

Abstract
Engineered crystallizable fragment (Fc) regions of antibody domains, which assume a unique and unprecedented asymmetric structure within the homodimeric Fc polypeptide, enable completely selective binding to the complement component C1q and activation of complement via the classical pathway without any concomitant engagement of the Fcγ receptor (FcγR). We used the engineered Fc domains to demonstrate in vitro and in mouse models that for therapeutic antibodies, complement-dependent cell-mediated cytotoxicity (CDCC) and complement-dependent cell-mediated phagocytosis (CDCP) by immunological effector molecules mediated the clearance of target cells with kinetics and efficacy comparable to those of the FcγR-dependent effector functions that are much better studied, while they circumvented certain adverse reactions associated with FcγR engagement. Collectively, our data highlight the importance of CDCC and CDCP in monoclonal-antibody function and provide an experimental approach for delineating the effect of complement-dependent effector-cell engagement in various therapeutic settings.
Therapeutic monoclonal antibodies (mAbs) ameliorate disease by two mechanisms that involve the binding and resultant modulation of the function of proteins associated with pathophysiology and the recruitment of effector mechanisms dependent on the crystallizable fragment (Fc) regions of antibody domains; these functions mediate, either directly or indirectly, the neutralization and clearance of targeted substrates, as well as the programming of adaptive immunity1,2. Effector functions arise from the binding of the Fc domain of immunoglobulin G (IgG) to Fcγ receptors (FcγRs) expressed on various leukocyte subsets and also from recruitment of the complement component C1q and the ensuing activation of the classical complement pathway. Human effector FcγRs include, in addition to the well-characterized ‘classical’ (type I) receptors (in humans, FcγRI, FcγRII, FcγRIII and their isoforms), the lectin-like type II receptors (CD23 and CD209), TRIM21 and members of the FCRL family of receptors3,4. The recruitment and signaling of type I receptors via immunocomplexes (ICs) are responsible for antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellmediated phagocytosis (ADCP), reactions that have been established clinically to contribute to the mechanism of action of many therapeutic antibodies5. Alternatively, activation of the classical complement pathway leads to target-cell clearance by two distinct processes6: first, direct cell lysis that results from insertion of the membrane attack complex into the cell membrane (complement-dependent cytotoxicity (CDC)); and second, the deposition of opsonins, such as C3b, that are covalently bound onto the cell surface and in turn are recognized by complement receptors (CRs) on effector cells. The CRs activated by the deposited opsonins trigger complement-dependent cell-mediated cytotoxicity (CDCC) and complement-dependent cell-mediated phagocytosis (CDCP)6,7. Additionally, activation of the classical pathway has been established to stimulate B cell and T cell adaptive immune responses8.
Determining in a quantitative way the relative roles of complementdependent and FcγR-dependent effector mechanisms in mAb function is critical for the development of improved therapeutics9,10. However, this has proven to be a very difficult problem to address experimentally, as evinced by the longstanding debate about the relative importance of complement in the clearance of CD20+ B cells by mAbs (such as rituximab (Rituxan)) to the B cell–specific surface antigen CD20 (refs. 11,12). IgG isotypes capable of activating complement also bind to FcγRs to varying degrees, especially after the formation of highly aggregated ICs on target cells or viruses13,14. As a result, it is not possible to distinguish, in the presence of serum, whether target-cell lysis by antibodies is dominated by ADCC or CDCC and, similarly, whether phagocytosis is due to ADCP or CDCP. While ADCC and ADCP can be readily studied by well-established in vitro assays15, there is no straightforward manner with which to quantify the effect of CDCC and CDCP on target-cell clearance by mAbs. Because the C1qand FcγR-binding sites on the Fc domain are proximal and partially overlap, amino-acid substitutions engineered to diminish the binding of FcγRs also eliminate the recruitment of C1q and vice versa16,17.
Among the cell-elimination pathways triggered by the classical complement pathway, CDC activity is by far the easiest to measure and has been studied in great detail11,15. In contrast, apart from the results of some very early, qualitative studies from more than 40 years ago, with polyclonal antibodies18, practically nothing is known about the kinetics and magnitude of target-cell elimination by CDCC and CDCP or their importance in mAb function. In the presence of serum, C3 fragments become deposited onto target cells as a result of activation of the classical pathway. Opsonized target cells are recognized by both CRs and FcγRs on effector cells. The different signaling pathways triggered by the activation of CRs and/or FcγRs ultimately result in killing of the target cells either through the release of cytotoxic proteins by effector cells or through phagocytosis. While synergism in the elimination of substrates when both CRs and FcγRs are activated has been inferred from some studies19, other reports have suggested antagonistic or opposing effects20, and the precise role of CDCC and CDCP in the absence of confound effects due to FcγR engagement is not known.
RESULTS
Engineering of aglycosylated C1q-selective IgG1 Fc domains
To delineate in detail the role of CDCC and CDCP in target-cell clearance, among diverse effector functions (Fig. 1a), we focused on engineering C1q-selective, aglycosylated antibodies that lacked the ubiquitous Asn297 glycan in the Fc domain. In aglycosylated mAbs, glycan-mediated effects such as signaling via type II receptors are not relevant and, from a technical standpoint, very large libraries of aglycosylated IgG1 antibody variants with mutant Fc domains can be readily screened via display by Escherichia coli and multicolor flow cytometry to achieve a desired ligand-binding selectivity profile21–23. Thus, we screened a library of approximately 1.5 × 109 E. coli transformant cells expressing full-length IgG1 antibodies with mutant Fc domains (Supplementary Fig. 1a,b and Supplementary Table 1) by flow cytometry via labeling with 10 nM phycoerythrin (PE)-labeled human C1q in the presence of a 100-fold excess (1 μM) of each human FcγR (FcγRI–FcγRIIIa) for counter-selection and in high-salt buffer to eliminate the nonspecific interaction of C1q with the E. coli surface (Supplementary Fig. 1c,d). Bacterial clones enriched after the seventh screening round were evaluated for binding to C1q-PE and lack of binding to high-avidity tetramers of streptavidin-tagged human FcγRIIIaV158-PE, one of two natural variants of FcγRIIIa (Supplementary Fig. 1e–h). IgG1 antibodies with mutant Fc domains from selected clones were constructed with the antigen-binding fragment (Fab) of Rituxan (mAb to human CD20) and were expressed and purified from mammalian cells either in glycosylated form or as aglycosylated antibodies through the introduction of a T299L substitution that disrupts the Asn297X298-Thr299 (where ‘X’ is any amino acid) glycosylation motif in the Fc (Supplementary Fig. 2). We evaluated binding to human C1q or to the human FcγRs by enzyme-linked immunosorbent assay and surface-plasmon-resonance analysis with dimerized high-avidity glutathione S-transferase–tagged human low-affinity FcγRs to detect even very weak interactions (Table 1 and Supplementary Fig. 3a,b). Two Rituxan variants with mutant Fc domains were studied in detail; clone 801, which contains the amino-acid substitutions K320E (in the second domain of the immunoglobulin heavy-chain γ-constant region (Cγ2)) and Q386R (in Cγ3), bound to C1q selectively in both the aglycosylated form (the Fc domain of aglycosylated clone 801 (A801) constructed with the Fab arms of Rituxan (RA801)) and the glycosylated form (the Fc domain of glycosylated clone 801 (G801) constructed with the Fab arms of Rituxan (RG801)). As analyzed by surface plasmon resonance, RA801 had no detectable binding to any FcγR, whereas RG801 bound very weakly only to the high-affinity receptor FcγRI (Table 1 and Supplementary Fig. 3a,b). Rituxan clone 802 (with the amino-acid substitutions L245K, G246M, G247R and L351Q) showed selective binding to C1q only when expressed as an aglycosylated protein (RA802) (Table 1 and Supplementary Fig. 3a,b). Its glycosylated form, RG802, showed complete loss of binding to C1q and to effector FcγRs (Table 1 and Supplementary Fig. 3a,b). Both RA801 and RA802 displayed pH-dependent binding to the human neonatal Fc receptor (FcRn), with an affinity at pH 6.0 comparable to that of wild-type glycosylated IgG1 (Supplementary Fig. 3c); this suggested that the substitutions in these antibodies were unlikely to have a negative effect on recycling by FcRn or pharmacokinetics in vivo.
Figure 1.
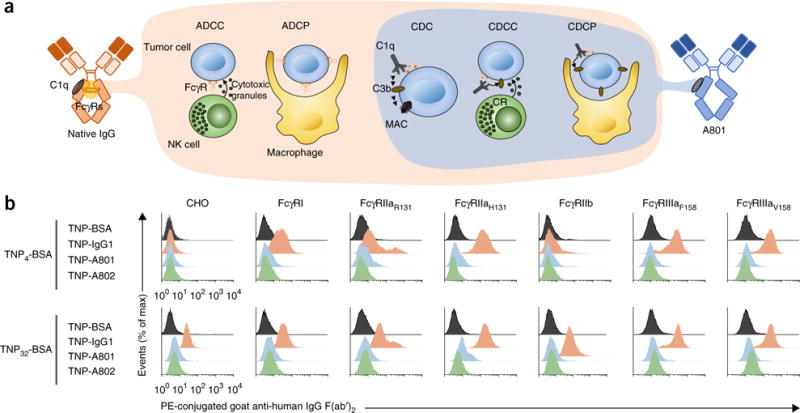
Biochemical and functional properties of antibodies with engineered Fc domains that bind to C1q with exquisite selectivity. (a) Antibodymediated FcγR mechanisms for cell killing (left: ADCC and ADCP) and complement-dependent mechanisms for cell killing (right: CDC, CDCC and CDCP). MAC, membrane attack complex. (b) Binding of lowand high-density ICs (left margin) formed by IgG1, A801 or A802 conjugated to TNP4-BSA (top) or TNP32-BSA (bottom) on CHO cells expressing human FcγRs14. Data are from one experiment representative of three experiments.
Table 1.
Binding properties of the engineered antibody variants
| N-gly | C1q | FcγRI | FcγRIIaH131 | FcγRIIaR131 | FcγRIIb | FcγRIIIaV158 | FcγRIIIaF158 | FcRn | |
|---|---|---|---|---|---|---|---|---|---|
| IgG1 | Yes | 23.0 | 1.5 | 120 | 310 | 1300 | 195 | 390 | 623 |
| IgG 801 | No (RA801) | 0.108 (213-fold) | NB | NB | NB | NB | NB | NB | 959 |
| Yes (RG801) | 0.385 (60-fold) | 648 (0.002 fold) | NB | NB | NB | NB | NB | 283. | |
| IgG 802 | No (RA802) | 0.145 (158-fold) | NB | NB | NB | NB | NB | NB | 110 |
| Yes (RG802) | NB | NB | NB | NB | NB | NB | NB | 289. |
Apparent dissociation constant (KD) values (nM) for the binding of RA801 and RA802 (far left) to human C1q, human FcγRs (two natural variants of FcγRIIa (His131 and Arg131) and FcγRIIIa (Phe158 and Val158), with variant amino acids subscripted) or human FcRn (assessed at a pH of 6.0) (top row), estimated with a global two-state binding model for C1q binding, the langmuir 1:1 binding model for the binding of the high-affinity receptor FcγRI or a bivalent binding model for dimeric forms of the low-affinity FcγRs, as described22; results were calculated by the following equation: KD (glycosylated IgG)/KD (IgG 801 or 802). NB, no binding detected. Data are representative of three experiments.
