

Abstract
Endothelial cell vascular cell adhesion molecule-1 (VCAM-1) regulates recruitment of lymphocytes, eosinophils, mast cells or dendritic cells during allergic inflammation. In this report, we demonstrated that, during allergic lung responses, there was reduced ZO-1 localization in lung endothelial cell junctions, whereas there was increased lung endothelial cell expression of VCAM-1, N-Cadherin, and angiomotin. In vitro, leukocyte binding to VCAM-1 reduced ZO-1 in endothelial cell junctions. Using primary human endothelial cells and mouse endothelial cell lines, antibody crosslinking of VCAM-1 increased serine phosphorylation of ZO-1 and induced dissociation of ZO-1 from endothelial cell junctions, demonstrating that VCAM-1 regulates ZO-1. Moreover, VCAM-1 induction of ZO-1 phosphorylation and loss of ZO-1 localization at cell junctions was blocked by inhibition of VCAM-1 intracellular signals that regulate leukocyte transendothelial migration, including NOX2, PKCα, and PTP1B. Furthermore, exogenous addition of the VCAM-1 signaling intermediate H2O2 (1 μM) stimulated PKCα-dependent and PTP1B-dependent serine phosphorylation of ZO-1 and loss of ZO-1 from junctions. Overexpression of ZO-1 blocked leukocyte transendothelial migration. In summary, leukocyte binding to VCAM-1 induces signals that stimulated ZO-1 serine phosphorylation and reduced ZO-1 localization at endothelial cell junctions during leukocyte transendothelial migration.
Keywords: endothelial cells, VCAM-1, ZO-1, lymphocyte migration
INTRODUCTION
Inflammatory and immune surveillance signals induce the migration of blood leukocytes across vascular endothelial cells. During allergic inflammation, vascular cell adhesion molecule -1 (VCAM-1) regulates transendothelial migration of eosinophils, lymphocytes, and mast cells from the blood into the tissue [1-10]. VCAM-1 is localized to the luminal surface of endothelial cells and endothelial cell junctions [11]. VCAM-1 also regulates recruitment of leukocytes during inflammation in atopic dermatitis [12], inflammatory bowel disease [13], atherosclerosis lessions [14], LCMV infections [15], hematopoietic stem cell recruitment to injured liver and melanoma metastasis to the liver [16-18].
Cell binding to VCAM-1 induces intracellular signals in endothelial cells that are required for cell recruitment in vivo and in vitro [1, 19-26] but it is not known whether VCAM-1 signal transduction regulates endothelial cell junction molecules. The VCAM-1-induced intracellular signals include endothelial cell NOX2-catalyzed production of nontoxic levels of reactive oxygen species (ROS) (1 μM H2O2) [1, 20, 22, 23, 27]. The 1 μM H2O2 generated during VCAM-1 signaling oxidizes and activates endothelial cell surface-associated matrix metalloproteinases (MMP) [23] and endothelial cell PKCα [24]. This active PKCα, then, stimulates serine phosphorylation and activation of protein tyrosine phosphatase 1B (PTP1B) [25]. Importantly, the stimulatory outcome of these low levels of H2O2 on MMPs, PKCα, and PTP1B is considerably different than the effects of high toxic levels of H2O2 which inhibit enzymes by oxidative denaturation and induce oxidative damage to endothelial cells [23-25, 28, 29]. The VCAM-1 signals through NOX2, MMPs, PKCα, and PTP1B are required for VCAM-1-dependent leukocyte migration in vitro [1, 23, 24] and in vivo [19, 20, 22, 23]. In in vivo studies, non-hematopoietic gp91phox knockout mice have reduced VCAM-1-dependent leukocyte recruitment in mice during allergic inflammation [20, 22, 23] and mice with an inducible endothelial cell-specific knockout of PTP1B have reduced recruitment of leukocytes to inflammatory sites during allergic inflammation [19]. It is not known whether VCAM-1 signals alter endothelial cell junction proteins during leukocyte transendothelial migration.
