

Abstract
Due to recent military conflicts and terrorist attacks, blast-induced traumatic brain injury (bTBI) presents a health concern for military and civilian personnel alike. Although secondary blast (penetrating injury) and tertiary blast (inertia-driven brain deformation) are known to be injurious, the effects of primary blast caused by the supersonic shock wave interacting with the skull and brain remain debated. Our group previously reported that in vitro primary blast exposure reduced long-term potentiation (LTP), the electrophysiological correlate of learning and memory, in rat organotypic hippocampal slice cultures (OHSCs) and that primary blast affects key proteins governing LTP. Recent studies have investigated phosphodiesterase-4 (PDE4) inhibition as a therapeutic strategy for reducing LTP deficits following inertia-driven TBI. We investigated the therapeutic potential of PDE4 inhibitors, specifically roflumilast, to ameliorate primary blast-induced deficits in LTP. We found that roflumilast at concentrations of 1 nM or greater prevented deficits in neuronal plasticity measured 24 h post-injury. We also observed a therapeutic window of at least 6 h, but <23 h. Additionally, we investigated molecular mechanisms that could elucidate this therapeutic effect. Roflumilast treatment (1 nM delivered 6 h post-injury) significantly increased total AMPA glutamate receptor 1 (GluR1) subunit expression, phosphorylation of the GluR1 subunit at the serine-831 site, and phosphorylation of stargazin at the serine-239/240 site upon LTP induction, measured 24 h following injury. Roflumilast treatment significantly increased PSD-95 regardless of LTP induction. These findings indicate that further investigation into the translation of PDE4 inhibition as a therapy following bTBI is warranted.
Keywords: Traumatic brain injury, Blast injury, Military injury, Memory, Long-term potentiation, Phosphodiesterase, Electrophysiology, Therapeutic, In vitro, Hippocampus
1. Introduction
Traumatic brain injury (TBI) is defined as a disruption of brain function due to mechanical forces acting on the head (Ommaya and Gennarelli, 1974). Since 2000, there have been approximately 350,000 diagnosed TBIs among U.S. military personnel, with 83% of these injuries considered mild TBI (mTBI) (DVBIC, 2016). Exposure to blast is the leading cause of TBIs for military personnel (Hoge et al., 2008; Rigg and Mooney, 2011). The biomechanics of blast-induced TBI (bTBI) are multi-phasic and can include penetrative injury and acceleration-based deformation (McIntosh et al., 1989; Tecoma et al., 1989); however, injury due to shockwave exposure, often referred to as primary blast injury, remains debated (Bass et al., 2012). Studying primary blast injury using in vivo models can be difficult due to the associated challenges of eliminating head motion and providing adequate thoracic protection (Gullotti et al., 2014). In comparison, our in vitro primary blast injury model isolates the shock wave component of blast from the other, confounding phases of injury (Effgen et al., 2012). The precisely controlled biomechanics of our injury model enables the study of neuronal dysfunction following primary blast injury in isolation (Effgen et al., 2012, 2016; Hue et al., 2014; Vogel et al., 2016b).
One common clinical symptom of bTBI is memory impairment (Kontos et al., 2013). Behavioral and ultrastructural changes in rodents following in vivo blast exposure suggest that the hippocampus is especially vulnerable to bTBI (Beamer et al., 2016; Cernak et al., 2001; Rubovitch et al., 2011). Long-term potentiation is the primary experimental model for investigating synaptic plasticity on a cellular level and is known to occur within the hippocampus (Bliss and Collingridge, 1993). It is well-documented that blast exposure in animals negatively affected hippocampal LTP, but this observation was not universal among preclinical models of blast TBI (Effgen et al., 2016; Goldstein et al., 2012; Huber et al., 2013; Vogel et al., 2016a; Yin et al., 2014). We have previously reported that 24 h post-injury, primary blast reduced the expression and phosphorylation of AMPA-GluR1 subunits (Vogel et al., 2016b), a key transmembrane receptor required for the induction and maintenance of LTP (Lee et al., 2000; Makino and Malinow, 2009; Mammen et al., 1997). We also observed that modulation of the second messenger cyclic adenosine monophosphate (cAMP) rescued blast-induced deficits in neuronal plasticity and the expression of key proteins involved in LTP maintenance (Vogel et al., 2016b). Those results suggested that modulation of the cAMP pathway could have therapeutic potential in preventing memory deficits following primary bTBI. Intriguingly, increasing cAMP through phosphodiesterase-4 inhibition was effective in improving outcome in some experimental models of TBI and also reduced cognitive impairments associated with Alzhemer’s disease, schizophrenia and aging (Gong et al., 2004; Maxwell et al., 2004; Smith et al., 2009; Titus et al., 2014; Wiescholleck and Manahan-Vaughan, 2012). Currently, there are no clinically-approved treatments for TBI (Silverberg et al., 2016).
This study examined the ability of PDE4 inhibitors, including roflumilast, to prevent primary blast-induced deficits in plasticity and the expression of key proteins necessary for LTP. Roflumilast is FDA-approved for treatment of chronic obstructive pulmonary disorder (COPD), making it an attractive therapeutic candidate. We observed that delivery of a PDE4 inhibitor immediately post-blast preserved neuronal plasticity measured 24 h following injury in vitro. When varying the time post-injury of drug delivery, the therapeutic window of PDE4 inhibitors following primary blast was 6 h post-injury. Phosphodiesterase-4 inhibition post-blast reversed blast-induced changes in protein expression/phosphorylation for key targets in the LTP pathway, including phosphorylation of AMPA-GluR1 subunits (pGluR1) at the serine-831 (Ser831) site, total GluR1 expression, and phosphorylation of stargazin (pStargazin) at the serine-239/240 (Ser239/240) site upon LTP induction. Roflumilast treatment significantly increased total postsynaptic density protein-95 (PSD-95) expression regardless of LTP induction. These findings indicate that further investigation into the therapeutic potential of PDE4 inhibition following bTBI is warranted.
2. Materials and methods
2.1. Organotypic hippocampal slice culture
All animal procedures were approved by the Columbia University Institutional Animal Care and Use Committee (IACUC). OHSCs were generated from P8-P10 Sprague Dawley rats as previously described (Effgen et al., 2012, 2014; Morrison et al., 2006; Vogel et al., 2016a). In brief, the hippocampus was excised, cut into 400 μm thick sections, and plated onto Millicell inserts (EMD Millipore, Billerica, MA) in Neurobasal medium supplemented with 2 mM GlutaMAX™, 1X B27 supplement, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 25 mM D-glucose (Life Technologies, Grand Island, NY). Following plating, cultures were fed every 2–3 days with full serummedium, containing 50%MEM, 25% Hank’s Balanced Salt Solution, 25% heat inactivated horse serum, 2 mM GlutaMAX, 25 mM D-glucose, and 10 mM HEPES (Sigma). Cultures were maintained for 10–14 days prior to blast injury.
2.2. Primary blast exposure
Blast injury methods have been described previously in detail (Effgen et al., 2012, 2014; Hue et al., 2014; Hue et al., 2013; Panzer et al., 2012; Vogel et al., 2016a). Cultures were placed into sterile bags, filled with pre-warmed, serum-free medium, pre-equilibrated with 5% CO2 at 37 °C. Any air bubbles were removed from the bag, which was sealed and placed into the receiver column. The receiver column was filled with pre-warmed water (37 °C), sealed with a silicone membrane, and the shock tube was fired. Piezoresistive pressure transducers (Endevco 8530B-500, San Juan Capistrano, CA, USA) recorded incident pressure at the shock tube exit and inside the fluid-filled receiver. Peak overpressure, duration, and impulse were recorded, processed, and quantified as previously described (Effgen et al., 2012, 2014; Hue et al., 2013; Vogel et al., 2016a). Sham cultures were treated identically except the shock tube was not fired.
