

Abstract
Covalent enzyme inhibitors are widely applied as biochemical tools and therapeutic agents. As a complement to categorization of these inhibitors by reactive group or modification site, we present a categorization by mechanism, which highlights common advantages and disadvantages inherent to each approach. Established categories for reversible and irreversible covalent inhibition are reviewed with representative examples given for each class, including covalent reversible inhibitors, slow substrates, residue-specific reagents, affinity labels (classical, quiescent, and photoaffinity), and mechanism-based inactivators. The relationships of these categories to proteomic profiling probes (activity-based and reactivity-based) as well as complementary approaches such as prodrug and soft drug design are also discussed. A wide variety of strategies are used to balance reactivity and selectivity in the design of covalent enzyme inhibitors. Use of a shared terminology is encouraged to clearly convey these mechanisms, to relate them to prior use of covalent inhibitors in enzymology, and to facilitate the development of more effective covalent inhibitors.
Graphical abstract
Covalent enzyme inhibitors are of significant interest as biochemical tools and therapeutic drugs. Many previous authors have summarized the distinctive advantages and disadvantages of covalent enzyme inhibition in drug and inhibitor design.(1-18) Briefly, some of the potential advantages of covalent inhibitors include: sustained duration of action leading to less frequent dosing, increased ligand efficiency, the ability to overcome competing endogenous ligands, enhanced capability of targeting shallow binding sites previously deemed “undruggable,” increased ability to evade resistant mutations, selective targeting of some clinically-isolated variants, and the ability to help identify the proteins targeted by covalent modification, among others. These advantages are offset by some potential disadvantages, including: the added difficulty of assessing and balancing reactivity and selectivity, the covalent modification of off-target proteins, nucleic acids, or small molecules through non-selective reaction, idiosyncratic adverse drug responses, inappropriate levels of inhibition of enzymes for which partial or short-duration inhibition is desired, the lack of some of the advantages noted above if the targeted enzyme is rapidly degraded and resynthesized, and the perceived difficulty of designing these inhibitors de novo.
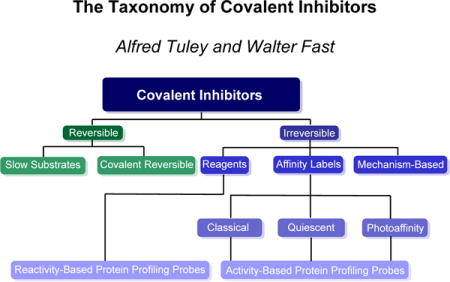
Prior reviews have categorized covalent inhibitors in terms of reaction type, functional group used for covalent modification, targeted residue type, strategies used for lead identification and development, opportunities for treating certain disease states, or application in clinical settings. Here, we recommend the use of a shared nomenclature for covalent inhibition mechanisms to bridge contemporary advances with the rich history of covalent enzyme inhibitor studies, and to spur further innovation in covalent inhibitor design. Toward that end, we provide a taxonomy of covalent inhibition mechanisms with representative contemporary examples of each. The prior introduction of new terminology has been helpful (e.g. Targeted Covalent Inhibitors, which target a poorly-conserved non-catalytic nucleophile),(12) but here we attempt to use only previously established categories as a framework to classify enzyme inhibitors that use covalent bond formation in their mechanisms (Figure 1).
Figure 1.

The Taxonomy of Covalent Inhibitors. Classification of covalent enzyme inhibitors by mechanism reveals different strategies used to balance reactivity and selectivity.
Broadly, covalent enzyme inhibitors are divided into two phenomenological categories according to whether inhibition is reversible or irreversible upon dialysis, competition with excess substrate, or extended incubation times. Even though different covalent inhibitors may show the same reversibility (or irreversibility) of inhibition, each can use very different strategies to selectively form and break bonds with the targeted enzyme. Identifying and classifying these strategies highlights advantages and disadvantages inherent to each mechanism.
Covalent Reversible Inhibition
This class of inhibitor uses covalent bond formation with the enzyme to facilitate inhibition, but enzyme activity is subsequently recovered due to cleavage of this bond. Covalent bond formation contributes to overall affinity, and subsequent bond cleavage can decrease the duration of inhibition and lower affinity. This approach mitigates some of the potential liabilities associated with formation of irreversible covalent adducts, but also makes identification of the proteins bound by these inhibitors more difficult.
Covalent Reversible Inhibitors
The most common type of covalent inhibitor that shows reversible inhibition also adopts the name of this category because “reversible” describes the underlying chemical mechanism. Formation of an initial non-covalent complex (E•I) between enzyme and inhibitor is followed by covalent bond formation (E-I), and both steps are readily reversible (Eq 1). Therefore, dialysis or dilution of the enzyme-inhibitor solution can result in reversible dissociation of the unmodified inhibitor, and excess substrate can compete for enzyme binding, each process resulting in recovery of activity. For the mechanism shown below, Ki is the dissociation constant for the non-covalent complex, and Ki* describes the overall dissociation constant encompassing both steps.(19) The chemical equations shown throughout are intended to only depict minimal kinetic mechanisms, highlighting differences between inhibitor types.
| (1) |
One example of reversible covalent inhibition is demonstrated by a series of acrylonitrile inhibitors designed to target a non-catalytic Cys residue in the ATP-binding pocket of ribosomal protein S6 kinase 2 (RSK2).(20, 21) The reactive acrylonitrile moiety is appended to a part of the inhibitor that provides non-covalent binding affinity to a particular site. Upon binding, the acrylonitrile is placed next to a Cys residue and the resulting high effective molarity(22) of this electrophile is sufficient to overcome the low nucleophilicity of the non-catalytic Cys (Figure 2). Conjugate addition subsequently affords the β-thioether covalent adduct. However, formation of the covalent bond is readily reversible as facilitated by the low pKa of the α-proton. The potencies of these inhibitors are not driven solely by covalent bond formation since changes in the remainder of the molecule significantly impact affinity and since proteolysis or denaturation of the inhibited kinase leads to facile release of the unmodified inhibitor. The reversible nature of covalent bond formation enhances selectivity by favoring the most thermodynamically stable product, stabilized through non-covalent interactions with the target’s binding site. Binding to off-target proteins is less favorable, even if they contain a more reactive nucleophile, due to the lack of stabilizing non-covalent interactions.
Figure 2.
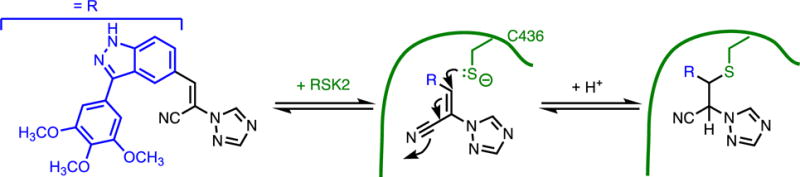
Reversible Covalent Inhibitor. A non-catalytic Cys of RSK2 is targeted by an acrylonitrile and reacts reversibly, presumably with the small fraction of ionized thiolate present at the reaction pH. In this figure and the following figures, the enzyme active site is depicted schematically in green, and for clarity, arrows are shown only for the initial attack leading to covalent bond formation.
