

Abstract
GGGGCC (G4C2) repeat expansion in C9ORF72 is the most common genetic cause of ALS and FTD. An important issue is how repeat RNAs and their translation products—various dipeptide repeat (DPR) proteins—cause neurodegeneration. Drosophila has been widely used to model G4C2 repeat RNA and DPR protein toxicity. Overexpression of disease molecules in flies has revealed important molecular insights. These have been validated and further explored in human neurons differentiated from induced pluripotent stem cells (iPSCs), a disease-relevant model in which expanded G4C2 repeats are expressed in their native molecular context. Approaches that combine the genetic power of Drosophila and the disease relevance of iPSC-derived patient neurons will continue to unravel the underlying pathogenic mechanisms and help identify potential therapeutic targets in C9ORF72-ALS/FTD.
Keywords: Autophagy, DNA Damage, DPR Protein, Nucleocytoplasmic Transport, RAN Translation, Repeat Expansion
C9ORF72 Repeat Expansion in ALS and FTD
Amyotrophic lateral sclerosis (ALS), or Lou Gehrig’s disease, is a fatal, progressive neurodegenerative disorder caused by degeneration of upper and lower motor neurons in the brain and spinal cord [1]. Although ALS has long been considered a motor neuron disease, some ALS patients have symptoms of frontotemporal dementia (FTD), and the two disorders share many pathological and genetic features and several dysfunctional molecular pathways downstream of many common disease-causing genetic mutations [2]. FTD, the second most common form of presenile dementia after Alzheimer’s disease, is accompanied by focal degeneration of the prefrontal and/or temporal cortex and changes in behavior, language, and executive functions [3]. FTD is a clinically heterogeneous disorder that includes behavioral variant FTD, semantic dementia, progressive nonfluent aphasia, FTD with motor neuron disease, and other conditions [3].
The most common genetic cause of both ALS and FTD is a GGGGCC (G4C2) repeat expansion in the noncoding region of a previously uncharacterized gene, C9ORF72 [4, 5]. How a single genetic mutation such as C9ORF72 repeat expansion causes distinct yet overlapping diseases is unclear. It is possible that different genetic modifiers and environmental influences also play a role. The identification of this unusual ALS/FTD-causing mutation in 2011 galvanized the field, leading to breathtaking progress in recent years. Much of this progress reflects the intellectual challenge posed by the multifaceted nature of the underlying pathogenic mechanisms. First, C9ORF72 expression is reduced by G4C2 repeat expansion, suggesting haploinsufficiency as a potential disease-causing mechanism. In cells and tissues of C9ORF72 patients, expression of C9ORF72 transcript variant 2 and total full-length C9ORF72 is reduced by expanded repeats in the promoter region [4, 6–11]. However, C9orf72 knockout mice have no detectable neurodegeneration [12–18], arguing against haploinsufficiency as a primary mechanism of disease, although partial loss of C9orf72 function in microglia may influence disease progression [16].
Additional pathogenic mechanisms of C9ORF72-ALS/FTD are suggested by other molecular hallmarks of the disease. Nuclear and cytoplasmic RNA foci formed by sense and antisense expanded G4C2 repeat transcripts imply that disease phenotypes are induced by RNA-mediated toxicity, since these foci might sequester RNA binding proteins and consequently alter RNA metabolism, as in other repeat expansion disorders [19]. Another distinctive feature of C9ORF72-ALS/FTD is the accumulation of dipeptide repeat (DPR) proteins, mostly in the cytoplasm of patient neurons [20–22]: poly(GR), poly(GA), and poly(GP) are synthesized from the G4C2 sense repeat transcripts, while poly(PR), poly(PA), and poly(PG) are produced from the antisense strand. Repeat-associated non-AUG (RAN) translation is proposed to be the mechanism responsible for DPR protein production [22]; however, other unconventional translational mechanisms may also be involved in neurons and other cell types of C9ORF72 patients [23].
The presence of both RNA foci and DPR proteins in the brains of C9ORF72-ALS/FTD patients [4, 20–22], in human neurons derived from C9ORF72-induced pluripotent stem cells (iPSCs) [7, 8, 24], and in C9ORF72 BAC transgenic mice [15, 25–27] raises important questions about their relative contributions to the disease pathogenesis: (1) Are G4C2 RNA foci toxic? If so, are they sufficient to cause disease? (2) Does unspliced C9ORF72 pre-mRNA or an intron containing spliced G4C2 repeats serve as a template for DPR protein synthesis? (3) Are DPR proteins necessary or sufficient to induce neurotoxicity? (4) Which DPR proteins are more toxic and what is the mechanism of their toxicity? (5) What cellular defects do endogenously expressed repeat RNA and DPR proteins cause in human neurons? To address these important questions, both Drosophila and C9ORF72-ALS/FTD patient-specific iPSCs have been used productively in recent years. Insights from these studies are discussed below.
Drosophila as a Genetic Model for Neurodegenerative Diseases
During the last two decades, Drosophila has emerged as a highly valuable in vivo genetic organism for modeling various neurodegenerative diseases, such as Huntington’s disease, spinocerebellar ataxia, and Parkinson’s disease [28, 29], in part because of evolutionary conservation of many gene functions and related molecular mechanisms between human and fly. Basic research in Drosophila has generated many genetic tools for analyzing both loss- and gain-of-function mutants. Moreover, the reduced complexity of the fly brain and the brief life span of flies make it feasible to conduct unbiased large-scale genetic screens to discover novel modifier pathways and mechanisms. Fly models are also used to test drugs and therapeutic compounds [e.g., 30]. On the other hand, disease phenotypes associated with cognitive and behavioral defects may not be truly recapitulated in Drosophila. Another limitation is the anatomical difference between human and fly brain, which makes it impossible to address in Drosophila the selective vulnerability seen in most neurodegenerative diseases. Moreover, most fly models rely on the overexpression of disease molecules, and many cellular phenotypes under investigation are developmental rather than late onset in the adult brain.
