

Abstract
Background
Lysine succinylation is a new kind of post-translational modification which plays a key role in protein conformation regulation and cellular function control. To understand the mechanism of succinylation profoundly, it is necessary to identify succinylation sites in proteins accurately. However, traditional methods, experimental approaches, are labor-intensive and time-consuming. Computational prediction methods have been proposed recent years, and they are popular because of their convenience and high speed. In this study, we developed a new method to predict succinylation sites in protein combining multiple features, including amino acid composition, binary encoding, physicochemical property and grey pseudo amino acid composition, with a feature selection scheme (information gain). And then, it was trained using SVM (Support Vector Machine) and an ensemble learning algorithm.
Results
The performance of this method was measured with an accuracy of 89.14% and a MCC (Matthew Correlation Coefficient) of 0.79 using 10-fold cross validation on training dataset and an accuracy of 84.5% and a MCC of 0.2 on independent dataset.
Conclusions
The conclusions made from this study can help to understand more of the succinylation mechanism. These results suggest that our method was very promising for predicting succinylation sites. The source code and data of this paper are freely available athttps://github.com/ningq669/PSuccE.
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2249-4) contains supplementary material, which is available to authorized users.
Keywords: Predict succinylation sites, Multiple features, Grey pseudo amino acid composition, Information gain, SVM, Ensemble learning algorithm
Background
As a type of widespread reversible post-translational modification, lysine succinylation plays a significant role in both eukaryotic and prokaryotic cells [1–3]. In succinylation procedure, the succinyl group (-CO-CH2-CH2-CO-) is covalent bonding to specific lysine residues in proteins which might lead to substantial chemistry changes to proteins [4]. Besides, lysine succinylation can induce mutations of charge in the environment with PH value (hydrogen ion concentration) range from − 1 to + 1 and promote structural and functional adjustment to substrate proteins [5]. It is extremely important to understand the molecular mechanism of succinylation in biological systems by identifying succinylated substrate proteins along with succinylation sites, so more and more focus is put on this field [6–23].
Many biological experimental methods have been developed to identify succinylated protein or succinylation sites, such as high performance liquid chromatography assays, spectrophotometric assays and liquid chromatography-mass spectrometry [24, 25]. However, these experimental approaches are inconvenient, time-consuming and costly, especially for large-scale data sets. Therefore, efficient computational prediction methods for the succinylated sites are urgently needed. Currently, numerous computational classifiers have been developed to identify PTM (Post Translation Modification) sites using various types of two-class machine learning algorithms [26–29]. We proposed a computational predictor, SucPred (2015), based on the combination of a kind of semi-supervised learning algorithm (Psol) and SVM classifier. This predictor took advantage of four types of sequence features, including autocorrelation function, encoding based on grouped weight, normalized van der Waals volume and position weight amino acids composition. Xu et al. (2015) built a predictor called iSuc-PseAAC based on SVM using Pseudo amino acid composition. And then, Xu et al. (2015) developed another predictor named SucFind. It was constructed based on SVM with k-spaced amino acid pairs and AAindex features. More recently, Hasan et al. (2016) proposed an approach SuccinSite based on Random Forest classifier. SucStruct predictor was built by Lopez et al. (2017) using structural properties of amino acids [30]. Thereafter, using profile bigram [31], PSSM-Suc [32] was introduced for identifying succinylation lysine sites by Lopez et al. (2017). Besides, they proposed Success predictor (2018) using evolutionary information of amino acids [33]. Thereafter, they (2018) used secondary structure information to further enhance the succinylation prediction [34]. Although these methods have already been developed to predict succinylation sites, there are some problems existing. First of all, the data set used in SucPred and iSuc-PseAAC was obtained from CPLM database [35] and the data set of SucFind was derived from several lysine modification databases and some relevant articles [36, 37], which are small and they didn’t cover novel succinylation data recently found. Besides, though the SuccinSite contains enough succinylation data, the performances of SuccinSite still have room for improvement.
To solve problems mentioned above, we developed a new predictor, which was proposed to predict succinylation sites in protein using the same data set with SuccinSite. We used multiple efficient feature descriptors to derive informative features, including amino acid composition (AAC), binary encoding (BE), physicochemical property (PCP) and grey pseudo amino acid composition (GPAAC) and we showed the flow chart in Fig. 1. Finally, we obtained promising results with an accuracy of 89.14% and a MCC of 0.79 using 10-fold cross validation on training data set and an accuracy of 84.5%, a MCC of 0.2 on independent test set. These results demonstrated that this predictor is promising to predict lysine succinylation sites and could serve as a helpful tool to the community.
Fig. 1.