Based on previous studies, a blast exposure intensity was utilized that produced consistent deficits in LTP, characterized by the peak pressure, duration, and impulse of the in-air shock wave (336 ± 8 kPa, 0.84 ± 0.01 ms, 87 ± 2 kPa·ms) and the in-fluid pressure transient (598 ±15 kPa, 1.85 ±0.30 ms, 440 ±13 kPa·ms) (Vogel et al., 2016a). Following blast or sham exposure, cultures were immediately removed from the receiver and returned to the incubator in fresh, full serum medium. Cultures were maintained in full serum medium until the indicated time points.
2.3. Drug treatment
A stock solution of roflumilast (SML1099, Sigma-Aldrich) was dissolved in DMSO at a final concentration of 200 μM. The drug was further diluted in fresh, full serum medium (≤0.07% DMSO) at indicated concentrations: 100 pM, 1 nM, 10 nM, and 100 nM. Cultures were placed into drug-containing medium immediately following blast injury. In a separate set of cultures, roflumilast (1 nM) or DMSO vehicle was delivered to OHSCs at varying times following blast exposure: 0 h, 1 h, 6 h, and 23 h. To further evaluate the effect of delayed roflumilast delivery, a separate set of cultures were treated with roflumilast 24 h following injury and function was evaluated 48 h following injury.
A stock solution of piclamilast (SML0585, Sigma-Aldrich), another PDE4 inhibitor, was dissolved in DMSO at a final concentration of 10 μM. The drug was further diluted in fresh, full serum medium (≤0.07% DMSO) to 5 nM, which was used to treat cultures at the indicated time points following blast exposure.
A stock solution of ibudilast (I0157, Sigma-Aldrich), another PDE4 inhibitor, was dissolved in DMSO at a final concentration of 200 mM. The drug was further diluted in fresh, full serum medium (≤0.07% DMSO) to 1 μM, which was used to treat cultures at the indicated time points following blast exposure.
A stock solution of papaverine (CDS021481, Sigma-Aldrich), a partially selective PDE10A inhibitor, was dissolved in DMSO to 400 μM. The drug was further diluted in fresh, full serum medium (≤0.07% DMSO) to 200 nM, which was used to treat cultures at the indicated time points following blast exposure.
For all experiments, an additional group of injured or sham cultures were treated with DMSO vehicle (0.07%) for comparison to drug-treated cultures.
2.4. Electrophysiology
Electrophysiological activity within the OHSC was recorded using 60-channel MEAs (8 × 8 electrode grid without the corners, 30 μm electrode diameter, 200 μm electrode spacing) at the indicated time points following blast injury (60MEA200/30iR-Ti-gr, Multi-Channel Systems, Reutlingen, Germany). OHSCs were perfused with aCSF (norm-aCSF) containing 125 mM NaCl, 3.5 mM KCl, 26 mM NaHCO3, 1.2 mM KH2PO4, 2.4 mM CaCl2, 1.3 mM MgCl2, 10 mM HEPES, and 10 mM glucose (pH=7.40), which was bubbled with 5% CO2/95% O2 and warmed to 37 °C, as previously described (Yu and Morrison, 2010). Recordings were acquired with an MEA1060-BC amplifier and data acquisition system (Multi-Channel Systems). Neural signals were recorded at 20 kHz with a 6 kHz analog, anti-aliasing filter and then further filtered in MATLAB, using an eighth-order, digital low-pass (1000 Hz) and a fourth-order, digital, high-pass (0.2 Hz) Butterworth filter.
2.5. Spontaneous activity
Spontaneous neural activity was measured by recording continuously for 3 min from all electrodes within the hippocampus, as previously described (Vogel et al., 2016a). In brief, neural event activity was detected based on the multi-resolution Teager energy operator (Choi et al., 2006; Kang et al., 2014; Kang and Morrison, 2014, 2015; Patel et al., 2015). Data from each electrode was segregated by anatomical ROI (dentate gyrus [DG], cornu ammonis 1 [CA1], cornu ammonis 3 [CA3]). The effect of blast injury and drug-treatment on spontaneous event rate, magnitude, and duration were analyzed by two-way ANOVA with statistical significance set as p < 0.05 (SPSS v22, IBM; Armonk, NY). It is important to note that there no significant difference between vehicle-treated cultures and roflumilast-treated cultures was observed for the number of electrodes per region.
Spontaneous network synchronization was also quantified using previously published methods (Kang et al., 2014; Li et al., 2007; Li et al., 2010; Patel et al., 2012; Vogel et al., 2016a). In brief, correlation between neural events was calculated for each electrode pair based upon neural event-timing, where two events occurring within 1.5 ms were considered synchronous, and the total number of events. A correlation matrix was constructed which represented the strength of correlation between electrode pairings. To determine statistical significance, this data was compared to randomized surrogate time-series data without correlated activity, but with an equal event-rate, to identify significantly synchronized clusters. The global synchronization index (GSI), ranging from 0 (random, uncorrelated activity) to 1 (perfectly synchronous, correlated activity on all electrodes), was calculated from the clusters of electrodes with the highest (significant) degree of synchronization. This analysis was based upon the eigenvalues of the correlation matrix, which represented correlation strength, and associated eigenvectors, which represented the cluster of electrodes. The effects of blast exposure and drug-treatment on GSI were analyzed by two-way ANOVA, with statistical significance set as p < 0.05.
2.6. Stimulus-response curves
Stimulus-response (SR) curves were generated by applying a constant current, bi-phasic, bi-polar stimulus (100 μs positive phase followed by 100 μs negative phase) of increasing magnitude (0–200 μA in 10 μA increments) to electrodes located in the Schaffer collateral (SC) pathway. As in previous studies, each electrode’s response was fit to a sigmoidal curve, and three parameters were quantified: Rmax, represented the maximum amplitude of the evoked response, I50 represented the current necessary to generate a half-maximal response, and the term m, represented the slope of the sigmoidal fit (Yu and Morrison, 2010). As before, data from each electrode was segregated by anatomical ROI. Each parameter (I50, m, Rmax) for an electrode was averaged within a region to determine that regional response for any given slice. Data reported for each region is the average across slices within a given experimental group. The effects of blast exposure and drug-treatment on individual SR parameters were analyzed by two-way ANOVA with statistical significance set as p < 0.05.
2.7. Long-term potentiation
Following SR recordings, the ability to induce LTP by electrical stimulus was measured as previously described (Vogel et al., 2016a). Baseline response was evoked by stimulating at I50 across the SC pathway once per minute for 30 min. LTP was induced by stimulating with a high-frequency stimulus (HFS) that consisted of three trains of 100 Hz pulses applied for 1 s at I50 magnitude, with each train separated by 10 s. Immediately following LTP induction, post-induction response was evoked by stimulating at I50 once every minute for 60 min. LTP induction was calculated as percent potentiation above baseline based on the last 10 min of recording in each recording window. To ensure only stable responses were included for analysis, electrodes were discounted if the coefficient of variance (pre or post-induction) was >20% (Heuschkel et al., 2002). The average number of discounted electrodes was 12%, and slices for which >50% of electrodes were discounted were removed from analysis. Only electrodes within the CA1 dendritic field and without spike-contamination were included in the analysis. The effect of drug treatment was analyzed by ANOVA, followed by Tukey HSD post hoc tests with statistical significance set as p < 0.05. For the dose response study, the effect of roflumilast concentration was analyzed by ANOVA, followed by Tukey HSD post hoc tests with statistical significance set as p < 0.05.