Other illustrative examples of reversible covalent inhibitors include several recently developed serine β-lactamase inhibitors. Avibactam is a diazabicyclooctane that covalently modifies the active-site Ser residue of TEM-1, resulting in a long-lived ester adduct, and this reaction can be slowly reversed to reform the original compound.(23) The related compound relebactam covalently inhibits Pseudomonas-derived cephalosporinase-3 (PDC-3), again through formation of an ester with the active-site Ser residue, and inhibition can be relieved by reversal of the reaction to reform the original compound.(24) Boron-containing inhibitors such as benzoxaboroles can also form reversible covalent bonds to result in inhibition, for example with the active-site Ser residue of OXA-10, although these compounds are not a priori covalent enzyme inhibitors since they have been shown to cause inhibition of other targets by forming covalent bonds with a second substrate rather than an enzyme residue.(25-27)
Reversible covalent bond formation also enables the interesting possibility of using abundant biological nucleophiles such as glutathione or nucleophiles in serum albumin as transport systems.(7, 28) For example, the acrylamide-containing anticancer drug neratinib forms a reversible covalent bond with a Lys (K190) in human serum albumin, and has been proposed to use this transient covalent complex to facilitate transport to, and subsequent release in, acidic tumor environments.(29, 30)
Slow Substrates
The second type of covalent inhibitor that shows reversible inhibition does not use a reversible mechanism to recover activity. Instead, the inhibitor is recognized as a substrate. Formation of an initial non-covalent complex (E•I) facilitates covalent bond formation, as catalyzed by the enzyme, to result in a long-lived covalent intermediate (E-I) that is primarily responsible for the observed inhibition (Eq 2). However, the covalent bond is not broken through the reverse reaction, but rather through a reaction further catalyzed by the enzyme, leading to release of the active enzyme and a product (P) that no longer serves as an inhibitor. In these cases, KM values are often lower than Kd values for the non-covalently bound substrate due to the contribution of the subsequent steps.(31) To differentiate this type of inhibitor from the covalent reversible inhibitors described above, they are termed “slow substrates” because they undergo turnover by the enzyme, although only at very attenuated rates. This type of reversible inhibition also mitigates some of the concerns associated with irreversible modification, but use of this strategy to design covalent inhibitors is limited to enzymes with a covalent mechanism. (Non-covalent slow substrate inhibitors are known, and inhibit through formation of non-covalent complexes.(32))
| (2) |
One example of covalent inhibition by a slow substrate is L-canavanine inhibition of arginine deiminase.(33, 34) This inhibitor closely resembles the L-arginine substrate of the enzyme and undergoes initial non-covalent binding followed by the first half reaction, attack of an active-site Cys and loss of ammonia, to form a covalent adduct (Figure 3). However, the substitution of a methylene in the L-arginine substrate by an oxygen in L-canavanine results in formation of a covalent intermediate in which the pseudothiourea has a lowered pKa, making the intermediate much less susceptible to hydrolysis in the second half reaction, and resulting in the observed inhibition. Subsequent hydrolysis through the normal catalytic mechanism (rather than non-enzymatic instability) leads to release of the O-ureido product and slow recovery of the active enzyme. For this example, inhibitor selectivity comes from similarity to substrate and susceptibility to the normal catalytic processing by the enzyme leading to a long-lived intermediate. The duration of inhibition is limited by the rate of intermediate decay.
Figure 3.

Slow Substrate As a Covalent Inhibitor. The catalytic Cys thiolate(35) of Pseudomonas aeruginosa arginine deiminase (ADI) attacks L-canavanine, resulting in a long-lasting covalent intermediate that causes the observed inhibition.
Other examples of slow substrates that act as covalent inhibitors include carbapenem β-lactams that make long lasting ester intermediates with the active-site Ser residue in serine β-lactamases. A tautomerization of the covalent intermediate formed between imipenem and TEM-1 slows subsequent hydrolysis by 50,000-fold compared to a non-inhibitory substrate.(36) The covalent intermediate formed between meropenem and SHV-1 is also stabilized, although through a different mechanism, by repositioning of the ester carbonyl away from the oxyanion hole that normally facilitates hydrolysis.(37) Each mechanism converts a substrate into a covalent inhibitor by stabilizing a covalent intermediate in the reaction.
Covalent Irreversible Inhibition
The second major category of covalent inhibitors includes those that cause irreversible inhibition (Figure 1). Such inhibitors are also called inactivators to emphasize the irretrievable loss in activity. Since covalent modification by these inhibitors is not reversible, the inhibitors cannot rely on thermodynamic equilibration to achieve selectivity and must instead use different strategies to achieve selective modification. Although forming an irreversible covalent adduct carries some inherent disadvantages, this type of inhibitor has the benefit of long-lasting inhibition that endures even after the concentration of inhibitor in solution decreases, and enables both target and off-target identification. Previous authors have noted the irony that the most objectionable aspect of irreversible inhibitors (covalent bond formation with unintended targets) also provides the exact means whereby the identity of modified proteins can be determined,(38) a feat not easily performed when searching for off-targets of non-covalent or covalent reversible inhibitors. With irreversible inhibitiors, the slow accumulation of inhibited enzyme over time, even in the presence of competing ligands, also adds to their effectiveness. One illustrative example is the drug vigabatrin. Although the non-covalent binding affinity of vigabatrin to its enzyme target, human brain γ-aminobutyric acid aminotransferase, is quite weak (KI = 1 mM)(39) compared to the typical goal of nanomolar Ki values sought in drug design, the selectivity and irreversibility of covalent bond formation (kinact = 0.35 min−1) leads to inhibition that is clinically effective as an antiepileptic treatment.
Briefly, we note that since inhibition by this category of inhibitors is time-dependent, the use of IC50 values alone to guide structure activity studies can be misleading.(18, 40) Parameters including KI to describe the non-covalent binding affinity (the subscript is given as a capital I to distinguish between this value and Ki values derived for reversible non-covalent inhibitors),(41) and kinact to describe the kinetics of covalent modification can be used to dissect the contributions of each to inhibition, and can be derived from fitting time dependent inhibition studies through a variety of methods described elsewhere.(41-45) Irreversible inhibitors can be classified into three major categories: residue-specific reagents, affinity labels and mechanism-based enzyme inactivators.
Residue-Specific Reagents
Residue-specific reagents used as covalent enzyme inhibitors are generally the least selective type of covalent irreversible inhibitor and are typically only used in vitro as biochemical tools. These reactive compounds rely on chemoselectivity for particular nucleophiles rather than non-covalent affinity to a particular binding site in order to achieve selective modification. The relative nucleophilicity and steric availability of the reacting residues in targeted proteins also influences selectivity. Many reagents used for residue-specific modification in proteins and peptides have been recently and extensively reviewed.(46-50) Since these reagents typically don’t have significant non-covalent affinity for a particular protein site, they usually display second order inactivation kinetics (Eq 3).
| (3) |
One relevant example of a residue-specific chemical modifying agent used as a covalent inhibitor describes the use of methyl methanethiosulfonate to react with Cys residues in soluble guanylate cyclase, resulting in covalent attachment of methanethiol moieties through disulfide bonds to multiple Cys residues throughout the protein (Figure 4).(51) Covalent modification of a Cys residue at a proposed allosteric site blocks enzyme activation that is otherwise achieved through heme binding of nitric oxide, and supports the hypothesis that Cys modification by reactive nitrogen species provides regulation. This example illustrates the nonselective nature of residue-specific reagents since high concentrations of methyl methanethiosulfonate result in nonspecific enzyme inhibition, presumably due to exhaustive Cys modification and disruption of enzyme structure. Lower concentrations of the reagent are able to react with a more discrete set of Cys residues in purified soluble guanylate cyclase to achieve selective inhibition at the allosteric site, presumably with the reacting and non-reacting Cys residues differentiated by either steric availability or differences in nucelophilicity. Studies with other enzymes have shown that methyl methanethiosulfonate can react with residues other than Cys, and serve as a reminder that the chemoselectivity of most residue-specific reagents is not absolute.(50) Although this reagent is sometimes referred to as a reversible thiol-modifying reagent, the covalent bond formed is not broken by the reverse reaction, but rather through a separate reaction with exogenously added thiols to regenerate the unmodified protein.(50)
Figure 4.
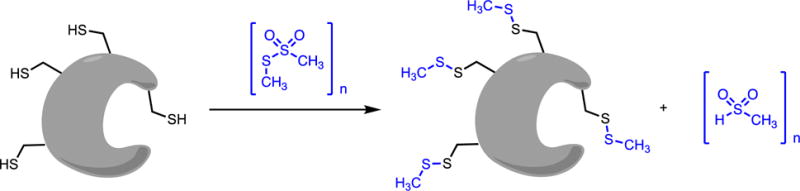
Residue-Specific Reagent As a Covalent Inhibitor. Methyl methanethiosulfonate reaction with multiple Cys residues results in inhibition of soluble guanylate cyclase activation. The figure does not reflect labeling stoichiometry of this particular experiment.