Molecular Contexts of Expanded G4C2 Repeats in Drosophila Models
Since the discovery of C9ORF72 repeat expansion in 2011 [4, 5], several Drosophila models expressing expanded G4C2 repeats have been established to analyze the gain of toxic function in a manner mostly similar to that used for other repeat expansion diseases [31–35]. These models have been useful for determining whether the RNA or DPR protein toxicity is the primary cause of disease and for discovering potential pathogenic mechanisms. However, each fly model has unique molecular features, and the length and molecular context of the G4C2 repeats in these models vary, which is critically important for the proper interpretation of the toxicity of repeats and the potential implications for our understanding of the pathogenic mechanisms of C9ORF72-ALS/FTD.
The first fly model of G4C2 repeat expansion was generated by cloning either 3 or 30 copies of repeats downstream of the upstream activating sequence (UAS) element in the pUAST vector, which allows spatial control of target gene expression in tissue-specific Gal4 driver lines [31]. The 30 G4C2 repeats were interrupted in the middle by a 6-nucleotide XhoI restriction site in the 5′ untranslated region (5′UTR) of the gene encoding EGFP followed by the SV40 3′ UTR containing a polyadenylation signal [(G4C2)30-EGFP, Figure 1A]. In this construct, it is predicted that poly(GP) is in frame with EGFP and that poly(GR) has an unnatural C-terminal polypeptide consisting of six amino acids. However, poly(GP) and poly(GR) were not detectable when the construct containing the interrupted 30 copies of G4C2 repeats was expressed with GMR-Gal4 to induce retinal degeneration [36]. On the other hand, poly(GA) is predicted to be in frame with an unnatural protein of 317 amino acids (P. Jin, personal communication). It is not known whether this poly(GA)-containing artificial protein is actually produced in this fly model.
Figure 1. Schematic Representation of Different Drosophila Models of G4C2 Repeat Expansion.

Major features of the DNA constructs used for each model are presented. In all Drosophila G4C2 models, the UAS-GAL4 system is used to overexpress G4C2 repeats with different lengthes, and these models share common SV40 3′UTR containing a polyadenylation signal. In the (G4C2)30-EGFP model, repeats are interrupted in the middle by a six base pair sequence and followed by the EGFP coding region containing the ATG initiation codon. The (G4C2)36 construct contains uninterrupted repeats, while the (G4C2)36-RO construct harbors stop codons between every 12 repeats. The intronic (G4C2)160 construct mimics the human C9ORF72 locus, as 160 copies of repeat sequence are flanked by C9ORF72 intronic sequences and adjacent exons. In the (G4C2)58-GFP model, 58 copies of repeats are upstream to the GFP codon region but without the ATG start codon. The (G4C2)48 construct contains stop codons in each frame 5′ to the repeat sequence. The three columns on the right list DPR proteins detected in these models when phenotypes are observed; the proposed mechanism of toxicity; and the respective references.
In another fly model of G4C2 repeat expansion, 3, 36, or 103 copies of uninterrupted G4C2 repeats were cloned into the pUASTattB vector between the UAS and an SV40 3′UTR containing a polyadenylation signal (Figure 1) [32]. In this model, as in the (G4C2)30-EGFP flies described above, G4C2 repeats are also expressed in the molecular context of poly(A)+ mRNA; thus, as expected, repeat RNA is mainly localized in the cytoplasm [33]. Each salivary gland cell expressing (G4C2)36 or (G4C2)103 has one nuclear RNA focus [32,33], which seems to be the site of repeat RNA transcription, as the focus colocalizes with RNA polymerase II and flies homozygous for the repeat transgene have two foci in each cell [33]. In this model, poly(GP) can be detected by western blot and poly(GR) by dot blot [32]. Unnatural C-terminal fragments are likely also synthesized from the vector sequence adjacent to the repeat RNA, but this has not yet been demonstrated experimentally. To block DPR production, 6 nucleotides containing stop codons in both sense and antisense directions were inserted periodically after every 12 repeats. This construct was named “RNA only (RO)” [(G4C2)36-RO, Figure 1) [32].
In human patients, expanded G4C2 repeats are located in the first intron of C9ORF72, and repeat RNA is mostly localized in the nucleus [4]. To recapitulate this genomic organization in a fly model, a mini C9ORF72 gene was constructed in which 5, 20, 40, 80, or 160 copies of G4C2 repeats are flanked by human intronic sequences of C9ORF72 and adjacent exons (Figure 1) [33]. This mini C9ORF72 gene was cloned into the pBID-UASC vector, which contains the same SV40 3′UTR as in fly models described above. In this model, the intron containing 160 G4C2 repeats is efficiently transcribed and spliced out, as in human C9ORF72 cells, and forms numerous nuclear RNA foci in neurons and glial cells [33]. Moreover, poly(GP) is detectable but expressed at roughly 1% of the level in (G4C2)36 flies [33], probably because (G4C2)36 in the context of poly(A)+ mRNA is exported from the nucleus and translated in the cytoplasm much more efficiently than intronic repeat RNA.
In 2015, two additional fly models of G4C2 repeat expansion were reported. In the (G4C2)58-GFP model, 58 copies of uninterrupted G4C2 repeats were placed 5′ to the coding region of GFP but without the AUG translation initiation site in the pUASTattB vector (Figure 1) [34]. In this model, G4C2 repeats are also part of poly(A)+ mRNA. Poly(GP) is in frame with GFP, and indeed a single band on western blot could be detected by poly(GP) or GFP antibodies [34], although the translation initiation site for this chimeric protein has not been mapped yet. Moreover, poly(GR) with a predicted short C-terminus, but not poly(GA) or poly(PR), was detected by dot blot in (G4C2)58-GFP flies [34]. Finally, in the (G4C2)48 model, 6 stop codons were engineered into 3 different reading frames 5′ adjacent to 48 copies of uninterrupted G4C2 repeats in the pUAST vector containing the same 3′UTR as all the other fly models mentioned above (Figure 1) [35]. The production of DPR proteins in this fly model was not examined [35].