The binding of ICs onto cells represents an exquisitely sensitive assay for the detection of physiologically relevant IgG–FcγR interactions13,14. To form ICs with defined stoichiometries, we used antibodies comprising the Fc domain of A801 or A802 fused to α–2-, 4-,6-trinitrophenyl (TNP) Fab arms (TNP-A801 and TNP-A802, respectively) and incubated those with bovine serum albumin (BSA) conjugated to an average of either 4 TNP molecules (TNP4-BSA; lowavidity IC) or 32 TNP molecules (TNP32-BSA; high-avidity ICs). ICs formed with wild-type antibody to TNP (anti-TNP) readily bound to Chinese hamster ovary (CHO) cells expressing each FcγR (Fig. 1b). In contrast, ICs of TNP-A801 and TNP-A802 of higher avidity (TNP32-BSA) or lower avidity (TNP4-BSA) showed no binding to FcγR-expressing CHO cells (Fig. 1b). We note that because of the higher surface hydrophobicity of TNP32-BSA, when mixed with IgG, TNP32-BSA ICs show greater background binding to CHO cells expressing no FcγRs than that of TNP4-BSA ICs.
RA801 and RA802 mediated potent CDC activity with three widely used CD20+ human Burkitt’s lymphoma cell lines (Raji, Ramos and Daudi), as well as with cells derived from patients with acute lymphocytic leukemia (Fig. 2a,b and Table 2). Complement activation of the classical pathway and opsonization of CD20+ cells by C3b was comparable to that achieved with Rituxan (Fig. 2c and Supplementary Fig. 4a,b). The lysis of Raji cells and Ramos cells via RA801 was characterized by EC50 values (effector concentration for a half-maximum response) 2.3-fold lower and 4.5-fold lower, respectively, than those obtained with Rituxan (Fig. 2a,b and Table 2). We also assessed ofatumumab, an approved second-generation mAb to CD20 whose enhanced clinical activity in chronic lymphocytic leukemia has been established to result from improved CDC24–26, and found that the greater CDC potency of RA801 compared favorably with that of ofatumumab: the EC50 values for Raji cells and Ramos cells obtained with ofatumumab were 1.9-fold lower and 4.1-fold lower, respectively, than those obtained with Rituxan (Fig. 2d).
Figure 2.
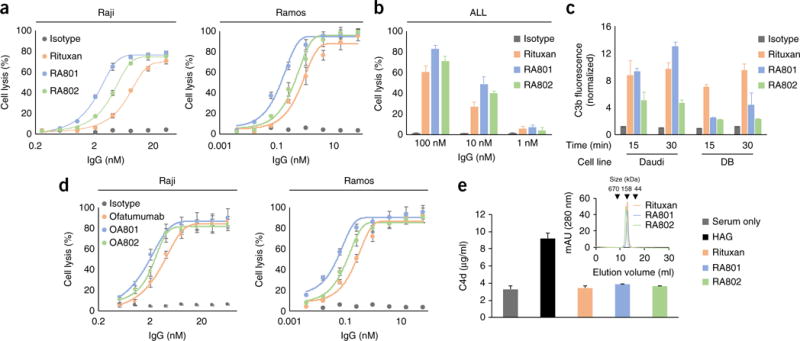
In vitro complement activation by C1q-specific antibodies to CD20. (a,d) Lysis (by CDC) of CD20+ Raji cells (left) or Ramos cells (right) opsonized by various concentrations (horizontal axes) of Rituxan or its variants RA801 or RA802 (a) or ofatumumab or its variants OA801 or OA802 (d), or isotype-matched control antibody (Isotype), plus 25% pooled human serum, assessed 1 h after the addition of cells and antibody. (b) Lysis of patient-derived acute lymphocytic leukemia cells (ALL) opsonized by antibodies as in a (key), assessed as in a,d. (c) Deposition of C3b on CD20+ Daudi (left) or DB (right) human B cell lymphoma cells opsonized by culture for 15 or 30 min (horizontal axis) with antibodies as in a (key); results are presented relative to those of cells opsonized with isotype-matched control antibody (clinical-grade trastuzumab). (e) Generation of C4d by serum alone, heat-aggregated IgG (HAG; positive control) or antibodies (as in a) in solution (key); inset, size-exclusion fast protein liquid chromatography of Rituxan, RA801 and RA802 (key). Data are from one experiment representative of three experiments (error bars, s.d.).
Table 2.
In vitro potency of the engineered antibody variants
| CDC | ADCC | ADCP | CDCC (or ADCC + CDCC) | CDCP | CMC (or ADCC + CMC) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Condition | PHS | +C9 | − | − | − | −C9 | +C9 | |||
| Effector | None | PBMC | PMN | MΦ | PBMC | PMN | MΦ | PBMC | PMN | |
| EC50 Raji | Isotype | No response | ||||||||
| Rituxan | 8.4 ± 1.7 | 9.5 ± 0.4 | 11.6 ± 0.8 | 0.04 ± 0.005 | 2.5 ± 0.1 | 1.9 ± 0.1 | 17.4 ± 0.2 | 1.0 ± 0.1 | 0.8 ± 0.02 | |
| RA801 | 3.7 ± 0.4 | – | 2.6 ± 0.2 | 2.1 ± 0.2 | 3.7 ± 0.04 | 0.5 ± 0.02 | 0.4 ± 0.02 | |||
| RA802 | 5.0 ± 1.0 | – | 6.2 ± 0.3 | 4.6 ± 0.2 | 4.6 ± 0.1 | 0.7 ± 0.03 | 0.6 ± 0.05 | |||
| EC50 Ramos | Isotype | No response | ||||||||
| Rituxan | 0.7 ± 0.1 | 0.8 ± 0.06 | 2.5 ± 0.2 | NA | 0.4 ± 0.03 | 0.3 ± 0.02 | NA | 0.2 ± 0.01 | 0.1 ± 0.01 | |
| RA801 | 0.2 ± 0.02 | – | NA | 0.3 ± 0.02 | 0.2 ± 0.01 | NA | 0.03 ± 0.002 | 0.02 ± 0.001 | ||
| RA802 | 0.4 ± 0.02 | – | NA | 0.5 ± 0.03 | 0.4 ± 0.02 | NA | 0.07 ± 0.005 | 0.06 ± 0.004 | ||
Lysis of antibody-opsonized Raji and Ramos cells (antibodies, second column) by various effector cells (third row), in the absence of serum (−) or in the presence of pooled human serum left undepleted (+C9) or depleted of C9 (−C9) (second row); results are presented as EC50 values (nM) and are ‘stratified’ by effector function (above table). MΦ, macrophage; NA, not assayed; CMC, complement-mediated cytotoxicity (including CDC and CDCC). Data are representative of three experiments (average ± s.d.).
The enhanced CDC killing potency of ofatumumab is due to its binding to a membrane-proximal epitope in CD20 that more optimally engages C1q27. Antibodies with the ofatumumab Fab fragments in combination with the clone 801 aglycosylated Fc domain (OA801) or clone 802 aglycosylated Fc domain (OA802) conferred on Raji cells and Ramos cells CDC of even greater potency than that conferred by ofatumumab (Fig. 2d and Supplementary Table 2).
After binding to antigens on the cell surface, IgG1 antibodies selfassociate via the Fc domain to form hexamers that, in turn, optimally engage C1q28. We investigated the possibility that the CDC activity of RA801 and RA802 might have been due to enhanced formation of hexamers. Certain amino-acid substitutions in the Fc region enhance the formation of hexamers (for example, IgG1 with three amino-acid substitutions: E345R, E430G and S440Y (RGY)), which results in the binding of C1q in solution and the activation of complement in the absence of antigen; this leads to the generation of the C4-complementcomponent cleavage product C4d28. In contrast to mutant IgG1 that is prone to form hexamers, RA801 and RA802 did not elicit C4d formation in solution or form oligomers that could be detected by size-exclusion chromatography (Fig. 2e). These results suggested that the activation of complement by RA801 and RA802 was probably not the result of an enhanced propensity to form hexamers, a conclusion also supported by the structural analysis reported below.
Analysis of RA801and RA802-mediated CDCC
In standard ADCC assays performed with culture medium in the absence of serum, RA801 and RA802 were completely unable to lyse CD20+ cells (Raji or Ramos) with either peripheral blood mononuclear cells (PBMCs) or polymorphonuclear cells (PMNs) as effector cells (Fig. 3a, Table 2 and Supplementary Fig. 4c). In contrast, in the presence of serum (25% of the total volume) that had been depleted of complement component C9 (to abolish the formation of membrane attack complexes and thus cell killing via CDC), RA801 and RA802 were very efficient in mediating target-cell lysis, with EC50 values slightly higher than those obtained with Rituxan under these conditions29 (Fig. 3b). In these experiments, cell lysis was strictly dependent on the presence of effector cells (PBMCs or PMNs) and was solely the outcome of CDCC, given that RA801 and RA802 were unable to bind to FcγRs and did not exhibit any CDC activity in serum depleted of C9 or any CDCC activity in in serum depleted of C1q (Fig. 3c,d). Thus, in serum, the efficacy of the lysis of CD20+ cells by RA801 and RA802, which perform only CDCC, was at least comparable to or greater than that of Rituxan, even though Rituxan executed target-cell killing via both FcγR-mediated mechanisms (ADCC) and CR-dependent mechanisms (CDCC). CR3 is a key receptor for the lysis of complement-opsonized target cells by effector cells26,30. The addition of blocking antibody to CR3 inhibited the lysis, by PBMCs and PMNs (as effector cells), of RA801-opsonized Raji cells in serum depleted of C9 (Fig. 3e and Supplementary Fig. 4e). Antibody blockade of CR4 also inhibited the lysis of RA801-opsonized cells by PBMCs and PMNs26,30 (Fig. 3e and Supplementary Fig. 4e). Thus, CR3 and CR4, which are expressed by a variety of myeloid cells, have an important role in CDCC. Finally, in assays performed in complete blood, a condition more relevant to in vivo settings, RA801 and RA802 were slightly more effective than Rituxan at achieving lysis of Raji cells and Ramos cells (Fig. 3f,g, Table 2 and Supplementary Fig. 4f). We concluded that at least within the time frame and conditions used in these experiments, complement activation made a major, if not dominant, contribution to the elimination of antibody-opsonized CD20+ cells.