Affinity Labels
Rather than relying solely on chemoselectivity, affinity labels increase their site selectivity by coupling a reactive group, typically a poor electrophile, to a second moiety that provides non-covalent binding affinity to a specific binding site.(52) Each of the three different types of affinity labels within this class increases the effective molarity of the reactive group near the site of enzyme modification by using a moiety to provide non-covalent binding. However, the strategy used to attenuate reactivity varies by each type and can impact selectivity.
Classical affinity labels
The simplest type of affinity label has been termed a “classical” affinity label,(16) and often couples an electrophile (usually with low intrinsic reactivity) to the non-covalent binding moiety. Non-covalent binding to the targeted site provides a very high effective molarity of the weak electrophile next to a nucleophile on the target protein and can thereby enable selective labeling at one particular residue over others with similar or greater nucleophilicity. Due to formation of the initial non-covalent complex (E•I), modification displays saturation kinetics (Eq 4), with KI defining the dissociation constant for the non-covalent complex, and kinact the rate constant for covalent bond formation.
| (4) |
An FDA-approved kinase inhibitor of epidermal growth factor receptor (EGFR), afatinib, is an example of a classical affinity label that uses an acrylamide as the reactive group, and is sufficiently selective for therapeutic use (Figure 5).(38, 44, 53) A non-reactive analog of afatinib, in which the double bond in the acrylamide is replaced with a single bond, inhibits EGFR signaling with similar potency (as compared using defined experimental conditions), but enzyme activity readily returns upon washout. Incubation of cells with supratherapeutic concentrations of afatinib analogs leads to covalent modification of many other proteins (> 400) and correlates with cytotoxicity. The off targets of similar inhibitors determined at lower concentrations are predominantly proteins that have Cys residues annotated as “functional” Cys residues. Taken together, these observations are illustrative of some of the common benefits and drawbacks of using classical affinity labels. Covalent inhibition provides a long lasting effect even upon removal of excess inhibitor. However, since classical affinity labels typically contain electrophiles as the reactive group, an increase in the concentration or incubation time of the inhibitor, or reaction with residues that have enhanced nucelophilicity, can lead to undesired covalent bond formation with other proteins.
Figure 5.
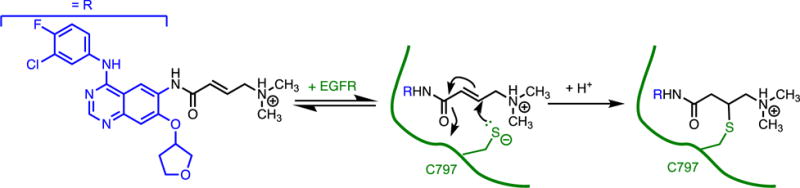
Classical Affinity Label As a Covalent Inhibitor. The high effective concentration of the acrylamide in afatinib enables selective reaction with a non-catalytic Cys (C797) in EGFR, presumably with the small fraction of ionized thiolate present at the reaction pH.
Although most classical affinity labels use electrophiles to target nucleophilic residues, it should be noted that nucleophilic moieties can also be used in affinity labels to target the relatively less abundant electrophilic sites found in some enzymes. For example, the amino group of α,α-difluoro-β-tyrosine has been used as a nucleophile to covalently modify the electrophilic 4-methylideneimidazole-5-one (MIO) cofactor of tyrosine aminomutase, resulting in inactivation.(54) This classification assumes a negligible off-rate; otherwise, the inhibitor would instead be classified as a covalent-reversible inhibitor.
Quiescent affinity labels
The next type of affinity label also uses non-covalent affinity to provide a high effective molarity of a reactive group near a nucleophilic residue. However, catalysis is also required to accelerate covalent bond formation (Eq 5), with KI defining the dissociation constant for the non-covalent complex and kinact the rate constant for covalent bond formation. The required catalysis does not include just lowering the pKa of the protein’s nucleophile, since that effect would also enhance reactivity with classical affinity labels, but instead catalysis is used to enhance the reactivity of the affinity label. Also, for inhibitors of this type, catalysis does not occur through the normal catalytic mechanism, but rather through a different “off pathway” mechanism, which differentiates them from mechanism-based inactivators (described below). To describe the dormant reactivity of the electrophile that is awakened by off-pathway catalysis, this class of covalent inhibitor has been named “quiescent” affinity labels.(55) In some previous publications, quiescent affinity labels have been defined simply as affinity labels that contain weak electrophilic groups.(18, 56) However, this definition does not clearly distinguish this class of inhibitors from classical affinity labels. We recommend instead using the definition of quiescent affinity labels described above, which includes a clear qualitative difference: the requirement for off-pathway catalysis.(16, 55, 57)
| (5) |
Fragment-sized 4-halopyridine derivatives can serve as an example of quiescent affinity labels for the enzyme dimethylarginine dimethylaminohydrolase.(58-60) A 4-chloropyridine reactive group has only weak non-enzymatic reactivity with thiol nucelophiles in solution, but can act as a “switchable electrophile” because formation of the charged pyridinium increases reactivity ~4500-fold. The catalysis that enhances electrophilicity of this reactive group is considered “off-pathway” since the protonated pyridinium is not formed through proton transfer from an active-site His residue that serves as a general acid during the normal reaction. Instead, the pyridinium form is stabilized by a neighboring Asp residue not used for that function in the normal mechanism (Figure 6). Attack of the active-site Cys residue and subsequent loss of the halide results in irreversible covalent inactivation of the enzyme. For quiescent affinity labels, the need for catalysis adds an additional criterion that limits modification of off-targets and increases the selectivity of covalent modification, but also limits the possible sites that can be targeted.
Figure 6.
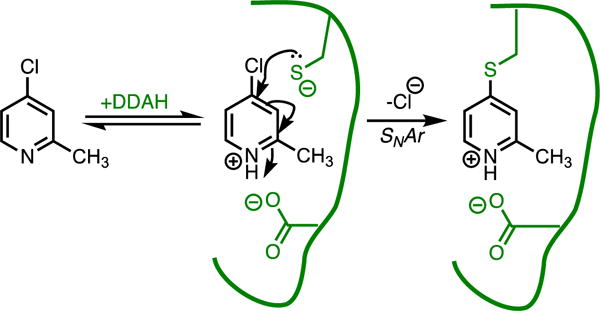
Quiescent Affinity Label As a Covalent Inhibitor. The reaction of 2-methyl-4-chloropyridine with the active-site Cys thiolate of dimethylarginine dimethylaminohydrolase (DDAH) is catalyzed by a neighboring Asp residue.
A similar enhancement in reactivity upon protonation is observed with epoxides, but if this protonation is facilitated by the same general acid used in the normal enzyme mechanism, as is likely the case with peptidyl epoxide inactivators of some cysteine proteases, designation as a mechanism-based inactivator would be more accurate.(61-63) The examples first described as quiescent affinity labels are peptidyl acyloxymethyl ketone inhibitors of cathepsin B, for which catalysis of an “aberrant” reaction pathway is required for inactivation.(56, 57)
Photoaffinity labels
The last type of affinity label, like the previous examples, uses non-covalent binding to provide a high effective molarity of a reactive group near a site on the enzyme that becomes covalently modified. However, the reactive group of photoaffinity labels is delivered in a latent unreactive form. Exposure to light initiates a non-enzymatic reaction that produces a highly reactive species that can readily form a covalent bond with a nearby residue at the binding site (Eq 6). The high reactivity of these species, once formed, also contributes to their selectivity since solvent can readily react and quench the group before it diffuses to non-selectively modify a different site. This technique is used most often to map ligand binding sites rather than for inhibition since the low labeling stoichiometry of many photoaffinity labels do not lead to effective inhibition.
| (6) |
One example of a photoaffinity label is given below. An analog of the Bruton’s tyrosine kinase inhibitor dasatinib is used as a moiety to provide non-covalent binding affinity, and a 2-aryl-5-carboxytetrazole is appended through an amide linkage as the latent reactive group (Figure 7).(64) After binding, the label is irradiated with 305 nm light, which leads to loss of N2 and formation of a reactive nitrile-imine intermediate. A neighboring Glu residue readily reacts with the label and a subsequent series of rearrangements result in formation of a stable covalently labeled enzyme. Selectivity of the labeling arises from the high effective molarity of the reactive group and the highly reactive nature of the nitrile-imine that can be quenched by solvent before diffusing and reacting at a more distant site.