Drosophila Models of DPR Protein Toxicity
Are DPR proteins sufficient to cause toxicity in C9ORF72-ALS/FTD? To answer this important question, several groups created Drosophila models of DPR protein toxicity by optimizing codons to disrupt RNA G-quadruplex structure and to express individual DPR proteins of different lengths in Drosophila through conventional AUG-initiated translation. These models include 36 and 100 copies of GA, GR, PA, and PR [32]; GFP-tagged 50 copies of GA, PA, and PR [37]; Flag-tagged 80 copies of GA, GR and PR [38]; GFP-tagged 47 copies of GP, and 50 copies of GA and GR [34]; and Flag-tagged 25 or 50 copies of GA, GR, PA, and PR [39]. Unlike the expanded G4C2 repeat models described above, these DPR protein models differ only in their length and tag. It is worth noting that in C9ORF72 patients, the exact length and composition of different DPR proteins remain unknown, and the expression level of each DPR protein may vary by anatomical region and in different neuronal cell types as a result of different rates of production and degradation. Moreover, sensitive ELISA assays to quantify the level of each DPR protein remain to be developed. Thus, Drosophila models overexpressing DPR proteins of different lengths have their limitations but are likely to be informative.
Insights from Drosophila Models of C9ORF72 Repeat Expansion
RNA foci versus DPR toxicity
The relative contributions of repeat-containing RNA foci versus DPR proteins to disease pathogenesis were one of the first issues addressed in the Drosophila models of G4C2 repeat expansion described above. Expression of (G4C2)36 or (G4C2)103 in the eye caused neurotoxicity and rough eye phenotypes, and regulated expression of these constructs in adult postmitotic neurons reduced life span [32]. On the other hand, stop codons inserted into G4C2 repeat sequences to stop DPR protein abolished repeat toxicity [32]. Although repeat RNA toxicity cannot be completely ruled out since these stop codons may change the secondary structure of the repeat-containing mRNA, these results suggest that DPR proteins generated from these repeats are the dominant toxic agents in this fly model [32]. Consistent with this notion, expression of poly(GR) and poly(PR), but not other DPR proteins, in the fly eye causes retinal degeneration [32, 34, 37, 38]. Moreover, interrupted antisense RNA containing about 100 copies of G2C4 repeats that cannot be translated into DPR proteins does not elicit toxicity in Drosophila adult neurons [40].
In contrast to (G4C2)103 flies, flies with intronic (G4C2)160 expression that were grown at 25°C had no obvious toxicity in the eye or reductions in dendritic branching, locomotor activity, or life span despite the abundance of nuclear RNA foci, consistent with very low level expression of poly(GP) (and presumably other DPR proteins as well) [33]. Thus, the subcellular localization and toxicity of G4C2 repeats are heavily influenced by their molecular context (Figure 2). In intronic (G4C2)160 flies grown at 29°C, however, life span was modestly reduced and correlated with a 4-fold increase in poly(GP) production; the number of nuclear G4C2 RNA foci in neurons or glial cells remained the same [33]. Thus, DPR proteins appear to be primarily responsible for disease phenotypes caused by intronic (G4C2)160.
Figure 2. The Effect of Poly(A) Tail on the Subcellualr Localization and Toxicity of Expanded G4C2 Repeats.
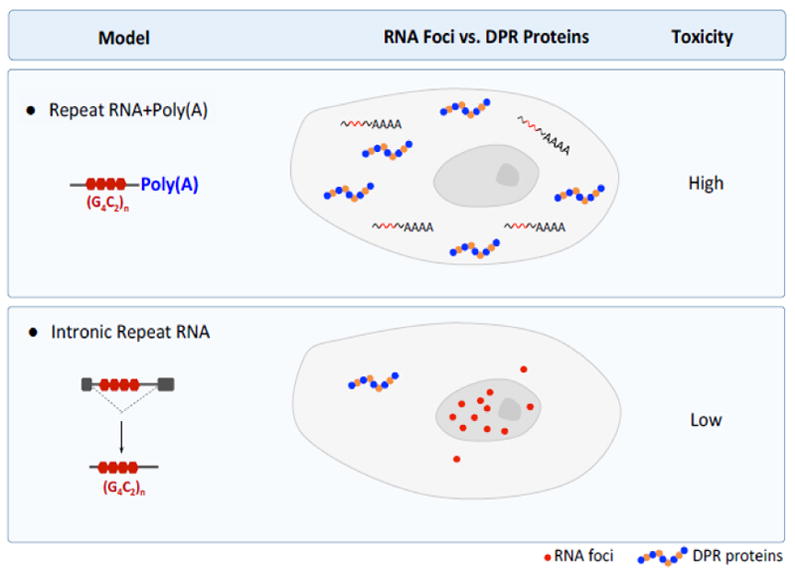
In (G4C2)n-polyA models (top panel), repeat RNA is transported to the cytoplasm, where it is translated into DPR proteins. Diffuse G4C2 RNA is detected mostly in the cytoplasm, and higher toxicity correlate with high levels of DPR protein production. In intronic (G4C2)n models (bottom panel), repeat RNA accumulates in the nucleus as RNA foci and less DPR protein is produced. Thus, only low levels of toxicity are observed.
Although results from Drosophila models cannot always be extrapolated to human patients, recent studies of postmortem tissues from C9ORF72 patients indicate that RNA foci are not the determinant of the clinicopathological phenotypes [41]. Moreover, among all DPR proteins, only poly(GR) seems to be significantly abundant in clinically related brain areas and correlates well with neurodegeneration [42]. Thus, it is critically important to further understand how poly(GR) alone causes neurodegeneration. On the other hand, it is highly likely that multiple DPR proteins are expressed in the same patient neuron. Indeed, poly(GA) can recruit poly(GR) into p62-positive aggregates in Drosophila cells [38]. To further investigate this issue, Drosophila will be an excellent model system for determining how different DPR proteins interact with each other.