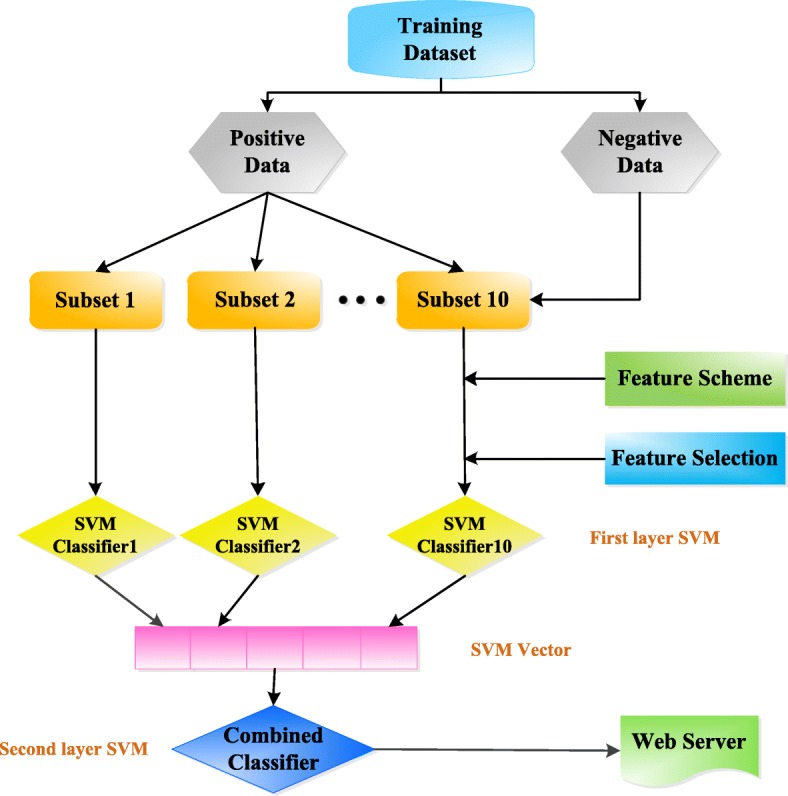
The flow chart of PSuccE
Methods
As demonstrated in compliance with Chou’s 5-step rule [38] in a series of recent publications [6–12], we should follow the following five guidelines to establish a useful sequence-based predictor for a biological system: (a) select or construct a valid benchmark data set to train and test the predictor; (b) formulate these protein sequence samples with an effective mathematical expression that can truly reflect their intrinsic correlation with the target to be predicted; (c) introduce or develop a powerful algorithm to operate the prediction; (d) properly perform cross-validation tests to objectively evaluate the anticipated accuracy of the predictor; (e) establish a user-friendly web-server for the predictor that is accessible to the public. Below, we are going to describe how to deal with these steps one-by- one.
Datasets
In this study, succinylation data was derived from UniProtKB/Swiss-Prot database and NCBI protein sequence database as Hasan et al. [29] did. After removing proteins that have more than 30% sequence identity to any other proteins in this dataset using CH-HIT, 2322 succinylation proteins including 5009 experimentally verified lysine succinylation sites were obtained. Then, 124 proteins were randomly separated from the 2322 proteins as an independent test set for testing, and the remaining proteins were training data set. We referred the experimentally verified lysine succinylation sites as positive sites, and all the lysine sites that lie on the same proteins as succinylation sites but don’t have any succinylation annotation were regarded as negative sites. Finally, 124 proteins with 254 succinylation sites and 2977 non-succinylation sites were obtained as independent test set, and 2198 proteins with 4755 succinylation sites and 50,565 non-succinylation sites as training set.
Information entropy
Initially, we extract positive fragment and negative fragment utilizing the sliding window strategy, just like some other PTM site predictors [39, 40] The window size was set to L = 2 l + 1, where l is the number of upstream residues or downstream residues of the central amino acid (lysine). And ‘X’ was used when the number of flanking residues was less than l.
Nevertheless, not all the position within the window are contributing to the prediction of succinylation sites and even play a negative role. So it’s necessary to take measure to filter useful positions around the center lysine. The information gain is a measure of the amount of information [41]. The more orderly a system is, the lower the information entropy values, on the contrary, the more chaotic a system is, the higher the information entropy values. Therefore, information entropy is also a measure of the degree of ordering. Consequently, we utilized information entropy to select efficient position within the sliding window. Information entropy can be calculated as follows:
| 1 |
where c represents the window size. xi represents a kind of amino acid, and n = 20 denotes 20 kinds of different amino acid. pc(xi) means the probability that amino acid xi appears at position c.
General Pseudo amino acid composition
With the rapid growth of the amount of biological sequences in the post-genome era, one of the most significant but also most difficult problems in computational biology is how to convert a biological sequence into a numerical vector, yet still retain significant sequence-order information or key pattern characteristic, which is because almost all the existing machine-learning algorithms can only handle vector instead of sequence samples [22]. However, a vector that is defined in a discrete model may completely lose all the sequence-order information. To avoid this, the pseudo amino acid composition or PseAAC [42] was proposed. Ever since the concept of Chou’s PseAAC [43, 44] was put forward, it has penetrated into nearly all the areas of computational proteomics [45–50], many biomedicine and drug development areas [51]. Because of its widely and increasingly usage, two powerful open access soft-wares, named ‘propy’ [43] and ‘PseAAC-General’ [50], were released recently. In addition, a very powerful web-server called Pse-in-One [52] has been established and it can generate any desired feature vectors for protein/peptide and DNA/RNA sequences according to the need of users’ studies.
Amino acid composition
Amino acid composition feature is common and widely used in prediction of protein sequences (such as phosphorylation and acetylation and so on) [53, 54] as one kind of the most popular coding methods. AAC describes the frequencies of amino acids in protein sequences. In this work, AAC is the fraction of each type of amino acid in a sequence fragment. We calculated amino acid occurrence frequencies in the sequence surrounding the query site (the center site itself is not counted). There are 21 types of amino acids (including ‘X’) in total, thus 21 frequencies are calculated as features, the sum of which equal 1.
Binary encoding
The information of the type and position of the amino acid residues are basic but important to a protein sequence. Binary encoding scheme is the most intuitive method to acquire the positional characteristics of amino acids for protein sequences. It has been used in many kinds of PTM site prediction. If 20 amino acids are ranked as ACDEFGHIKLMNPQRSTVWY, it enciphered each kind of amino acid into a 20-dimension binary vector according to its position in this array. For example, A is replaced by 10,000,000,000,000,000,000, and Y is converted into 00000000000000000001. Especially, X is represent as 00000000000000000000.