Figure 7.
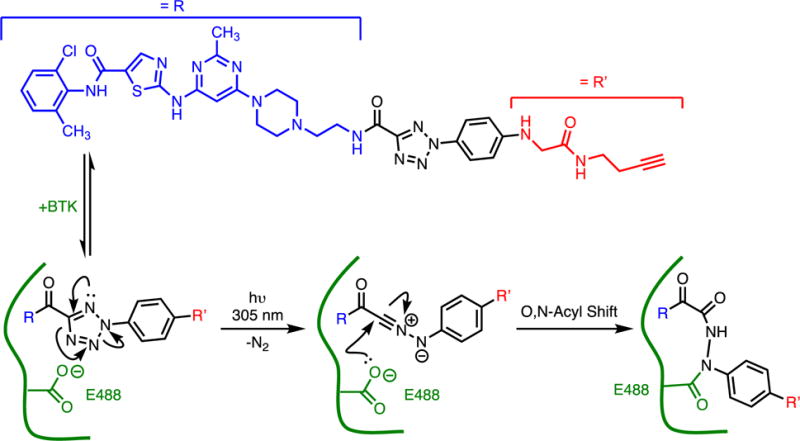
Photoaffinity Label As a Covalent Inhibitor. Non-covalent binding provides a high effective molarity of an imine nitrile, formed in situ through irradiation with light, that facilitates selective reaction with a particular Glu side chain.
Covalent Mechanism-Based Enzyme Inactivators
Of all covalent enzyme inhibitors, those classified as mechanism-based enzyme inactivators are potentially the most selective because they start as unreactive molecules. (Mechanism-based inactivators that use non-covalent inhibition mechanisms are not discussed here.) These inhibitors bind to the active site of enzymes and are processed by the normal catalytic mechanism to produce a reactive species that results in covalent bond formation (Eq 7), with KI and kinact representing a mixture of individual rate constants (Eqs 8, 9), which in some cases (e.g. k4 = 0, rapid equilibrium, k2 is rate limiting) can be simplified to represent the dissociation constant for the non-covalent complex and k2, respectively.(65) If the reactive species dissociates from the active site before covalent bond formation, it can react with solvent or other biological nucleophiles. The ratio of this turnover to inactivation (k4/k3) gives the partition ratio.(41) However, if the dissociated species is responsible for inactivation of a second enzyme molecule, the inhibitor is instead considered a prodrug (see below) rather than a mechanism-based inactivator. Of each of the categories of covalent inhibitors, this name is probably most often misapplied to inhibitors that do not meet the specific criteria defined elsewhere for true mechanism-based enzyme inactivators.(65) One potential benefit of mechanism-based inactivators is that they target active-site residues, which are most likely to be conserved, even when mutations arise in clinical settings. However, mechanism-based inactivators are often perceived as difficult to design de novo, and many examples have been discovered post hoc through investigation of the mechanisms of known inhibitors.
| (7) |
| (8) |
| (9) |
One recent example of a rationally designed, covalent, mechanism-based enzyme inactivator is that of isocitrate lyase inhibition by 2-vinyl-D-isocitrate.(66) The starting inhibitor is unreactive to nucleophiles in solution, but is bound and processed as a substrate by the enzyme (Figure 8). A retro-aldol cleavage generates reactive 2-vinylglyoxylate in situ at the active site of the enzyme. The other product, aci-succinate, is proposed to abstract a proton from the active-site Cys, enhancing the nucelophilicity of this residue and quenching the succinate enolate formed in the previous step. Subsequent dissociation of the resulting succinate allows the deprotonated Cys thiolate to react with the newly unmasked Michael acceptor 2-vinylglyoxylate, forming a covalent adduct that inhibits any further catalysis. Some reversibility of the covalent adduct is measured, but only with a small rate constant.
Figure 8.
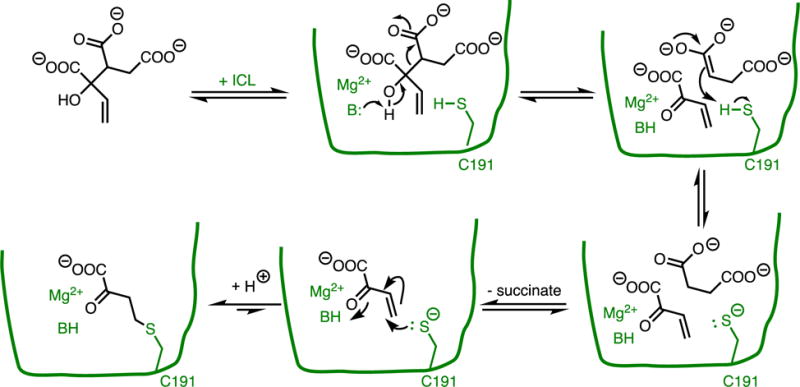
Mechanism-Based Enzyme Inactivator As a Covalent Inhibitor. The unreactive 2-vinyl-D-isocitrate is processed by the normal catalytic reaction of isocitrate lyase to produce a reactive species that covalently modifies Cys191.
Other notable examples among many reported mechanism-based inactivators include those that use reactions catalyzed by the enzyme cofactor pyridoxal 5′-phosphate to generate reactive species leading to covalent bond formation. A comprehensive collection of mechanism-based inactivators using such strategies to target γ-aminobutyric acid amino transferase has been recently and extensively reviewed.(67)
Taken together, the strategies described above summarize the spectrum of mechanisms used by covalent enzyme inhibitors (Table 1).(16) We note that each category describes not only the inhibitor, but also the mechanism of its interaction with the targeted enzyme. Therefore, a single compound may act through different mechanisms on different targets, and thus be placed in a separate category for each. One example of dual assignment is the antiplatelet drug clopidogrel. This compound starts as an unreactive prodrug (see below) that is metabolized into a reactive sulfenic acid, which acts as a classical affinity label for selective covalent modification of an extracellular Cys (C97) residue in the P2Y12 receptor of ADP on platelet membranes, irreversibly blocking ADP-mediated platelet aggregation.(68, 69) However the same compound, clopidogrel, acts instead as a mechanism-based inactivator of cytochrome P450 2B6, causing irreversible inactivation of this enzyme without prior dissociation of the responsible reactive species.(70) Therefore, correct categorization of a covalent inhibitor embeds information about both the inactivator and how it interacts with its targeted enzyme.
Table 1.
Summary of Covalent Inhibition Strategies
| Reversibility of Inhibition | Class of Inhibitor | Scheme | Major Selectivity Determinant | |
|---|---|---|---|---|
| Reversible | Covalent Reversible |
|
Thermodynamic equilibrium | |
| Slow Substrate |
|
Enzymatic catalysis | ||
| Irreversible | Residue-Specific Reagent |
|
Chemical reactivity | |
| Affinity Label: Classical |
|
Effective molarity & chemical reactivity | ||
| Affinity label: Quiescent |
|
Effective molarity & “off pathway” cat. | ||
| Affinity label: Photoaffinity |
|
Effective molarity & light activation | ||
| Mechanism-Based |
|
Enzymatic catalysis |
Proteomic Profiling Probes
The revolutionary combination of irreversible covalent inhibitors with chemical tags used for detection and purification has enabled proteome-wide determination of selectivity, reactivity, and target identification, as well as other applications.(71) Rather than selectively targeting one enzyme, these probes covalently label a family of related enzymes, or a specific residue type, proteome-wide. Proteomic profiling probes typically consist of an irreversible covalent modifier that uses one of the strategies outlined above, and have a relaxed selectivity that allows labeling of a broad set of related proteins. In addition, these probes also contain a moiety, often introduced through bioorthogonal chemistry, that allows covalently modified proteins to be purified or selectively detected. A number of powerful techniques have been developed using proteomic-profiling probes with broad selectivity in parallel with more specific covalent inhibitors to assess selectivity of inhibitors on a proteomic scale. In general, these probes can be divided into two different categories: activity-based and reactivity-based.