Figure 3.
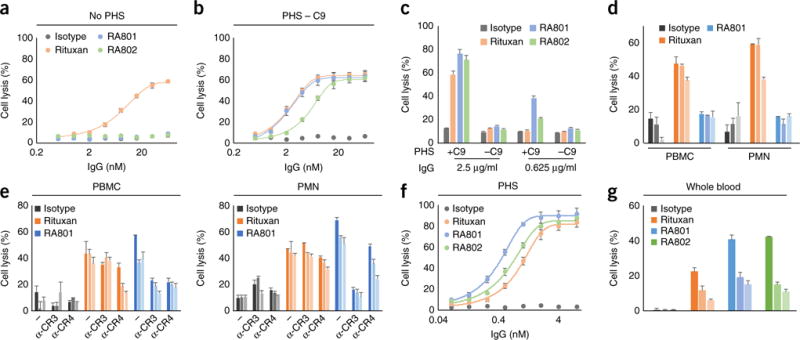
Killing of CD20+ cells by CDCC. (a,b) Lysis of Raji tumor cells (opsonized by various concentrations (horizontal axes) of isotype-matched control antibody (as in Fig. 1d), Rituxan, RA801 or RA802 (key)) by PBMCs (as effector cells), at an effector cell/tumor cell ratio of 10:1, in RPMI1640 medium without pooled human serum (No PHS) (a) or supplemented with 25% pooled human serum depleted of C9 (PHS – C9) (b); results are presented relative to those obtained by incubation in SDS lysis buffer. (c) Lysis (by CDC) of Raji cells opsonized by various concentrations (horizontal axes) of IgG antibodies as in a (key), assessed in the presence of undepleted serum (+C9) or serum depleted of C9 (−C9). (d) Lysis of Raji cells (opsonized by 20 μg/ml, 4 μg/ml or 0.8 μg/ml (dark to light bar shading) of isotype-matched control antibody, Rituxan or RA801 (key)) by PBMCs or PMNs in RPMI-1640 medium supplemented with 25% serum depleted of C1q. (e) Lysis of Raji cells (opsonized by 4 μg/ml, 0.8 μg/ml or 0.16 μg/ml (shading as in d) of antibodies as in d (key)) by CDCC via PBMCs or PMNs (above plots) coated with no antibody (−) or 10 μg/ml of anti-CR3 (α-CR3) or anti-CR4 (α-CR4) (below plots), in RPMI-1640 medium supplemented with 25% serum depleted of C9. (f) Lysis of Raji cells (opsonized as in a) by PBMCs, in RPMI-1640 medium supplemented with 25% pooled human serum. (g) Lysis of Raji cells (opsonized with 10 μg/ml, 2.5 μg/ml or 0.6 μg/ml (shading as in d) of antibodies as in a (key)) by PBMCs, in RPMI-1640 medium supplemented with 25% pooled whole blood. Data are from one experiment representative of three experiments (a–d,f,g) or two experiments (e) (error bars, s.d.).
Quantitative analysis of NK cell–mediated CDCC
Natural killer (NK) cells are by far the most effective cytolytic leukocyte population among PBMCs in vitro. NK cells can form stable conjugates with antibody-opsonized target cells via either FcγRIIIa (CD16a) or CRs26,30–32; this results in the release of cytotoxic granules, which leads to apoptosis and target-cell lysis. We used high-throughput single-cell killing assays to evaluate, at high resolution, details of the kinetics of the killing of Raji cells by human NK cells. We used single-cell assays because the time constants for processes such as the formation of synapses between effector and target cells, time required for cell lysis and the proficiency with which effector cells perform multiple killings cannot be determined by conventional macroscopic assays. In brief, fluorescence-labeled NK cells from two healthy donors were incubated in nanowells together with an average of one to three Raji (target) cells that had been opsonized with either Rituxan or RA801. Cell motility and lysis were monitored by time-lapse imaging microscopy in nanowell grids (TIMING)33 (Supplementary Fig. 5). The time to establish stable conjugation, the duration of conjugation before tumor-cell apoptosis and the time to tumor-cell apoptosis were determined for >150 single-cell killing events per sample (Fig. 4a,b). We observed no significant difference, following opsonization with Rituxan or RA801, in the frequency of oneor two-target-cell killing events (Fig. 4b). Mechanistically, killing via RA801 involves the formation of synapses between CRs and opsonins deposited on target cells, whereas with Rituxan, synapses can form both as a result of opsonin deposition and via the binding of antibody to FcγRIIIa on NK cells. The similar overall NK cell–killing activity or serial killing of more than one cell induced by the two mAbs indicated that either CR engagement had a dominant role in cytotoxic synapse formation or that, alternatively, in the presence of serum, FcγRIIIa-mediated cell killing was suppressed. Consistent with the latter hypothesis, complement activation has been shown to inhibit FcγRIIIadependent ADCC20.
Figure 4.
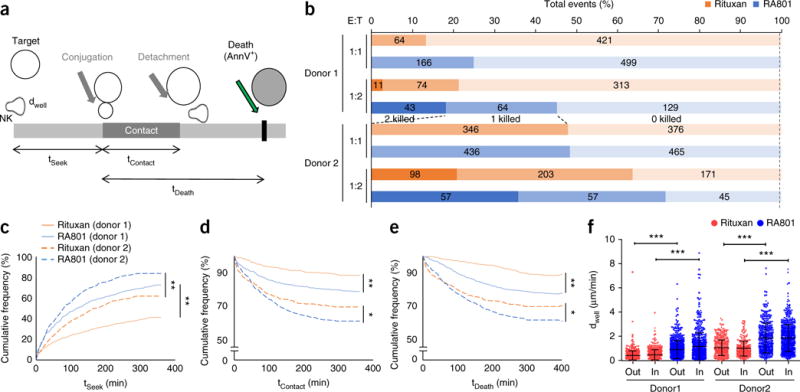
Quantitative analysis of RA801-mediated CDCC. (a) Features of single-cell analysis of target-cell killing by TIMING33. AnnV, annexin V. (b) Frequency of the killing of Raji cells (target cells opsonized by Rituxan or RA801 (key)) by human NK cells (effector cells from two donors; far left), at various effector cell/target cell (E:T) ratios (left margin), in the presence of 25% serum depleted of C9, monitored by TIMING and presented as 0, 1 or 2 target cells killed (bar shading; middle of plot); numbers in bars indicate percent for each bar section. (c–e) Kaplan-Meier curves of the time to establish stable conjugation (tSeek) (c), total duration of conjugation before tumor-cell apoptosis (tContact) (d) and time between first contact and tumor-cell apoptosis (tDeath) (e) for NK cells (from two donors; key) incubated with Raji tumor cells (at a ratio of 1:1) opsonized with Rituxan or RA801 (key). (f) Motility of NK cells (donor 1, >840 cells; donor 2, >400 cells) in the presence of Raji cells opsonized with Rituxan or RA801 (key), with cells at a ratio of 1:1, presented as displacement (dWell) between two time-lapse images (displacement out of contact and in contact with the target cell computed separately33). Each symbol represents an individual cell; small horizontal lines indicate the average (± s.d.). *P ≤ 0.05, **P ≤ 0.0001 and ***P ≤ 0.00001 (log-rank test (c–e) or one-way analysis of variance test (f)). Data are from two independent experiments (n ͥ≥ 350 cells).
Notably, for both donors noted above and at an effector cell/target cell ratio of either 1:1 or 1:2, the times noted above (for establishment of conjugation, duration of conjugation before apoptosis and tumor-cell apoptosis) were all shorter for cells opsonized with RA801 than for those opsonized with Rituxan (Fig. 4c–e). Additionally, by quantifying the displacement of cells while out of or in contact with the target cells, we found that NK cells displayed greater motility in the presence of RA801-coated Raji cells than in the presence of Rituxan-coated Raji cells (Fig. 4f). This effect might have been related to the somewhat more efficient complement activation by RA801 and the ensuing deposition of inactivated C3b, whose receptor, CR3, is expressed on human NK cells26,30–32. Collectively, our data revealed that CDCC could be a major mechanism in the cytotoxic effect of NK cells on antibody-opsonized target cells.
Analysis of CDCP by RA801 and RA802
In the absence of serum, RA801or RA802-opsonized CD20+ cells were not phagocytosed by classically activated (M1) macrophages, even at the highest concentration of antibody tested (400 nM) (Fig. 5a). However, RA801 and RA802 were effective in performing phagocytosis by macrophages via complement when human serum depleted of C9 was added to the assay (Fig. 5b,c). The finding that phagocytosis with RA801 or RA802 occurred only in the presence of serum unequivocally demonstrated that it was mediated by complement deposition; i.e., it was due solely to CDCP. In the presence of serum, phagocytosis of RA801or RA802-opsonized cells was slightly more effective than that of Rituxan-opsonized cells, with EC50 values of 3.7 ± 0.04 nM, 4.6 ± 0.1 nM and 17.4 ± 0.2 nM (mean ± s.d.), respectively (Fig. 5b,c). Notably, at saturating concentrations of antibody, the presence of serum markedly increased the magnitude of phagocytosis and resulted in >80% clearance of Raji cells with either RA801 or Rituxan, compared with 30% cell clearance observed with ADCP (Fig. 5b,c). However, consistent with published reports34, serum increased the EC50 for phagocytosis via Rituxan (17.4 ± 0.2 nM with serum, compared with 37 ± 5 pM for medium only (mean ± s.d.); Fig. 5b,c).
Figure 5.
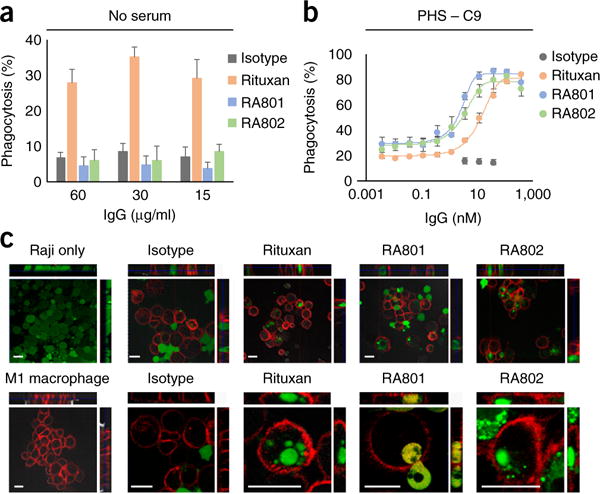
CDCP of CD20+ cells. (a,b) Phagocytosis of antibody-opsonized Raji cells (antibodies in key; concentration, horizontal axes) by monocytederived human M1-macrophages in RPMI-1640 medium without serum (a) or with serum depleted of C9 (b). (c) Fluorescence microscopy analyzing the phagocytosis of Raji cells, stained with the cell-permeant dye calcein-AM (green) by M1 macrophages, stained with anti-CD14 and anti-CD11b with allophycocyanin (red), showing Raji cells without macrophages (top left) or macrophages without Raji cells (bottom left), with opsonization by isotype-matched control antibody, Rituxan, RA801 or RA802 in the presence of serum depleted of C9 (top row, low magnification; bottom row, high magnification); top and right ‘strips’ show different views (xz and yz) of the same cells at a confocal plane. Scale bars, 20 μm. Data are from one experiment representative of three experiments (error bars (a,b), s.d. of technical triplicates).