Physicochemical property
AAindex is a database that includes numerical indices representing various physicochemical and biochemical properties of amino acids and pairs of amino acids [55]. Now it contains 544 PCPs in the version 9.0. An amino acid index is a set of 20 numerical values on behalf of various PCPs of amino acids. PCP has ever been successfully used in prediction of many protein modifications, such as S-glutathionylation and acetylation [56, 57]. In this work, we ranked these PCPs according to their abilities to distinguish between succinylation and non succinylation sites and used following top ten physicochemical properties: (1) consensus normalized hydrophobicity scale; (2) positive charge; (3) partition energy; (4) net charge; (5) conformational preference for all beta-strands; (6) conformational preference for antiparallel beta-strands; (7) mean polarity; (8) principal property value z3; (9) apparent partition energies calculated from Wertz-Scheraga index; (10) weights from the IFH scale.
Grey Pseudo amino acid composition
We combined Chou’s PseAAC [58, 59] and the grey model (GM (1,1)) [60] to convey protein fragments. It has already been successfully used in previous study [61–65]. GM (1,1) is an important and generally used approach in GM which can generate a series of regular data sequence by identifying difference between the trend of system factors, which also called correlation analysis. Assume that we have a known array
| 2 |
which is irregular. Then, calculate the first-order accumulative generation operation (1-AGO) series for X(0):
| 3 |
in which x(1)(k) is computed by following equation:
| 4 |
Next, an albinism differential equation can be gained according to X(1):
| 5 |
-α is the developing coefficient and -β is the influence coefficient. α and β are two elements of parameter vector θ.
| 6 |
θ can be calculated using a least square estimator.
| 7 |
Where.
| 8 |
| 9 |
In view of this, some important information are covered in coefficients. In this work, we incorporated PseAAC into these coefficients to reflect the difference between the positive data and negative data. The first arrays X(0) were obtained from the physicochemical property which is described above. Each kind of AAindex corresponds to a series of X(0) and works out a pair of coefficients.
Totally, we obtained 791 dimensions of features, including 21 dimensions for AAC (Amino Acid Composition), 500 dimensions for BE (Binary Encoding), 250 dimensions for PCP (Physicochemical Property) and 20 dimensions for GPAAC (Grey Pseudo Amino Acid Composition).
Feature selection scheme
Not all features are equally important. Some features may not be relevant to the prediction of succinylation sites or they could be redundant with each other. Therefore, we performed a feature selection method IG (Information Gain) to remove the irrelevant and redundant features [66]. IG indicates the quantity of information a feature can bring to the classification system. The more information a feature brings, the more important it is. Thus the information gain can be utilized to evaluate the contribution of each feature to the classification. The formula of IG is as follows.
| 10 |
where x means a dimension of feature, and E(x) is the information entropy value of x. V means the amount of different values in each dimension feature x, and xv (v = 1,2,...,V) indicates the probable value in feature x, and E(xv) is the corresponding information entropy value to xv.
Ensemble learning
Ensemble Learning is one of the four main research directions in the field of machine learning. It uses multiple classifiers to solve the same problem, significantly improving the generalization ability of learning system. In our training data set, the amount of negative data (50565) is much larger than the amount of positive data (4755), so we adopted ensemble learning to resolve the unbalance between them.
We used Bootstrap Sampling to extract different subset data [67, 68]. It gets the difference of the base classifier through the difference of the training set. First, ten subsets with 4750 data were randomly selected from negative training data, and there is no coincidence between any two subsets. Then, combine every subset with the whole positive training data, respectively. Now, we have ten training data subsets with 9510 data, and we make a feature selection for each data subset using independent test set. After selecting the optimal feature group for every train data set, 10 SVM classifiers were obtained as the first layer classifiers. Next, we collected the results from the first layer classifiers and combined them as the feature of the second layer classifier. Finally, we predicted with the second layer classifier.
Performance assessment
Independent test, subsampling test, and jackknife test are three commonly used cross validation methods to examine a predictor [69]. The jackknife test is deemed as the most reliable one among them [70]. However, n-fold cross validation test is commonly used instead of jackknife test because it can save much time. This method divides dataset into n equal subsets randomly, every n-1 of which are used for training and the rest one for testing. The procedure repeats several times and final result is calculated by averaging the accuracy of the n testing subsets. In this study, independent test and 10-fold cross validation were both used for evaluating the predictor.
Four measurements are generally used to evaluate the predictor: sensitivity (Sn), specificity (Sp), accuracy (Acc) and Mattew’s correlation coefficient (MCC). They are defined as follows:
| 11 |
| 12 |
| 13 |
| 14 |
where TP, TN, FP and FN means the number of true positive, true negative, false positive and false negative, respectively.
This set of metrics is valid for the single-label systems instead multi-label systems. As for the multi-label systems, which exists frequently in system biology and system medicine [11, 71, 72], match with another completely diverse set of metrics as showed in [73].
Result and discussion
Optimal choice of positions
In this study, we used information entropy (IE) to evaluate the importance of positions. Firstly, we chose 51 as the initial window size, with 25 amino acid residues upstream and 25 amino acid residues downstream. And then the entropy of each position was calculated by the formula (1). Entropy values are shown in Fig. 2.
Fig. 2.