Activity-based protein profiling probes
This set of proteomic probes uses affinity labels that target the active site of enzymes, or sites functionally linked to the active site, as the covalent bond forming moiety. By targeting the active site, these probes act as inhibitors, but are most typically used instead as reporters of whether an enzyme active site is available for binding and reaction, thus serving as a proxy for enzyme activity. Several different types of affinity labels have been used in activity-based protein profiling probes, including classical affinity labels and photoaffinity labels. For example, some of the first activity-based protein profiling probes rely on a fluorophosphonate moiety, shared with the widely-used diisopropylfluorphosphonate serine protease inhibitor,(52) to mimic the substrate and provide binding affinity, and to provide a reactive site that covalently modifies the active-site Ser in this family of proteins.(72) One version of this classical affinity label is converted into an activity based protein profiling probe by tethering it to a biotin molecule that facilitates purification and/or detection (Figure 9).
Figure 9.
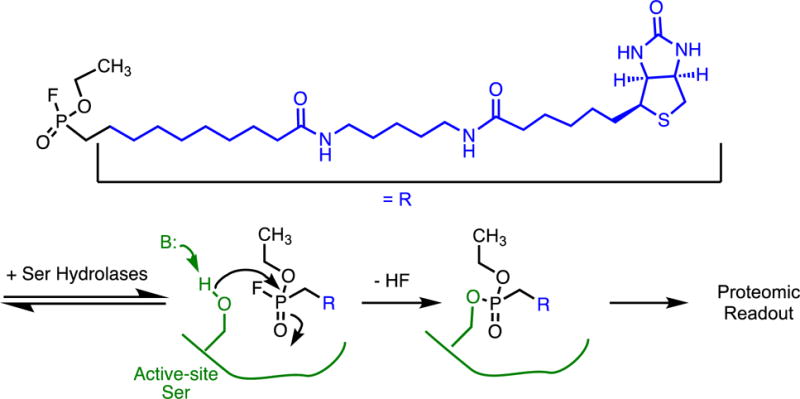
Activity-Based Proteomic Profiling Probe As a Covalent Inhibitor. A classical affinity label (a fluorophosphonate) tethered to a purification/detection tag (biotin) broadly targets the active-site Ser of serine proteases for covalent modification.
Reactivity-based protein profiling probes
This set of probes are similar to the activity-based probes, but instead substitute a residue-specific reagent for the broad spectrum affinity label in order to make covalent bonds with a specific residue type, proteome wide. An example of a reactivity-based protein profiling probe is given in a recent report that describes sulfotetrafluorophenyl esters that chemoselectively modify Lys residues (Figure 10).(73) This reactive ester was chosen from a set of amine-reactive groups to balance stability against hydrolysis and broad reactivity with Lys in proteomic extracts, resulting in the modification of > 9000 Lys residues (modification of some Ser residues is also noted). An alkyne is incorporated into this probe to serve as a latent purification/detection tag. Although many Lys modifications do not result in inhibition, some Lys residues modified by this and similar probes are found in enzyme active sites (e.g. pyridoxamine-5′phosphate oxidase), and modification results in covalent inhibition. Other enzymes are inhibited by modification of Lys residues found in allosteric sites (e.g. platelet phosphofructokinase).
Figure 10.
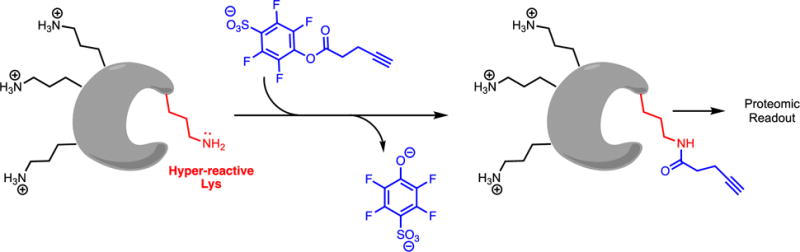
Reactivity-Based Protein Profiling Probe As a Covalent Inhibitor. Proteome-wide, residue selective modification by activated sulfotetrafluorophenyl esters produces Lys residues labeled with a latent purification/detection tag. Hyper-reactive Lys residues (e.g. Lys residues with enhanced nucleophilicity due to lowered pKa values) can be preferentially modified under certain conditions (depicted above).
Another notable recent example of reactivity-based protein profiling probes is the design of a matched pair of thiol-reactive reagents that couple an iodoacetamide moiety to an alkyne that serves as a latent purification/detection tag, but also includes a benzyl substituent. (74) The phenyl ring in one partner of this pair is selectively enriched in 13C, resulting in isotopically distinct probes that enable quantitative proteome-wide monitoring of Cys modification.
Complementary Approaches
The examples of covalent inhibition mechanisms given above describe different strategies that have been used to achieve or assess selective covalent modification, many achieving selectivity through attenuation of the reactive group and/or use of non-covalent affinity to provide a high effective molarity nearby a reactive site on the targeted enzyme. In addition to these strategies, other complementary approaches can be combined with covalent inhibitors to further increase selectivity. Two examples of such complementary approaches include prodrug and soft drug design. Each of these strategies works by limiting the time proteins are exposed to reactive groups, although they achieve this end by using opposite mechanisms.
Prodrug Forms of Covalent Inhibitors
To limit non-selective modification of proteins by reactive chemical species, a prodrug approach can be used to generate covalent inhibitors only near the site where modification is desired. Prodrugs are pharmacologically inactive compounds that are metabolically or non-enzymatically transformed into active species after administration.(75) These differ from mechanism-based inactivators, which cause inactivation before dissociation from the activating enzyme. One common drug used to treat symptoms of gastroesophageal reflux disease, omeprazole, is an example of a prodrug strategy used to mask a covalent enzyme inhibitor (Figure 11).(17, 76-78) The starting compound is pharmacologically inactive and chemically unreactive. However, when this prodrug encounters low pH conditions produced by acid secreting cells lining the stomach wall, protonation triggers a series of rearrangements and loss of water to form a sulfenamide bond that serves as the reactive group of what has essentially become a classical affinity label. The sulfenamide formed by omeprazole reacts with two different accessible Cys residues in the H+,K+-ATPase responsible for generating gastric acid, with modification of Cys813 likely leading to inhibition. The related prodrugs lansoprazole and rabeprazole also form a sulfenamide with similar reactivity, yet these inhibitors also label a third Cys residue, suggesting that the structure of omeprazole does provide some selectivity for the site of modification. Even though a very reactive species is used in this classical affinity label, the prodrug approach imparts sufficient selectivity to allow clinical use because the reactive group is only formed in a very localized space (the acidic canaliculi of parietal cells).
Figure 11.