2.8. Cell death measurement
Propidium iodide (PI) fluorescence was used to observe the effect of roflumilast on cell viability. Cell death was measured immediately prior and 24 h following injury using 2.5 μM PI (Life Technologies) in serum-free medium. Previous studies with this injury model have reported that blast injury caused minimal cell death (Effgen et al., 2014; Vogel et al., 2016a). Cultures were treated with full serum medium containing either 1 nM roflumilast or DMSO (0.07%) vehicle at 6 h post-injury. Cell death was determined for ROI, as previously described, using MetaMorph (Molecular Devices, Downingtown, PA), and reported as percentage area (Cater et al., 2006; Effgen et al., 2012, 2014; Vogel et al., 2016a). To confirm OHSC viability, a subset of cultures were exposed to blast and either roflumilast or vehicle treatment, and subsequently subjected to an excitotoxic exposure of glutamate (10 mM for 3 h) 24 h following blast exposure (18 h following drug delivery). OHSC were returned to fresh serum-free medium following excitotoxic exposure, and cultures were imaged for cell death 24 h later. Cell death was analyzed by ANOVA, followed by Tukey HSD post hoc tests with statistical significance set at p < 0.05.
2.9. Chemically-induced LTP (chemLTP)
In a separate set of cultures, LTP was chemically induced. The chemLTP protocol replaced electrical LTP induction with a 3 min perfusion with a modified aCSF solution (gly-aCSF) (Lu et al., 2001): norm-aCSF containing 200 μM glycine and 0 mM MgCl (reduced from 1.3 mM). Perfusate was then switched back to norm-aCSF for 20 min prior to assessing LTP induction. LTP was quantified as described above, including procedures to include only stable recordings from the CA1 dendritic field that did not contain spikes. The effect of drug-treatment was analyzed by ANOVA with statistical significance set as p < 0.05.
2.10. Western blotting
For each condition tested by Western blotting, 8 slice cultures from 2 different animals were collected for protein extraction at the indicated time points. Protein concentrations were determined by the BCA assay according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA), and 50 μg of protein per sample was separated on a 4–12% Bis-Tris gel (Life Technologies) and transferred to a nitrocellulose membrane by semi-dry transfer, as previously described (Vogel et al., 2016a). The membrane was blocked in TBS (pH 7.4) with 1% BSA for 90 min. Membranes were incubated overnight at 4 °C with primary Ab (total GluR1 [RRID:AB_1977216, 1:1000], phosphorylated GluR1-Ser831 [RRID:AB_1977218, 1:500], total PSD-95 [RRID:AB_ 2092361, 1:500], phosphorylated stargazin-Ser239/240 [RRID:AB_ 10807145, 1:500], β-actin [RRID: AB_476692, 1:2000]) in TBS with Tween (TBS-T, 0.1% Tween-20, pH 7.4) and 0.25% BSA. To detect protein bands, the membranes were labeled with a corresponding secondary Ab (Donkey anti-Rabbit Alexa Fluor 488 or Goat anti-Mouse Alexa Fluor 647, Life Technologies). Fluorescence was detected using a CRi Maestro 2 Imaging System (Perkin Elmer). The bands were quantified using ImageJ software. Average fluorescence was quantified from, at minimum, 4 lysates per exposure group (8 slices per lane, 32 slices in total). All cultures were exposed to blast injury and received either 1 nM roflumilast or DMSO vehicle at 6 h post-injury. At 24 h post-injury, cultures were exposed to aCSF containing 200 μM glycine (LTP induction) or DMSO vehicle (No LTP induction) prior to cell lysis. The effects of drug-treatment and chemLTP treatment were analyzed by two-way ANOVA with statistical significance set as p < 0.05.
3. Results
3.1. PDE4 inhibitors prevented LTP deficits after blast
Our group previously observed that 1 μM roflumilast delivered immediately post-blast prevented LTP deficits measured 24 h following injury (Vogel et al., 2016b). When delivered immediately following blast exposure, roflumilast (concentrations ≥1 nM) was efficacious in preventing blast-induced LTP deficits 24 h post-injury (Fig. 1), as compared to blast-injured cultures treated with vehicle or 100pM roflumilast. Alternative PDE4 inhibitors, ibudilast and piclomilast, were similarly effective in preventing blast-induced LTP deficits.
Fig. 1.

Effect of PDE inhibitors on LTP measured 24 h following primary blast injury. A) Cultures were treated with either DMSO (vehicle), roflumilast (RFM), piclamilast (PIC), ibudilast (IBD), or papaverine (PAP) immediately following blast exposure. LTP was significantly increased in cultures that received roflumilast (1 nM or greater), piclamilast, or ibudilast compared to those receiving vehicle. LTP in cultures that received 100 pM roflumilast or papverine was not significantly different from those receiving vehicle. Potentiation was evaluated 60 min following HFS. B) Examples of field potentials from a single CA1 dendritic field electrode prior to and following LTP induction in a slice culture exposed to blast and subsequently treated with 1 nM RFM. C) Average electrode response over time for cultures exposed to blast and immediately treated with either 1 nM roflumilast or DMSO vehicle. High-frequency LTP was induced at 30 min. (Mean ± S.E.M.; n ≥ 6; *p < 0.05, as compared to blast + vehicle, #p < 0.05, as compared to blast + 100 pM RFM.)
PDE4 enzymes are present in abundance across the hippocampus, including dendrites of CA1 neurons, and have been attributed with maintaining basal levels of cAMP (Ahmed and Frey, 2003; Cherry and Davis, 1999; Perez-Torres et al., 2000). This contrasts with PDE10A which also inhibits degradation of cAMP; however, basal expression of PDE10A within the hippocampus is limited to cell bodies, with no discernable expression in the dendritic or axonal processes (Seeger et al., 2003). We observed that treatment with the PDE10A inhibitor papaverine immediately following blast did not prevent LTP deficits measured 24 h post-injury.
3.2. Delayed roflumilast treatment prevented LTP deficits after blast
We varied the time of roflumilast delivery, using the minimum therapeutic roflumilast concentration (1 nM), to determine the duration of the therapeutic window. Roflumilast delivered at 1 and 6 h post-injury prevented LTP deficits 24 h following blast exposure (Fig. 2). In contrast, roflumilast delivered 23 h post-injury did not prevent LTP deficits measured 1 h later. To verify that inefficacy of 23-h delayed drug delivery was not due to minimal exposure time, we delivered the drug 23 h post-injury in a separate set of cultures and observed an LTP deficit when recording after an additional 24 h (47 h post-injury).
Fig. 2.
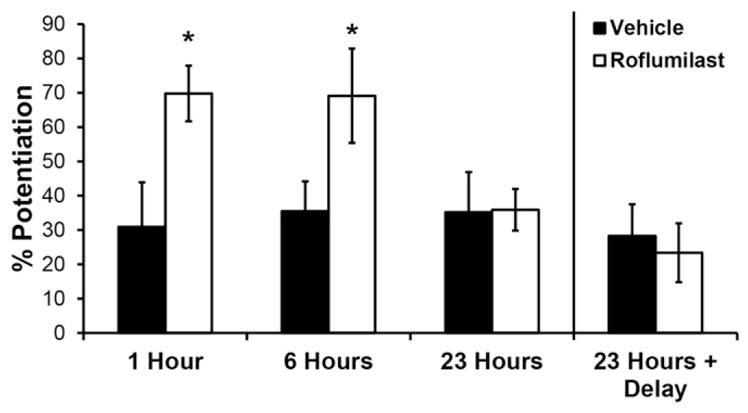
Effect of roflumilast delivery time on LTP measured after primary blast injury. Cultures were treated with either DMSO (vehicle) or 1 nM roflumilast at increasing times post-blast exposure (1, 6 or 23 h). LTP was significantly increased in cultures that received roflumilast at 1 or 6 h post-blast, as compared to time-matched vehicle treatment. Cultures that received vehicle or roflumilast at 23 h post-blast were unable to significantly potentiate. When cultures were treated with vehicle or roflumilast at 23 h post-injury and LTP was evaluated after an additional 24 h (47 h post-blast), potentiation was not increased by treatment. Potentiation was evaluated 60 min following HFS. (Mean ± S.E.M.; n ≥ 6; *p < 0.05, as compared to time-matched blast + vehicle, #p < 0.05, as compared to blast + roflumilast delivered at 23 h.)