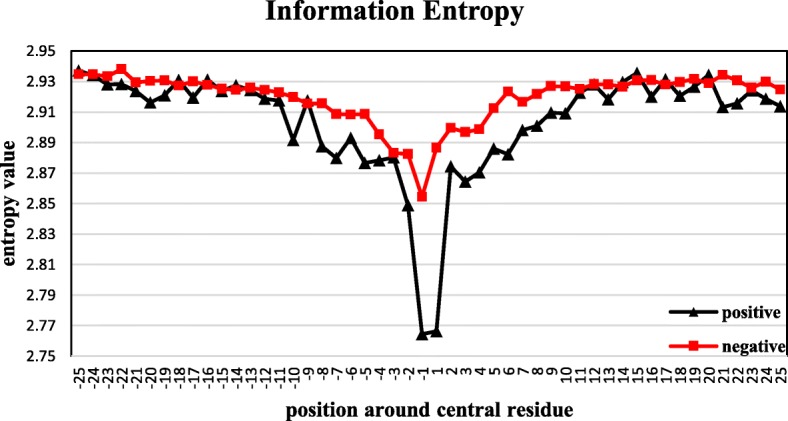
The information entropy value of positions around the central residue
As we can see in Fig. 2, nearly all the information entropy values for positive data are lower than the values for negative data, which indicates that information entropy can be beneficial to distinguish succinylation sites and non-succinylation sites. The closer to the central residues, the lower the entropy values are, especially for the position 1 and − 1 which corresponds to the difference between succinylaiton and non-succinylation according to the two sample logo [74]. We can speculate from this appearance that succinylation may enhance the conservation of the target lysine and its surroundings which is consistent with Fig. 3. Eventually, we chose 25 positions which have greater difference between positive information entropy values and negative information entropy values, including − 20, − 17, − 10, − 8, − 7, − 6, − 5, − 4, − 2, − 1, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 16, 21, 22, 24, 25.
Fig. 3.

Two sample logos of the compositional biases around succinylation sites compared to non-succinylation sites. Statistically significant symbols are plotted using the size of the symbol that is proportional to the difference between the two samples. Residues are separated in two groups: (1) enriched in the positive samples; and (2) depleted in the positive samples. Color of the symbols was classified according to the polarity of the residue side chain
Analysis of optimal features
Not all positions and features are equally important in a protein. In this study, information gain was employed to acquire an optimal feature subset. For each subset, feature selection was processed respectively. Table 1 shows the final number of features in every training dataset, and the MCC curves of the succinylation prediction on ten training datasets for different dimensions of features are shown in Additional file 1: Figure S1.
Table 1.
The number of features in every training dataset
| Training Datasets | Number of features | ACC | BE | PCP | GPAAC |
|---|---|---|---|---|---|
| Subset1 | 186 | 14 | 38 | 123 | 11 |
| Subset2 | 191 | 14 | 34 | 133 | 10 |
| Subset3 | 158 | 13 | 27 | 107 | 11 |
| Subset4 | 112 | 12 | 14 | 77 | 9 |
| Subset5 | 194 | 14 | 38 | 131 | 11 |
| Subset6 | 177 | 14 | 30 | 122 | 11 |
| Subset7 | 194 | 14 | 37 | 134 | 9 |
| Subset8 | 88 | 9 | 6 | 64 | 9 |
| Subset9 | 66 | 7 | 5 | 45 | 9 |
| Subset10 | 45 | 4 | 5 | 30 | 6 |
| Common features | 34 | 4 | 5 | 19 | 6 |
As we can see, after feature selection, the numbers of features for ten training datasets are different. It strongly proves that there is otherness between these ten training datasets even though they are separated from one negative dataset, and otherness is the requirement for using the ensemble learning. In spite of the difference, there are also many common features in ten feature vectors, including 4 AAC features, 5 BE features, 19 PCP features and 6 GPAAC features. We also evaluate the performance change between before feature selection and after feature selection for ten subsets (Additional file 1: Figure S2 and Table S1). As we can see in Additional file 1: Figure S2 and Table S1, the value of Sn, Sp, Acc and MCC are larger after feature selection, and the value of AUC (area below ROC curve) obviously increase.
Comparison between ensemble learning and single SVMs
Ensemble learning train combinations of base models, which may be decision trees, neural networks, SVM, or others traditionally used in supervised learning. In this study, Bootstrap Sampling was used to extract different subset data. There are 50,565 negative sites and 4755 positive sites in our training dataset, nearly 10:1 for ratio of negative and positive data, so we randomly select 4755 data from negative data for ten times and there is no coincidence between any two subsets. Therefore, we have 10 separate training data subsets, which contains 4755 positive samples and 4755 negative samples, respectively (1:1 ratio of positive and negative data).
To verify if ensemble models perform consistently better than the single SVMs, we evaluate the performance of 10-fold cross validation on training dataset, and the results are shown in Table 2 and Fig. 4. As listed in Table 2, single SVMs always predict a lower Sp value and the Acc value are also not outstanding. After ensemble the training result from ten single SVMs, all the performances are obviously increased, especially for Sp, MCC and AUC.
Table 2.
10-fold cross validation performance of 10 subsets and ensemble classifier on training dataset
| Training dataset | Sn (%) | Sp (%) | Acc | MCC |
|---|---|---|---|---|
| Subset1 | 72.29 | 66.91 | 0.6961 | 0.3926 |
| Subset2 | 72.15 | 66.39 | 0.6927 | 0.3861 |
| Subset3 | 72.21 | 66.33 | 0.6927 | 0.3861 |
| Subset4 | 72.83 | 65.73 | 0.6929 | 0.3867 |
| Subset5 | 71.69 | 67.24 | 0.6948 | 0.3898 |
| Subset6 | 72.12 | 66.46 | 0.6930 | 0.3865 |
| Subset7 | 71.94 | 65.64 | 0.6881 | 0.3767 |
| Subset8 | 72.07 | 65.53 | 0.6880 | 0.3768 |
| Subset9 | 72.97 | 63.52 | 0.6824 | 0.3665 |
| Subset10 | 72.36 | 62.48 | 0.6742 | 0.3502 |
| Ensemble | 84.31 | 93.97 | 0.89136 | 0.7864 |
Fig. 4.
