

Ionized calcium (Ca2+) has long been known to be a major controller of cell function including pancreatic acinar cell secretion; in fact, research on pancreatic acinar cells in the 1980’s was instrumental in establishing the mechanisms by which sequestered calcium is released into the cytoplasm to act as an intracellular messenger in response to hormones and neurotransmitters (1). In acinar cells this signaling consists of transient Ca2+ elevations in the apical pole of the cell that triggers exocytosis of zymogen granules (2). Hyperstimulation of acinar cells especially by CCK led to a large single peak of Ca2+ followed by a plateau of elevated calcium. Since hyperstimulation by the CCK analog cerulein has long been known to induce experimental pancreatitis (3), it was a logical step forward to show using isolated pancreatic acini that a high level of intracellular Ca2+ was associated with and probably caused premature trypsin activation, cell vacuolization, and necrosis which are considered the in vitro equivalent of acute pancreatitis (AP) (4-6).
Animal models of AP have been shown to involve two major pathways, the premature activation of trypsin and the development of local and systemic inflammation triggered by activation of NFκ-B in the acinar cell (7,8). Both pathways are activated in part by Ca2+ but targeting Ca2+ signaling to treat pancreatitis had to await a detailed knowledge of Ca2+ signaling (2,9,10).
Figure 1 shows some of the molecules involved in regulating intracellular Ca2+ in acinar cells. The inositol 1, 4, 5-trisphosphate receptor (IP3R) and the ryanodine receptor (RyR) release Ca2+ from the ER into the cytoplasm while PMCA and SERCA are ATPases pumping Ca2+ out of the cytoplasm and into extracellular space or back into the ER respectively. When the ER calcium store is depleted, the protein stromal interaction molecule (STIM) 1 and 2 present in the ER membrane aggregates and moves to the plasma membrane to activate calcium influx through the calcium channels Orai1 and TRPC3. Agents targeting the IP3 receptor, the RyR, the Ca2+ influx mechanism mediated by Orai1, the calcium activated phosphatase calcineurin, and the plasma membrane calcium efflux pump PMCA are all under study as ways to diminish pancreatitis. Because of its central position in Ca2+ signaling and relevance to this Editorial some additional information on the IP3R will be presented. IP3Rs are coded for by 3 genes (Type 1, 2 and 3) in vertebrates that are closely related. Each IP3R is a large protein of about 2,700 amino acids and they assemble into homo or heterotetramers which contain a central pore that functions as a gated Ca2+ channel (11). IP3 binds to a segment termed the IP3-binding core in the N terminal portion which leads to an opening of the transmembrane channel. This event is also regulated by Ca2+, ATP, phosphorylation of the IP3R, by various kinases but most importantly the cyclic adenosine monophosphate (cAMP) activated kinase, and the binding of a large number of proteins (12,13). The Ca2+ released by the IP3R channel can also activate the RyR, a related calcium channel prominent in muscle that is activated by Ca2+ but not IP3. RyRs are also present in acinar cells and play a supporting role in Ca2+ mobilization under physiological conditions.
Figure 1.