Anti-tumor effects by complement in an in vivo model
We first established that RA801 did not bind to mouse FcγRs but was able to activate mouse complement and mediate CDC (Supplementary Fig. 6a–c). We evaluated the anti-tumor effect of RA801 in the treatment of established subcutaneous tumors formed by Ramos cells in outbred nude mice, which lack a thymus and mature T cells but produce macrophages and NK cells. When the tumors reached a diameter of 40–45 mm2, the mice were treated three times with mAb (10 mg per kg body weight), administered intraperitoneally. Nearly complete inhibition of tumor growth was observed with both Rituxan and RA801, despite the fact RA801 could not bind to FcγRs or perform ADCC or ADCP (Fig. 6a).
Figure 6. In vivo.
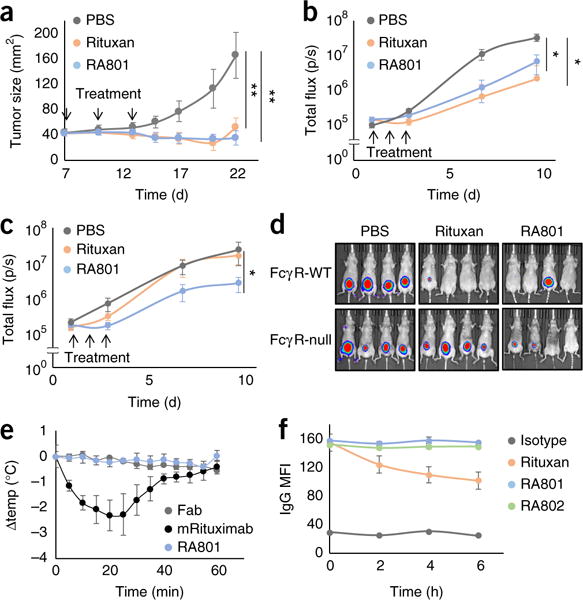
activity of RA801. (a) Growth of Ramos cell xenografts (1 × 106 cells) implanted subcutaneously into nude mice (n = 9) treated (downward arrows) with PBS or 200 μg of Rituxan or RA801 (key) administered after palpable tumors had developed, at day 7, 10 and 13 after tumor implantation. **P < 0.0001, versus PBS treatment (Student’s t-test). (b,c) Growth of luciferase-expressing EL4-hCD20 cell xenografts (1 × 105 cells) in NMRI-Foxn1−/− nude mice (FcγR-WT; n = 10 per group) (b) or NMRI-Foxn1−/− FcγR-null mice (n = 10 per group) (c) treated (upward arrows) with PBS or 100 μg of Rituxan or RA801 (key) administered on day s1, 2 and 3 after implantation; results are presented as tumor flux over time. *P < 0.01, versus PBS treatment (Mann-Whitney test). (d) Pseudocolor images of tumors from NMRIFoxn1−/− nude mice (FcγR-WT) and NMRI-Foxn1−/− FcγR-null nude mice (left margin) as in b,c, showing tumor intensity (high (red) to low (blue)) at day 10 after implantation. (e) Change in body temperature (Δtemp) in C57BL/6J mice (n = 3) at various times (horizontal axis) after intraperitoneal injection of 600 μg of heat-aggregated mouse Fab, IgG2aRituximab or RA801 (key). (f) Internalization of IgG into CD20+FcγRIIb+ Raji cells incubated for 0, 2, 4 or 6 h (horizontal axis) with 100 nM isotype-matched control antibody, Rituxan, RA801 or RA802 (key), assessed by flow cytometry and presented as the mean fluorescence intensity (MFI) of surface-bound IgG. Data are from one experiment representative of three independent experiments (a,e,f; error bars, s.d.) or are from two independent experiments (b–d; error bars (b,c), s.d.).
The clearance of EL4 mouse lymphoma cells expressing human CD20 (EL4-hCD20 cells) by Rituxan has been reported by some investigators to be completely dependent on FcγRs35, while others have found that either complement alone or both complement and FcγRs are important, in a manner that depends on tumor-cell burden and location26. We implanted 1 × 105 luciferase-expressing EL4-hCD20 cells subcutaneously into nude mice of the NMRI strain that were deficient in the transcription factor Foxn1 (NMRI-Foxn1−/− mice) and then treated the mice three times with RA801 or Rituxan (5 mg per kg body weight), administered intravenously. No significant difference between Rituxan-treated mice and RA801-treated mice was observed in terms of tumor growth, with each antibody being more effective than vehicle alone (Fig. 6b,d). In NMRI-Foxn1−/− mice lacking genes encoding all effector IgG receptors (Fcgr1, Fcgr2b, Fcgr3 and Fcgr4; called ‘FcγRnull’ here), the anti-tumor effect of Rituxan was abolished (Fig. 6c,d). Rituxan has weak complement activity with mouse serum36,37, and in mice in which FcγR-dependent effector mechanisms were abolished, complement activation alone was not sufficient to retard tumor growth (Fig. 6c,d). In contrast, administration of RA801 had a significant effect in slowing tumor growth in FcγR-null mice (Fig. 6c,d).
The presence of micro-aggregates in antibody preparations can trigger systemic, FcγR-mediated anaphylactic responses. It has been reported that intravenous administration of heat-aggregated Rituxan results in a rapid reduction in body temperature38. In contrast, we observed no change in body temperature in mice given injection of heat-aggregated RA801 (Fig. 6e). Separate from that, the engagement of FcγRs by anti-CD20 has been shown to lead to decreased surface expression of CD20 on B cells, a process that is mediated in part by the co-ligation of FcγRIIb and internalization of antibody–antigen complexes in cis39,40. Consistent with those observations, Raji cells incubated with Rituxan showed a significant decrease in surface expression of CD20, with a reduction of >40% by 6 h (Fig. 6f and Supplementary Fig. 6d). In contrast, cells incubated with RA801 showed no decreasing expression of surface CD20 (Fig. 6f and Supplementary Fig. 6d). Collectively, these findings revealed that mAbs that activated only complement had a strong anti-tumor effect in mouse models and, in addition, were able to circumvent detrimental effects of IgG1 antibodies known to be dependent on FcγR binding.
Structural analysis of the clone 801 Fc
To understand the molecular basis of the unique selectivity of the clone 801 Fc fragment for C1q, we solved the crystal structures of the Fc domain of both the aglycosylated clone 801 (A801-Fc) and glycosylated clone 801 (G801-Fc) at 2.3 Å and 3.2 Å, respectively (Table 3). The most striking feature of the A801-Fc structure was the extreme asymmetric orientation of Cγ2 in chain B (Cγ2B), relative to that of Cγ3 (Fig. 7a). Topologically, the orientation of Cγ2 relative to Cγ3 is described by the Cγ2–Cγ3 dihedral angle, which is defined from the Cα atoms of Tyr300 and Tyr319 in Cγ2, and Gln362 and Met428 in Cγ3 (ref. 41). Chain A of A801-Fc had a dihedral angle of −20.5°, which was slightly smaller than yet comparable to the dihedral angles of chain A observed in the ten glycosylated Fc (G-Fc) structures (−27° ± 4.1° (mean ± s.d.)) or the comparable angle (−25° ± 9.4°) in the four aglycosylated or deglycosylated human Fc proteins (A-Fc) in the Protein Data Bank (PDB) (Fig. 7a and Supplementary Table 3). However, chain B of A801-Fc showed an extreme twist and packed against Cγ2A, which resulted in a dihedral angle of 136° (Fig. 7a). As a comparison, in all other Fc domains in the PDB, chains A and B have similar dihedral angles (−23° ± 3.8° for ten G-Fc structures, and −25° ± 1.9° for four A-Fc structures). This extreme twist was observed only for A801-Fc, but G801-Fc showed similar dihedral angles for both chain A and chain B, comparable to those in other Fc structures (Supplementary Table 3). Although it is well established that the two homodimeric Fc chains crystallize with distinct temperature factors, dynamics and geometries for each chain41, the different orientation of Cγ2A and Cγ2B relative to that of Cγ3 in A801-Fc is unprecedented, to our knowledge. No direct physical contract of Cγ2 with other molecules in the asymmetric unit was observed (Supplementary Fig. 7a), which suggested that the dihedral angle in chain B of A801-Fc was probably not the result of crystal-packing artifacts. We used cross-linking with the carbodiimide EDC and liquid chromatography–tandem mass spectrometry to determine whether the observed crystal structure reflected the conformation of A801-Fc in solution. We observed a cross-linked product in A801-Fc between a peptide with Lys334 in chain A and a second peptide with Glu269 in chain B (Fig. 7b). In the highly asymmetric A801-Fc structure, the side chains of Lys334 in chain A and Glu269 in chain B were only 3.6 Å apart (Fig. 7b), a distance compatible with the formation of cross-linkage via EDC (~4 Å). In contrast, Lys334 in chain A and Glu269 in chain B were 24.1 Å apart in G-Fc (PDB accession code 3AVE) and were 24.1 Å apart in A-Fc (PDB accession code 3S7G) (Fig. 7b). Thus, the results of the crosslinking experiments (Fig. 7c) were fully consistent with, and provided independent support for, the proposal that the crystal structure of A801-Fc reflected the conformation of the molecule in solution.
Table 3.