3.3. Roflumilast treatment prevented deficits in glycine-induced LTP after blast
The gly-aCSF activates synaptic NMDA receptors, allowing Ca2+ ions to enter the dendritic spine, similar to electrically-induced LTP (Lu et al., 2001). Roflumilast (1 nM) delivered 6 h post-blast exposure significantly enhanced glycine-induced potentiation 24 h post-blast, as compared to vehicle (Fig. 3). We had previously reported that blast exposure significantly reduced glycine-induced potentiation 24 h post-blast, as compared to a sham exposure, which was confirmed in the current study (Vogel et al., 2016b).
Fig. 3.
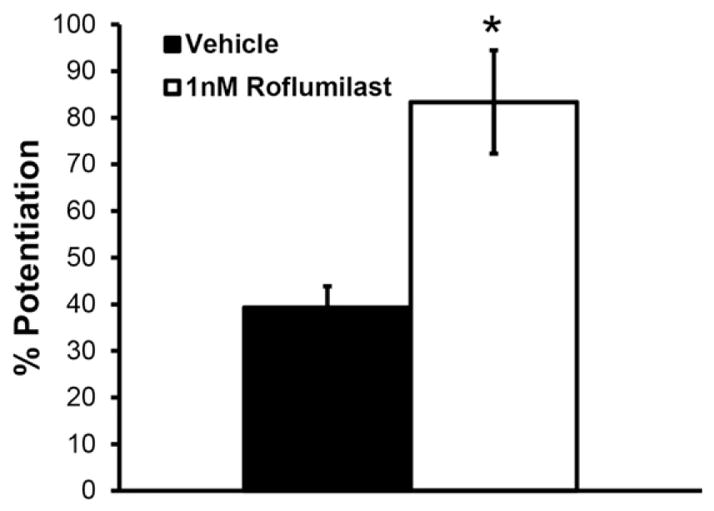
Roflumilast treatment significantly increased glycine-induced LTP 24 h following blast exposure. LTP was significantly increased in injured, roflumilast-treated (1 nM delivered 6 h post-injury) cultures when induced by gly-aCSF at 24 h following injury, as compared to injured, vehicle-treated cultures also induced by gly-aCSF. Potentiation was evaluated 60 min following end of chemical LTP induction protocol. (Mean ± S.E.M.; n ≥ 7, *p < 0.05, as compared to blast+ vehicle.)
3.4. Roflumilast treatment prevented deficits in spontaneous activity after blast
Blast exposure significantly decreased spontaneous event rate across all ROI for vehicle-treated cultures as compared to vehicle-treated sham cultures (Fig. 4). Roflumilast (1 nM) delivered 6 h post-blast, prevented the decrease in spontaneous event rate across all ROI at 24 h, as compared to vehicle treated cultures. There was no effect of blast or drug exposure on spontaneous event magnitude for any ROI. Roflumilast significantly reduced event duration in all ROI, as compared to sham + vehicle, and in CA1 and CA3, as compared to blast + vehicle. Roflumilast also rescued blast-induced deficits in GSI.
Fig. 4.

Roflumilast treatment affected spontaneous activity measured 24 h following blast exposure. A) Event rate (Hz) was significantly depressed across all regions for injured, vehicle-treated (6 h) cultures, as compared to sham + vehicle treatment. Roflumilast (1 nM) treatment 6 h following blast significantly increased event rate, as compared to injured, vehicle-treated cultures, returning it to sham levels. B) Event magnitude (μV) was unaffected by either blast exposure or roflumilast treatment. C) Blast exposure did not significantly affect event duration (ms); however, roflumilast exposure 6 h following blast significantly depressed event duration, as compared to vehicle-treated sham or blast cultures in CA1 and CA3 regions (only was significantly depressed in DG for blast + vehicle). D) GSI was significantly depressed for injured, vehicle-treated cultures, as compared to sham + vehicle treated slices; however, roflumilast treatment following blast injury significantly increased GSI, as compared to injured, vehicle-treated cultures, returning it to sham levels. E) An example of an identified spontaneous field event. (Mean ± S.E.M.; n ≥ 7, *p < 0.05, as compared to sham + vehicle; #p < 0.05, as compared to blast + vehicle.)
3.5. Roflumilast treatment did not affect basal evoked function or cell viability
We previously reported that blast exposure had minimal effect on basal evoked function, measured through SR (Vogel et al., 2016a; Vogel et al., 2016b). We observed no significant effect of blast exposure or roflumilast delivery in any ROI on SR parameters 24 h post-injury (data not shown). We observed no significant effect of blast exposure or roflumilast in any ROI on cell viability; exposure of cultures to excitotoxic concentrations of glutamate verified the presence of viable cells after blast (data not shown).
3.6. After blast, roflumilast preserved protein transduction pathways necessary for LTP induction
Phosphorylation of GluR1-Ser831 has been shown to be a key molecular step for induction of LTP (Lee et al., 2000). We previously reported that blast exposure significantly reduced phosphorylation of GluR1-Ser831 and expression of total GluR1 after glycine-exposure 24 h postblast (Vogel et al., 2016b). We found in this study that after blast exposure and LTP-induction, increased phosphorylation of GluR1-Ser831 was eliminated (Fig. 5A). However, post-injury treatment with roflumilast restored GluR1-Ser831 phosphorylation after LTP induction. In the absence of LTP induction, roflumilast did not affect levels of phosphorylation for GluR1-Ser831.
Fig. 5.

Roflumilast treatment 6 h following blast injury altered protein expression measured 24 h post-injury. Protein expression (normalized to loading control β-actin)was evaluated at 24 h following blast for four protein targets critical to LTP: pGluR1-Ser831 (A), total GluR1 (B), total PSD-95 (C), and pStargazin-Ser239/240 (D). Cultures were exposed to blast and treated with either DMSO (vehicle) or 1 nM roflumilast 6 h following injury. At 24 h post-injury, cultured were treated with either aCSF+DMSO (No LTP Induction) or aCSF+200 μMglycine (LTP induction) prior to cell lysis. Roflumilast treatment significantly increased pGluR1-Ser831, total GluR1, total PSD-95, and pStargazin-Ser239/240 when LTP was induced, as compared to injured, vehicle-treated cultures. Roflumilast treatment significantly increased total PSD-95 when LTP was not induced, as compared to injured, vehicle-treated cultures. LTP induction significantly increased pGluR1-Ser831 and pStargazin-Ser239/240 in injured, roflumilast-treated cultures. Representative bands from each group are shown below the graphs, along with the size of the identified protein. (Mean ± S.E.M.; n ≥ 4, *p < 0.05, as compared to LTP-matched vehicle treatment, #p < 0.05 as compared to treatment-matched, non-LTP induction.)
Blast exposure eliminated the increase of total GluR1 expression normally observed after LTP induction (Vogel et al., 2016b). However, post-injury treatment with roflumilast increased total GluR1 expression after LTP induction (Fig. 5B). In the absence of LTP induction, roflumilast did not affect total expression of GluR1.
Our group has previously observed that primary blast exposure significantly reduced expression of PSD-95 and phosphorylation of stargazin at the Ser239/240 site 24 h post-injury (Vogel et al., 2016b). In this study, roflumilast significantly increased total expression of PSD-95 regardless of LTP induction (Fig. 5C). PSD-95 expression after LTP induction was the same for either vehicle-treated or roflumilast-treated cultures. LTP induction was previously shown to not affect total PSD-95 expression (Kim et al., 2007). Post-blast, phosphorylation of stargazin-Ser239/240 was not increased after LTP induction (Fig. 5D). However, post-injury treatment with roflumilast restored stargazin-Ser239/240 phosphorylation after LTP induction. In the absence of LTP induction, roflumilast did not affect levels of phosphorylation for GluR1-Ser831.