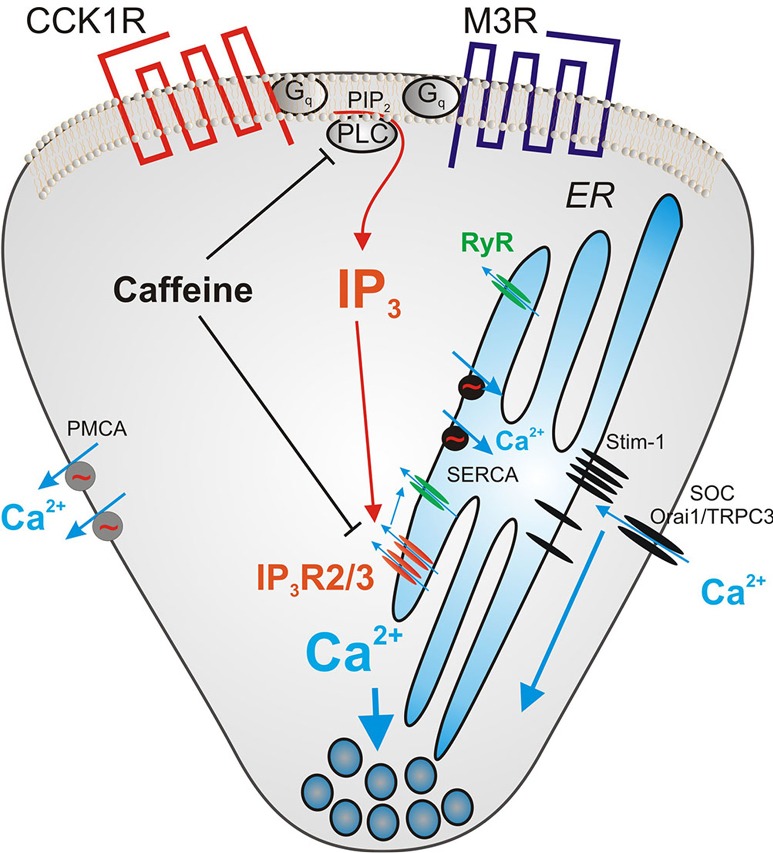
Pathways of calcium signaling through IP3 in pancreatic acinar cells initiated by CCK and acetylcholine. The sites at which caffeine has been shown to inhibit signaling, the Gq activated phospholipase C (PLC) that produces IP3 and the IP3 R are shown. Figure simplified from the Pancreapedia with permission. For more detail on acinar cell Ca2+ signaling see the Pancreapedia review (10).
A study recently published in Gut by Huang, Sutton and colleagues on the use of caffeine to protect against experimental pancreatitis provides hope that targeting Ca2+ may provide a therapeutic mechanism for human pancreatitis (14). Caffeine had previously been shown to inhibit intracellular Ca2+ signaling mediated by IP3 (1,15). In pancreatic acinar cells it is not clear whether caffeine was blocking IP3 production or action (16). Huang et al. utilized both isolated acini and intact mice as well as measuring caffeine metabolites and the intracellular concentration of caffeine (14). They showed that when intracellular caffeine levels reached 2 mM in mouse acinar cells that Ca2+ signaling induced by acetylcholine was inhibited and that dosage of 25 mg/kg caffeine every hour to mice led to similar serum levels of caffeine. They also showed that caffeine blocked the effect of uncaged IP3 although higher concentrations of caffeine seem to have been required. Most importantly, they showed that caffeine given after the toxic insult significantly ameliorated three models of experimental AP, those induced by cerulein, bile salts and ethanol plus fatty acid. While related xanthines and caffeine metabolites could inhibit Ca2+ signaling, only caffeine inhibited pancreatitis. The work is comprehensive, well carried out and a significant step forward in developing a therapeutic protocol that can be applied to human disease.
Caffeine is chemically 1, 3, 7-trimethylxanthine and is structurally related to adenosine (Figure 2). Its major actions are as a CNS stimulant, a relaxer of smooth muscle, a stimulant of cardiac muscle and a stimulant of urinary diuresis. These and other overall effects result from a number of biochemical actions which include inhibition of adenosine receptors, inhibition of cyclic nucleotide phosphodiesterase which increases cAMP levels and as a modulator of intracellular Ca2+ handling. Caffeine is metabolized primarily in the liver and its major metabolites are paraxanthine, theobromine and theophylline, each of which lacks one specific methyl group. The metabolism is carried out by cytochrome P450 enzymes particularly CYP1A2 and the metabolites are excreted in the urine (17). The plasma half-life of caffeine is 4–6 hours in humans and 1 hour in mouse and can be affected by polymorphisms in CYP enzymes. It is important to note that drinking a cup of coffee results in plasma caffeine of at most 10 µM which is a hundred times lower than the concentrations studied by Huang et al. (14).