Nucleocytoplasmic transport
Expression of (G4C2)28-GFP and (G4C2)58-GFP in a tissue-specific manner causes repeat-length- and dosage-dependent retinal degeneration in the fly eye and morphological and functional deterioration in motor neurons [34]. A collaborative large-scale genetic screen in this fly model showed that several components of the nuclear pore complex, including Nup50, Nup152, Nup107, and Nup160, enhance or suppress G4C2 repeat toxicity [34]. Moreover, other genes that function in RNA export and protein import were identified as genetic modifiers, highlighting the central role of nucleocytoplasmic transport in G4C2 repeat toxicity (Figure 3). Indeed, (G4C2)58-GFP expression compromises the integrity of the nuclear envelope of Drosophila salivary gland cells [34]. Because poly(GR), but not poly(GA) or poly(GP), also causes retinal degeneration in the fly eye [32, 34, 37, 38], it is likely that poly(GR) detected in (G4C2)58-GFP is at least partly responsible for the nucleocytoplasmic transport defects. Indeed, independent studies in yeast and fly by other groups also identified proteins involved in nucleocytoplasmic transport as strong genetic modifiers of toxicity induced by arginine-containing DPR proteins [39, 43] (Figure 3).
Figure 3. Overview of the Nucleocytoplasmic Transport Defects in C9ORF72-ALS/FTD.
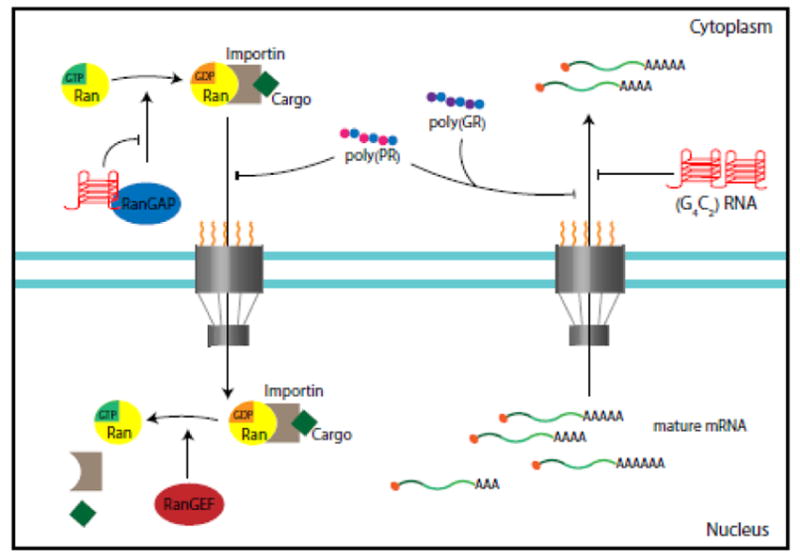
This cartoon summarizes the identified nuclear cytoplasmic transport defects in Drosophila, yeast and C9ORF72 iPSC-derived neurons. G4C2 quadruplex RNA disrupts the RAN gradient and the nuclear import of proteins by binding to RanGAP, which catalyzes the conversion of Ran-GTP to Ran-GDP. Impairment of nuclear export caused by repeat RNA or DPR proteins leads to nuclear retention of RNA.
In a study of proteins that bind to G4C2 repeat RNA in vitro, overexpression of Ran GTPase-activating protein (RanGAP) suppressed the rough eye phenotype induced by (G4C2)30-EGFP, and several other nucleocytoplasmic transport genes also genetically modified repeat toxicity [36]. RanGAP is a guanine exchange factor that catalyzes the hydrolysis of Ran-GTP to Ran-GDP, which mediates the import of proteins into the nucleus [44]. In Drosophila salivary gland cells expressing (G4C2)30-EGFP, the nuclear:cytoplasmic ratio of shuttling proteins such as Ran and TDP-43 is decreased [36]. Since RanGAP directly binds to G4C2 quadruplex RNA in an in vitro assay, and poly(GP) and poly(GR) were not detected in (G4C2)30-EGFP flies grown at ambient temperature, it was proposed that G4C2 repeat RNA is toxic in part because it sequesters RanGAP [36] (Figure 3). It is not known whether poly(GA)-containing protein is expressed in this fly model or why the RNA toxicity in this model, in which 30 copies of G4C2 repeats are interrupted by the XhoI restriction site, differs so drastically from that in flies expressing G4C2 repeat RNA interrupted by stop codons [(G4C2)36-RO] [32].
RNP granules
Like RanGAP [36], Pur-alpha—a DNA/RNA binding protein that participates in stress granule formation and mRNA transport and translation in neuronal dendrites [45–47]—interacts with G4C2 repeat RNA in vitro [31]. In (G4C2)30-EGFP flies, the rough eye phenotype was mitigated by overexpression of Pur-alpha, suggesting that it, too, modifies repeat toxicity [31]. A potential detrimental role for G4C2 repeats in dendrites was suggested by the finding that expression of (G4C2)48 in Drosophila dendritic arborization neurons led to branching defects that were modified by RNA transport proteins such as FMR1 and Orb2 [35]. Since poly(GR) can itself decrease dendritic branching [38], poly(GR) produced through RAN translation might conceivably be at least partly responsible for dendritic defects, in addition to toxicity caused by G4C2 repeat RNA in neuronal transport granules.
Independent proteomics analyses show that arginine-containing DPR proteins bind preferentially to many RNA binding proteins, including components of stress granules (SGs) [48–52]. Genes encoding many of these DPR-interacting proteins enhance or suppress poly(GR) or poly(PR) toxicity in the Drosophila eye [48]. Similarly, a study using APEX proximity labeling and mass spectrometry identified ~150 previously unknown human SG proteins, some of which were stress- and cell type-dependent [53]. Moreover, fly homologues of some of these newly identified SG genes genetically modified poly(GR) toxicity in Drosophila cells [53]. Thus, dysregulation of SG dynamics and function may be a central pathogenic mechanism in C9ORF72-ALS/FTD.