Data collection and refinement statistics for G801-Fc and A801-Fc
| Data collection | G801-Fc | A801-Fc |
|---|---|---|
| Space group | P 1 21 1 | P 65 |
| Cell dimensions | ||
| a, b, c (Å) | 93.6, 141.0, 98.9 | 66.3, 66.3, 370.4 |
| α, β, γ (°) | 90.0, 117.7, 90.0 | 90.0, 90.0, 120.0 |
| Resolution (Å) | 40.0–3.22 (3.28–3.22) | 50.0–2.25 (2.29–2.25) |
| Rsym or Rmerge | 0.150 (0.808) | 0.089 (0.826) |
| I/σI | 9.69 (1.55) | 19.27 (1.24) |
| Completeness (%) | 99.90 (99.80) | 99.30 (96.40) |
| Redundancy | 3.6 (3.6) | 5.1 (3.7) |
| Refinement | ||
| Resolution (Å) | 40.0–3.22 (3.28–3.22) | 50.0–2.25 (2.29–2.25) |
| No. reflections | 36958 | 81432 |
| Rwork/ Rfree | 0.252/0.292 | 0.197/0.243 |
| No. atoms | 11844 | 6713 |
| Protein | 11351 | 6588 |
| Sugar atoms | 493 | 0 |
| Water | 0 | 125 |
| B-factors (Å2) | 84 | 64 |
| Protein | 84 | 64 |
| Sugar atoms | 101 | NA |
| Water | NA | 51 |
| r.m.s. deviations | ||
| Bond lengths (Å) | 0.002 | 0.005 |
| Bond angles (°) | 0.61 | 0.82 |
Shell of highest resolution is in parenthesis. NA, not applicable.
Figure 7.
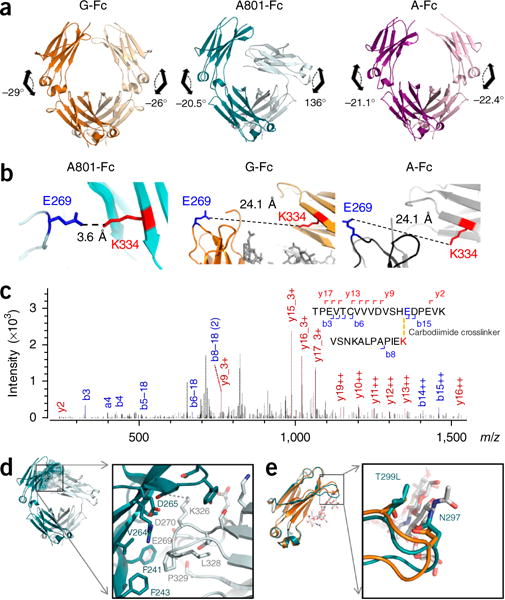
Structural features of A801-Fc and G801-Fc. (a) Dihedral angles of Cγ2–Cγ3 in G-Fc (PDB accession code 3AVE; left; orange), A801-Fc (PDB accession code 5V43; middle; teal) and G801 Fc (PDB accession code 5V4E; right; purple). (b) Distance from Glu269 in chain B (E269) to Lys334 in chain A (K334) in the crystal structures of A801Fc (PDB accession code 5V43), G-Fc (PDB accession code 3AVE) and A-Fc (PDB accession code 3S7G). (c) Tandem mass spectrometry of tryptic fragments of A801-Fc after crosslinking with EDC. (d) The chain A (teal)–chain B (gray) interface of A801-Fc. (e) Overlaid Cγ2 domain and the C’E loop (arrow) of A801-Fc (teal) and G-Fc (orange) structures; white, glycan in PDB accession code 3AVE. Data are representative of three independent experiments.
In the A801-Fc structure, loops BC and FG in Cγ2B formed extensive interactions with β-strands (A-B) in Cγ2A and buried 404 Å2 at the interaction interface42 (Fig. 7d). The principal contacts at this surface were dominated by hydrophobic interactions between residues Phe241, Phe243 and Val264 of chain A with backbone atoms of residues Glu269, Asp270, Lys326 and Pro329 in chain B. Furthermore, a salt bridge was formed between Asp265 of chain A and Lys326 of chain B.
The A801-Fc structure suggested a molecular basis for the selective binding to C1q and lack of binding to FcγRs. The K320E amino-acid substitution lay within the predicted C1q-binding region43–46 and probably mediated binding to C1q by electrostatic interactions. The binding of A801-Fc to C1q was probably the result of more subtle effects probably related to differences in the conformational flexibility of Cγ2. Within the A801-Fc structure, the apex of Cγ2B folded over to support and stabilize Cγ2A at the region normally occupied by the glycan (Supplementary Fig. 7b). The B factor of Cγ2A was substantially lower than that of Cγ2B (63 Å2 versus 93 Å2; Supplementary Fig. 7b). By comparison, Cγ2 in A-Fc structures23, which showed no binding to C1q, exhibited high B factors for both chain A and chain B (for example, 98 Å2 and 98 Å2, respectively, in PDB accession code3S7G), which was much larger than the B factors of Cγ3 in the respective structures (Supplementary Fig. 7b). Other biochemical data has also shown that removal of the Asn297 glycan increases the conformational flexibility of Cγ2 (ref. 23). We postulate that the enthalpic contribution from the K320E substitution, probably coupled with the lower flexibility of Cγ2A, was a key reason for the efficient binding of A801-Fc to C1q.
X-ray structures also provided an explanation for the complete loss of binding of FcγR to A801-Fc. With IgG1, FcγRs dock on both Cγ2A and Cγ2B and interact with two subsites centered near the C’E loop of one chain and the hinge LLPP motif of the other47. Interactions with both subsites are needed for FcγR binding, which results in an interaction surface of ~1,000 Å2, of which about 600 Å2 of interface is formed with the LLPP motif48. However, the change in the orientation of Cγ2B in A801-Fc precluded the simultaneous interaction of FcγRs with both subsites (Supplementary Fig. 7c).
The two FcγR-binding subsites of G801-Fc were sterically accessible; however, they seemed to be highly flexible and/or disordered. Specifically, in three of the four molecules within each asymmetric unit, either the LLPP motif or the C’E loop was disordered (Fig. 7e and Supplementary Fig. 7d,e). In the only molecule within the asymmetric unit in which these regions were sufficiently ordered to be to traced, the C’E loop assumed a ‘soft’ configuration observed only in A-Fc, which does not bind to FcγRs23,47 (Fig. 7e and Supplementary Fig. 7d,e). The high flexibility of the C’E loop of this ‘soft’ conformation would probably ‘penalize’ FcγR binding with a high entropic cost. Collectively, the X-ray crystal structures of A801-Fc and G801-Fc provided a structural explanation for the enhanced C1q binding and the lack of recognition by FcγRs.
DISCUSSION
Effector cells express both FcγRs and CRs; therefore, in the presence of serum, ADCC (or ADCP) and CDCC (or CDCP) occur at the same time13–15. Whereas ADCC and ADCP can be assayed readily without CDCC or CDCP taking place, simply through the use of medium depleted of serum and C1q, the reverse is not the case. For analysis of CDCC or CDCP alone, without ADCC or ADCP also taking place, it is necessary to block the binding of ICs to the multitude of FcγRs expressed by effector cells. We reasoned that a much more experimentally tractable approach for the study of CDCC and CDCP would be to engineer human IgG1 Fc domains that bound only to C1q and activated complement without engaging FcγRs. For this purpose, it was critical to ‘silence’ FcγR binding, since even very low-affinity interactions between IgGs and FcγRs can trigger effector phenotypes13,14. The engineering of Fc domains with absolute or nearly absolute C1q selectivity involved starting with aglycosylated antibodies in which binding to both C1q and FcγRs was substantially, albeit not completely, attenuated, followed by screening of very large mutant libraries for both binding to fluorescence-labeled C1q and the simultaneous absence of binding to high-avidity FcγRs. Using this approach, we isolated A801-Fc; of its two amino-acid substitutions, K320E in Cγ2 conferred substantial affinity for C1q, and Q386R in Cγ3 resulted in a radical conformational change that abolished any binding to FcγRs.
Overwhelmingly, the effector function of therapeutic antibodies is thought to be dominated by FcγR-dependent processes and/or by CDC. We demonstrated in various assays that effector-cellmediated substrate-clearance mechanisms triggered by activation of the classical complement pathway resulted in very extensive and rapid target-cell lysis and phagocytosis. Our results suggest that the extent and kinetics of the clearance of CD20+ cells via CDCC or CDCP are at least comparable to those due to ADCC or ADCP, respectively. Consequently, CDCC and CDCP deserve careful consideration and optimization in studies of the mechanism of action of mAbs.
The relative importance of CDCC and CDCP probably depends heavily on the target and disease setting. First, optimal activation of complement is critically dependent on the density and organization of antigens on the surface of target cells. Second, many pathogenic cells have high expression of complement-inhibitory proteins, and clearly, in these cases, activation of the classical pathway is compromised11. Third, it is well established that in vivo extensive activation of complement due to a considerable burden of target cells and antibody can rapidly lead to the depletion of either C1q or downstream complement components, such as C2 (ref. 49). Clearly, in such circumstances, ADCC and ADCP are probably the dominant antibody effector mechanisms. In conclusion, mAbs with absolute C1q-binding selectivity, as described here, might be useful for the eradication of pathogenic cells in certain disease states while circumventing FcγRdependent adverse effects, as well as the lower responsiveness to mAbs in patients with low-affinity FcγR polymorphisms50.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Cells and reagents
All cancer cell lines were tested for mycoplasma contamination before cytotoxicity assays. Burkitt’s lymphoma Raji (ATCC CCL-86), Ramos (ATCC CRL-1596), Daudi (ATCC CCL-213) and DB cancer cells (human B cell lymphoma, ATCC CRL2289) were obtained from American Type Culture Collection. TMD8 cells (human ABC-DLBCL cell line) and HBL-1 cells (human diffuse large B-cell lymphoma cell line) were obtained from P. Tucker (University of Texas at Austin). Patient-derived primary acute lymphocytic leukemia cells were obtained from D. Lee (MD Anderson Cancer Center). All cancer cells were cultured in complete RPMI with 10% FBS (FBS). The collection of CHO cells expressing human and mouse FcγRs was described previously13,14,51.
Human serum depleted of C1q or C9 was purchased from CompTech. Human FcγR and antibody proteins were produced in Expi293 cells (Invitrogen) as described previously22. All primers were synthesized by IDT (Supplementary Table 1).
Library construction and screening
IgG polypeptides were displayed on the inner membrane of Escherichia coli (E. coli) as described previously22. Random mutations were introduced on the Fc domain by three different methods (methods details, Supplementary Fig. 1b). As described previously22, the libraries were made into spheroplasts after induction with 1 mM of isopropyl1-thio-d-galactopyranoside (IPTG) and 2% d-arabinose, and were labeled with 10 nM C1q-PE in the presence of 1 μM of FcγRs as competitors and screened on a FACSAria (BD Biosciences) for seven rounds. 23 randomly selected clones were analyzed by flow cytometry using 10 nM C1q-PE or 10 nM of a tetramer of FcγRIIIaV158 (human FcγRIIIa of an allotype of higher affinity) and SA-PE.
Detection of lipopolysaccharide on spheroplasts of E. coli
Antibody M18 to PA domain 4 (10 μg/ml) was incubated with cells in spheroplasts expressing PA domain 4. In this manner, the spheroplasts are coated with full-length glycosylated IgG to enable monitoring of the binding of C1q or FcγRs onto cells in spheroplasts. Antibody-expressing cells in spheroplasts were incubated with 10 nM C1q-PE for 1 h in PBS, pH 7.4, or with high-salt phosphate buffer (50 mM phosphate and 330 mM NaCl, pH 7.4). Binding intensity of C1q-PE was detected by flow cytometry.