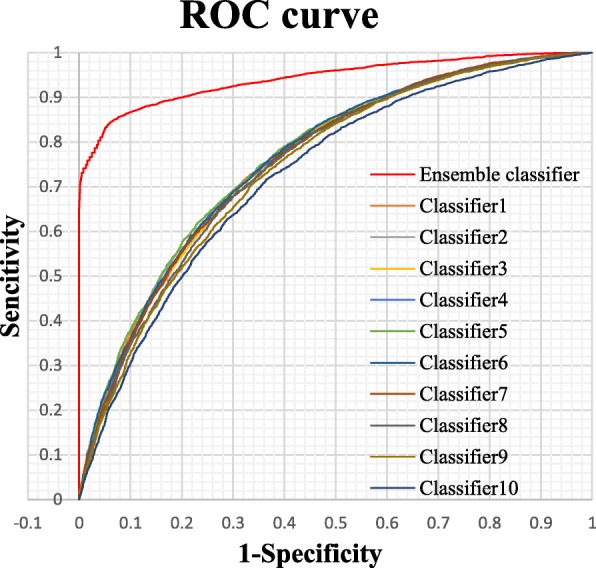
ROC curves (AUC) of predictions based on 10-fold cross validation
Comparison between our method and existing methods
To further evaluate the performance of our method, we compared our method with four other existing predictors, SucPred, iSuc-PseAAC, SuccFind, SuccinSite and Success, using independent test dataset, including 254 succinylation sites and 2977 non-succinylation sites. Sn, Sp, Acc and MCC are used to measure the performance (Table 3). Because of the limitation of amount of independent test set, the result of independent test is not as good as 10-fold cross validation. However, when we control the threshold as 0.9 for these predictor, SucPred only obtain 67.3, 27.1% and 0.643 for Sp, Sn, and Acc, and the MCC value was only − 0.03. iSuc-PseAAC and Success have satisfying values of Sp, but the Sn and MCC values are lower. SuccFind and SuccinSite are favorable, while our method achieve a Sp of 88.6%, a Sn of 37.5%, an Acc of 84.5% and a MCC of 0.204, which were much better than SuccFind’s and SuccinSite’s performance. Because of the high value of threshold to guarantee the prediction of positive samples, the sensitivity values are less than the specificity value. The promising performance demonstrated that the this predictor was particularly useful for protein succinylation prediction.
Table 3.
A comparison of PSuccE with existing predictors using an independent test set
| Measurement* | SucPred | iSuc-PseAAC | SuccFind | SuccinSite | Success | PSuccE |
|---|---|---|---|---|---|---|
| Sp(%) | 67.3 | 88.7 | 79.2 | 88.2 | 86.8 | 88.6 |
| Sn(%) | 27.2 | 12.2 | 25.2 | 37.1 | 14.2 | 37.5 |
| Acc | 0.643 | 0.827 | 0.750 | 0.842 | 0.811 | 0.845 |
| MCC | −0.030 | 0.013 | 0.029 | 0.199 | 0.007 | 0.204 |
* The threshold value was controlled as 0.9 for these predictors
Conclusion
Here, we implement an application of Ensemble learning to protein succinylation prediction problem. Results show that our method is helpful to identification of succinylation sites. This work also indicated that Ensemble learning was a useful technique for combining weak classifiers and improving performance. We are looking forward that our method will give a powerful help for further studies of succinylation process.
Additional file
Figure S1. The MCC score of the optimal feature subsets. Figure S2. AUC (area below ROC curve) change between before feature selection and after feature selection for ten subsets. Table S1. The performance change between before feature selection and after feature selection for ten subsets. (DOCX 2693 kb)
Acknowledgements
This study is partially funded by National Natural Science Foundation of China (61403077), the China Postdoctoral Science Foundation funded project (2014 M550166, 2015 T80285).
Funding
This research is partially supported by National Natural Science Foundation of China (61403077), the China Postdoctoral Science Foundation funded project (2014 M550166, 2015 T80285).
Availability of data and materials
All data used in this paper can be downloaded from https://github.com/ningq669/PSuccE.
Abbreviations
- AAC
Amino acid composition
- Acc
Accuracy
- BE
Binary encoding
- FN
False negative
- FP
False positive
- GM
Grey model
- GPAAC
Grey pseudo amino acid composition
- IG
Information gain
- MCC
Matthew correlation coefficient
- PCP
Physicochemical property
- PH value
Hydrogen ion concentration
- PTM
Post translation modification
- Sn
Sensitivity
- Sp
Specificity
- SVM
Support vector machine
- TN
True negative
- TP
True positive
Authors’ contributions
ZM and XZ conceived and designed the experiments. QN performed the experiments. LB and XZ analyzed the data. QN wrote the manuscript with revision by XZ. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All authors approval and consent to participate.
Consent for publication
All authors read and consent to publish the manuscript.
Competing interests
The authors declare that they no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2249-4) contains supplementary material, which is available to authorized users.
Contributor Information
Qiao Ning, Email: ningq669@nenu.edu.cn.
Xiaosa Zhao, Email: zhaoxs686@nenu.edu.cn.
Lingling Bao, Email: baoll601@nenu.edu.cn.
Zhiqiang Ma, Phone: +86-0431-84536338, Email: zhiqiang.ma967@gmail.com.
Xiaowei Zhao, Phone: +86-0431-84536338, Email: zhaoxw303@nenu.edu.cn.