Figure 2.
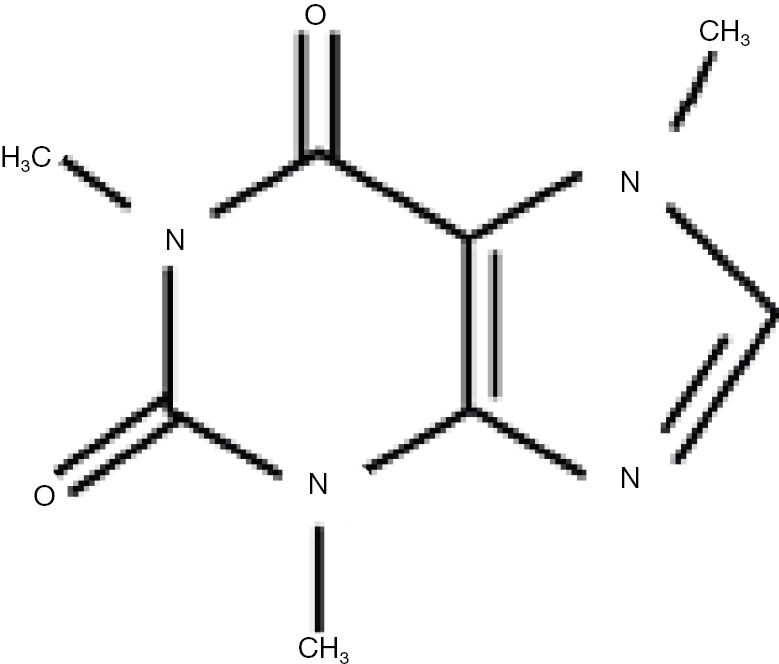
Structure of the caffeine molecule. The core is xanthine while the methyl groups are on nitrogen atoms in position 1, 3, and 7.
Caffeine has both advantages and disadvantages as a potential therapeutic agent. It is cell permeant, inexpensive and acts orally as attested by millions of users. A disadvantage is that the therapeutic dose may be close to the toxic dose. The other disadvantage is that caffeine has multiple actions throughout the body and the actual target affecting pancreatitis is unclear. Huang et al. assume that caffeine is an IP3R antagonist (14). The current state-of-the-art for studying IP3 receptors is to express normal or modified IP3R in DT40 cells which lack endogenous IP3R. Saleem et al. studied DT40 cells expressing IP3R type 1, 2 and 3 individually and found that caffeine was a low affinity antagonist of IP3R1 without affecting binding of IP3 (18). However, caffeine had no effect on IP3 induced calcium release by IP3R type 2 and 3, the most abundant forms in pancreatic acinar cells and whose compound gene deletion blocks acinar cell secretion (19). Thus caffeine may have other and possibly multiple targets in acinar cells. Epidemiological data on caffeine consumption and the occurrence of pancreatitis is mixed (20,21) and such chronic consumption almost certainly leads to lower plasma levels of caffeine then used by Huang et al.
Of the other mechanisms to ameliorate pancreatitis by inhibiting Ca2+ signaling, the best studied is to block the calcium release-activated calcium channel whose major channel component is the protein Orai1. This process of Ca2+ entry is also known as store-operated calcium entry and is activated by decreases in endoplasmic reticulum calcium stores and mediated by STIM1 and STIM2 which activate distinct channels composed of Orail1 and TRPC3 by physical interaction (22,23). Two Orai channel blockers have been developed, GSK-7975A by GlaxoSmithKline and CM_128 by CalciMedica. Gerasimenko et al. showed in isolated mouse acini that GSK-7975A inhibited store-operated calcium entry induced by depleting ER calcium stores with thapsigargin, a SERCA inhibitor and by palmitoleic ethyl ester, a toxic metabolite of alcohol induced pancreatitis (24). Wen et al. then showed that both of the Orai1 inhibitors reduced pancreatitis in mice induced by cerulein, bile salts or ethanol plus palmitoleic acid when given 1 hour after induction of pancreatitis and to a lesser extent after 6 hours (25). The Orai inhibitors were also shown to prevent necrosis in vitro in isolated human as well as mouse pancreatic cells. Another approach to inhibit Ca2+ signaling is to increase Mg2+ as it can act to attenuate Ca2+ signaling in some physiological situations. In particular, high extracellular Mg2+ has been reported to reduce store operated Ca2+ entry in isolated mouse acini (26). Mg2+ administration to rats and mice has been shown to reduce experimental pancreatitis (27) and is under study in clinical trials. Mg2+ has the advantage that it is safe and easy to administer although the exact mechanism of action is not fully understood.
