

Abstract
We report a new class of redox-active vertex-differentiated dodecaborate clusters featuring pentafluoroaryl groups. These [B12(OR)11NO2] clusters share several unique photophysical properties with their [B12(OR)12] analogues, while exhibiting significantly higher (+0.5 V) redox potentials. This work describes the synthesis, characterization, and isolation of [B12(O-CH2C6F5)11NO2] clusters in all 3 oxidation states (dianion, radical, and neutral). Reactivity to post-functionalization with thiol species via SNAr on the pentafluoroaryl groups is also demonstated.
Graphical abstract
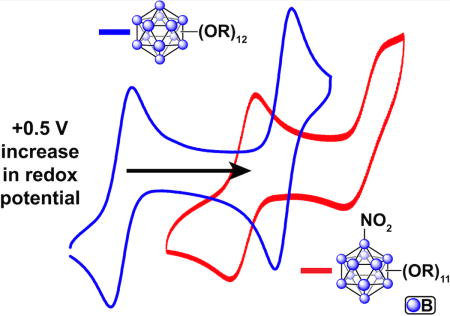
The dodecaborate cluster closo-[B12H12]2− is a unique three-dimensional molecule which can be functionalized in a manner reminiscent of classical organic aromatic compounds (e.g. benzene).1–3 Three-dimensional aromaticity in this icosahedral cluster is manifested in its exceptional thermal and chemical stability.4 The closo-[B12H12]2− cluster has been previously subjected to various homoperfunctionalization strategies leading to molecules decorated with a diverse array of functional groups including halogens,5,6 ethers, esters, carbonates, and carbamates.7–12 While the parent closo-[B12H12]2− cluster does not undergo a reversible electrochemical transformation, homoperfunctionalization via substitution of all 12 B-H vertices can engender reversible redox behavior for some of the aforementioned derivatives.11–19 This redox activity can provide access to isolable compounds in hypo- and hypercloso- forms. These additional redox states are accessed via sequential one-electron oxidation of the parent dianionic closo-[B12X12]2− species to form stable radical hypocloso-[B12X12]1− molecules, followed by the consecutive one–electron oxidation allowing isolation of neutral hypercloso-[B12X12] clusters. Importantly, the redox potential of the 2-/1- and 1-/0 transitions for these clusters can be tuned as a function of the substituent by well over 1 V for the same redox event.11,12,17 Among the redox active B12-based clusters, homoperfunctionalized [B12(OR)12]2-/1-/0 compounds represent, perhaps, the most modular class of redox active species, for which the electrochemical potential window was previously shown to be tunable in the range between −1.1 – +0.67 V vs. Fc/Fc+.11 This tunability is achieved by rationally changing an organic substituent (R) attached to the cluster via ether linkage. However, it remains unclear whether highly reversible redox behavior is inherent to only B12(OR)12 species and whether electronic perturbations on the cluster core with mixed substituents other than the alkoxy group can produce molecules with similar properties.20–26 Here we present a new approach to further tune the redox properties of the perfunctionalized B12-based clusters by substituting one of the OR moieties with an NO2 group, thus introducing an additional structural handle via heterofunctionalization. As a result, a new class of [B12(OR)11NO2]2-/1-/0 clusters can be isolated which feature pronouncedly different electrochemical properties when compared to those of their [B12(OR)12] analogues.
Commercially available Cs2[B12H12] undergoes selective monoamination in 60% yield on a decagram scale.20 This compound can be safely subjected to the perhydroxylation conditions using 30% H2O2 (safety note – see SI) as was previously reported by Hawthorne and co-workers20 (Figure 1, [1]). Based on this protocol, one can isolate [B12(OH)11(NO2)]2− as a tetrabutylammonium (TBA) salt [1] on a multigram scale in 70% yield (see SI). Compound [1] is highly soluble in polar organic solvents, which allowed us to subject this compound perbenzylation conditions similar to those developed for the perfunctionalization of closo-[B12(OH)12]2−.11 The reaction of [1] with pentafluorobenzyl bromide dissolved in acetonitrile (MeCN) in the presence of an amine base under microwave conditions at 140 ⁰C resulted in a nearly quantitative conversion of the parent cluster molecule as judged by the 11B NMR spectroscopy and mass spectrometry taken in situ after 1 hour.