4. Discussion
We previously reported that a PDE4 inhibitor, roflumilast at high concentration, prevented neuronal dysfunction after primary blast exposure (Vogel et al., 2016b), and herein, we report molecular mechanisms by which PDE4 inhibitors may be preserving LTP induction. Although we focused on changes in proteins critical for LTP, additional mechanisms may also be responsible, in part, for disruption of neuronal plasticity after blast, including changes to cable properties of dendrites, electrotonic coupling, or ion channels in dendrites altering excitatory postsynaptic potential-spike (E-S) coupling. We observed that roflumilast (1 nM) increased LTP when treatment was delayed by up to 6 h post-injury but was not effective when delivered 23 h post-injury. We also observed that roflumilast increased protein expression and phosphorylation caused by LTP induction that was previously lost when slices were exposed to blast (Vogel et al., 2016b). Roflumilast restored the increase in pGluR1-Ser831, expression of total GluR1, and pStargazin-Ser239/240 upon LTP induction 24 h following blast exposure. Additionally, roflumilast increased PSD-95 expression, regardless of LTP induction, at 24 h following blast exposure. These observations suggest that modulation of the cAMP pathway through inhibition of PDE4 can prevent primary blast-induced alterations in proteins critical for synaptic plasticity.
Investigation of blast-induced alteration of cAMP has been limited. One study observed that 3′,5′-cAMP did not significantly change in pre-frontal cortex using one rodent mild blast injury model (Kochanek et al., 2013). Conversely, FPI studies observed hippocampal cAMP levels decreased between 15 min and 24 h following injury (Atkins et al., 2007; Dhillon et al., 1995; Titus et al., 2013a). Increased hippocampal PDE4 expression was observed between 1 and 24 h following FPI in rats (Wilson et al., 2016). Studies using PDE4 inhibitors have demonstrated that a cAMP-dependent mechanism is a part of the pathogenesis of TBI and modulation of this pathway can prevent electrophysiological deficits measured in vitro and cognitive deficits measured in vivo after blast (Eakin et al., 2013; Titus et al., 2013b).
Previous work showed PDE4 inhibitors can enhance hippocampal-dependent memory and LTP in animals (Randt et al., 1982; Rutten et al., 2008; Wang et al., 2012). The current study observed that roflumilast concentrations of at least 1 nM preserved plasticity after blast (Fig. 1). Alternative PDE4 inhibitors (piclamilast, ibudilast) similarly preserved plasticity. Although no other study has investigated the therapeutic potential of PDE4 inhibition after blast, several studies have observed that PDE4 inhibition preserved LTP measured 2 weeks and 12 weeks following FPI in rats (Titus et al., 2013b; Titus et al., 2016). The effectiveness of either ibudilast or piclamilast following TBI is unknown; however, ibudilast prevented LTP-deficits in cortical cell cultures after microglia activation (Mizuno et al., 2004). Similar findings across biomechanically distinct models provide greater confidence in the therapeutic potential of PDE4 inhibition following injury (Dixon et al., 1987; Panzer et al., 2012; Thibault et al., 1992). Conversely, we found that the PDE10A inhibitor papaverine was unable to rescue plasticity after blast injury. A previous study observed that PDE10A expression decreased at 1 h post-FPI in rats, whereas PDE4 expression increased (Wilson et al., 2016). It is possible that this change in expression leads to the observed deficits in in CA1 synaptic plasticity (Rutten et al., 2008). The contrasting abilities of PDE4 and PDE10A inhibitors to prevent blast-induced deficits in hippocampal LTP suggests that there may be an important subcellular effect and regulation of recovery following blast.
Previous studies found that PDE4 inhibition at 2 weeks (Titus et al., 2013b) and 3 months (Titus et al., 2016) post-FPI improved both LTP in vitro and cognition in vivo. This contrasts with our observation that delayed PDE4 inhibition (23 h post-injury) did not prevent blast-induced LTP deficits. One possible explanation is that our study delivered the PDE4 inhibitor at acute time points following injury, whereas the aforementioned non-blast studies delivered PDE4 inhibitors at subacute time points after injury and during LTP induction with electrical stimulation. Our findings showing efficacy with a 6 h delayed treatment exceed the current target for an extended therapeutic window within clinical trials (3 h post-TBI) (Wang et al., 2006). PDE4 inhibition may provide an extended delivery window making it an exciting prospect.
PDE4 inhibition by roflumilast reversed negative effects of blast on network synchronization and spontaneous event rate measured 24 h following injury (Fig. 3). In cortical neurons, PDE4 inhibition increased spontaneous spike-rate (Castro et al., 2010). Increasing cAMP concentration increased spontaneous spike rate in the hippocampus, but decreased burst duration (Niedringhaus et al., 2013). Niedringhaus and colleagues suggested collective network activity contracted and reorganized into shorter episodes following PKA activation, which could explain the observed changes in event rate and duration in our study. We observed that blast did not alter spontaneous event duration, but roflumilast treatment decreased duration. More research is needed to confirm if the protective effect of PDE4 inhibition on spontaneous neural function following TBI will translate to animal studies.
We observed that measures of basal evoked function were unaffected by PDE4 inhibition. Several studies observed that TBI-induced deficits in hippocampal input/output (I/O) curves were improved following PDE4 inhibition in rats (Titus et al., 2013b). There was no observable effect on I/O curves or PPF in PDE4D genetic KO mice (Rutten et al., 2008). These data suggest that PDE4 inhibition can repair deficits in synaptic plasticity without negatively affecting basal evoked function.
We observed that roflumilast significantly increased glycine-induced LTP measured 24 h following blast exposure (Fig. 4). PDE4 inhibitors’ effect on chemical LTP is unclear. One study observed that in vivo blast injury decreased chemical LTP (via rolipram/forskolin) in hippocampal slices measured 2 weeks following injury (Goldstein et al., 2012). Our group previously found that rolipram/forskolin-induced LTP was not decreased by blast at 24 h following in vitro primary blast (Vogel et al., 2016b). The varied responses may be due to several differences between these studies, in terms of injury model, blast injury mechanics, and observation time post-injury.
This study is the first to report that PDE4 inhibitors prevented changes in protein expression after primary blast. Roflumilast treatment significantly increased phosphorylation of GluR1-Ser831 when LTP was induced, as compared to injured, vehicle-treated cultures. Several studies show that increasing cAMP concentration promotes AMPA receptor function, both directly and indirectly (Blitzer et al., 1998; Oh et al., 2006). We observed that when LTP was induced after injury, roflumilast treatment significantly increased expression of total GluR1 subunits compared to vehicle-treated cultures. However, this finding contrasts with another study in which rolipram did not increase total GluR1 expression in rat hippocampal neuronal cultures (Andreetta et al., 2016). More studies are needed to understand the influence of PDE4 inhibitors on AMPARs following injury.
The proteins PSD-95 and stargazin are critical for anchoring AMPARs at the postsynaptic membrane during LTP (Bats et al., 2007; Colledge et al., 2003). Here we observed that roflumilast treatment significantly increased expression of total PSD-95 after injury regardless of LTP induction. Forskolin activation of the cAMP pathway was found to prevent NMDA-induced loss of PSD-95, implicating cAMP in the regulation of PSD-95 (Colledge et al., 2003). We also observed that roflumilast significantly increased pStargazin-Ser239/240 after injury when LTP was induced. Although the effect of PDE4 inhibitors on stargazin phosphorylation has not been studied previously, ghrelin, an activator of both PKA and protein kinase C (PKC), increased pStargazin-Ser239/240 (Ribeiro et al., 2013). It is important to note that the Ser239/240 site is phosphorylated by PKC, but there is evidence of crosstalk between the PKA and PKC pathways (Mao et al., 2007; Roberson et al., 1999). Our working hypothesis for blast-induced LTP deficits is that degradation of PSD-95 led to a reduced ability of GluR1-containing AMPARs to immobilize at the PSD, which prevented phosphorylation of GluR1-Ser831 and thus reduced potentiation (Fig. 6). It is possible that PDE4 inhibition restored LTP induction by preventing blast-induced PSD-95 degradation which enables immobilization and phosphorylation of GluR1-containing AMPARs at the PSD and, ultimately, increased potentiation.