References
- 1.Weinert B, Schölz C, Wagner S, Iesmantavicius V, Su D, Daniel J, Choudhary C. Lysine Succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation [J] Cell Rep. 2013;4(4):842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke J, Zhao Y. Lysine Succinylation and lysine Malonylation in histones [J] Mol Cell Proteomics Mcp. 2012;11(5):100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan M, Peng C, Anderson K, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, Ro J, Wagner GR, Green MF, Madsen AS, Schmiesing J, Peterson BS, Xu G, Ilkayeva OR, Muehlbauer MJ, Braulke T, Mühlhausen C, Backos DS, Olsen CA, McGuire PJ, Pletcher SD, Lombard DB, Hirschey MD, Zhao Y. Lysine Glutarylation is a protein posttranslational modification regulated by SIRT5 [J] Cell Metab. 2014;19(4):605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papanicolaou KN, O'Rourke B, Foster DB. Metabolism leaves its mark on the powerhouse: recent progress in post-translational modifications of lysine in mitochondria [J] Front Physiol. 2013;5(5):301. doi: 10.3389/fphys.2014.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao T. Identification of lysine succinylation as a new post-translational modification [J] Nat Chem Biol. 2011;7(1):58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia J, Liu Z, Xiao X, Liu B. pSuc-Lys: predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J Theor Biol. 2016;394:223–230. doi: 10.1016/j.jtbi.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 7.Jia J, Liu Z, Xiao X. iCar-PseCp: identify carbonylation sites in proteins by Monto Carlo sampling and incorporating sequence coupled effects into general PseAAC. Oncotarget. 2016;7:34558–34570. doi: 10.18632/oncotarget.9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia J, Zhang L, Liu Z. pSumo-CD: predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics. 2016;32:3133–3141. doi: 10.1093/bioinformatics/btw387. [DOI] [PubMed] [Google Scholar]
- 9.Qiu WR, Sun BQ, Xiao X, Xu D. iPhos-PseEvo: identifying human phosphorylated proteins by incorporating evolutionary information into general PseAAC via grey system theory. Mol Inf. 2016; 10.1002/minf.201600010. [DOI] [PubMed]
- 10.Qiu WR, Sun BQ, Xu ZC. iHyd-PseCp: identify hydroxyproline and hydroxylysine in proteins by incorporating sequence-coupled effects into general PseAAC. Oncotarget. 2016;7:44310–44321. doi: 10.18632/oncotarget.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu WR, Sun BQ, Xiao X. iPTM-mLys: identifying multiple lysine PTM sites and their different types. Bioinformatics. 2016;32:3116–3123. doi: 10.1093/bioinformatics/btw380. [DOI] [PubMed] [Google Scholar]
- 12.Qiu WR, Xiao X, Xu ZH. iPhos-PseEn: identifying phosphorylation sites in proteins by fusing different pseudo components into an ensemble classifier. Oncotarget. 2016;7:51270–51283. doi: 10.18632/oncotarget.9987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y, Ding J, Wu LY. iSNO-PseAAC: predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS One. 2013;8:e55844. doi: 10.1371/journal.pone.0055844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Y, Shao XJ, Wu LY. iSNO-AAPair: incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ. 2013;1:e171. doi: 10.7717/peerj.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu WR, Xiao X, Lin WZ. iMethyl-PseAAC: identification of protein methylation sites via a Pseudo amino acid composition approach. Biomed Res Int (BMRI) 2014;2014:947416. doi: 10.1155/2014/947416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Zhao X, Sun P, Ma Z. PSNO: predicting cysteine S-Nitrosylation sites by incorporating various sequence-derived features into the general form of Chou's PseAAC. Int J Mol Sci. 2014;15:11204–11219. doi: 10.3390/ijms150711204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia C, Lin X, Wang Z. Prediction of protein S-Nitrosylation sites based on adapted normal distribution bi-profile Bayes and Chou's Pseudo amino acid composition. Int J Mol Sci. 2014;15:10410–10423. doi: 10.3390/ijms150610410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y, Wen X, Shao XJ. iHyd-PseAAC: predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. Int J Mol Sci (IJMS) 2014;15:7594–7610. doi: 10.3390/ijms15057594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y, Wen X, Wen LS, Wu LY, Deng NY. iNitro-Tyr: prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. PLoS One. 2014;9:e105018. doi: 10.1371/journal.pone.0105018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu WR, Xiao X, Lin WZ. iUbiq-Lys: prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a grey system model. J Biomol Struct Dyn (JBSD) 2015;33:1731–1742. doi: 10.1080/07391102.2014.968875. [DOI] [PubMed] [Google Scholar]
- 21.Jia J, Liu Z, Xiao X. iSuc-PseOpt: identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal Biochem. 2016;497:48–56. doi: 10.1016/j.ab.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Chou KC. Impacts of bioinformatics to medicinal chemistry. Med Chem. 2015;11:218–234. doi: 10.2174/1573406411666141229162834. [DOI] [PubMed] [Google Scholar]
- 23.Xu Y. Recent progress in predicting posttranslational modification sites in proteins. Curr Top Med Chem. 2016;16:591–603. doi: 10.2174/1568026615666150819110421. [DOI] [PubMed] [Google Scholar]
- 24.Machida Y, Chiba T, Takayanagi A, Tanaka Y, Asanuma M, Ogawa N, Koyama A, Iwatsubo T, Ito S, Jansen PH, Shimizu N, Tanaka K, Mizuno Y, Hattori N. Corrigendum to “common anti-apoptotic roles of parkin and α-synuclein in human dopaminergic cells” [J] Biochem Biophys Res Commun. 2005;332(1):233–240. doi: 10.1016/j.bbrc.2005.04.124. [DOI] [PubMed] [Google Scholar]