Fig. 1.
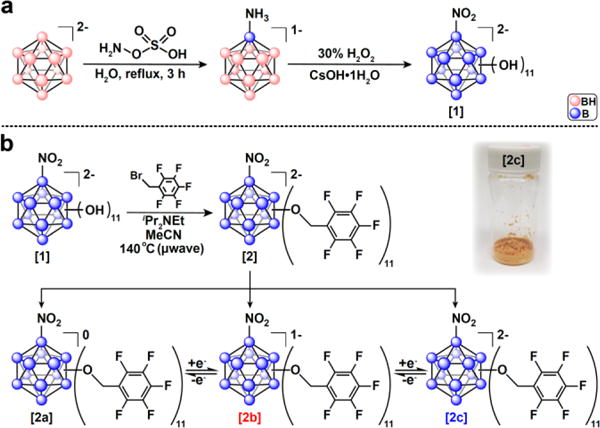
(a) Synthetic route to produce [1] from Cs2[B12H12]2−. (b) Microwave synthesis of the B12(OR)11NO2 cluster [2] studied in this work, showing all three oxidation states of [2] isolated (neutral [2a]0, radical [2b]1−, and the dianionic [2c]2−).
The desired product [2c] was isolated via purification on a silica gel column followed by ion exchange to the Na+ salt using a cation-exchange resin (see SI). The identity of Na2[2c] was confirmed by 11B, 19F, and 1H NMR spectroscopy, mass spectrometry, and elemental analysis. Performing the same synthesis followed by oxidation of the unpurified crude [2c]2− obtained directly from the microwave reaction with FeCl3•6H2O in 9:1 [v/v] mixture of ethanol:MeCN yielded a dark purple solution. This mixture was subjected to silica gel column chromatography resulting in the isolation of analytically pure hypocloso species [2b]1− as a TBA salt (see SI for characterization). In constrast, when [2c]2− was exposed to NOBF4, a stronger oxidant, in dry MeCN, neutral [2a]0 formed and was subsequently isolated.
All three species exhibit distinctive splitting patterns in both their 11B and 19F NMR spectra arising from the unique symmetry of the molecule. Unlike homoperfunctionalized [B12(OR)12] species, which possesses a nearly perfect icosahedral structure (hence appearing as a singlet in 11B NMR spectrum), the introduction of the nitro (NO2) group in [2] breaks the symmetry of the cluster. The 11B NMR spectra obtained confirm the asymmetry of the molecule, with both [2c]2− and [2a]0 exhibiting four distinct signals with relative integrations of 1:5:5:1 (Fig. 2a). The 11B NMR of the radical [2b]1− shows no visible signals consistent with the paramagnetic nature of the cluster in this oxidation state and is further corroborated by EPR spectroscopy (Fig. 2a, inset). Specifically, the broad singlet in the EPR spectrum of [2b]1− showcases a highly delocalized doublet state, where an electron is shared by all twelve boron atoms in the cluster. This feature as well as the g-factor observed in the EPR spectrum of [2b]1− are similar to other [B12(OR)12]1− species (Figure 2).
Fig. 2.