Fig. 6.

Hypothesized mechanism for the prevention of primary blast-induced long-term potentiation (LTP) deficits via phosphodiesterase-4 (PDE4) inhibition. On the left is a dendritic spine exposed to blast, and on the right, is a dendritic spine that has been treated with roflumilast after blast. Our findings suggest that primary blast exposure reduces expression of PSD-95, which in turn prevents immobilization and phosphorylation of glutamate receptor-1 (GluR1)-containing α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) at the PSD, a necessary step for potentiation. We hypothesize that PDE4 inhibition maintains expression of PSD-95 (by a mechanism to be determined), which then allows for the downstream immobilization and phosphorylation of GluR1 subunits necessary for LTP induction.
Although we report that PDE4 inhibition following primary blast injury preserves LTP, there are some limitations associated with this study. The use of an in vitro culture model prevents direct comparisons between electrophysiology and cognitive function observed in vivo (Goldstein et al., 2012; Patel et al., 2014). A benefit of our injury model is that it provides precise control over the injury biomechanics, which is difficult to achieve with in vivo injury models. Another limitation of this study is the induction of LTP via glycine exposure in cultures lysed for Western blotting. This study required that chemical induction be employed due to the number of slices needed for Western blotting. Glycine-induced LTP generated hallmark signs of electrically-induced LTP, including increased electrical response, phosphorylation of AMPA-GluR1 subunits, and activation of CaMKII (Lu et al., 2001). Another limitation of this study is that we did not evaluate total stargazin expression. As phosphorylation of stargazin is necessary to bind AMPARs to PSD-95 at the postsynaptic density, we viewed phosphorylated stargazin as a surrogate for AMPAR translocation, which was the mechanism we wished to investigate. As such, we felt that total stargazin levels were not critical to the mechanism for blast-induced LTP dysfunction and chose not to include total stargazin. Another limitation of this study is that it is possible that roflumilast mediated its preservative effect via alternative mechanisms other than those explored here. These alternative mechanisms could include changes to dendritic cable or ion channel properties or electrotonic coupling that could influence E-S coupling (Reeves et al., 1995; Smith et al., 1993; Witgen et al., 2005). Additionally, roflumilast may influence other synaptic plasticity proteins outside of those studied here (Atkins et al., 2009; Bell et al., 2009; D’Ambrosio et al., 1998). Finally, this study may be limited by the use of a pan-PDE4 inhibitor, like roflumilast, over a subtype-specific inhibitor. Pan-PDE4 inhibitors can induce emetic effects in patients, limiting their clinical effectiveness (Titus et al., 2016); however, a recent study observed that non-emetic doses of roflumilast improved memory in rodents (Vanmierlo et al., 2016). In that study, free brain concentrations of roflumilast were estimated to be 10.37 nM, which is greater than our lowest effective concentration tested (1 nM). Time scaling between slice cultures and humans may also be a necessary consideration moving forward (Panzer et al., 2014).
In summary, we report that PDE4 inhibitors preserved LTP induction in the hippocampus after primary blast. We found the minimal therapeutic concentration of roflumilast was 1 nM and the therapeutic window was at least 6 h, but <23 h. Roflumilast treatment postblast increased phosphorylation or expression of proteins critical for LTP induction including AMPAR-GluR1 (total and pGluR1-Ser831), and pStargazin-Ser239/240 when LTP was induced. Regardless of LTP induction, roflumilast treatment post-blast increased total PSD-95 expression. Future studies will investigate the potential of PDE4 inhibitors to prevent cognitive deficits after in vivo blast injury.
Acknowledgments
Funding
This work was supported in part by a Multidisciplinary University Research Initiative from the Army Research Office (W911NF-10-1-0526) and by a National Defense Science & Engineering Graduate Fellowship from the Department of Defense (EWV-2012).
Abbreviations
- bTBI
blast-induced traumatic brain injury
- mTBI
mild traumatic brain injury
- GSI
global synchronization index
- pGluR1
phosphorylated glutamate receptor subunit 1
- pStargazin
phosphorylated stargazin
- PDE4
phosphodiesterase-4
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- Ahmed T, Frey JU. Expression of the specific type IV phosphodiesterase gene PDE4B3 during different phases of long-term potentiation in single hippocampal slices of rats in vitro. Neuroscience. 2003;117:627–638. doi: 10.1016/s0306-4522(02)00838-2. [DOI] [PubMed] [Google Scholar]
- Andreetta F, Carboni L, Grafton G, Jeggo R, Whyment AD, van den Top M, Hoyer D, Spanswick D, Barnes NM. Hippocampal 5-HT7 receptors signal phosphorylation of the GluA1 subunit to facilitate AMPA receptor mediated neurotransmission in vitro and in vivo. Br J Pharmacol. 2016;173:1438–1451. doi: 10.1111/bph.13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Oliva AA, Jr, Alonso OF, Pearse DD, Bramlett HM, Dietrich WD. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp Neurol. 2007;208:145–158. doi: 10.1016/j.expneurol.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Falo MC, Alonso OF, Bramlett HM, Dietrich WD. Deficits in ERK and CREB activation in the hippocampus after traumatic brain injury. Neurosci Lett. 2009;459:52–56. doi: 10.1016/j.neulet.2009.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass CR, Panzer MB, Rafaels KA, Wood G, Shridharani J, Capeheart BP. Brain injuries from blast. Ann Biomed Eng. 2012;40:185–202. doi: 10.1007/s10439-011-0424-0. [DOI] [PubMed] [Google Scholar]
- Bats C, Groc L, Choquet D. The interaction between stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Beamer M, Tummala SR, Gullotti D, Kopil K, Gorka S, Abel T, Bass CR, Morrison B, III, Cohen AS, Meaney DF. Primary blast injury causes cognitive impairments and hippocampal circuit alterations. Exp Neurol. 2016;283:16–28. doi: 10.1016/j.expneurol.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JD, Park E, Ai J, Baker AJ. PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma. Cell Death Differ. 2009;16:1665–1680. doi: 10.1038/cdd.2009.106. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science. 1998:280. doi: 10.1126/science.280.5371.1940. [DOI] [PubMed] [Google Scholar]
- Castro LRV, Gervasi N, Guiot E, Cavellini L, Nikolaeva VO, Paupardin-Tritsch D, Vincent P. Type 4 phosphodiesterase plays different integrating roles in different cellular domains in pyramidal cortical neurons. J Neurosci. 2010;30:6143–6151. doi: 10.1523/JNEUROSCI.5851-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cater HL, Sundstrom LE, Morrison B., III Temporal development of hippocampal cell death is dependent on tissue strain but not strain rate. J Biomech. 2006;39:2810–2818. doi: 10.1016/j.jbiomech.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Cernak I, Wang Z, Jiang J, Bian X, Savic J. Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J Trauma. 2001;50:695–706. doi: 10.1097/00005373-200104000-00017. [DOI] [PubMed] [Google Scholar]
- Cherry JA, Davis RL. Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated wiht reinforcement, movement, and affect. J Comp Neurol. 1999;407:287–301. [PubMed] [Google Scholar]
- Choi JH, Jung HK, Kim T. A new action potential detector using the MTEO and its effects on spike sorting systems at low signal-to-noise ratios. IEEE Trans Biomed Eng. 2006;53:738–746. doi: 10.1109/TBME.2006.870239. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. Selective loss of hippocampal long-term potentiation, but not depression, following fluid percussion injury. Brain Res. 1998;786:64–79. doi: 10.1016/s0006-8993(97)01412-1. [DOI] [PubMed] [Google Scholar]