Induced Pluripotent Stem Cell (iPSC) Models of C9ORF72-ALS/FTD
In addition to Drosophila, induced pluripotent stem cell (iPSC) lines derived from skin fibroblasts or other cell types is a complementary and powerful approach, since these iPSC lines can be differentiated into different cell types such as cortical or motor neurons or glial cells, and disease-causing mutations are expressed in their native genetic and molecular contexts [54, 55]. Human neurons differentiated from iPSCs can be used to reveal many disease-relevant molecular and cellular phenotypes, including some age-related phenotypes. On the other hand, the selective vulnerability of different brain regions is challenging to address in iPSC models of neurodegenerative diseases. Moreover, most if not all iPSC differentiation protocols lead to the production of relatively immature human neurons that may not fully recapiculate the aging process of mature neurons in vivo [56]. Nonetheless, shortly after C9ORF72 repeat expansion was discovered, three laboratories independently generated iPSC lines from C9ORF72-ALS/FTD patients [7–9]. These and additional iPSC lines generated by other labs have been differentiated into cortical or motor neurons, and studies of these cells have provided important insights into disease mechanisms.
RNA Foci Toxicity in C9ORF72 iPSC-Derived Human Neurons
Both nuclear RNA foci and DPR proteins are found in human cortical and motor neurons differentiated from multiple C9ORF72 iPSC lines [7, 8, 24, 49, 57, 58]. Although RNase H-mediated degradation of repeat RNAs by antisense oligonucleotides appears to be beneficial in C9ORF72 iPSC-derived neurons and C9ORF72 BAC transgenic mice, treatment with antisense oligonucleotides reduces the levels of both repeat RNA and DPR protein production in neuronal and non-neuronal cells [8, 14, 24, 59, 60]. It is unclear therefore whether the beneficial effects are mediated by loss of either repeat RNA or DPR proteins. Some small molecules that bind to expanded G4C2 RNA and reduce the number of RNA foci also decrease DPR production in C9ORF72 iPSC-derived neurons or neurons directly transdifferentiated from fibroblasts [58, 61]. Thus, the relative toxicities of RNA foci and DPR proteins in human neurons are still unknown.
In vitro biochemical studies have identified a large number of proteins that can bind to expanded G4C2 repeat RNA in cell lysates [7, 8, 31, 62–68]. C9ORF72 iPSC-derived human neurons have been widely used to examine the extent of sequestration of some of these proteins in G4C2 RNA foci. For instance, proteins reported to partially colocalize with RNA foci in C9ORF72 iPSC-derived human neurons and patient brain tissues include ADARB2 [8], hnRNP-A1 and Pur-α [9], nucleolin [67], RanGAP [36], and hnRNP-H [63, 65, 68]. It is not known whether any of these proteins play a key role in neurodegeneration mediated by repeat RNA toxicity as shown for Muscleblind in myotonic dystrophy [19].
Nucleocytoplasmic Transport Defects in C9ORF72 iPSC-Derived Human Neurons
Nucleocytoplasmic transport defects reported in several Drosophila models of C9ORF72-ALS/FTD have been confirmed in C9ORF72 iPSC-derived neurons and patient brain samples, further supporting the role of this gene in disease pathogenesis. Nuclear retention of RNAs has been observed in C9ORF72 iPSC-derived cortical neurons, indicating the impairment of nuclear RNA export [34]. In C9ORF72 iPSC-derived neurons or neurons trandifferentiated from C9ORF72 fibroblasts, the nuclear:cytoplasmic ratio of Ran and RanGEF (RCC1) is decreased, leading to defects in nuclear import of proteins [36, 43]. Some defects related to nucleocytoplasmic transport were observed in C9ORF72 iPSC-derived neurons but not in neurons generated from direct conversion [36, 43], probably because of differences in differentiation protocols and/or the age of the neurons. The exact mechanism of nuclear transport defects in C9ORF72 neurons is unclear, although both G4C2 RNA-mediated RanGAP depletion and DPR-mediated disruption of nuclear pore complex have been suggested as potential mechanisms (Figure 3). Altered nucleocytoplasmic transport in iPSC-derived motor neurons from sporadic ALS cases and ALS patients with TDP-43 mutations has been attributed to TDP-43 pathology [69], providing further evidence that nucleocytoplasmic transport defects are a common pathogenic mechanism in ALS/FTD. In fact, nucleocytoplasmic transport is also impaired in iPSC-derived neurons from patients with Huntington disease and in mouse models of the disease [70, 71].
Other Insights from C9ORF72 iPSC-Derived Human Neurons
Autophagy and Lysosomes
One of the first cellular defects revealed in C9ORF72 iPSC-derived human neurons was compromise of the autophagy pathway [7]. Autophagy is a cytoplasmic bulk degradation pathway in which misfolded proteins and damaged organelles are transported to lysosomes for degradation, a process altered in many neurodegenerative diseases [72]. C9ORF72 iPSC-derived cortical neurons are more prone to cell death induced by autophagy inhibitors, and the level of the autophagy adaptor protein p62 is elevated in these cells [7]. This cellular defect could in part reflect partial loss of C9ORF72 function, since C9ORF72 protein has been implicated in the regulation of autophagy through binding to SMCR8 and ULK1 [17, 73–76]. Under starvation conditions, C9ORF72 is also localizes to the surface of lysosomes [75], and C9orf72 knockout mice have enlarged lysosomes, especially in microglia [17]. Although C9orf72 knockout mice do not exhibit obvious neurodegeneration [12–18], partial loss of C9orf72 function, such as in microglia, may contribute to disease progression [16]. Indeed, C9ORF72 is required for lysosomal biogenesis in iPSC-derived motor neurons, and partial loss of C9ORF72 impairs the clearance of endogenous DPR proteins [77]. More importantly, ectopic C9ORF72 expression in motor neurons derived from patient iPSCs improves survival, indicating that C9ORF72 haploinsufficiency contributes to disease in the presence of toxic DPR proteins [77].