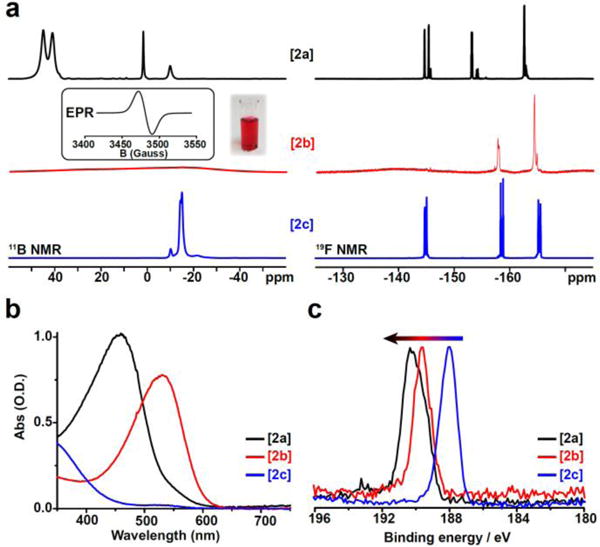
(a) 11B and 19F NMR spectra of all redox states of [2] with EPR (inset, g = 2.00674) and photo of [2b]1− in MeCN; (b) UV-vis spectra of [2a]0 and [2b]1− (50 mM) and [2c]2− (100 mM) in CH2Cl2; (c) XPS spectra for all redox states of [2] showing increasing B-B binding energy as redox state increases.
An unexpected but very interesting phenomenon is observed in the 19F NMR spectra of [2] series. Normally, the spectra for all previously reported [B12(OR)12] species containing fluorine atoms in the close vicinity from the boron cluster core show a single signal for each set of equivalent atoms.11,18,19 However, [2c]2− exhibits three distinct (though partially overlapping) fluorine signals for each of the equivalent pentafluoroaryl fluorine atoms, which show the same 1:5:5 relative integrations stemming from the symmetry of the boron cluster core. The ortho-fluorine signal for [2b]1− is difficult to observe due to the significant paramagnetic broadening that normally affects the atoms closest to the paramagnetic center (boron cluster core). Since the meta- and para-fluorine atoms are located further away from the paramagnetic cluster in [2b]1−, their 19F signals could be resolved and also exhibit a 1:5:5 relative pattern similar to the one observed in [2c]2− (Figure 2a). Likewise, a 1:5:5 pattern can be well resolved for the diamagnetic [2a]0 species. Furthermore, the two methylene proton resonances (1:10 integration) can be resolved in the 1H NMR spectrum of [2a]0 consistent with the introduction of asymmetry by the NO2 group.
Consistent with the [B12(OR)12]2− clusters synthesized to date, pure dianionic [2c]2− cluster is essentially colourless and shows little visible absorption in its UV-vis spectrum. There is a small absorbance in the UV-vis spectrum of [2c]2−around 350 – 400 nm stemming from the presence of the NO2 group, while the UV-vis spectrum for [3]2− ([B12(OR)12] when R = CH2C6F5) shows negligible absorption across the entire visible region (See SI). On the other hand, the radical [2b]1− exhibits a deep pink/purple color when dissolved in CH2Cl2, and the neutral species [2a]0 appears as a bright red-orange when dissolved in the same solvent (Fig. 2b). These light absorption properties are similar to those of homoperfunctionalized [B12(OR)12] clusters in the corresponding oxidation states. X-ray photoelectron spectroscopy analysis of [2] series also reveals a similar trend to that observed with [B12(OR)12] clusters, where the B-B binding energy increases with higher oxidation states sequentially from [2c]2− to [2b]1− and [2a]0 (Fig. 2c).
A single crystal suitable for X-ray diffraction analysis was obtained for the radical [NnBu4][2b] from a chloroform/pentane solution of [2b]1−, and the crystal structure (Fig. 3a) shows a structural similarity to that of the radical [3]1− (Fig. 3c, see SI). Overlaying the two structures (Fig. 3b) effectively illustrates these similarities, with only the rotation of a few pentafluorobenzyl rings nearest the NO2 group differing significantly, while the core bonding metrics (B-B and B-O bond lengths and angles) remain indistinguishable within the margin of error produced by the experiment. Despite the remarkably clear effects visible in the 19F NMR spectra of the asymmetry introduced into the molecule by the lone NO2 group in solution, the two different clusters retain very similar structures in the solid-state. This structural similarity, especially in the radical state, is consistent with the EPR evidence that the environment of the unpaired electron in [2b]1− is largely unchanged from [3]1−.