Preparation of antibodies, C1q and FcγR
Genes encoding 801-Fc and 802-Fc were cloned in-frame into the mammalian expression vector pcDNA3.4 with (or without) Fab of Rituximab or ofatumumab using Gibson Assembly cloning (NEB)52. Expression plasmids encoding histidine-tagged mouse FcγRI-IV (His-mFcγRI-IV) were constructed by cloning gene blocks of the respective genes synthesized by IDT into pcDNA3.4 vector (Invitrogen) using HindIII and Xho I sites. IgG proteins, human and mouse FcγRs were produced as described previously22. Human C1q protein was purchased from CompTech and was labeled with R-phycoerythrin (R-PE) using an EasyLink R-PE Conjugation Kit (Abcam) according to the manufacturer’s instructions.
Liquid chromatography–mass spectrometry of 801-Fc
The 801-Fc proteins were analyzed by liquid chromatography–mass spectrometry on an Orbitrap Fusion mass spectrometer (ThermoFisher). A linear gradient of 0.1% formic acid in water and 0.1% formic acid in acetonitrile over 10 min was used to elute the intact proteins from an OPTI-TRAPTM protein microtrap (Optimize Technologies). The Orbitrap Fusion was operated in Intact Protein Mode at 60,000 resolution from 400–2,000 m/z. Following acquisition, the data were deconvoluted using MagTran software.
Tandem mass spectrometry of crosslinked A801-Fc
A801-Fc (10 μM) and EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride) (1 mM) were mixed in a buffer containing 20 mM MES, pH 6.0, and 50 mM NaCl. Samples were incubated for 1 d at 4 °C before being separated by SDS-PAGE. The dimer bands on the SDS-PAGE gel were cut and trypsinized, and tryptic digests were analyzed by liquid chromatography–tandem mass spectrometry on an Orbitrap Elite (Thermo Fischer). Published methods were used for analysis of data obtained by mass spectrometry and tandem mass spectrometry53 and assignment of peptide spectral matches. Crosslinked peptides were identified using Protein Metrics Byonic software custom crosslinking with EDC on glutamic acid, aspartic acid and lysine residues.
Surface-plasmon-resonance analysis
To determine the affinities of antibody variants with C1q, FcγRs and FcRn, a Biacore 3000 instrument (GE Healthcare) was used with HBS-EP running buffer (GE Healthcare). Bovine serum albumin (BSA) was immobilized in the reference channel of the CM5 sensor chip, and each antibody was immobilized on the CM5 sensor chip by amine coupling at a pH of 5.0. All FcγRs (400 nM) or the serially diluted C1q proteins (1–40 nM) were injected into the CM5 chip at 30 μl/min for 2 min, followed by 10 min of dissociation. The chip was regenerated after each binding event with 10 mM glycine, pH 3.0, with a contact time of 1 min. FcRn–β2-microglobulin– glutathione S-transferase (730 nM) was injected in the CM5 chip at 30 μl/min in HBS-EP, pH 7.4, for 90 s, and the chip was regenerated after each binding event with 10 mM glycine, pH 3.0, with a contact time of 1 min. Monomeric FcRn–β2-microglobulin (50–400 nM) was injected into the CM5 chip at 30 μl/min in PBS, pH 6.0, for 90 s and the chip was regenerated after each binding event with 10 mM glycine, pH 3.0, with a contact time of 1 min. The resulting sensorgrams were fit with a global two-state binding model for C1q, and with a 1:1 Langmuir isotherm model for monomeric FcγRI and FcRn using Biaevaluation 3.0 software. Because C1q is hexameric protein, the measured KD values do not correspond to the true equilibrium dissociation constants for the interaction of the C1q globular head with IgG and are used here only as a measure of the relative affinity of the C1q for each antibody.
C3b deposition assays
Cells cultured in complete RPMI-1640 medium were mixed with an equal volume of normal human serum (NHS), and then mAbs were added to a final concentration of 10 μg/ml. After incubation for 30 min at 37 °C, the cells were washed twice with 1% BSA in PBS and developed with FITC-conjugated mouse mAb 3E7 to C3b (BioLegend; 1:50 dilution). Flow cytometry was performed on a FACSCalibur flow cytometer (BD Biosciences), and mean fluorescence intensity was converted into molecules of equivalent soluble fluorochrome with calibrated beads (Spherotech).
Solution-phase complement-activation assay
Complement activation in the absence of tumor cells was determined by measurement of the concentration of C4d, a product of the classical complement activation pathway, after incubation of 100 μg antibody in 1 ml 90% NHS for 1 h at 37 °C. C4d concentrations were measured by enzyme-linked immunosorbent assay (MicroVue C4d EIA kit, Quidel Corporation) according to the manufacturer’s instructions.
Size-exclusion chromatography (SEC)
SEC was performed on Agilent 1100 high-performance liquid chromatography system using a SuperdexTM200 10/300GC, (GE Healthcare), with a mobile phase of PBS, pH 7.4, at a flow rate of 0.5 ml/min. Proteins were detected by monitoring the absorbance at 280 nm. The injection amount was 100 μg protein in a volume of 100 μl.
Enzyme-linked immunosorbent assay
To measure binding of antibodies to FcγRs and C1q, 96-well plates (Qiagen) were coated with 1 μg/well of each antibody at 4 °C for 16 h and were washed three times with PBS containing 0.05% Tween20 (PBST). The plates were blocked for 1 h at 25 °C with 3% skim milk in PBS and were washed three times with PBST. FcγRs (50 nM or 500 nM) was added in plates. After 1 h of incubation at 25 °C, the plates were washed with PBST and incubated with 50 μl of PBS containing 1:5,000 goat antihistidine (Abcam; Cat# ab1269) or antibody to glutathione S-transferase IgG conjugated to HRP (Rockland; Cat# 600-103-200) for 1 h. After washing three times with PBST, 50 μl TMB substrate was added per well (Thermo Scientific), 50 μl of 1 M H2SO4 was added for neutralization, and the absorbance at 450 nm was recorded.
Crystallization and data collection
Crystallization conditions for the 801-Fc domain were screened initially by sparse-matrix solutions using sitting dropvapor diffusion on Phenix instrument (Art Robins Instruments). Crystals for A801-Fc were obtained by mixing protein at a concentration of 20 mg/ml with buffer containing 50 mM sodium citrate, pH 5.0, as a pH buffer, and 0.44 M NaCl with 24% wt/vol PEG 4000 as precipitant at 1:1 ratio at 25 °C. G801-Fc at a concentration of 10 mg/ml crystallized in 40 mM potassium phosphate, 16% wt/vol PEG 8000 and 20% vol/vol glycerol. The crystals were cryo-protected in 20% glycerol before vitrified in liquid nitrogen for data collection54.
Crystal diffraction data were collected at the Advanced Light Source beamline BL5.0.3 (Berkeley, CA) and Advanced Photo Source BL 23-IDD (Chicago, IL). The data were processed using the program HKL2000 (ref. 54). The diffractions were scaled to 2.3 Å for A801-Fc and 3.2 Å for G801-Fc. The statistics for data collection are summarized in Table 3.
Structure determination, refinement and analysis
The structures of A801 was determined by molecular replacement using wild-type Fc as the search model (PDB code: 3AVE). The first round of molecular refinement using program Phaser in CCP4 suite55,56 identified two dimers per asymmetric unit and revealed that the Cγ2-Cγ3 dihedral angle varied considerably from those in all other Fc structures in the database. Therefore, the Cγ2 and Cγ3 were searched separately by Phaser55,56, and the initial solution was identified and then confirmed with electron-density-fitting the model. Rigid-body and CNS refinement were carried out in Phenix.refine57. The model was optimized by iterative cycles of manual model building using COOT58 followed by refinement with Phenix.refine57 to improve the quality of the model. Prior to the refinement, 5% of the diffraction data were reserved as an unbiased test set for cross-validation (Rfree)59. The final model exhibits an Rwork of 20% and an Rfree of 24%.
The structure of G801-Fc was also determined by molecular replacement using the structure of glycosylated human Fc as the search model (PDB code: 3AVE). A total of four Fc molecules were found per asymmetric unit. The fitted model was rebuilt using COOT58, and several iterative cycles of optimization were carried out with manual building and refinement. The final model showed an Rwork of 25% and an Rfree of 29%.
Both structures were evaluated by MolProbity60 and Procheck. For A801, the MolProbity Score was 1.71 and placed this at the 99th percentile of all structures for the similar resolution range in database. For G801-Fc, the MolProbity Score was 1.47 and reached 100th percentile. Detailed refinement statistics for A801-Fc and G801-Fc are summarized in Table 3. Figures were prepared using PyMol (DeLano, 2002)61.
C1q cell-surface-binding assay
Raji cells were resuspended in 5% NHS and then incubated with mAbs at a final concentration of 10 μg/ml at 37 °C for various times (0–30 min). Classical complement activation reactions were then quenched with 20 volumes of ice-cold 1% BSA-PBS. The cells were pelleted and washed once and then were probed with FITC anti-C1q (Dako; Cat# F0254; 1:200 dilution) for 30 min at 25 °C. The samples were washed and then analyzed by flow cytometry (FACSCalibur flow cytometer; BD Biosciences), and mean fluorescence intensity was converted to molecules of equivalent soluble fluorochrome with calibrated beads (Spherotech).
Binding of ICs to FcγRs expressed on CHO cells
ICs were generated by co-incubation of 5 μg/ml of variants of anti-TNP IgG (clone 7B4; provided by F.N.) and 2.5 μg/ml of TNP-coupled BSA (TNP4-BSA or TNP32-BSA; Biosearch Technologies) for 3 h at 25 °C (ref. 14). The relative size of TNP4BSA and TNP32-BSA ICs was analyzed by polyethylene glycol (PEG) precipitation. ICs were then incubated with 1 × 105 CHO cells stably expressing human FcγRs for 1 h with gentle shaking at 4 °C, followed by detection of bound ICs by flow cytometry using a PE-conjugated goat anti-human IgG F(ab′)2 fragment at 0.5 mg/ml (Jackson Laboratories; Cat# 109-096-006). ICs were also generated by mixing 10 μg/ml of antibodies and 5 μg/ml of PE-conjugated F(ab′)2 goat anti-human IgG F(ab′)2 (identified above) for 30 min at 37 °C13. CHO cells expressing mouse FcγRs were incubated with these ICs for 1 h on ice and were analyzed by flow cytometry with the method described above. Data were analyzed with Flow Cytometry Analysis Software (FlowJo).