Migrating leukocytes encounter paracellular endothelial cell-cell junctions. In the endothelial cell junctions, several transmembrane junction proteins bind to members of the zonula occludens (ZO) protein family [30-42]. Dissociation of ZO-1 binding to cell junction proteins reduces junction protein affinity. The dissociation of ZO-1 from junctions in epithelial and endothelial cells is induced by phosphorylation of ZO-1 [42-44]. Recruitment of ZO-1 to cell junction molecules is regulated by angiomotin [45] and ZO-1 localizes with angiomotin in cell junctions in CHO cells [45-47]. Interestingly, asthmatic patient lung endothelial cells have increased angiomotin [48]. Lung endothelial cells also exhibit increased expression of N-Cadherin during endotoxin stimulation [49], but it is not known whether allergic inflammation upregulates lung endothelial cell N-cadherin expression. Thus, whether induction of allergic inflammation or VCAM-1 signaling alters expression or localization of ZO-1, angiomotin or N-Cadherin in endothelial cells is not known.
Here, we demonstrate that, during VCAM-1-dependent allergic lung inflammation [3], there is an increase in N-cadherin, an increase in angiomotin, and a decrease in ZO-1 in mouse lung endothelial cell junctions. Moreover, we demonstrate that VCAM-1 signals through ROS, PKCα, and PTP1B induce serine phosphorylation of ZO-1 and loss of ZO-1 from endothelial cell junctions during VCAM-1-dependent leukocyte transendothelial migration.
METHODS
Animals
Male 6-8 week old BALB/c mice (Harlan Industries, Indianapolis, IN) were the source of resting splenic lymphocytes. All animal procedures were reviewed and approved by the Animal Care and Use Committee at Northwestern University.
Inhibitors and Antibodies
Inhibitors were as follows: CinnGEL-2ME and Gö-6976 from Biomol, apocynin from Acros Organics. Inhibitors and antibody crosslinking of VCAM-1 did not affect cell viability during the time courses of these studies, consistent with our previous reports [1, 24, 25]. Antibodies were as follows: Rat anti-mouse VCAM-1 (clone MVCAM.A), mouse anti-human VCAM-1 (clone 51-10C9), rat anti-mouse VE-Cadherin (CD144, cat#550548), rat IgG (isotype antibody, clone R35-95), and FITC-conjugated goat anti-rabbit Ig (cat#554020) [PharMingen]; zenon Alexa Fluor 568-labeled (cat#Z25106, Molecular Probes) rat anti-mouse CD45 (clone I3/2.3), goat anti-mouse IgG1 (cat#1070-01) and goat anti-rat IgG (cat#3050-01) [Southern Biotech]; mouse anti-phosphotyrosine (cat#9411) [Cell Signaling Technology]; Texas Red-conjugated goat anti-rat Ig (cat#R40005) [Caltag]; rabbit anti-human ZO-1 (cat# 41090971), FITC-conjugated rabbit anti-ZO-1 (cat#bs-1329R-FITC)[Bioss], and rabbit anti-phosphoserine (cat#61-8100) [Zymed Lab, Inc]; Alexa 594-conjugated donkey anti rabbit N-Cadherin (cat#150080) [Abcam]; rabbit anti-N-Cadherin (cat#PA5-19486)[Thermo Scientific]; rabbit anti-N-terminal region of angiomotin (cat# ARP34660_P050)[Aviva Systems Biology]; HRP-conjugated donkey anti-rabbit antibody [Amersham Pharmacia]; HRP-conjugated goat anti-mouse IgG [Bio-Rad].