Fig. 3.
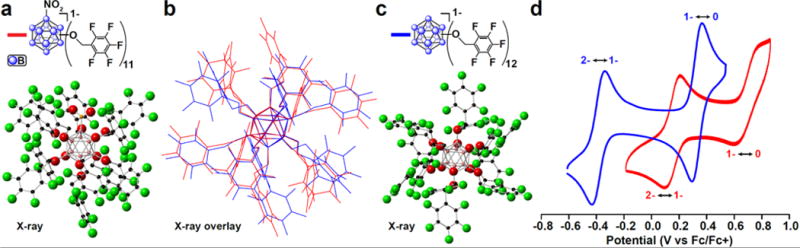
(a) Single crystal X-ray structure of [2b]1− (hydrogens and [NnBu4]+ counter ion omitted for clarity); (b) Overlay of [2b]1− and [3]1−; (c) Single crystal X-ray structure of [3]1– (hydrogens and [NnBu4]+ counter ion omitted for clarity); (d) Cyclic voltammogram for [2b]1− and [3] in acetonitrile with glassy carbon working electrode, Pt wire counter electrode, and Ag/AgCl reference electrode (in sat. KCl) internally referenced to the Fc/Fc+ couple.
Despite all these similarities, perhaps the most striking difference between the two species is the exceptional increase in redox potential of [2] compared to that of [3]. The significant difference between the redox potential of [2] and [3] of over +0.5 V for the same redox couple (Fig. 3b) is remarkable given the aforementioned similarities between most of the photophysical properties discussed. For the 2-/1- redox event, [3] displays a reversible wave with an E1/2 of −0.38 V vs Fc/Fc+, while the same couple for [2c]2−/[2b]1− has an E1/2 of +0.15 V (Fig. 3 d). The increase in the 1-/0 redox couple is less extreme, but still significant, going from an E1/2 of +0.33 V for [3]1−/[3]0 up to an E1/2 of 0.69 V (vs Fc/Fc+) for [2b]1−/[2a]0. The substitution of a single boron vertex with a NO2 group instead of an ether moiety therefore provides a dramatic shift in the electrochemical potential in this class of clusters.
Consistent with the reactivity previously observed for [3],18 [2] undergoes a facile SNAr reaction with thiols enabling dense functionalization of this class of three-dimensional scaffolds (Fig. 4). Under similar conditions to those described for [3] previously,18 full substitution of the para-fluorines on [2c]2− with mercaptoethanol was achieved using only 1.05 equiv. of the thiol per pentafluoroaryl group at room temperature within 19 hours (Fig. 4). The quantitative conversion observed under these mild conditions to form [2c]-ME indicates that the introduction of the nitro group does not interfere with the SNAr chemistry and [2] is potentially well-suited for building atomically precise hybrid nanomolecule assemblies.18
Fig. 4.
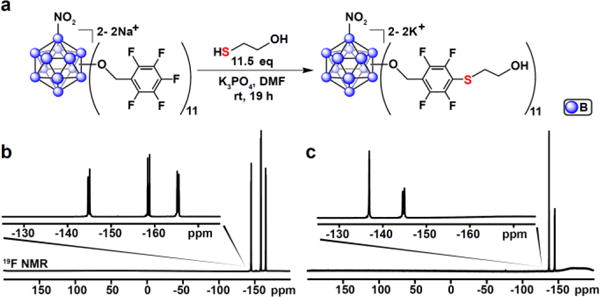
(a) Scheme showing the SNAr reaction between [2c]2− and mercaptoethanol; (b) 19F NMR spectrum of the starting material [2c]2−; (c) 19F NMR spectrum of the reaction product showing full conversion to [2c]-ME based upon the disappearance of the para-fluorine signal.