Several issues need to be overcome before inhibiting Ca2+ signaling will become a useful therapeutic protocol. First, inhibiting Ca2+ signaling is expected to have effects throughout the body. At this point there is no way to target only pancreas. Second, all in vivo studies to date have been carried out in mice. While there are good reasons for the ascendancy of mice as experimental animals, larger animals show a slower time course of events and sometimes a qualitative difference. To date the success of translating therapeutic approaches for pancreatitis from mice to humans has been poor. Current research standards require multiple experimental models but they are all generally in mice. Whether studies in large animals such as pigs would help is unknown. Finally, the calcium signaling events occur early in the course of pancreatitis and patients, especially in referral centers, come with established disease. It will be important to compare efficacy in rapidly treated cases seen in the ER within a limited number of hours after the initiation of symptoms with patients seen after 24–48 hours. In this regard, studies evaluating ERCP induced pancreatitis have an advantage even though the pancreatitis is usually mild. Patients requiring treatment after pancreatitis is established may not benefit as much and may require other treatments.
Overall, blocking the sustained increase in intracellular free Ca2+ has the potential to reduce the cellular damage in pancreatitis. Both caffeine and the calcium influx blockers are worthy of further study.
Acknowledgements
None.
Provenance: This is a Guest Editorial commissioned by Section Editor Ali A. Aghdassi, MD (Department of Medicine A, University Medicine Greifswald, Greifswald, Germany).
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Berridge MJ. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol Rev 2016;96:1261-96. 10.1152/physrev.00006.2016 [DOI] [PubMed] [Google Scholar]
- 2.Petersen OH, Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol 2008;70:273-99. 10.1146/annurev.physiol.70.113006.100618 [DOI] [PubMed] [Google Scholar]
- 3.Lampel M, Kern HF. Acute interstitial pancreatitis in the rat induced by excessive doses of a pancreatic secretagogue. Virchows Arch A Pathol Anat Histol 1977;373:97-117. 10.1007/BF00432156 [DOI] [PubMed] [Google Scholar]
- 4.Saluja AK, Bhagat L, Lee HS, et al. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol 1999;276:G835-42. [DOI] [PubMed] [Google Scholar]
- 5.Krüger B, Albrecht E, Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Pathol 2000;157:43-50. 10.1016/S0002-9440(10)64515-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raraty M, Ward J, Erdemli G, et al. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci U S A 2000;97:13126-31. 10.1073/pnas.97.24.13126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sah RP, Dawra RK, Saluja AK. New insights into the pathogenesis of pancreatitis. Curr Opin Gastroenterol 2013;29:523-30. 10.1097/MOG.0b013e328363e399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gukovskaya AS, Pandol SJ, Gukovsky I. New insights into the pathways initiating and driving pancreatitis. Curr Opin Gastroenterol 2016. [Epub ahead of print]. 10.1097/MOG.0000000000000301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerasimenko JV, Gerasimenko OV, Petersen OH. The role of Ca2+ in the pathophysiology of pancreatitis. J Physiol 2014;592:269-80. 10.1113/jphysiol.2013.261784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yule DI. Ca2+ signaling in pancreatic acinar cells. Pancreapedia: Exocrine Pancreas Knowledge Base 2015. Available online: https://www.pancreapedia.org/reviews/ca2signaling-in-pancreatic-acinar-cells