OVA administration in vivo
Mice (8-10 mice/group) received intraperitoneal (i.p.) injections (200 μl) of chicken egg ovalbumin fraction V (OVA, catalog #5503, Sigma) (10 μg)/alum or saline/alum on days 1 and 8 [20]. OVA grade V was used for sensitization because it contains low endotoxin levels which are required for adequate OVA sensitization [50]; in contrast, high levels of endotoxin suppress the OVA response [50]. On days 15, 18, and 20, mice received intranasal endotoxin-free OVA fraction VI (catalog #A2512, Sigma) (150 μg) in saline or saline alone. Then, 24-hrs after the last treatment, lungs were lavaged and then lungs in OCT were stored at −80°C. Bronchoalveolar lavage (BAL) cells were counted and cytospun for differential counts [20]. Cytospins of BAL cells were differentially stained using the Diff-Quick staining kit (Dade Behring) and neutrophils, eosinophils, leukocytes, and monocytes were identified and counted according to morphological criteria. Frozen lung tissue sections from these OVA-treated or saline-treated mice were used for immunolabeling of ZO-1, VCAM-1, N-Cadherin, and angiomotin. Lung tissue sections were fixed in 100% ice-cold methanol for 15 minutes, rehydrated in 1× PBS for one hour, and blocked for 30 minutes in 300 μl PBS/BSA/Azide containing 3 μl goat and rat serums. Sections were labeled with rabbit anti-N-Cadherin, rabbit anti-angiomotin or polyclonal rabbit antibodies as an isotype control (no. ab27478 Abcam), washed, and labeled with polyclonal goat anti-rabbit antibodies conjugated with Alexa Fluor 594. N, sections were washed, labeled with FITC-conjugated rat IgG anti-mouse anti-ZO-1 antibodies, washed, and coverslipped with ProLong Gold Anti Fade reagent with DAPI (Life Technologies).
Cells
The well characterized mouse endothelial cell line mHEVa that has an activated phenotype and support leukocyte transendothelial migration on VCAM-1 has been previously described [3, 51, 52]. HEV culture medium consisted of RPMI-1640 containing 1 mM HEPES (pH 7.2, Sigma), 10 mM NaHCO 3 (Sigma), 2 mM glutamine (Sigma), 100 units/ml penicillin (Fisher), 100 μg/ml streptomycin (Sigma), 50 μg/ml gentamicin (GIBCO Laboratories, Grand Island, NY), and 20% heat-inactivated fetal calf serum. Human microvascular endothelial cells from the lung (HMEC-Ls) passage 1-4 were from Clonetics. Spleen cells from BALB/c mice were minced and pressed through a 100 μm mesh screen as previously described [1]. Spleen red blood cells were lysed by hypotonic shock and counted with a hemacytometer (cat#0267110, Fisher Scientific) [51].
Immunoprecipitation [53]
mHEV cells in 12 well plates were pretreated with the PTP1B inhibitor 10 μM CinnGel (Biomol) or the vehicle control DMSO (0.1%) for 30 minutes and then stimulated in duplicates with stimulated with 27 μg/ml anti-VCAM-1 antibody or an isotype control antibody plus 15 μg/ml a secondary antibody for 15 minutes (optimal time point, data not shown) or 1mM H2O2 for the time indicated. The cells were washed with PBS and lysed with 100mL/well radioimmunoprecipitation (RIPA) buffer (1% Triton X-100, 0.5% sodium deoxycholate, 0.2% SDS, 150 mM NaCl, 10 mM, HEPES, pH 7.3, 2 mM EDTA, 10 mg/ml leupeptin, 10 mg/ml aprotinin, 100 mg/ml iodoacetamide, and 1 mM phenylmethylsulfonlyfluoride (PMSF)). Duplicate samples were combined, and lysates were incubated on ice for 20 minutes. Lysates were homogenized by passing 20 times through a 22-gauge needle and then centrifuged for 10 min at 15,000×g at 4°C. Total protein concentration was determined by a protein assay kit (Bio-Rad), and protein concentration was adjusted to 1 mg/ml. Equal volumes of lysate were then precleared with 50 μL protein G beads with rabbit serum for 1 h at 4°C with gentle rocking. Lysates were incubated with 50 μl protein G beads and polyclonal rabbit anti-mouse ZO-1 (2.5 μg/mL) or rabbit anti-mouse PTP1B (2.5 μg/mL) overnight at 4°C with gentle rocking. The beads were washed 3 times with RIPA buffer and 1 time each with 0.5 M NaCl followed by 10 mM Tris, pH 7.4. Protein was eluted from the beads by heating for 5 min at 100°C in SDS sample buffer.