Neuronal excitability
Another cellular phenotype found in C9ORF72 iPSC-derived neurons is abnormal neuronal excitability. C9ORF72 motor neurons fire fewer action potentials after 2 months in culture, and the decrease correlates with altered expression of several genes including KCNQ3, a regulator of neuronal excitability [24, 78]. However, hyperexcitability is observed in 2-week-old iPSC-derived motor neurons with C9ORF72 repeat expansion or other ALS mutations, in part because of reduced delayed-rectifier potassium current amplitudes [78, 79], suggesting an age-dependent transition from hyper- to hypoexcitiability, at least in this cellular model of C9ORF72-ALS/FTD. Hyperexcitability was proposed to induce calcium imbalance and subsequent excitotoxic cell death. Indeed, C9ORF72 iPSC-derived motor neurons are more susceptible to glutamate toxicity [8] and show increased Ca2+-permeable AMPA receptor expression [80]. Because iPSC-derived motor neurons are more similar to human fetal than adult spinal tissues [81], additional studies are needed to further investigate the misregulation of neuronal excitability in adult neurons of C9ORF72-ALS/FTD patients [82].
Nucleolar stress
The nucleolus is the site for ribosome biogenesis and also serves as a sensor for different types of stress. As a central hub for coordinated stress response, the nucleolus shows significant changes in morphology and composition [83]. Nucleolar stress in C9ORF72-ALS/FTD was first suggested by the aberrant subcellular localization of nucleolin in C9ORF72 iPSC-derived motor neurons and patient motor cortex [68]. Subsequently, nucleolin mislocalization was found in C9ORF72 BAC transgenic mice but without significant changes in ribosomal RNA processing or splicing [25]. DPR toxicity is likely at least partly responsible for nucleolar stress in C9ORF72-ALS/FTD since overexpressed poly(GR) or poly(PR) causes nucleolar stress such as nucleolar enlargement in both Drosophila and mammalian neuronal or nonneuronal cells [37, 38, 84, 85]. However, poly(GR) or poly(PR) does not seem to localize within the nucleolus in C9ORF72 patient brains [86], suggesting an indirect effect on nucleolar stress. Moreover, the extent of nucleolar stress in C9ORF72 patient brains remains to be further clarified [85, 86].
DNA damage
In many neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and ALS, DNA damage is increased and the DNA damage response is altered [87]. Indeed, increased DNA double-strand breaks are found in 4-month-old but not 2-week-old C9ORF72 iPSC-derived motor neurons [50]. Similar observations have been made in spinal motor neurons from C9ORF72 ALS patients [87]. Expression of poly(GR) in human control iPSC-derived motor neurons or poly(GR) and poly(PR) in human neuroblastoma cells can increase DNA damage [50, 88], in part because inhibiting ROS production partially decreases poly(GR)-induced DNA damage and increases oxidative stress in human motor neurons or poly(GR) toxicity in flies [50]. Moreover, increased DNA damage results in elevated P53 in C9ORF72 motor neurons, and partial reduction of P53 activity suppresses poly(GR) toxicity in flies [50]. The R-loop structure formed by G4C2 DNA-RNA hybrid and poly(GA) might also help increase DNA damage [89]. It remains to be determined how an altered DNA damage response (DDR) pathway contributes to eventual neurodegeneration in C9ORF72-ALS/FTD.
Concluding Remarks
Disease-relevant cellular phenotypes have been identified in C9ORF72 iPSC-derived motor or cortical neurons, and these cells have been increasingly used to confirm many phenotypes first observed in other experimental systems (Figure 4). For instance, these neurons also show vesicle trafficking defects [90], mislocalization of poly(GR)-interacting splicing factors [91], and changes in expression levels of genes of interest [38, 92, 93]. C9ORF72 iPSC-derived neurons have also been used to confirm cell-to-cell transmission of DPR proteins [94] and to test beneficial effects of therapeutic drugs [95]. However, iPSC-derived motor neurons are more similar to human fetal spinal tissues than adult spinal tissues [56], and C9ORF72 promoter methylation status may change during reprogramming [96]. Thus, novel experimental strategies are needed to establish better iPSC models of C9ORF72-ALS/FTD, such as use of three-dimensional organoid cultures [97]. Nonetheless, approaches combining the power of genetic analyses in Drosophila and the disease relevance of iPSC-derived human neurons will undoubtedly continue to be useful both for addressing many scientific challenges (see Outstanding Questions) and for revealing important insights into pathogenic mechanisms and therapeutic targets of C9ORF72-ALS/FTD and other neurodegenerative diseases.
Figure 4. Insights from C9ORF72 Patient-Derived Human Neurons.

Currently, patient-derived human neurons can be obtained by direct conversion of patient cells to neurons or by reprograming of the patient cells into induced pluripotent stem cells (iPSCs) followed by differentiation into neurons (iPSC-derived neurons). Several disease-relevant cellular phenotypes have been identified in studies of neurons derived from C9ORF72 patient. These include defects in autophagy and lysosome biogenesis, glutamate excitotoxicity, abnormalities in neuronal excitability, increased DNA damage, dysregulation of gene expression networks, vesicle trafficking defects, and nucleolar stress. In other instances, patient-derived neurons were used to confirm phenotypes observed first in other experimental systems. Examples include nucleocytoplasmic transport defects discovered in fly models, as well as pre-mRNA splicing and RNP granule transport defects and cell-to-cell transmission of di-peptide repeat (DPR) proteins discovered in other cell models.
Outstanding Questions.
What are the major toxic molecules in each Drosophila or mouse model of G4C2 repeat expansion and iPSC-derived neurons? How does partial loss of C9ORF72 function contribute to disease pathogenesis?
What is the composition of the G4C2 repeats in different C9ORF72 patients? Are they pure repeats or interrupted by other nucleotide sequences? How are repeats expanded or retracted in somatic cells?