Complement-dependent cytotoxicity (CDC) assays
CDC assays were performed as described previously52. In brief, serially diluted antibodies were incubated with 25% pooled human serum and cancer cells loaded with calcein AM (Life Technologies) at 37 °C for 1 h in 96-well plates. The plates were centrifuged at 1,000g for 10 min and then the supernatants were collected. The released calcein-AM was detected by spectrophotometry using an excitation wavelength of 485 nm and emission wavelength of 535 nm. The percent of tumor cell lysis was calculated relative to SDS lysis buffer. The detailed equation is provided in Supplementary Figure 4. CDC assays with serum depleted of C9 or pooled mouse serum were performed exactly as described above.
PBMC or PMN purification
All in vitro assays were performed under a protocol approved by the UT Austin Institutional Review Board (IRB). Human peripheral blood mononuclear cells (PBMCs) and polymorphonuclear leukocytes (PMNs) were isolated from human blood from healthy donors on the day before the ADCC, CDCC and CMC assays. In brief, 50 ml of human blood was collected in heparinized vials (BD biosciences). 25 ml of blood was layered over 25 ml of Histopaque (Sigma-Aldrich) in 50-ml conical tubes. The tubes were centrifuged at 1,000g for 30 min in a swing-out bucket with no brakes. PBMCs were aspirated in the interphase between Histopaque and medium, and PMNs were collected from the pellet. Both human PBMCs and PMNs were resuspended with red-blood-cell lysis buffer (155 mM NH4Cl, 12 mM NaHCO3 and 0.1 mM EDTA) and were washed twice with PBS.
Complementor FcγR-mediated cellular cytotoxicity assays
For ADCC assays, 5 × 104 human PBMCs or PMNs were mixed with various concentrations of IgG variants and 5 × 103 of calcein-AM-loaded cancer cells. For complement-mediated cell cytotoxicity assays, 25% of serum depleted of C9 or pooled human serum was added in wells containing PBMCs (or activated-PMNs), antibodies and cancer cells. Before use, PMNs were stimulated by GM-CSF (10 ng/ml) for 24 h. After 4 h at 37 °C, the fraction of lysed tumor cells was determined by the same method as CDC assay. For all assays, an E:T ratio of 10:1 was used.
Complement-receptor-inhibition assays
Calcein-AM-loaded cancer cells were pre-incubated with antibodies for 30 min. Antibody-opsonized Raji cells were incubated with effector cells (PBMC or PMN) in RPMI-1640 medium supplemented with 25% serum depleted of C1q. After 4 h at 37 °C, the fraction of lysed tumor cells was detected as described for CDC assays above. For all assays, an E:T ratio of 10:1 was used.
Blockade assays for CR3 or CR4 were also performed. 10 μg/ml of anti-CR3 or anti-CR4 (BioLegend; Cat# 301302 or Cat# 301616) was pre-incubated with effector cells for 30 min. Calcein-AM-loaded and antibody-opsonized CD20+ cancer cells were then incubated with anti-CR3or anti-CR4-coated effector cells in RPMI-1640 medium supplemented with 25% serum depleted of C9. After 4 h at 37 °C, the fraction of lysed tumor cells was detected as described for CDC assays above. For all assays, an E:T ratio of 10:1 was used.
Phagocytosis assays (ADCP or CDCP)
CD14+ monocytes were first isolated from PBMCs by EasySep (STEMCELL Inc). Monocytes were differentiated into M1 macrophages by culture for 7 d in RPMI-1640 medium containing 10% FBS and 100 ng/ml GM-CSF in 5% CO2 incubator. On the day of the assay, adherent macrophages were detached with HyQtase (GE Healthcare). For ADCP assays, 1 × 105 M1 macrophages were mixed with various concentrations of IgG variants and 1 × 104 of calcein-AM-loaded cancer cells. For CDCP assays, 25% serum depleted of C9 was added to wells containing M1 macrophage, antibodies, and cancer cells. After 2 h, M1 macrophages were labeled with anti-human-CD11b-APC (BioLegend; Cat# 301310) and antihuman-CD14-APC (BioLegend ; Cat# 301807). Phagocytosis was evaluated by flow cytometry on an LSRFortessa (BD Bioscience), and results are reported as the ratio of cells positive for both calein-AM and CD11b-CD14 to the total number of tumor cells in the sample.
Fluorescent images of macrophages phagocytosing Raji cells were obtained by confocal microscopy using calcein-AM-loaded Raji cells opsonized with 5 μg/ml of Rituxan, RA801 and RA802 and incubated with serum depleted of C9 and M1 macrophages at 37 °C for 2 h. Approximately 1 × 106 labeled Raji cells and 1 × 106 macrophages were co-incubated in a total volume of 1 ml. Subsequently, the co-incubated cells were labeled with anti-human-CD11b-APC (BioLegend; Cat# 301310) and anti-human-CD14-APC (BioLegend; Cat# 301807). Phagocytosis was visualized by confocal microscopy using a Zeiss LSM 710/Elyra S.1.
TIMING assays
Thin Nanowell arrays were fabricated following an adaptation of previously described protocol62, and a piece 2-cm × 2-cm in size was cut and fixed on a #1.5 glass-bottomed, 50-mm diameter Petri dish (Ted Pella). The single-cell cytotoxicity assay was performed as described before33. In brief, Raji cells (targets) and freshly isolated blood NK cells (effectors)33 were labeled for 2 min with 1 μM of red PKH26 and green PKH67 (Sigma-Aldrich), respectively. Except for the control in which Raji cells were not opsonized with antibodies, Raji cells were incubated with 1 μg/ml of RA801 or rituximab for 30 min at 4 °C in complete medium containing serum depleted of C9. NK cells and Raji cells were loaded sequentially onto Nanowell arrays at a concentration of 1 × 106 cells/ml, allowed to settle for 3 min, and the entire chip was immersed in phenol-red-free complete medium containing 1/60 (vol/vol) annexin V-AlexaFluor-647 (Life Technologies) and, depending on the condition, the antibodies. Images were acquired on an AxioObserver mounted with a 5% CO2, 37 °C and 100% humidity incubator, piloted with Zen software (Carl Zeiss) and fitted with a Hamamatsu EM-CCD camera and a 20× 0.8 NA objective. TIMING images were taken for 6 h at intervals of 5 min, and data were processed and analyzed as described previously63. In brief, a semi-automated pipeline of image analysis takes care of image renaming and timelapse stacking, spectral overlap ‘un-mixing’, background subtraction, cropping of images around the wells and detection of cells by binarization of cell fluorescence, tracking of cell along time, and feature computation.
Animal studies
All animal experiments were performed under a protocol approved by UT Austin institutional Animal Care and Use Committee (IACUC). 1 × 106 Ramos cells in 100 μl PBS with 50% Matrigel (BD Bioscience) were injected subcutaneously in the right flank of athymic nude mice (Jackson Laboratory). Administration of antibodies (10 mg/kg) or PBS was begun when the tumor area reached to an average of 30 mm2 and was repeated a total three times on day 7, day 10 and day 13. Tumor diameters were measured every 3–4 d with a caliper, and tumor areas were calculated by the formula (length) × (width) × π. Mice were euthanized when the tumor size reached 2,500 mm3 in volume.
1 × 105 EL-hCD20-Luc2 cells in culture medium were injected subcutaneously into either NMRI-Foxn1nullFoxn1null mice (n = 10, Janvier Labs) or FcγR-null NMRI-Foxn1nullFoxn1null mice generated by intercross of FcγRnull mice (Fcgr1, Fcgr2b, Fcgr3 and Fcgr4) on the 129/C57BL/6 background and NMRI-Foxn1nullFoxn1null mice. Antibodies (5 mg/kg) or PBS was intravenously administrated three times on days 1, 2 and 3. For measurement of tumor growth by bioluminescence (IVIS SpectrumCT, PerkinElmer), 750 μg of d-luciferin (PerkinElmer) was intraperitoneal injected after anesthesia. Total photon flux (photons/seconds) was calculated using Living-Imagev4.5 software.
Anaphylaxis assay
Heat aggregated IgG variants, Fab as negative control, mouse Rituximab as positive control and RA801 were heat aggregated by incubation at 63 °C for 1 h. 600 μg of each heat-aggregated antibodies was injected into C57BL/6J mice (The Jackson Laboratory) intravenously. Mouse core body temperature was measured every 5 min using a rectal thermoprobe.
FcγRIIb-mediated internalization assay
CD20+FcγRIIb+ Raji, HBL-1 or TMD8 cells were incubated with 10 μg of antibodies for 0, 2, 4 or 6 h in RPMI1640 medium supplemented with 10% FBS (Invitrogen). The level of cell-surface bound antibodies was detected by goat anti-human Fc with FITC (Abcam; Cat# ab97224; 1:200 dilution).
Data analysis and statistics
Data were post-processed using Excel and GraphPad Prism. Statistical testing was run using Fisher’s exact test when contingency numbers were compared, log-rank test when survival curves were compared, and elsewhere we used analysis of variance and t-tests.
Data availability
All the other data that support the findings of this study are available from the corresponding author upon request. PDB accession codes for the protein structural data are 5V43 for A801-Fc and 5V4E for G801-Fc.
Supplementary Material
Acknowledgments
We thank Y. Tanno for assistance with protein expression; A. Bui for assistance with liquid chromatography–tandem mass spectrometry; P. Tucker (University of Texas at Austin) for cancer cell lines; D. Lee (MD Anderson Cancer Center) for patient-derived primary acute lymphocytic leukemia cells; the Macromolecular Crystallography Facility of the University of Texas at Austin; the Berkeley Center for Structural Biology; the Advanced Light Source (supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under contract DE-AC02-05CH11231); the Proteomics Facility at the University of Texas at Austin (supported by grant RP110782 from the Cancer Prevention Research Training Program); and A. Nicola and the Plate-Forme d’Imagerie Dynamique (Institut Pasteur, Paris) for help with the bioluminescence experiments. Supported by the Clayton Foundation, the Institut Pasteur (P.B. laboratory), the Institut National de la Santé et de la Recherche Médicale (P.B. laboratory), the European Research CouncilSeventh Frame-work Program (ERC-2013-CoG 616050 for the P.B. laboratory), the Pasteur–Paris University International PhD program (B.B.), the Cancer Prevention Research Training Program (RP140108 to M.D.; RP160015 to H.T.; and RP130570 to N.V.), the American Cancer Society (123506-PF-13-354-01-CDD to N.M.), Uehara Memorial Foundation (H.T.), Japan Society for the Promotion of Science (H.T.), Deutsche Forschungsgemeinschaft (CRC1181-A07 to F.N.), the US National Institutes of Health (R01CA174385 to N.V.; and R01 GM104896 to Y.J.Z.) and the Welch Foundation (F-1778 to Y.J.Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Cancer Prevention and Research Institute of Texas.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
C.-H.L. and G.G. conceived of and designed the research; C.-H.L., G.R., W.Y., M.W., W.C., B.T., J.L., K.T., M.D., A.L., N.M., M.A.L., O.R.-L.G., B.B., T.H.K., H.T., G.D. and C.A. performed experiments; C.-H.L, G.R., W.Y., O.I.L., R.P.T., F.N., N.V., P.B., Y.J.Z. and G.G. analyzed data; and C.-H.L, G.R., N.V., P.B., Y.J.Z., and G.G. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
In the version of this article initially published online, the labels identifying each plot in Figure 1b were missing. The labels are as follows (left to right): CHO, FcγRI, FcγRIIaR131, FcγRIIaH131, FcγRIIb, FcγRIIIaF158 and FcγRIIIaV158. Also, the reference cited in the accompanying legend (ref. 21) is incorrect. The correct reference is ref. 14. The errors have been corrected in the print, PDF and HTML versions of this article.