- Dhillon HS, Yang L, Padmaperuma B, Dempsey RJ, Fiscus RR, Prasad MR. Regional concentrations of cyclic nucleotides after experimental brain injury. J Neurotrauma. 1995;12:1035–1043. doi: 10.1089/neu.1995.12.1035. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- DVBIC. Department of Defense Numbers for Traumatic Brain Injury. Armed Forces Health Surveillance Center, Defense and Veterans Brain Injury Center (DVBIC); 2016. [Google Scholar]
- Eakin K, Li Y, Chiang YH, Hoffer BJ, Rosenheim H, Greig NH, Miller JP. Exendin-4 ameliorates traumatic brain injury-induced cognitive impairments in rats. PLoS One. 2013;8:82016. doi: 10.1371/journal.pone.0082016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effgen GB, Hue CD, Vogel EW, III, Panzer MB, Bass CR, Meaney DF, Morrison B., III Amultiscale approach to blast neurotrauma modeling: part II: methodology for inducing blast injury to in vitro models. Front Neurol. 2012;3:1–10. doi: 10.3389/fneur.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effgen GB, Vogel EW, III, Lynch KA, Lobel A, Hue CD, Meaney DF, Bass CR, Morrison B., III Isolated primary blast alters neuronal function with minimal cell death in organotypic hippocampal slice cultures. J Neurotrauma. 2014;31:1202–1210. doi: 10.1089/neu.2013.3227. [DOI] [PubMed] [Google Scholar]
- Effgen GB, Ong T, Nammalwar S, Ortuno AI, Meaney DF, Bass CR, Morrison B., III Primary blast exposure increases hippocampal vulnerability to subsequent exposure: reducing long-term potentiation. J Neurotrauma. 2016 doi: 10.1089/neu.2015.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra160. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–1634. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullotti DM, Beamer M, Panzer MB, Chia Chen Y, Patel TP, Yu A, Jaumard N, Winkelstein B, Bass CR, Morrison B, Meaney DF. Significant head accelerations can influence immediate neurological impairments in a murine model of blast-induced traumatic brain injury. J Biomech Eng. 2014;136:091004. doi: 10.1115/1.4027873. [DOI] [PubMed] [Google Scholar]
- Heuschkel MO, Fejtl M, Raggenbass M, Bertrand D, Renaud P. A three-dimensional multi-electrode array for multi-site stimulation and recording in acute brain slices. J Neurosci Methods. 2002;114:135–148. doi: 10.1016/s0165-0270(01)00514-3. [DOI] [PubMed] [Google Scholar]
- Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. soldiers returning from Iraq. N Engl J Med. 2008;358:453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, Cernak I, Petrie EC, Emery MJ, Swenson ER, Mayer C, Mehic E, Peskind ER, Cook DG. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis. 2013;37:309–323. doi: 10.3233/JAD-130182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue CD, Cao S, Haider SF, Vo KV, Effgen GB, Vogel EW, III, Panzer MB, Bass CR, Meaney DF, Morrison B., III Blood-brain barrier dysfunction after primary blast injury in vitro. J Neurotrauma. 2013;30:1652–1663. doi: 10.1089/neu.2012.2773. [DOI] [PubMed] [Google Scholar]
- Hue CD, Cao S, Bass CR, Meaney DF, Morrison B., III Repeated primary blast injury causes delayed recovery, but not additive disruption, in an in vitro blood–brain barrier model. J Neurotrauma. 2014;31:951–960. doi: 10.1089/neu.2013.3149. [DOI] [PubMed] [Google Scholar]
- Kang WH, Morrison B., III Functional tolerance to mechanical deformation developed from organotypic hippocampal slice cultures. Biomech Model Mechanobiol. 2014 doi: 10.1007/s10237-014-0622-4. [DOI] [PubMed] [Google Scholar]
- Kang WH, Morrison B., III Predicting changes in cortical electrophysiological function after in vitro traumatic brain injury. Biomech Model Mechanobiol. 2015:1–12. doi: 10.1007/s10237-015-0652-6. [DOI] [PubMed] [Google Scholar]
- Kang WH, Cao W, Graudejus O, Patel TP, Wagner S, Meaney DF, Morrison B., III Alterations in hippocampal network activity after in vitro traumatic brain injury. J Neurotrauma. 2014 doi: 10.1089/neu.2014.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron. 2007;56:488–502. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Kochanek PM, Dixon CE, Shellington DK, Shin SS, Bayir H, Jackson EK, Kagan VE, Yan HQ, Swauger PV, Parks SA, Ritzel DV, Bauman R, Clark RSB, Garman RH, Bandak F, Ling G, Jenkins LW. Screening of biochemical and molecular mechanisms of secondary injury and repair in the brain after experimental blast-induced traumatic brain injury in rats. J Neurotrauma. 2013;30:920–937. doi: 10.1089/neu.2013.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontos AP, Kotwal RS, Elbin RJ, Lutz RH, Forsten RD, Benson PJ, Guskiewicz KM. Residual effects of combat-related mild traumatic brain injury. J Neurotrauma. 2013;30:680–686. doi: 10.1089/neu.2012.2506. [DOI] [PubMed] [Google Scholar]
- Lee H, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Li X, Cui D, Jiruska P, Fox JE, Yao X, Jefferys JG. Synchronization measurement of multiple neuronal populations. J Neurophysiol. 2007;98:3341–3348. doi: 10.1152/jn.00977.2007. [DOI] [PubMed] [Google Scholar]
- Li X, Ouyang G, Usami A, Ikegaya Y, Sik A. Scale-free topology of the CA3 hippocampal network: a novel method to analyze functional neuronal assemblies. Biophys J. 2010;98:1733–1741. doi: 10.1016/j.bpj.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: The role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the aamino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- Mao L, Tang Q, Wang JQ. Protein kinase C-regulated cAMP response element-binding protein phosphorylation in cultured rat striatal neurons. Brain Res Bull. 2007;72:302–308. doi: 10.1016/j.brainresbull.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell CR, Kanes SJ, Abel T, Siegel SJ. Phosphodiesterase inhibitors: a novel mechanism for receptor-independent antipsychotic medications. Neuroscience. 2004;129:101–107. doi: 10.1016/j.neuroscience.2004.07.038. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL. Traumatic brain injury in the rat: characterization of a lateral fluid-percusssion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kurotani T, Komatsu Y, Kawanokuchi J, Kato H, Mitsuma N, Suzumura A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004;46:404–411. doi: 10.1016/j.neuropharm.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Morrison B, 3rd, Cater HL, Benham CD, Sundstrom LE. An in vitro model of traumatic brain injury utilising two-dimensional stretch of organotypic hippocampal slice cultures. J Neurosci Methods. 2006;150:192–201. doi: 10.1016/j.jneumeth.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Niedringhaus M, Chen X, Conant K, Dzakpasu R. Synaptic potentiation facilitates memory-like attractor dynamics in cultured in vitro hippocampal networks. PLoS One. 2013;8:e57144. doi: 10.1371/journal.pone.0057144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Ommaya AK, Gennarelli TA. Cerebral concussion and traumatic unconsciousness. Brain. 1974;97:633–654. doi: 10.1093/brain/97.1.633. [DOI] [PubMed] [Google Scholar]