Western blotting
Cell lysates or immunoprecipitates in SDS sample buffer were analyzed by SDS-PAGE (7.5%) and transferred to PVDF membranes according to manufacturer’s instructions (Bio-Rad) (300 mA for 3h). Membranes were blocked in 5% non-fat dried milk or in 5% BSA for the anti-phosphoserine antibodies in Tris-buffered saline plus 0.1% Tween 20 (TBS-T) or for 1 h at RT. Primary antibodies rabbit anti-mouse ZO-1 (1:200), rabbit anti-mouse PTP1B (1:1000), rabbit anti-phospho PKCα Thr638, mouse anti-phosphotyrosine, or mouse anti-phosphoserine were incubated with the membranes in TBS-T plus 5% BSA overnight and then washed 3 times for 5 minutes in TBS-T. HRP-conjugated secondary antibodies (1:1000) were then incubated with the membrane for 1 h at RT and washed 3 times for 5 minutes in TBS-T. Protein expression was detected by enhanced chemiluminescence (ECL, Amersham) and autoradiography visualization using x-ray film.
Transient transfection with dominant negative PKCα (DN PKCα)
Transient transfection with DN PKCα was performed and did not affect cell viability as previously described [24, 25]. Briefly, cells grown on slides were transfected with 1 μg of dominant negative PKCα or the vector pCMV per well with 2 μl/well LipofectAMINE 2000 Reagent (Invitrogen, Carlsbad, CA) for 3.5 hours and then medium replaced.
Lymphocyte Migration assay
A parallel plate flow chamber was used to examine migration under conditions of laminar flow at 2 dynes/cm2 for 15 minutes as we described previously [24] Briefly, spleen cells were used as a source of cells contiguous with the blood stream that could then migrate across endothelial cells. Spleen cell migration across the mHEV cell lines is stimulated by mHEV cell constitutive production of the chemokine MCP-1 [54] and is dependent on adhesion to VCAM-1 [1]. We have previously reported that, after migration across the mHEV cells, the spleen cells are 65-70% B cells, 12-15% CD4+ cells and 5-8% CD8+ cells [27]. After 15 minutes at 2 dynes/cm2, the cells are washed at 2 dynes/cm2 with PBS supplemented with 0.2 mM CaCl2 and 0.1 mM MgCl2 since cations are required for cell adhesion. These cells were fixed with 3% paraformaldehyde for 1 h. To quantify migrated spleen cells, phase contrast microscopy was used to count migrated cells that are phase dark [55].
Permeability of Endothelial Monolayer
mHEV cells were grown to confluence in phenol red-free HEV medium in transwell chambers (8 μm pores, 48 well plates, catalog #3422, Corning Incorporated). Control transwells had no cells. The mHEV cells were stimulated with 27 μg/ml anti-VCAM-1 antibody or an isotype control antibody plus 15 μg/ml a secondary antibody and then, FITC-albumin (37 μg, catalog #A23015, Fisher Scientific) was added to the upper chamber. At the indicated time points, 15 μl of medium were collected from the bottom chamber of the transwells and fluorescence measured using a fluorescence plate reader and a FITC-albumin standard curve.