- 25.Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Löwenhielm HB, Holmgren A, Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis [J] Arch Biochem Biophys. 2002;406(2):229–240. doi: 10.1016/S0003-9861(02)00468-X. [DOI] [PubMed] [Google Scholar]
- 26.Zhao X, Qiao N, Chai H, Ma Z. Accurate in silico identification of protein succinylation sites using an iterative semi-supervised learning technique [J] J Theor Biol. 2015;374:60–65. doi: 10.1016/j.jtbi.2015.03.029. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y, Ding YX, Ding J, Lei Y, Wu L, Deng N. iSuc-PseAAC: predicting lysine succinylation in proteins by incorporating peptide position-specific propensity [J] Sci Rep. 2015;5:10184. doi: 10.1038/srep10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu HD. SuccFind: A novel succinylation sites online prediction tool via enhanced characteristic strategy [J] Bioinformatics. 2015;31(23):3748–3750. doi: 10.1093/bioinformatics/btv439. [DOI] [PubMed] [Google Scholar]
- 29.Hasan MM, Yang S, Zhou Y, Mollah MN. SuccinSite: a computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties [J] Mol BioSyst. 2016;12(3):786–795. doi: 10.1039/C5MB00853K. [DOI] [PubMed] [Google Scholar]
- 30.López Y, Dehzangi A, Lal SP, Taherzadeh G, Michaelson J, Sattar A, Tsunoda T, Sharma A. SucStruct: prediction of succinylated lysine residues by using structural properties of amino acids [J] Anal Biochem. 2017;527:24–32. doi: 10.1016/j.ab.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 31.Sharma A, Lyons J, Dehzangi A, Paliwal KK. A feature extraction technique using bi-gram probabilities of position specific scoring matrix for protein fold recognition [J] J Theor Biol. 2014;13(1):41–46. doi: 10.1016/j.jtbi.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 32.Dehzangi A, López Y, Lal SP, Taherzadeh G, Michaelson J, Sattar A, Tsunoda T, Sharma A. PSSM-Suc: accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction [J] J Theor Biol. 2017;425:97. doi: 10.1016/j.jtbi.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 33.López Y, Sharma A, Dehzangi A, Lal SP, Taherzadeh G, Sattar A, Tsunoda T. Success: evolutionary and structural properties of amino acids prove effective for succinylation site prediction [J] BMC Genomics. 2018;19(1):923. doi: 10.1186/s12864-017-4336-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dehzangi A, López Y, Lal SP, Taherzadeh G, Sattar A, Tsunoda T, Sharma A. Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams [J] PLoS One. 2018;13(2):e0191900. doi: 10.1371/journal.pone.0191900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z, Wang Y, Gao T, Pan Z, Cheng H, Yang Q, Cheng Z, Guo A, Ren J, Xue Y. CPLM: a database of protein lysine modifications [J] Nucleic Acids Res. 2014;42(Database issue):531–536. doi: 10.1093/nar/gkt1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Hu X, Wan Y, Xie G, Li X, Chen D, Cheng Z, Yi X, Liang S, Tan F. Systematic identification of the lysine Succinylation in the protozoan parasite toxoplasma gondii [J] J Proteome Res. 2014;13(12):6087–6095. doi: 10.1021/pr500992r. [DOI] [PubMed] [Google Scholar]
- 37.Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, Lombard DB, Zhao Y. SIRT5-mediated lysine Desuccinylation impacts diverse metabolic pathways [J] Mol Cell. 2013;50(6):919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition (50th anniversary year review) J Theor Biol. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu L, Li Z, Wang K, Niu S, Shi X, Cai Y, Li H. Prediction and analysis of protein methylarginine and methyllysine based on multisequence features [J] Biopolymers. 2011;95(11):763–771. doi: 10.1002/bip.21645. [DOI] [PubMed] [Google Scholar]
- 40.Zhao XW, Li XT, Ma ZQ, Yin MH. Prediction of lysine Ubiquitylation with ensemble classifier and feature selection. Int J Mol Sci. 2011;12(12):8347–8361. doi: 10.3390/ijms12128347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shannon C. Part III: A mathematical theory of communication [J] M.D.Comput Comput Med Pract. 1997;14(4):306–317. [PubMed] [Google Scholar]
- 42.Chou KC. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005;21:10–19. doi: 10.1093/bioinformatics/bth466. [DOI] [PubMed] [Google Scholar]
- 43.Cao DS, Xu QS, Liang YZ. Propy: a tool to generate various modes of Chou's PseAAC. Bioinformatics. 2013;29:960–962. doi: 10.1093/bioinformatics/btt072. [DOI] [PubMed] [Google Scholar]
- 44.Lin SX, Lapointe J. Theoretical and experimental biology in one —A symposium in honour of Professor Kuo-Chen Chou’s 50th anniversary and Professor Richard Giegé’s 40th anniversary of their scientific careers. J Biomed Sci Eng (JBiSE) 2013;6:435–442. doi: 10.4236/jbise.2013.64054. [DOI] [Google Scholar]
- 45.Kabir M, Hayat M. iRSpot-GAEnsC: identifing recombination spots via ensemble classifier and extending the concept of Chou's PseAAC to formulate DNA samples. Mol Gen Genomics. 2016;291:285–296. doi: 10.1007/s00438-015-1108-5. [DOI] [PubMed] [Google Scholar]
- 46.Behbahani M, Mohabatkar H, Nosrati M. Analysis and comparison of lignin peroxidases between fungi and bacteria using three different modes of Chou's general pseudo amino acid composition. J Theor Biol. 2016;411:1–5. doi: 10.1016/j.jtbi.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 47.Khan M, Hayat M, Khan SA, Iqbal N. Unb-DPC: Identify mycobacterial membrane protein types by incorporating un-biased dipeptide composition into Chou's general PseAAC. J Theor Biol. 2016;415:13–19. doi: 10.1016/j.jtbi.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 48.Rahimi M, Bakhtiarizadeh MR, Mohammadi-Sangcheshmeh A. OOgenesis_Pred: a sequence-based method for predicting oogenesis proteins by six different modes of Chou's pseudo amino acid composition. J Theor Biol. 2016;414:128–136. doi: 10.1016/j.jtbi.2016.11.028. [DOI] [PubMed] [Google Scholar]
