of hepatitis C infections. Pharmacol. Ther. 2015;145:92–102. doi: 10.1016/j.pharmthera.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 3.Liang TJ. Current progress in development of hepatitis C virus vaccines. Nat. Med. 2013;19:869–878. doi: 10.1038/nm.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N. Engl. J. Med. 2013;368:1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Francesco R, Migliaccio G. Challenges and successes in developing new therapies for hepatitis C. Nature. 2005;436:953–960. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- 6.Liang TJ, Ghany MG. Therapy of hepatitis C–back to the future. N. Engl. J. Med. 2014;370:2043–2047. doi: 10.1056/NEJMe1403619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Z, Lan K-H, He S, Swaroop M, Hu X, Southall N, Zheng W, Liang TJ. Novel Cell-Based Hepatitis C Virus Infection Assay for Quantitative High-Throughput Screening of Anti-Hepatitis C Virus Compounds. Antimicrob. Agents Chemother. 2014;58:995–1004. doi: 10.1128/AAC.02094-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho C-H, Neuenswander B, Lushington GH, Larock RC. Parallel Synthesis of a Multi-Substituted Benzo[b]furan Library. J. Comb. Chem. 2008;10:941–947. doi: 10.1021/cc800120y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ismail MAH, Adel M, Ismail NSM, Abouzid KAM. Molecular Design, Synthesis and Cell Based HCV Replicon Assay of Novel Benzoxazole Derivatives. Drug Res. (Stuttgart, Ger.) 2013;63:109–120. doi: 10.1055/s-0032-1331765. [DOI] [PubMed] [Google Scholar]
- 10.Zupan M, Iskra J, Stavber S. Room Temperature Regioselective Iodination of Aromatic Ethers Mediated by Select-fluorTM Reagent F-TEDA-BF4. Tetrahedron Lett. 1997;38:6305–6306. [Google Scholar]
- 11.Valadon P, Dansette PM, Girault J-P, Amar C, Mansuy D. Thiophene Sulfoxides as Reactive Metabolites: Formation upon Microsomal Oxidation of a 3-Aroylthiophene and Fate in the Presence of Nucleophiles in Vitro and in Vivo. Chem. Res. Toxicol. 1996;9:1403–1413. doi: 10.1021/tx9601622. [DOI] [PubMed] [Google Scholar]
- 12.Smith GF. Prog. Med. Chem. 2011;50:1–47. doi: 10.1016/B978-0-12-381290-2.00001-X. [DOI] [PubMed] [Google Scholar]
- 13.Treiber A, Dansette PM, El Amri H, Girault J-P, Ginderow D, Mornon J-P, Mansuy D. Chemical and Biological Oxidation of Thiophene: Preparation and Complete Characterization of Thiophene S-Oxide Dimers and Evidence for Thiophene S-Oxide as an Intermediate in Thiophene Metabolism in Vivo and in Vitro. J. Am. Chem. Soc. 1997;119:1565–1571. [Google Scholar]
- 14.He S, Lin B, Chu V, Hu Z, Hu X, Xiao J, Wang AQ, Schweitzer CJ, Li Q, Imamura M, Hiraga N, Southall N, Ferrer M, Zheng W, Chayama K, Marugan JJ, Liang TJ. Repurposing of the Antihistamine Chlorcyclizine and Related Compounds for Treatment of Hepatitis C Virus Infection. Sci. Transl. Med. 2015;7:282ra49. doi: 10.1126/scitranslmed.3010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. virol. 2007;81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.