What is the template for endogenous DPR production, spliced intron RNA containing repeats or unspliced mRNA? How are sense and antisense repeat RNAs translated in patient cells? Accordingly, what are the N-terminus, C-terminus, and size of different DPR proteins in patient cells?
Are all DPR proteins expressed in the same patient neuron or glial cell? What is the relative contribution of each DPR protein to disease pathogenesis? How do different DPR proteins interact with each other? What molecular events trigger TDP-43 pathology?
Several cellular pathways have been implicated in the pathogenesis of C9ORF72-ALS/FTD. What is the relative contribution of alterations in each pathway in disease initiation and progression? Which potential disease mechanism triggers the pathogenic events? How do different dysregulated molecular pathways interact with each other during disease progression?
How does the repeat expansion in C9ORF72 cause ALS in some carriers and FTD in others? Do other genetic or environmental factors contribute to the different manifestations of the same mutation?
What are the most predictive biomarkers for C9ORF72-ALS/FTD? Which therapeutic approaches are the most effective?
Highlights.
The toxicity of expanded G4C2 repeats in Drosophila models is influenced by their molecular context.
DPR proteins but not RNA foci are major toxic molecules in Drosophila models of G4C2 repeat expansion.
Nucleocytoplasmic transport defects are found in both Drosophila models of G4C2 repeat expansion and neurons derived from C9ORF72 iPSCs.
Multiple interconnected molecular pathways are dysregulated in C9ORF72-ALS/FTD, such as autophagy, stress granule dynamics, oxidative stress DNA damage, nucleolar stress, and neuronal excitability.
Experimental approaches that combine the genetic power of Drosophila and the disease relevance of iPSC-derived patient neurons are excellent strategies to help uncover pathogenic mechanisms in C9ORF72-ALS/FTD.
Acknowledgments
We thank Li Qiu for help with EndNote. We are also grateful for financial support from the National Institutes of Health (R37NS057553, R01NS101986 and R01NS093097 to F.-B.G.), the Packard Center for ALS Research, the MDA Foundation, and the Target ALS Foundation (F.-B.G.), as well as the Frick Foundation for ALS Research and the Alzheimer’s Association (NIRG-396129) (S.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hardiman O, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. doi: 10.1038/nrdp.2017.71. [DOI] [PubMed] [Google Scholar]
- 2.Gao FB, et al. Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 2017;36:2931–2950. doi: 10.15252/embj.201797568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olney NT, et al. Frontotemporal dementia. Neurol Clin. 2017;35:339–374. doi: 10.1016/j.ncl.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gijselinck I, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 7.Almeida S, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donnelly CJ, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belzil VV, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126:895–905. doi: 10.1007/s00401-013-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waite AJ, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;3:1779e5–1779.e13. doi: 10.1016/j.neurobiolaging.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao S, et al. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015;78:568–583. doi: 10.1002/ana.24469. [DOI] [PubMed] [Google Scholar]
- 12.Atanasio A, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204. doi: 10.1038/srep23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burberry A, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8:347ra93. doi: 10.1126/scitranslmed.aaf6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang J, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 Is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90:535–550. doi: 10.1016/j.neuron.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koppers M, et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015;78:426–438. doi: 10.1002/ana.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Rourke JG, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351:1324–1329. doi: 10.1126/science.aaf1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan PM, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016;4:51. doi: 10.1186/s40478-016-0324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ugolino J, et al. Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet. 2016;12:e1006443. doi: 10.1371/journal.pgen.1006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohan A, et al. RNA-protein interactions in unstable microsatellite diseases. Brain Res. 2014;1584:3–14. doi: 10.1016/j.brainres.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori K, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 21.Ash PE, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zu T, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110:E4968–E4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao FB, et al. Rethinking unconventional translation in neurodegeneration. Cell. 2017;(171):994–1000. doi: 10.1016/j.cell.2017.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sareen D, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Rourke JG, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88:892–901. doi: 10.1016/j.neuron.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peters OM, et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron. 2015;88:902–909. doi: 10.1016/j.neuron.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, et al. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron. 2016;90:521–534. doi: 10.1016/j.neuron.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 28.McGurk L, et al. Drosophila as an in vivo model for human neurodegenerative disease. Genetics. 2015;201:377–402. doi: 10.1534/genetics.115.179457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Senturk M, Bellen HJ. Genetic strategies to tackle neurological diseases in fruit flies. Curr Opin Neurobiol. 2017;50:24–32. doi: 10.1016/j.conb.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Costa MDC, et al. Unbiased screen identifies aripiprazole as a modulator of abundance of the polyglutamine disease protein, ataxin-3. Brain. 2016;139:2891–2908. doi: 10.1093/brain/aww228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Z, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110:7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizielinska S, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran H, et al. Differential toxicity of nuclear RNA foci versus dipeptide repeat proteins in a Drosophila model of C9ORF72 FTD/ALS. Neuron. 2015;87:1207–1214. doi: 10.1016/j.neuron.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freibaum BD, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–313. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burguete AS, et al. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife. 2015;4:e08881. doi: 10.7554/eLife.08881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang K, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wen X, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72 ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84:1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang D, et al. FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. 2015;130:525–535. doi: 10.1007/s00401-015-1448-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boeynaems S, et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016;6:20877. doi: 10.1038/srep20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moens TG, et al. Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathol. 2018;135:445–457. doi: 10.1007/s00401-017-1798-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeJesus-Hernandez M, et al. In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol. 2017;134:255–269. doi: 10.1007/s00401-017-1725-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saberi S, et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2017 doi: 10.1007/s00401-017-1793-8. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jovicic A, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–1229. doi: 10.1038/nn.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore MS. Ran and nuclear transport. J Biol Chem. 1998;273:22857–22860. doi: 10.1074/jbc.273.36.22857. [DOI] [PubMed] [Google Scholar]