- 49.Chou KC. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr Proteomics. 2009;6:262–274. doi: 10.2174/157016409789973707. [DOI] [Google Scholar]
- 50.Du P, Gu S, Jiao Y. PseAAC-general: fast building various modes of general form of Chou's pseudo amino acid composition for large-scale protein datasets. Int J Mol Sci. 2014;15:3495–3506. doi: 10.3390/ijms15033495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong WZ, Zhou SF. Molecular science for drug development and biomedicine. Int J Mol Sci. 2014;15:20072–20078. doi: 10.3390/ijms151120072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu B, Liu F, Wang X, Chen J, Fang L. Pse-in-one: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015;43:W65–W71. doi: 10.1093/nar/gkv458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM. Identification, analysis, and prediction of protein ubiquitination sites [J] Proteins Struct Funct Bioinformatics. 2010;78(2):365–380. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee T, Chen S, Hung H, Ou Y. Incorporating distant sequence features and radial basis function networks to identify ubiquitin conjugation sites [J] PLoS One. 2010;6(3):e17331. doi: 10.1371/journal.pone.0017331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suo S, Qiu J, Shi S, Sun X, Huang S, Chen X, Liang R. Position-specific analysis and prediction for protein lysine acetylation based on multiple features [J] PLoS One. 2012;7(11):e49108. doi: 10.1371/journal.pone.0049108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kawashima S, Ogata H, Kanehisa M. AAindex: Amino acid index database [J] Nucleic Acids Res. 1999;27(1):368–369. doi: 10.1093/nar/27.1.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao X, Ning Q, Ai M, Chai H, Yin M. PGluS: prediction of protein S-glutathionylation sites with multiple features and analysis. Mol BioSyst. 2015;11:923–929. doi: 10.1039/C4MB00680A. [DOI] [PubMed] [Google Scholar]
- 58.Chou K. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes [J] Bioinformatics. 2005;21(1):10–19. doi: 10.1093/bioinformatics/bth466. [DOI] [PubMed] [Google Scholar]
- 59.Chou K. Prediction of protein cellular attributes using pseudo-amino acid composition [J]. Proteins structure function. Bioinformatics. 2001;43(3):246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- 60.Deng J. Introduction to Grey system theory. J Grey Syst. 1989;1:1–24. [Google Scholar]
- 61.Lin W, Xu D. Imbalanced Multi-label Learning for identifying antimicrobial peptides and their functional types [J] Bioinformatics. 2016;32:3745–3752. doi: 10.1093/bioinformatics/btw560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin WZ, Fang JA, Xiao X. iDNA-Prot: identification of DNA binding proteins using random Forest with Grey model. PLoS One. 2011;6:e24756. doi: 10.1371/journal.pone.0024756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin WZ, Fang JA, Xiao X. Predicting secretory proteins of malaria parasite by incorporating sequence evolution information into Pseudo amino acid composition via Grey system model. PLoS One. 2012;7:e49040. doi: 10.1371/journal.pone.0049040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin WZ, Fang JA, Xiao X. iLoc-animal: a multi-label learning classifier for predicting subcellular localization of animal proteins. Mol BioSyst. 2013;9:634–644. doi: 10.1039/c3mb25466f. [DOI] [PubMed] [Google Scholar]
- 65.Xiao X, Min JL, Wang P. iGPCR-drug: a web server for predicting interaction between GPCRs and drugs in cellular networking. PLoS One. 2013;8:e72234. doi: 10.1371/journal.pone.0072234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jing H, Berger SL. The emerging field of dynamic lysine methylation of non-histone proteins [J] Curr Opin Genet Dev. 2008;18(2):152–158. doi: 10.1016/j.gde.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 67.Efron B. Bootstrap Methods: Another Look at the Jackknife [J]. 1979;7(1):1–26.
- 68.Efron B. Monographs on statistics and applied probability An Introduction to the Bootstrap, vol. 57: Chapman[C]//SCIENCE DIRECT. Uncorrected proof YJMBI 55132—26/2/2003—AMADEN—65243/GH article in; 1993.
- 69.Chou KC, Zhang CT. Prediction of protein structural classes [J] Crit Rev Biochem Mol Biol. 1995;30(4):275–349. doi: 10.3109/10409239509083488. [DOI] [PubMed] [Google Scholar]
- 70.Chou K, Shen H. Cell-PLoc: a package of web servers for predicting subcellular localization of proteins in various organisms [J] Nat Protoc. 2008;3(2):153–162. doi: 10.1038/nprot.2007.494. [DOI] [PubMed] [Google Scholar]
- 71.Chen W, Ding H, Feng P. iACP: a sequence-based tool for identifying anticancer peptides. Oncotarget. 2016;7:16895–16909. doi: 10.18632/oncotarget.7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu ZC, Xiao X. iLoc-hum: using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol BioSyst. 2012;8:629–641. doi: 10.1039/C1MB05420A. [DOI] [PubMed] [Google Scholar]
- 73.Chou KC. Some remarks on predicting multi-label attributes in molecular Biosystems. Mol Biosyst. 2013;9:1092–1100. doi: 10.1039/c3mb25555g. [DOI] [PubMed] [Google Scholar]
- 74.Vacic V, Iakoucheva LM, Radivojac P. Two sample logo: a graphical representation of the differences between two sets of sequence alignments [J] Bioinformatics. 2006;22(12):1536–1537. doi: 10.1093/bioinformatics/btl151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The MCC score of the optimal feature subsets. Figure S2. AUC (area below ROC curve) change between before feature selection and after feature selection for ten subsets. Table S1. The performance change between before feature selection and after feature selection for ten subsets. (DOCX 2693 kb)
Data Availability Statement
All data used in this paper can be downloaded from https://github.com/ningq669/PSuccE.