In summary, a new class of perfunctionalized, vertex-differentiated boron clusters was developed and fully characterized. While several routes exist to achieve vertex-differentiated perfunctionalization with other boron cluster molecules with inherent asymmetry such as monocarborane,21–25 significantly fewer synthetic pathways have been developed to form vertex-differentiated perfunctionalized dodecaborate clusters.20,26 These NO2-substituted dodecaborate clusters feature higher redox potentials while retaining key beneficial traits found in their perfunctionalized [B12(OR)12] analogues, including three distinct accessible redox states. The readily-accessible isolable radical cluster also represents a new addition to the boron-centered radical molecules.27–35 The pentafluoroaryl groups decorating the cluster were demonstrated to be amenable to facile post-functionalization with thiol-containing groups via SNAr chemistry, thus enabling the possibility of orthogonal substitution without affecting the B-NO2 vertex. Overall, this chemistry provides another unique entry towards robust and redox-active 3D aromatic building blocks for potentially designing new photoredox reagents19 and hybrid materials.36
Supplementary Material
Acknowledgments
A.M.S. acknowledges the University of California, Los Angeles (UCLA) Department of Chemistry and Biochemistry for start-up funds, 3M for a Non-Tenured Faculty Award, the Alfred P. Sloan Foundation for a research fellowship in chemistry and the NIGMS for the Maximizing Investigators Research Award (MIRA, R35GM124746). The authors thank the MRI program of the National Science Foundation (NSF grant no. 1532232 and no. 1625776). E.A.Q. thanks the US Public Health Service of the National Institutes of Health (NIH) for the Predoctoral Training Fellowship through the UCLA Chemistry-Biology Interface Training Program under the National Research Service Award (T32GM008496). We thank UCLA Molecular Instrumentation Center for mass spectrometry and NMR spectroscopy (NIH grant 1S10OD016387-01).
Footnotes
Electronic Supplementary Information (ESI) available: Full synthetic details, NMR and IR spectra, and cif files for the radical species. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Grimes RN. J Chem Educ. 2004;81:657. [Google Scholar]
- 2.Hawthorne MF. J Chem Educ. 2009;86:1131. [Google Scholar]
- 3.(a) Spokoyny AM. Pure Appl Chem. 2013;85:903–919. doi: 10.1351/PAC-CON-13-01-13. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sivaev IB, Bregadze VI, Sjöberg S. Collect Czech Chem Commun. 2002;67:679–727. [Google Scholar]
- 4.Muetterties EL, Balthis JH, Chia YT, Knoth WH, Miller HC. Inorg Chem. 1964;3:444–451. [Google Scholar]
- 5.Knoth WH, Miller HC, Sauer JC, Balthis JH, Chia YT, Muetterties EL. Inorg Chem. 1964;3:159–167. [Google Scholar]