- 11.Foskett JK, White C, Cheung KH, et al. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 2007;87:593-658. 10.1152/physrev.00035.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner LE, 2nd, Yule DI. Differential regulation of the InsP receptor type-1 and -2 single channel properties by InsP, Ca2+ and ATP. J Physiol 2012;590:3245-59. 10.1113/jphysiol.2012.228320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prole DL, Taylor CW. Inositol 1,4,5-trisphosphate receptors and their protein partners as signalling hubs. J Physiol 2016;594:2849-66. 10.1113/JP271139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang W, Cane MC, Mukherjee R, et al. Caffeine protects against experimental acute pancreatitis by inhibition of inositol 1,4,5-trisphosphate receptor-mediated Ca2+ release. Gut 2017;66:301-13. 10.1136/gutjnl-2015-309363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker I, Ivorra I. Caffeine inhibits inositol trisphosphate-mediated liberation of intracellular calcium in Xenopus oocytes. J Physiol 1991;433:229-40. 10.1113/jphysiol.1991.sp018423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toescu EC, O’Neill SC, Petersen OH, et al. Caffeine inhibits the agonist-evoked cytosolic Ca2+ signal in mouse pancreatic acinar cells by blocking inositol trisphosphate production. J Biol Chem 1992;267:23467-70. [PubMed] [Google Scholar]
- 17.Tassaneeyakul W, Birkett DJ, McManus ME, et al. Caffeine metabolism by human hepatic cytochromes P450: contributions of 1A2, 2E1 and 3A isoforms. Biochem Pharmacol 1994;47:1767-76. 10.1016/0006-2952(94)90304-2 [DOI] [PubMed] [Google Scholar]
- 18.Saleem H, Tovey SC, Molinski TF, et al. Interactions of antagonists with subtypes of inositol 1,4,5-trisphosphate (IP3) receptor. Br J Pharmacol 2014;171:3298-312. 10.1111/bph.12685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Futatsugi A, Nakamura T, Yamada MK, et al. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 2005;309:2232-4. 10.1126/science.1114110 [DOI] [PubMed] [Google Scholar]
- 20.Morton C, Klatsky AL, Udaltsova N. Smoking, coffee, and pancreatitis. Am J Gastroenterol 2004;99:731-8. 10.1111/j.1572-0241.2004.04143.x [DOI] [PubMed] [Google Scholar]
- 21.Oskarsson V, Sadr-Azodi O, Orsini N, et al. A prospective cohort study on the association between coffee drinking and risk of non-gallstone-related acute pancreatitis. Br J Nutr 2016;115:1830-4. 10.1017/S0007114516000866 [DOI] [PubMed] [Google Scholar]
- 22.Lur G, Haynes LP, Prior IA, et al. Ribosome-free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP(3) receptors. Curr Biol 2009;19:1648-53. 10.1016/j.cub.2009.07.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim MS, Hong JH, Li Q, et al. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 2009;137:1509-17. 10.1053/j.gastro.2009.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerasimenko JV, Gryshchenko O, Ferdek PE, et al. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci U S A 2013;110:13186-91. 10.1073/pnas.1300910110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen L, Voronina S, Javed MA, et al. Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 2015;149:481-92.e7. 10.1053/j.gastro.2015.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mooren FC, Turi S, Gunzel D, et al. Calcium-magnesium interactions in pancreatic acinar cells. FASEB J 2001;15:659-72. 10.1096/fj.00-0172com [DOI] [PubMed] [Google Scholar]
- 27.Schick V, Scheiber JA, Mooren FC, et al. Effect of magnesium supplementation and depletion on the onset and course of acute experimental pancreatitis. Gut 2014;63:1469-80. 10.1136/gutjnl-2012-304274 [DOI] [PubMed] [Google Scholar]