- 45.Kanai Y, et al. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 46.Johnson EM, et al. Role of Pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res. 2006;83:929–943. doi: 10.1002/jnr.20806. [DOI] [PubMed] [Google Scholar]
- 47.Daigle JG, et al. Pur-alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathol. 2016;131:605–620. doi: 10.1007/s00401-015-1530-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin Y, et al. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell. 2016;167:789–802. e12. doi: 10.1016/j.cell.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee KH, et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell. 2016;167:774–788. e17. doi: 10.1016/j.cell.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lopez-Gonzalez R, et al. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron. 2016;92:383–391. doi: 10.1016/j.neuron.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boeynaems S, et al. Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol Cell. 2017;65:1044–1055. e5. doi: 10.1016/j.molcel.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin S, et al. Evidence that C9ORF72 dipeptide repeat proteins associate with U2 snRNP to cause mis-splicing in ALS/FTD patients. Cell Rep. 2017;19:2244–2256. doi: 10.1016/j.celrep.2017.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markmiller S, et al. Context-dependent and disease-specific diversity in stress granules formed from pre-existing protein interactions. Cell. 2018;164:487–498. doi: 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 55.Mertens J, et al. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat RevNeurosci. 2016;17:424–437. doi: 10.1038/nrn.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho R, et al. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat Neurosci. 2016;19:1256–1267. doi: 10.1038/nn.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dafinca R, et al. C9orf72 Hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral aclerosis and frontotemporal dementia. Stem Cells. 2016;34:2063–2078. doi: 10.1002/stem.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simone R, et al. G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol Med. 2017;10:22–31. doi: 10.15252/emmm.201707850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lagier-Tourenne C, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110:E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gendron TF, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aai7866. pii: eaai7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Su Z, et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83:1043–1050. doi: 10.1016/j.neuron.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reddy K, et al. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem. 2013;288:9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee YB, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5:1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mori K, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125:413–423. doi: 10.1007/s00401-013-1088-7. [DOI] [PubMed] [Google Scholar]
- 65.Cooper-Knock J, et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 2014;137:2040–2051. doi: 10.1093/brain/awu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haeusler AR, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rossi S, et al. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J Cell Sci. 2015;128:1787–1799. doi: 10.1242/jcs.165332. [DOI] [PubMed] [Google Scholar]
- 68.Conlon EG, et al. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife. 2016;5:e317820. doi: 10.7554/eLife.17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chou, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. doi: 10.1038/s41593-017-0047-3. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grima JC, et al. Mutant huntingtin disrupts the nuclear pore complex. Neuron. 2017;94:93–107. e6. doi: 10.1016/j.neuron.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gasset-Rosa F, et al. Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron. 2017;94:48–57. e4. doi: 10.1016/j.neuron.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Menzies FM, et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–1034. doi: 10.1016/j.neuron.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 73.Sellier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016;35:1276–1297. doi: 10.15252/embj.201593350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang M, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. 2016;2:e1601167. doi: 10.1126/sciadv.1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Amick J, et al. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell. 2016;27:3040–3051. doi: 10.1091/mbc.E16-01-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jung J, et al. Multiplex image-based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. Elife. 2017;6:E23063. doi: 10.7554/eLife.23063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shi Y, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24:313–325. doi: 10.1038/nm.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Devlin AC, et al. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun. 2015;6:5999. doi: 10.1038/ncomms6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wainger BJ, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Selvaraj BT, et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMAP receptor-mediated excitotoxicity. Nat Commun. 2018;9:347. doi: 10.1038/s41467-017-02729-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ho R, et al. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat Neurosci. 2016;19:1256–1267. doi: 10.1038/nn.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schanz O, et al. Cortical hyperexcitability in patients with C9ORF72 mutations: Relationship to phenotype. Muscle Nerve. 2016;54:264–269. doi: 10.1002/mus.25047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boulon S, et al. The nucleolus under stress. Mol Cell. 2010;40:216–227. doi: 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kwon I, et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mizielinska S, et al. Bidirectional nucleolar dysfunction in C9orf72 frontotemporal lobar degeneration. Acta Neuropathol Commun. 2017;5:29. doi: 10.1186/s40478-017-0432-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schludi MH, et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015;130:537–555. doi: 10.1007/s00401-015-1450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Madabhushi R, et al. DNA damage and its links to neurodegeneration. Neuron. 2014;83:266–282. doi: 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Farg MA, et al. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet. 2017;26:2882–2896. doi: 10.1093/hmg/ddx170. [DOI] [PubMed] [Google Scholar]
- 89.Walker C, et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat Neurosci. 2017;20:1225–1235. doi: 10.1038/nn.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aoki Y, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017;140:887–897. doi: 10.1093/brain/awx024. [DOI] [PubMed] [Google Scholar]
- 91.Yin S, et al. Evidence that C9ORF72 dipeptide repeat proteins associate with U2 snRNP to cause mis-splicing in ALS/FTD patients. Cell Rep. 2017;19:2244–2256. doi: 10.1016/j.celrep.2017.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sivadasan R, et al. C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci. 2016;19:1610–1618. doi: 10.1038/nn.4407. [DOI] [PubMed] [Google Scholar]
- 93.Coyne AN, et al. Post-transcriptional inhibition of Hsc70-4/HSPA8 expression leads to synaptic vesicle cycling defects in multiple models of ALS. Cell Rep. 2017;21:110–125. doi: 10.1016/j.celrep.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Westergard T, et al. Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 2016;17:645–652. doi: 10.1016/j.celrep.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Imamura K, et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci Transl Med. 2016;9 doi: 10.1126/scitranslmed.aaf3962. pii: eaaf3962. [DOI] [PubMed] [Google Scholar]
- 96.Esanov R, et al. C9orf72 promoter hypermethylation is reduced while hydroxymethylation is acquired during reprogramming of ALS patient cells. Exp Neurol. 2016;277:171–177. doi: 10.1016/j.expneurol.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kawada J, et al. Generation of a motor nerve organoid with human stem cell-derived neurons. Stem Cell Reports. 2017;9:1441–1449. doi: 10.1016/j.stemcr.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]