- Panzer MB, Matthews KA, Yu AW, Morrison B, III, Meaney DF, Bass CR. A Multiscale approach to blast neurotrauma modeling: part I – development of novel test devices for in vivo and in vitro blast injury models. Front Neurol. 2012:3. doi: 10.3389/fneur.2012.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzer MB, Wood GW, Bass CR. Scaling in neurotrauma: how do we apply animal experiments to people? Exp Neurol. 2014;261:120–126. doi: 10.1016/j.expneurol.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Patel TP, Ventre SC, Meaney DF. Dynamic changes in neural circuit topology following mild mechanical injury in vitro. Ann Biomed Eng. 2012;40:23–36. doi: 10.1007/s10439-011-0390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TP, Gullotti DM, Hernandez P, O’Brien WT, Capeheart BP, Morrison B, III, Bass CR, Eberwine JH, Abel T, Meaney DF. An open-source toolbox for automated phenotyping of mice in behavioral tasks. Front Behav Neurosci. 2014;8:1–16. doi: 10.3389/fnbeh.2014.00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TP, Man K, Firestein BL, Meaney DF. Automated quantification of neuronal networks and single-cell calcium dynamics using calcium imaging. J Neurosci Methods. 2015;243:26–38. doi: 10.1016/j.jneumeth.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Torres S, Miro X, Palacios JM, Cortes R, Puigdomenech P, Mengod G. Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and [3H]rolipram binding autoradiography comparison with monkey and rat brain. J Chem Neuroanat. 2000;20:349–374. doi: 10.1016/s0891-0618(00)00097-1. [DOI] [PubMed] [Google Scholar]
- Randt CT, Judge ME, Bonnet KA, Quatermain D. Brain cyclic AMP and memory in mice. Pharmacol Biochem Behav. 1982;17:677–680. doi: 10.1016/0091-3057(82)90344-6. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Lyeth BG, Povlishock JT. Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp Brain Res. 1995;106:248–256. doi: 10.1007/BF00241120. [DOI] [PubMed] [Google Scholar]
- Ribeiro LF, Catarino T, Santos SD, Benoist M, van Leeuwen JF, Esteban JA, Carvalho AL. Ghrelin triggers the synaptic incorporation of AMPA receptors in the hippocampus. Proc Natl Acad Sci U S A. 2013;111:149–158. doi: 10.1073/pnas.1313798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigg JL, Mooney SR. Concussions and the Military: Issues Specific to Service Members. PM R. 2011;3:S380–S386. doi: 10.1016/j.pmrj.2011.08.005. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubovitch V, Ten-Bosch M, Zohar O, Harrison CR, Tempel-Brami C, Stein E, Hoffer BJ, Balaban CD, Schreiber S, Chiu W, Pick CG. A mouse model of blast-induced mild traumatic brain injury. Exp Neurol. 2011;232:280–289. doi: 10.1016/j.expneurol.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutten K, Misner DL, Works M, Blokland A, Novak TJ, Santarelli L, Wallace TL. Enhanced long-term potentiation and impaired learning in phosphodiesterase 4D-knockout (PDE4D−/−) mice. Eur J Neurosci. 2008;28:625–632. doi: 10.1111/j.1460-9568.2008.06349.x. [DOI] [PubMed] [Google Scholar]
- Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL, Lanfear J, Ryan AM, Schmidt CJ, Strick CA, Varghese AH, Williams RD, Wylie PG, Menniti FS. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003;985:113–126. doi: 10.1016/s0006-8993(03)02754-9. [DOI] [PubMed] [Google Scholar]
- Silverberg ND, Crane PK, Dams-O’Connor K, Holdnack J, Ivins BJ, Lange RT, Manley GT, McCrea M, Iverson GL. Developing a cognition endpoint for traumatic brain injury clinical trials. J Neurotrauma. 2016 doi: 10.1089/neu.2016.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Okiyama K, Thomas MJ, McIntosh TK. Effects of the excitatory amino acid receptor antagonists kynurenate and indole-2-carboxylic acid on behavioral and neurochemical outcome following experimental brain injury. Neuroscience. 1993;13:5383–5392. doi: 10.1523/JNEUROSCI.13-12-05383.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DL, Pozueta J, Gong B, Arancio O, Shelanski M. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc Natl Acad Sci U S A. 2009;106:16877–16882. doi: 10.1073/pnas.0908706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecoma ES, Monyer H, Goldberg MP, Choi DW. Traumatic neuronal injury in vitro is attenuated by NMDA antagonists. Neuron. 1989;2:1541–1545. doi: 10.1016/0896-6273(89)90042-1. [DOI] [PubMed] [Google Scholar]
- Thibault LE, Meaney DF, Anderson BJ, Marmarou A. Biomechanical aspects of a fluid percussion model of brain injury. J Neurotrauma. 1992;9:311–322. doi: 10.1089/neu.1992.9.311. [DOI] [PubMed] [Google Scholar]
- Titus DJ, Furones C, Kang Y, Atkins CM. Age-dependent alterations in cAMP signaling contribute to synaptic plasticity deficits following traumatic brain injury. Neuroscience. 2013a;231:182–194. doi: 10.1016/j.neuroscience.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus DJ, Sakurai A, Kang Y, Furones C, Jergova S, Santos R, Sick TJ, Atkins CM. Phosphodiesterase inhibition rescues chronic cognitive deficits induced by traumatic brain injury. J Neurosci. 2013b;33:5216–5226. doi: 10.1523/JNEUROSCI.5133-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus DJ, Oliva AA, Wilson NM, Atkins CM. Phosphodiesterase inhibitors as therapeutics for traumatic brain injury. Curr Pharm Des. 2014;21:332–342. doi: 10.2174/1381612820666140826113731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus DJ, Wilson NM, Freund JE, Carballosa MM, Sikah KE, Furones C, Dietrich WD, Gurney ME, Atkins CM. Chronic cognitive dysfunction after traumatic brain injury is improved with a phoshodiesterase 4B inhibitor. J Neurosci. 2016;36:7095–7108. doi: 10.1523/JNEUROSCI.3212-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmierlo T, Creemers P, Akkerman S, van Duinen M, Sambeth A, De Vry J, Uz T, Blokland A, Prickaerts J. The PDE4 inhibitor roflumilast improves memory in rodents at non-emetic doses. Behav Brain Res. 2016;303:26–33. doi: 10.1016/j.bbr.2016.01.031. [DOI] [PubMed] [Google Scholar]
- Vogel EW, III, Effgen GB, Patel TP, Meaney DF, Bass CR, Morrison B., III Isolated primary blast inhibits long-term potentiation in organotypic hippocampal slice cultures. J Neurotrauma. 2016a;33:652–661. doi: 10.1089/neu.2015.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel EW, III, Rwema SH, Meaney DF, Bass CR, Morrison B., III Primary blast injury depressed hippocampal long-termpotentiation through disruption of synaptic proteins. J Neurotrauma. 2016b doi: 10.1089/neu.2016.4578. (Available online) [DOI] [PubMed] [Google Scholar]
- Wang KKW, Larner SF, Robinson G, Hayes RL. Neuroprotection targets after traumatic brain injury. Curr Opin Neurol. 2006;19:514–519. doi: 10.1097/WCO.0b013e3280102b10. [DOI] [PubMed] [Google Scholar]
- Wang C, Yang X, Zhuo Y, Zhou H, Lin H, Cheng Y, Xu J, Zhang H. The phosphodiesterase-4 inhibitor rolipram reverses Aβ-induced cognitive impairment and neuroinflammatory and apoptotic responses in rats. Int J Neuropsychopharmacol. 2012;15:749–766. doi: 10.1017/S1461145711000836. [DOI] [PubMed] [Google Scholar]
- Wiescholleck V, Manahan-Vaughan D. PDE4 inhibition enhances hippocampal synaptic plasticity in vivo and rescues MK801-induced impairment of long-term potentiation and object recognition memory in an animal model of psychosis. Transl Psychiatry. 2012:2. doi: 10.1038/tp.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NM, Titus DJ, Oliva AA, Jr, Furones C, Atkins CM. Traumatic brain injury upregulates phosphodiesterase expression in the hippocampus. Front Syst Neurosci. 2016:10. doi: 10.3389/fnsys.2016.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witgen BM, Lifshitz J, Smith ML, Schwarzbach E, Liang SL, Grady MS, Cohen AS. Regional hippocampal alteration associated with cognitive deficit following experimental traumatic brain injury: a systems, network, and cellular evaluation. Neuroscience. 2005;133:1–15. doi: 10.1016/j.neuroscience.2005.01.052. [DOI] [PubMed] [Google Scholar]
- Yin TC, Britt JK, Ready JM, Pieper AA. P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Rep. 2014;8:1731–1740. doi: 10.1016/j.celrep.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Morrison B., III Experimental mild traumatic brain injury induces functional alteration of the developing hippocampus. J Neurophysiol. 2010;103:499–510. doi: 10.1152/jn.00775.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]