Localization of ZO-1
Endothelial cells grown to confluence on glass slides were nontreated or pretreated with 2.3 nM Gö-6976 for 30 minutes and washed 5 times. The monolayers were subjected to 2 dynes/cm2 laminar flow in the presence of either lymphocytes or 1 μM H2O2 for 15 minutes. The cells were washed with PBS-Ca-Mg, fixed for 15 minutes with cold methanol, rehydrated with PBS for 1 hour, and labeled with rabbit anti-ZO-1 and FITC-conjugated goat anti-rabbit Ig (mouse adsorbed), rat anti-mouse VCAM-1 and Texas Red-conjugated goat anti-rat Ig (mouse adsorbed), Zenon Alexa Fluor 568-labeled rat anti-mouse CD45, Alexa 594-conjugated donkey anti-rabbit N-Cadherin, or rabbit anti-angiomotin followed by texas red-conjugated secondary antibody. The cells were coverslipped using 1 μg DAPI/ml Vectashield mounting medium for fluorescence (cat#H1200, Vector Laboratories, Burlingame, CA). Images were collected using a Leica TCS4D confocal microscope (Heidelberg, Germany) equipped with an Omnichrome krypton-argon laser (Chino, CA) and INNOVA Interprise UV Ion Laser, Coherent Inc. (Santa Clara, CA). Total immunolabeled ZO-1 fluorescent intensity in the Z-stack of an 9 × 25 μm area behind the leukocyte was analyzed by Slidebook Software (Intelligent Imaging Innovations, Inc).
Over expression of ZO-1-GFP
Stable transfections of mHEV cells with enhanced green fluorescent protein (EGFP) vector pEGFP-C1 (BD Biosciences Clontech, Palo Alto, CA) or vector with ZO-1-EGFP (kind gift from Dr Alan Fanning, University of North Carolina) were generated. Briefly, mHEV cells were grown to 90% confluence in 6 well plates and transfected with 5 μg enhanced green fluorescent protein (EGFP) vector pEGFP-C1 (BD Biosciences Clontech, Palo Alto, CA) alone, or vector with ZO-1-EGFP (kind gift from Dr. Alan Fanning). Each of these transfections were performed in the presence of 7 or 15 μl/well LipofectAMINE 2000 Reagent (Invitrogen, Carlsbad, CA) according to instructions in culture medium without gentamicin, penicillin or streptomycin. After 6 hours, the medium was removed and replaced with medium containing antibiotics. At 24 hours, the cells were suspended by treatment with 0.03% EDTA in PBS, washed, and plated in 100 mm petri dishes. At 48 hours, the medium was removed and cells were treated with 0.3 μg geneticin/ml medium (Invitrogen, Carlsbad, CA), a concentration that is completely toxic to nontransfected mHEV cells in 72 hours. After grown to 75% confluence, the cells were suspended with 0.03% EDTA, washed and fluorescence examined by flow cytometry. Approximately 20% of these cells were EGFP+ as determined by flow cytometry. Therefore, the cells were subcloned twice at 1/3 of a cell/well to generate clones with homogeneous expression of EGFP+ and then subcloned once more to determine whether all subclones were EGFP+ as determined by flow cytometry (data not shown).
Statistics
Data were analyzed by a one way ANOVA followed by Tukey’s multiple comparisons test (SPSS, Jandel Scientific, San Ramon, CA).
RESULTS
OVA challenge increases allergic inflammation, reduces endothelial ZO-1 and increases endothelial VCAM-1, Angiomotin, and N-Cadherin in the lung
To examine leukocyte recruitment and adhesion molecules in allergic lung inflammation, we used the OVA mouse model because in this model, recruitment of eosinophil and lymphocytes is mediated by binding to VCAM-1 [1-10]. Endothelial cells express VCAM-1 on the cell surface and in cell junctions and express angiomotin and VE-cadherin in cell junctions; also, the junction molecule N-cadherin can be induced on endothelial cells [42-47, 49]. OVA-challenged mice had increased expression of VCAM-1, angiomotin and N-Cadherin by lung venule endothelial cells as compared to saline-challenged lungs (Figure 1A-C, F-I), whereas there was no change in expression of the VE-cadherin with OVA challenge (Figure 1D, J). In contrast, there was reduced expression of ZO-1 in venule endothelial cells as compared to saline-challenged lungs bg(Figure 1A-C, F), indicative of either a change in localization of ZO-1 or a change in total ZO-1; this is addressed in the next figures. In the bronchoalveolar lavage, numbers of eosinophils, lymphocytes, monocytes and neutrophils were increased (Figure 1E). In the lung tissue sections (Figure 1A-D), the increase in blue nuclei in OVA-challenged lungs is consistent with the increase in inflammatory cells.