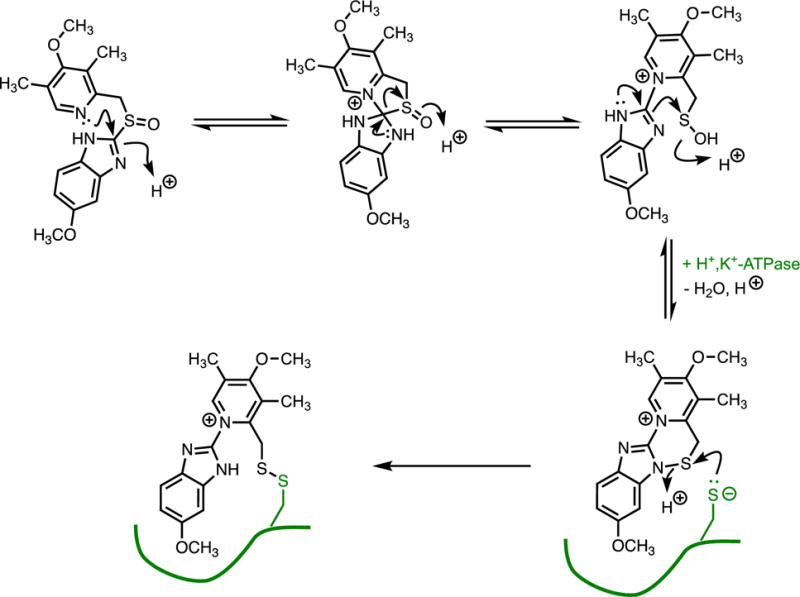
Prodrug Form of a Covalent Inhibitor. In an acidic cell compartment, the prodrug omeprazole is converted into a classical affinity label containing a sulfenamide group that reacts with non-catalytic Cys residues, presumably with the small fraction of ionized thiolate.
Soft Drug Forms of Covalent Inhibitors
Soft drugs are often described as the opposite of prodrugs because soft drugs instead begin as pharmacologically active species that are designed to be rapidly metabolized or non-enzymatically converted into inactive, non-toxic species.(79) This approach has also been applied to covalent inhibitors to improve selectivity.(80, 81) A clinically-approved affinity label for Bruton’s tyrosine kinase, ibrutinib, is a classical affinity label that uses an acrylamide as the reactive moiety to modify a non-catalytic Cys near the active site of the enzyme. An alkyne-containing analog of this drug was used to show that extended treatment times lead to increased modification of numerous off target proteins, similar to what has been seen with other classical affinity labels. To make a soft-drug version of this covalent inhibitor, the acrylamide moiety was replaced with a fumarate ester, resulting in an inhibitor that is still capable of covalent inhibition, although presumably through Cys attack at the position alpha to the amine rather than beta as occurs with the acrylamide, since the ester is a better electron withdrawing group (Figure 12). Notably, installation of the ester also now allows rapid hydrolysis by cellular carboxylesterases to yield the acid, which renders the compound much less electrophilic due to the newly formed anion, thus preventing modification of unintended targets during longer incubation times. In contrast to covalent reversible inhibitors (described above), which use thermodynamic control to achieve selectivity, this soft drug approach instead uses kinetic control to achieve selectivity, allowing covalent inhibition of the intended target, which reacts quickly, but then enabling rapid metabolism of the inhibitor to inactive products that minimize labeling of off-targets at longer incubation times.
Figure 12.
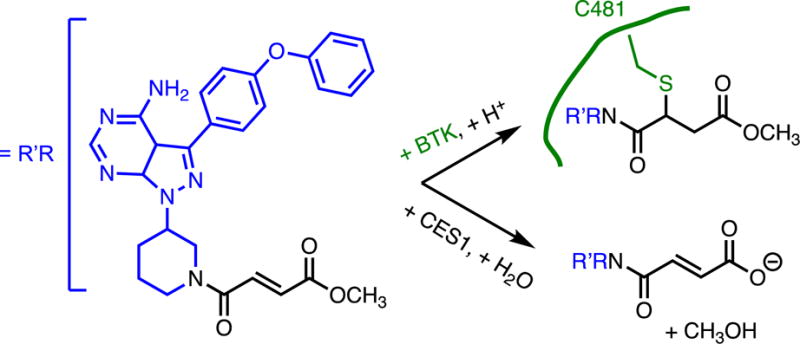
Soft Drug Form of a Covalent Inhibitor. This classical affinity label bearing a fumarate ester reactive group covalently modifies a Cys in Bruton’s tyrosine kinase (BTK), but hydrolysis of the ester by human carboxylesterase-1 (CES1) results in rapid deactivation of the reactive group, improving selectivity.
Summary
Optimization of covalent enzyme inhibitors for diverse applications involves balancing reactivity and selectivity. The mechanisms used to achieve the balance of these attributes can vary widely, as illustrated above by the two therapeutic drugs afatinib and omeprazole. Each acts as a classical affinity label and uses non-covalent affinity to provide a high effective concentration of their reactive group at a specific site on their respective targets. Yet, one uses a weak electrophile, and the other instead masks a highly reactive group in prodrug form. The categories described above, covalent reversible inhibitors, slow substrates, residue-specific reagents, affinity labels (classical, quiescent, and photoaffinity), and mechanism-based enzyme inactivators, as well as related approaches including proteomic profiling (activity and reactivity-based) and prodrug/soft drug design describe the spectrum of mechanisms used to achieve and assess this balance. We recommend categorizing covalent enzyme inhibitors by mechanism to provide a shared terminology that bridges contemporary advances with the long history of covalent inhibitors used in enzymology, to clearly convey the strategies used to optimize each inhibitor, and to spur the innovation of new strategies for covalent inhibition of enzymes.
Acknowledgments
Work in the authors’ lab was supported in part by the Robert A. Welch Foundation (F-1572) and NIH (GM111926). A.T. acknowledges a postdoctoral fellowship from the NIH (1K12 GM102745).
Footnotes
ORCID
Walter Fast: 0000-0001-7567-2213
Notes
The authors declare no competing financial Interest.
References
- 1.De Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N. Covalent inhibitors design and discovery. Eur J Med Chem. 2017;138:96–114. doi: 10.1016/j.ejmech.2017.06.019. [DOI] [PubMed] [Google Scholar]
- 2.Jackson PA, Widen JC, Harki DA, Brummond KM. Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J Med Chem. 2017;60:839–885. doi: 10.1021/acs.jmedchem.6b00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu S, Zhang J. Designed covalent allosteric modulators: an emerging paradigm in drug discovery. Drug Discov Today. 2017;22:447–453. doi: 10.1016/j.drudis.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 4.Visscher M, Arkin MR, Dansen TB. Covalent targeting of acquired cysteines in cancer. Curr Opin Chem Biol. 2016;30:61–67. doi: 10.1016/j.cbpa.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bandyopadhyay A, Gao J. Targeting biomolecules with reversible covalent chemistry. Curr Opin Chem Biol. 2016;34:110–116. doi: 10.1016/j.cbpa.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez-Bello C. Designing Irreversible Inhibitors–Worth the Effort? ChemMedChem. 2016;11:22–30. doi: 10.1002/cmdc.201500469. [DOI] [PubMed] [Google Scholar]
- 7.Baillie TA. Targeted Covalent Inhibitors for Drug Design. Angew Chem. 2016;55:13408–13421. doi: 10.1002/anie.201601091. [DOI] [PubMed] [Google Scholar]
- 8.Bauer RA. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov Today. 2015;20:1061–1073. doi: 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Mah R, Thomas JR, Shafer CM. Drug discovery considerations in the development of covalent inhibitors. Bioorg Med Chem Lett. 2014;24:33–39. doi: 10.1016/j.bmcl.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Niphakis MJ, Cravatt BF. Enzyme inhibitor discovery by activity-based protein profiling. Ann Rev Biochem. 2014;83:341–377. doi: 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- 11.Kalgutkar AS, Dalvie DK. Drug discovery for a new generation of covalent drugs. Exp Opin Drug Discov. 2012;7:561–581. doi: 10.1517/17460441.2012.688744. [DOI] [PubMed] [Google Scholar]
- 12.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 13.Johnson DS, Weerapana E, Cravatt BF. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med Chem. 2010;2:949–964. doi: 10.4155/fmc.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Potashman MH, Duggan ME. Covalent modifiers: an orthogonal approach to drug design. J Med Chem. 2009;52:1231–1246. doi: 10.1021/jm8008597. [DOI] [PubMed] [Google Scholar]
- 15.Smith AJ, Zhang X, Leach AG, Houk KN. Beyond picomolar affinities: quantitative aspects of noncovalent and covalent binding of drugs to proteins. J Med Chem. 2009;52:225–233. doi: 10.1021/jm800498e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krantz A. A Classification of Enzyme Inhibitors. Bioorg Med Chem Lett. 1992;2:1327–1334. [Google Scholar]