- 6.(a) Knoth WH, Miller HC, England DC, Parshall GW, Muetterties EL. J Am Chem Soc. 1962;84:1056–1057. [Google Scholar]; (b) Ivanov SV, Miller SM, Anderson OP, Solntsev KA, Strauss SH. J Am Chem Soc. 2003;125:4694–4695. doi: 10.1021/ja0296374. [DOI] [PubMed] [Google Scholar]
- 7.Hawthorne MF. Pure Appl Chem. 2003;75:1157–1164. [Google Scholar]
- 8.Farha OK, Julius RL, Lee MW, Huertas RE, Knobler CB, Hawthorne MF. J Am Chem Soc. 2005;127:18243–18251. doi: 10.1021/ja0556373. [DOI] [PubMed] [Google Scholar]
- 9.Maderna A, Knobler CB, Hawthorne MF. Angew Chem Int Ed. 2001;40:1661–1664. [PubMed] [Google Scholar]
- 10.Jalisatgi SS, Kulkarni VS, Tang B, Houston ZH, Lee MW, Hawthorne MF. J Am Chem Soc. 2011;133:12382–12385. doi: 10.1021/ja204488p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wixtrom AI, Shao Y, Jung D, Machan CW, Kevork SN, Qian EA, Axtell JC, Khan SI, Kubiak CP, Spokoyny AM. Inorg Chem Front. 2016;3:711–717. doi: 10.1039/c5qi00263j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Axtell JC, Saleh LMA, Qian EA, Wixtrom AI, Spokoyny AM. Inorg Chem. 2018;57:2333–2350. doi: 10.1021/acs.inorgchem.7b02912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brezesinski T, Wang J, Tolbert SH, Dunn B. Nat Mater. 2010;9:146–151. doi: 10.1038/nmat2612. [DOI] [PubMed] [Google Scholar]
- 14.Boeré RT, Kacprzak S, Keßler M, Knapp C, Riebau R, Riedel S, Roemmele TL, Rühle M, Scherer H, Weber S. Angew Chem Int Ed. 2011;50:549–552. doi: 10.1002/anie.201004755. [DOI] [PubMed] [Google Scholar]
- 15.Boeré RT, Derendorf J, Jenne C, Kacprzak S, Keßler M, Riebau R, Riedel S, Roemmele TL, Rühle M, Scherer H, Vent-Schmidt T, Warneke J, Weber S. Chem—Eur J. 2014;20:4447–4459. doi: 10.1002/chem.201304405. [DOI] [PubMed] [Google Scholar]
- 16.Peymann T, Knobler CB, Khan SI, Hawthorne MF. Angew Chem Int Ed. 2001;40:1999–2002. [PubMed] [Google Scholar]
- 17.Lee MW, Farha OK, Hawthorne MF, Hansch CH. Angew Chem Int Ed. 2007;46:3018–3022. doi: 10.1002/anie.200605126. [DOI] [PubMed] [Google Scholar]
- 18.Qian EA, Wixtrom AI, Axtell JC, Saebi A, Jung D, Rehak P, Han Y, Moully EH, Mosallaei D, Chow S, Messina MS, Wang JY, Royappa AT, Rheingold AL, Maynard HD, Král P, Spokoyny AM. Nat Chem. 2017;9:333–340. doi: 10.1038/nchem.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Messina MS, Axtell JC, Wang Y, Chong P, Wixtrom AI, Kirlikovali KO, Upton BM, Hunter BM, Shafaat OS, Khan SI, Winkler JR, Gray HB, Alexandrova AN, Maynard HD, Spokoyny AM. J Am Chem Soc. 2016;138:6952–6955. doi: 10.1021/jacs.6b03568. [DOI] [PubMed] [Google Scholar]
- 20.Bondarev O, Khan AA, Tu X, Sevryugina YV, Jalisatgi SS, Hawthorne MF. J Am Chem Soc. 2013;135:13204–13211. doi: 10.1021/ja4069613. [DOI] [PubMed] [Google Scholar]
- 21.Asay M, Kefalidis CE, Estrada J, Weinberger DS, Wright J, Moore CE, Rheingold AL, Maron L, Lavallo V. Angew Chem Int Ed. 2013;52:11560–11563. doi: 10.1002/anie.201306764. [DOI] [PubMed] [Google Scholar]
- 22.Lavallo V, Wright JH, Tham FS, Quinlivan S. Angew Chem Int Ed. 2013;52:3172–3176. doi: 10.1002/anie.201209107. [DOI] [PubMed] [Google Scholar]