References
- 1.Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 2015;15:361–370. doi: 10.1038/nrc3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nimmerjahn F, Ravetch JV. Translating basic mechanisms of IgG effector activity into next generation cancer therapies. Cancer Immun. 2012;12:13. [PMC free article] [PubMed] [Google Scholar]
- 3.Pincetic A, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. 2014;15:707–716. doi: 10.1038/ni.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li FJ, et al. Emerging roles for the FCRL family members in lymphocyte biology and disease. Curr Top Microbiol Immunol. 2014;382:29–50. doi: 10.1007/978-3-319-07911-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van de Donk NWCJ, et al. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev. 2016;270:95–112. doi: 10.1111/imr.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 8.Hess C, Kemper C. Complement-mediated regulation of metabolism and basic cellular processes. Immunity. 2016;45:240–254. doi: 10.1016/j.immuni.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sondermann P, Szymkowski DE. Harnessing Fc receptor biology in the design of therapeutic antibodies. Curr Opin Immunol. 2016;40:78–87. doi: 10.1016/j.coi.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Bruhns P, Jönsson F. Mouse and human FcR effector functions. Immunol Rev. 2015;268:25–51. doi: 10.1111/imr.12350. [DOI] [PubMed] [Google Scholar]
- 11.Taylor RP, Lindorfer MA. The role of complement in mAb-based therapies of cancer. Methods. 2014;65:18–27. doi: 10.1016/j.ymeth.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 12.Di Gaetano N, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581–1587. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 13.Bruhns P, et al. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 14.Lux A, Yu X, Scanlan CN, Nimmerjahn F. Impact of immune complex size and glycosylation on IgG binding to human FcγRs. J Immunol. 2013;190:4315–4323. doi: 10.4049/jimmunol.1200501. [DOI] [PubMed] [Google Scholar]
- 15.Golay J, Introna M. Mechanism of action of therapeutic monoclonal antibodies: promises and pitfalls of in vitro and in vivo assays. Arch Biochem Biophys. 2012;526:146–153. doi: 10.1016/j.abb.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Vafa O, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65:114–126. doi: 10.1016/j.ymeth.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 17.Arduin E, et al. Highly reduced binding to high and low affinity mouse Fcγ receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol Immunol. 2015;63:456–463. doi: 10.1016/j.molimm.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 18.Schreiber AD, Frank MM. Role of antibody and complement in the immune clearance and destruction of erythrocytes. II. Molecular nature of IgG and IgM complement-fixing sites and effects of their interaction with serum. J Clin Invest. 1972;51:583–589. doi: 10.1172/JCI106847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ackerman M, Nimmerjahn F. Antibody Fc: Linking Adaptive and Innate Immunity. Academic Press; 2013. [Google Scholar]
- 20.Wang SY, Racila E, Taylor RP, Weiner GJ. NK-cell activation and antibodydependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood. 2008;111:1456–1463. doi: 10.1182/blood-2007-02-074716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harvey BR, et al. Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc Natl Acad Sci USA. 2004;101:9193–9198. doi: 10.1073/pnas.0400187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung ST, et al. Effective phagocytosis of low Her2 tumor cell lines with engineered, aglycosylated IgG displaying high FcγRIIa affinity and selectivity. ACS Chem Biol. 2013;8:368–375. doi: 10.1021/cb300455f. [DOI] [PubMed] [Google Scholar]
- 23.Borrok MJ, Jung ST, Kang TH, Monzingo AF, Georgiou G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem Biol. 2012;7:1596–1602. doi: 10.1021/cb300130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pawluczkowycz AW, et al. Binding of submaximal C1q promotes complementdependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J Immunol. 2009;183:749–758. doi: 10.4049/jimmunol.0900632. [DOI] [PubMed] [Google Scholar]
- 25.Teeling JL, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 2004;104:1793–1800. doi: 10.1182/blood-2004-01-0039. [DOI] [PubMed] [Google Scholar]
- 26.Boross P, Leusen JHW. Mechanisms of action of CD20 antibodies. Am J Cancer Res. 2012;2:676–690. [PMC free article] [PubMed] [Google Scholar]
- 27.Teeling JL, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol. 2006;177:362–371. doi: 10.4049/jimmunol.177.1.362. [DOI] [PubMed] [Google Scholar]
- 28.Diebolder CA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–1263. doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beum PV, et al. Complement activation on B lymphocytes opsonized with rituximab or ofatumumab produces substantial changes in membrane structure preceding cell lysis. J Immunol. 2008;181:822–832. doi: 10.4049/jimmunol.181.1.822. [DOI] [PubMed] [Google Scholar]
- 30.Gorter A, Meri S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol Today. 1999;20:576–582. doi: 10.1016/s0167-5699(99)01537-6. [DOI] [PubMed] [Google Scholar]
- 31.Min X, et al. Expression and regulation of complement receptors by human natural killer cells. Immunobiology. 2014;219:671–679. doi: 10.1016/j.imbio.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 32.Ramos OF, Sármay G, Klein E, Yefenof E, Gergely J. Complement-dependent cellular cytotoxicity: lymphoblastoid lines that activate complement component 3 (C3) and express C3 receptors have increased sensitivity to lymphocyte-mediated lysis in the presence of fresh human serum. Proc Natl Acad Sci USA. 1985;82:5470–5474. doi: 10.1073/pnas.82.16.5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romain G, et al. Antibody Fc engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood. 2014;124:3241–3249. doi: 10.1182/blood-2014-04-569061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bologna L, et al. Ofatumumab is more efficient than rituximab in lysing B chronic lymphocytic leukemia cells in whole blood and in combination with chemotherapy. J Immunol. 2013;190:231–239. doi: 10.4049/jimmunol.1202645. [DOI] [PubMed] [Google Scholar]
- 35.DiLillo DJ, Ravetch JV. Differential Fc-receptor engagement drives an antitumor vaccinal effect. Cell. 2015;161:1035–1045. doi: 10.1016/j.cell.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klaus GG, Pepys MB, Kitajima K, Askonas BA. Activation of mouse complement by different classes of mouse antibody. Immunology. 1979;38:687–695. [PMC free article] [PubMed] [Google Scholar]
- 37.Neuberger MS, Rajewsky K. Activation of mouse complement by monoclonal mouse antibodies. Eur J Immunol. 1981;11:1012–1016. doi: 10.1002/eji.1830111212. [DOI] [PubMed] [Google Scholar]
- 38.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119:5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- 39.Lim SH, et al. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood. 2011;118:2530–2540. doi: 10.1182/blood-2011-01-330357. [DOI] [PubMed] [Google Scholar]
- 40.Vaughan AT, et al. Activatory and inhibitory Fcγ receptors augment rituximabmediated internalisation of CD20 independent of signalling via the cytoplasmic domain. J Biol Chem. 2015:jbc.M114.593806. doi: 10.1074/jbc.M114.593806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frank M, Walker RC, Lanzilotta WN, Prestegard JH, Barb AW. Immunoglobulin G1 Fc domain motions: implications for Fc engineering. J Mol Biol. 2014;426:1799–1811. doi: 10.1016/j.jmb.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 43.Gaboriaud C, et al. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J Biol Chem. 2003;278:46974–46982. doi: 10.1074/jbc.M307764200. [DOI] [PubMed] [Google Scholar]
- 44.Duncan AR, Winter G. The binding site for C1q on IgG. Nature. 1988;332:738–740. doi: 10.1038/332738a0. [DOI] [PubMed] [Google Scholar]
- 45.Idusogie EE, et al. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol. 2000;164:4178–4184. doi: 10.4049/jimmunol.164.8.4178. [DOI] [PubMed] [Google Scholar]
- 46.Schneider S, Zacharias M. Atomic resolution model of the antibody Fc interaction with the complement C1q component. Mol Immunol. 2012;51:66–72. doi: 10.1016/j.molimm.2012.02.111. [DOI] [PubMed] [Google Scholar]
- 47.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol. 2003;325:979–989. doi: 10.1016/s0022-2836(02)01250-0. [DOI] [PubMed] [Google Scholar]
- 48.Lu J, et al. Structure of FcγRI in complex with Fc reveals the importance of glycan recognition for high-affinity IgG binding. Proc Natl Acad Sci USA. 2015;112:833–838. doi: 10.1073/pnas.1418812112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kennedy AD, et al. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol. 2004;172:3280–3288. doi: 10.4049/jimmunol.172.5.3280. [DOI] [PubMed] [Google Scholar]
- 50.Cartron G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. 2002;99:754–758. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 51.Mancardi DA, et al. FcγRIV is a mouse IgE receptor that resembles macrophage FcεRI in humans and promotes IgE-induced lung inflammation. J Clin Invest. 2008;118:3738–3750. doi: 10.1172/JCI36452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kelton W, et al. IgGA: a “cross-isotype” engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol. 2014;21:1603–1609. doi: 10.1016/j.chembiol.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 53.Lee CF, Paull TT, Person MD. Proteome-wide detection and quantitative analysis of irreversible cysteine oxidation using long column UPLC-pSRM. J Proteome Res. 2013;12:4302–4315. doi: 10.1021/pr400201d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 55.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 60.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liadi I, Roszik J, Romain G, Cooper LJN, Varadarajan N. Quantitative high-throughput single-cell cytotoxicity assay for T cells. J Vis Exp. 2013;72:e50058. doi: 10.3791/50058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Merouane A, et al. Automated profiling of individual cell-cell interactions from high-throughput time-lapse imaging microscopy in nanowell grids (TIMING) Bioinformatics. 2015;31:3189–3197. doi: 10.1093/bioinformatics/btv355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the other data that support the findings of this study are available from the corresponding author upon request. PDB accession codes for the protein structural data are 5V43 for A801-Fc and 5V4E for G801-Fc.