- 17.Silverman RB, Holladay MW. The Organic Chemistry of Drug Design and Drug Action. Third. Academic Press, Elsevier; Amsterdam: 2014. [Google Scholar]
- 18.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery. Wiley-Interscience; Hoboken, N.J: 2005. [Google Scholar]
- 19.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 20.Krishnan S, Miller RM, Tian B, Mullins RD, Jacobson MP, Taunton J. Design of reversible, cysteine-targeted Michael acceptors guided by kinetic and computational analysis. J Am Chem Soc. 2014;136:12624–12630. doi: 10.1021/ja505194w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. J Am Chem Soc. 2013;135:5298–5301. doi: 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menger FM. On the Source of Intramolecular and Enzymatic Reactivity. Acc Chem Res. 1985;18:128–134. [Google Scholar]
- 23.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Nat Acad Sci USA. 2012;109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnes MD, Bethel CR, Alsop J, Becka SA, Rutter JD, Papp-Wallace KM, Bonomo RA. Inactivation of the Pseudomonas-Derived Cephalosporinase-3 (PDC-3) by Relebactam. Antimicrob Agents Chemother. 2018 doi: 10.1128/AAC.02406-17. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu CT, Tomsho JW, Benkovic SJ. The unique chemistry of benzoxaboroles: current and emerging applications in biotechnology and therapeutic treatments. Bioorg Med Chem. 2014;22:4462–4473. doi: 10.1016/j.bmc.2014.04.065. [DOI] [PubMed] [Google Scholar]
- 26.McKinney DC, Zhou F, Eyermann CJ, Ferguson AD, Prince DB, Breen J, Giacobbe RA, Lahiri S, Verheijen JC. 4,5-Disubstituted 6-Aryloxy-1,3-dihydrobenzo[c][1,2]oxaboroles Are Broad-Spectrum Serine β-Lactamase Inhibitors. ACS Infect Dis. 2015;1:310–316. doi: 10.1021/acsinfecdis.5b00031. [DOI] [PubMed] [Google Scholar]
- 27.Rock FL, Mao W, Yaremchuk A, Tukalo M, Crepin T, Zhou H, Zhang YK, Hernandez V, Akama T, Baker SJ, Plattner JJ, Shapiro L, Martinis SA, Benkovic SJ, Cusack S, Alley MR. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 28.Baillie TA, Slatter G. Glutathione: A Vehicle for the Transport of Chemically Reactive Metabolites in Vivo. Acc Chem Res. 1991;24:264–270. [Google Scholar]
- 29.Chandrasekaran A, Shen L, Lockhead S, Oganesian A, Wang J, Scatina J. Reversible covalent binding of neratinib to human serum albumin in vitro. Drug Metab Lett. 2010;4:220–227. doi: 10.2174/187231210792928206. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Li-Chan XX, Atherton J, Deng L, Espina R, Yu L, Horwatt P, Ross S, Lockhead S, Ahmad S, Chandrasekaran A, Oganesian A, Scatina J, Mutlib A, Talaat R. Characterization of HKI-272 covalent binding to human serum albumin. Drug Metab Dispos. 2010;38:1083–1093. doi: 10.1124/dmd.110.032292. [DOI] [PubMed] [Google Scholar]
- 31.Fersht A. Structure and Mechanism in Protein Science. W.H. Freeman; New York: 1999. [Google Scholar]
- 32.Martin MT, Holmquist B, Riordan JF. An angiotensin converting enzyme inhibitor is a tight-binding slow substrate of carboxypeptidase A. J Inorg Biochem. 1989;36:39–50. doi: 10.1016/0162-0134(89)80011-x. [DOI] [PubMed] [Google Scholar]
- 33.Lu X, Li L, Feng X, Wu Y, Dunaway-Mariano D, Engen JR, Mariano PS. L-Canavanine is a time-controlled mechanism-based inhibitor of Pseudomonas aeruginosa arginine deiminase. J Am Chem Soc. 2005;127:16412–16413. doi: 10.1021/ja056226p. [DOI] [PubMed] [Google Scholar]
- 34.Li L, Li Z, Chen D, Lu X, Feng X, Wright EC, Solberg NO, Dunaway-Mariano D, Mariano PS, Galkin A, Kulakova L, Herzberg O, Green-Church KB, Zhang L. Inactivation of microbial arginine deiminases by L-canavanine. J Am Chem Soc. 2008;130:1918–1931. doi: 10.1021/ja0760877. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Li Z, Wang C, Xu D, Mariano PS, Guo H, Dunaway-Mariano D. The electrostatic driving force for nucleophilic catalysis in L-arginine deiminase: a combined experimental and theoretical study. Biochemistry. 2008;47:4721–4732. doi: 10.1021/bi7023496. [DOI] [PubMed] [Google Scholar]
- 36.Taibi P, Mobashery S. Mechanism of Turnover of Imipenem by the TEM β-Lactamase Revisited. J Am Chem Soc. 1995;117:7600–7605. [Google Scholar]
- 37.Nukaga M, Bethel CR, Thomson JM, Hujer AM, Distler A, Anderson VE, Knox JR, Bonomo RA. Inhibition of class A β-lactamases by carbapenems: crystallographic observation of two conformations of meropenem in SHV-1. J Am Chem Soc. 2008;130:12656–12662. doi: 10.1021/ja7111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lanning BR, Whitby LR, Dix MM, Douhan J, Gilbert AM, Hett EC, Johnson TO, Joslyn C, Kath JC, Niessen S, Roberts LR, Schnute ME, Wang C, Hulce JJ, Wei B, Whiteley LO, Hayward MM, Cravatt BF. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol. 2014;10:760–767. doi: 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeon SG, Bahn JH, Jang JS, Park J, Kwon OS, Cho SW, Choi SY. Human brain GABA transaminase tissue distribution and molecular expression. Eur J Biochem. 2000;267:5601–5607. doi: 10.1046/j.1432-1327.2000.01626.x. [DOI] [PubMed] [Google Scholar]
- 40.Holdgate GA, Meek TD, Grimley RL. Mechanistic enzymology in drug discovery: a fresh perspective. Nat Rev Drug Discov. 2018;17:115–132. doi: 10.1038/nrd.2017.219. [DOI] [PubMed] [Google Scholar]
- 41.Silverman RB. Mechanism-based enzyme inactivation: chemistry and enzymology. CRC Press; Boca Raton, FL: p. 1988. [Google Scholar]
- 42.Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 43.Tsou CL. Kinetics of substrate reaction during irreversible modification of enzyme activity. Adv Enzymol Rel Areas Mol Biol. 1988;61:381–436. doi: 10.1002/9780470123072.ch7. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz PA, Kuzmic P, Solowiej J, Bergqvist S, Bolanos B, Almaden C, Nagata A, Ryan K, Feng J, Dalvie D, Kath JC, Xu M, Wani R, Murray BW. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc Nat Acad Sci USA. 2014;111:173–178. doi: 10.1073/pnas.1313733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson KA. Fitting enzyme kinetic data with KinTek Global Kinetic Explorer. Methods Enzymol. 2009;467:601–626. doi: 10.1016/S0076-6879(09)67023-3. [DOI] [PubMed] [Google Scholar]
- 46.So WH, Zhang Y, Kang W, Wong CTT, Sun H, Xia J. Site-selective covalent reactions on proteinogenic amino acids. Curr Opin Biotech. 2017;48:220–227. doi: 10.1016/j.copbio.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 47.deGruyter JN, Malins LR, Baran PS. Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry. 2017;56:3863–3873. doi: 10.1021/acs.biochem.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shannon DA, Weerapana E. Covalent protein modification: the current landscape of residue-specific electrophiles. Curr Opin Chem Biol. 2015;24:18–26. doi: 10.1016/j.cbpa.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 49.Basle E, Joubert N, Pucheault M. Protein chemical modification on endogenous amino acids. Chem Biol. 2010;17:213–227. doi: 10.1016/j.chembiol.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Lundblad RL. Chemical Reagents for Protein Modification. Fourth. CRC Press; Boca Raton, FL: p. 2014. [Google Scholar]