- 23.Körbe S, Schreiber PJ, Michl J. Chem Rev. 2006;106:5208–5249. doi: 10.1021/cr050548u. [DOI] [PubMed] [Google Scholar]
- 24.Gu W, McCulloch BJ, Reibenspies JH, Ozerov OV. Chem Commun. 2010;46:2820. doi: 10.1039/c001555e. [DOI] [PubMed] [Google Scholar]
- 25.(a) Xie Z, Tsang CW, Sze ETP, Yang Q, Chan DTW, Mak TCW. Inorg Chem. 1998;37:6444–6451. doi: 10.1021/ic9805628. [DOI] [PubMed] [Google Scholar]; (b) Duttwyler S. Pure Appl Chem. 2018;90:733–744. [Google Scholar]; (c) Alleman O, Duttwyler S, Romanato P, Baldridge KK, Siegel JS. Science. 2011;332:574–577. doi: 10.1126/science.1202432. [DOI] [PubMed] [Google Scholar]; (d) Douvris C, Ozerov OV. Science. 2008;321:1188–1190. doi: 10.1126/science.1159979. [DOI] [PubMed] [Google Scholar]
- 26.Recent contributions:; (a) Zhang Y, Liu J, Duttwyler S. Eur J Inorg Chem. 2015;31:5158–5162. [Google Scholar]; (b) Pluntze AM, Bukovsky EV, Lacroix MR, Newell BS, Rithner CD, Strauss SH. J Fluor Chem. 2018;209:33–42. [Google Scholar]; (c) Bertocco P, Derendorf J, Jenne C, Kirsch C. Inorg Chem. 2017;56:3459–3466. doi: 10.1021/acs.inorgchem.6b03006. [DOI] [PubMed] [Google Scholar]
- 27.Ueng S-H, Solovyev A, Yuan X, Geib SJ, Fensterbank L, Lacôte E, Malacria M, Newcomb M, Walton JC, Curran DP. J Am Chem Soc. 2009;131:11256–11262. doi: 10.1021/ja904103x. [DOI] [PubMed] [Google Scholar]
- 28.Braunschweig H, Dyakonov V, Jimenez-Halla JOC, Kraft K, Krummenacher I, Radacki K, Sperlich A, Wahler J. Angew Chem Int Ed. 2012;51:2977–2980. doi: 10.1002/anie.201108632. [DOI] [PubMed] [Google Scholar]
- 29.Kaim W, Hosmane NS, Záliš S, Maguire JA, Lipscomb WN. Angew Chem Int Ed. 2009;48:5082–5091. doi: 10.1002/anie.200803493. [DOI] [PubMed] [Google Scholar]
- 30.Thomas JC, Peters JC. Inorg Chem. 2003;42:5055–5073. doi: 10.1021/ic034150x. [DOI] [PubMed] [Google Scholar]
- 31.Hudnall TW, Chiu C-W, Gabbaï FP. Acc Chem Res. 2009;42:388–397. doi: 10.1021/ar8001816. [DOI] [PubMed] [Google Scholar]
- 32.Staubitz A, Robertson APM, Sloan ME, Manners I. Chem Rev. 2010;110:4023–4078. doi: 10.1021/cr100105a. [DOI] [PubMed] [Google Scholar]
- 33.Stephan DW. Angew Chem Int Ed. 2017;56:5984–5992. doi: 10.1002/anie.201700721. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Zhang J, Lin Z, Xie Z. Organometallics. 2016;35:2579–2582. [Google Scholar]
- 35.Martin CD, Soleilhavoup M, Bertrand G. Chem Sci. 2013;4:3020–3030. doi: 10.1039/C3SC51174J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung D, Saleh LMA, Berkson ZJ, El-Kady MF, Hwang JY, Mohamed N, Wixtrom AI, Titarenko E, Shao Y, McCarthy K, Guo J, Martini IB, Kraemer S, Wegener EC, Saint-Cricq P, Ruehle B, Langeslay RR, Delferro M, Brosmer JL, Hendon CH, Gallagher-Jones M, Rodriguez J, Chapman KW, Miller JT, Duan X, Kaner RB, Zink JI, Chmelka BF, Spokoyny AM. Nat Mater. 2018;17:341–348. doi: 10.1038/s41563-018-0021-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.