- 51.Fernhoff NB, Derbyshire ER, Marletta MA. A nitric oxide/cysteine interaction mediates the activation of soluble guanylate cyclase. Proc Nat Acad Sci USA. 2009;106:21602–21607. doi: 10.1073/pnas.0911083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plapp BV. Application of affinity labeling for studying structure and function of enzymes. Methods Enzymol. 1982;87:469–499. doi: 10.1016/s0076-6879(82)87027-4. [DOI] [PubMed] [Google Scholar]
- 53.Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 54.Christianson CV, Montavon TJ, Festin GM, Cooke HA, Shen B, Bruner SD. The mechanism of MIO-based aminomutases in β-amino acid biosynthesis. J Am Chem Soc. 2007;129:15744–15745. doi: 10.1021/ja0762689. [DOI] [PubMed] [Google Scholar]
- 55.Krantz A. Some Thoughts on Enzyme Inhibition and the Quiescent Affinity Label Concept. Adv Med Chem. 1992;1:235–261. [Google Scholar]
- 56.Smith RA, Copp LJ, Coles PJ, Pauls HW, Robinson VJ, Spencer RW, Heard SB, Krantz A. New Inhibitors of Cysteine Proteinases. Peptidyl Acyloxymethyl Ketones and the Quiescent Nucleofuge Strategy. J Am Chem Soc. 1988;110:4429–4431. [Google Scholar]
- 57.Krantz A, Copp LJ, Coles PJ, Smith RA, Heard SB. Peptidyl (acyloxy)methyl ketones and the quiescent affinity label concept: the departing group as a variable structural element in the design of inactivators of cysteine proteinases. Biochemistry. 1991;30:4678–4687. doi: 10.1021/bi00233a007. [DOI] [PubMed] [Google Scholar]
- 58.Schardon CL, Tuley A, Er JAV, Swartzel JC, Fast W. Selective Covalent Protein Modification by 4-Halopyridines through Catalysis. ChemBioChem. 2017;18:1551–1556. doi: 10.1002/cbic.201700104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson CM, Monzingo AF, Ke Z, Yoon DW, Linsky TW, Guo H, Robertus JD, Fast W. On the mechanism of dimethylarginine dimethylaminohydrolase inactivation by 4-halopyridines. J Am Chem Soc. 2011;133:10951–10959. doi: 10.1021/ja2033684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson CM, Linsky TW, Yoon DW, Person MD, Fast W. Discovery of halopyridines as quiescent affinity labels: inactivation of dimethylarginine dimethylaminohydrolase. J Am Chem Soc. 2011;133:1553–1562. doi: 10.1021/ja109207m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pocker Y, Ronald BP, Anderson KW. A Mechanistic Characterization of the Spontaneous Ring Opening Process of Epoxides in Aqueous Solution: Kinetic and Product Studies. J Am Chem Soc. 1988;110:6492–6497. [Google Scholar]
- 62.Albeck A, Kliper S. Mechanism of cysteine protease inactivation by peptidyl epoxides. Biochem J. 1997;322(Pt 3):879–884. doi: 10.1042/bj3220879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arafet K, Ferrer S, Gonzalez FV, Moliner V. Quantum mechanics/molecular mechanics studies of the mechanism of cysteine protease inhibition by peptidyl-2,3-epoxyketones. Phys Chem Chem Phys. 2017;19:12740–12748. doi: 10.1039/c7cp01726j. [DOI] [PubMed] [Google Scholar]
- 64.Herner A, Marjanovic J, Lewandowski TM, Marin V, Patterson M, Miesbauer L, Ready D, Williams J, Vasudevan A, Lin Q. 2-Aryl-5-carboxytetrazole as a New Photoaffinity Label for Drug Target Identification. J Am Chem Soc. 2016;138:14609–14615. doi: 10.1021/jacs.6b06645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Silverman RB. Mechanism-based enzyme inactivators. Methods Enzymol. 1995;249:240–283. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- 66.Pham TV, Murkin AS, Moynihan MM, Harris L, Tyler PC, Shetty N, Sacchettini JC, Huang HL, Meek TD. Mechanism-based inactivator of isocitrate lyases 1 and 2 from Mycobacterium tuberculosis. Proc Nat Acad Sci USA. 2017;114:7617–7622. doi: 10.1073/pnas.1706134114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silverman RB. Design and Mechanism of GABA Aminotransferase Inactivators. Treatments for Epilepsies and Addictions. Chem Rev. 2018;118:4037–4070. doi: 10.1021/acs.chemrev.8b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maffrand JP. The Story of Clopidogrel and its Predecessor, Ticlopidine: Could These Major Antiplatelet And Antithrombotic Drugs Be Discovered and Developed Today? Comptes Rendus Chimie. 2012;15:737–743. [Google Scholar]
- 69.Savi P, Zachayus JL, Delesque-Touchard N, Labouret C, Herve C, Uzabiaga MF, Pereillo JM, Culouscou JM, Bono F, Ferrara P, Herbert JM. The active metabolite of Clopidogrel disrupts P2Y12 receptor oligomers and partitions them out of lipid rafts. Proc Nat Acad Sci USA. 2006;103:11069–11074. doi: 10.1073/pnas.0510446103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang H, Amunugama H, Ney S, Cooper N, Hollenberg PF. Mechanism-based inactivation of human cytochrome P450 2B6 by clopidogrel: involvement of both covalent modification of cysteinyl residue 475 and loss of heme. Mol Pharm. 2011;80:839–847. doi: 10.1124/mol.111.073783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Ann Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 72.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Nat Acad Sci USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, Cravatt BF. Global profiling of lysine reactivity and ligandability in the human proteome. Nat Chem. 2017;9:1181–1190. doi: 10.1038/nchem.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abo M, Li C, Weerapana E. Isotopically-Labeled Iodoacetamide-Alkyne Probes for Quantitative Cysteine-Reactivity Profiling. Mol Pharm. 2018;15:743–749. doi: 10.1021/acs.molpharmaceut.7b00832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Testa B. Prodrugs: bridging pharmacodynamic/pharmacokinetic gaps. Curr Opin Chem Biol. 2009;13:338–344. doi: 10.1016/j.cbpa.2009.04.620. [DOI] [PubMed] [Google Scholar]
- 76.Lambrecht N, Munson K, Vagin O, Sachs G. Comparison of covalent with reversible inhibitor binding sites of the gastric H,K-ATPase by site-directed mutagenesis. J Biol Chem. 2000;275:4041–4048. doi: 10.1074/jbc.275.6.4041. [DOI] [PubMed] [Google Scholar]
- 77.Besancon M, Simon A, Sachs G, Shin JM. Sites of reaction of the gastric H,K-ATPase with extracytoplasmic thiol reagents. J Biol Chem. 1997;272:22438–22446. doi: 10.1074/jbc.272.36.22438. [DOI] [PubMed] [Google Scholar]
- 78.Hersey SJ, Steiner L, Mendlein J, Rabon E, Sachs G. SCH28080 prevents omeprazole inhibition of the gastric H+/K+-ATPase. Biochim Biophy Acta. 1988;956:49–57. doi: 10.1016/0167-4838(88)90296-8. [DOI] [PubMed] [Google Scholar]
- 79.Bodor N, Buchwald P. Soft drug design: general principles and recent applications. Med Res Rev. 2000;20:58–101. doi: 10.1002/(sici)1098-1128(200001)20:1<58::aid-med3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 80.Zaro BW, Whitby LR, Lum KM, Cravatt BF. Metabolically Labile Fumarate Esters Impart Kinetic Selectivity to Irreversible Inhibitors. J Am Chem Soc. 2016;138:15841–15844. doi: 10.1021/jacs.6b10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, Jeffery DA, Spoerke JM, Honigberg LA, Young PR, Dalrymple SA, Palmer JT. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]