Figure 1. Allergic lung inflammation induces a decrease in ZO-1 in lung endothelial cells and increased lung endothelial cell expression of VCAM-1, N-Cadherin and angiomotin.
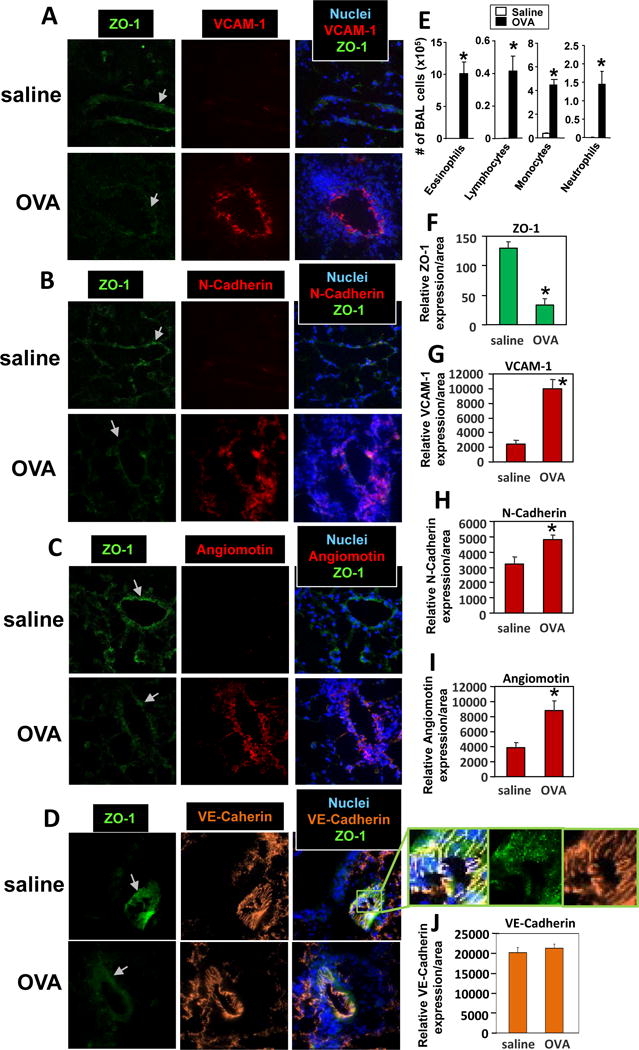
Mice were sensitized and challenged with OVA or saline and then lungs were lavaged and lungs in OCT were stored frozen at -80°C. A-D) Lung tissue sections were prepared and labeled with rat anti-mouse VCAM-1, rabbit anti-mouse N-Cadherin, rabbit anti-mouse angiomotin or rat-anti-mouse VE-Cadherin followed by Texas red-conjugated secondary antibodies. Then the sections were labeled with FITC-conjugated anti-mouse ZO-1. Representative wide-field fluorescence micrographs are shown. Panel D has an enlarged image depicting a slice of the vessel in the section that is at a slight oblique angle through the vessel because this nicely encompassed and thus depicted more of the cell junctions. The vascular structure in the lung tissue sections was confirmed by characteristic morphology of vessels in the lung tissue sections (data not shown). Arrow indicates venule. E) Lung lavage cells. F-J) Relative sum of fluorescence pixel intensities per area of the venule endothelial cells. Isotype control antibody-labeled cells were negative (data not shown). N=8-10 mice. Presented as mean ±SEM. *, p<0.05.
VCAM-1 signals induce serine phosphorylation of ZO-1 in HMEC-L cells
To further examine ZO-1 expression, we determined whether ZO-1 was regulated by ligand binding to VCAM-1 in primary human endothelial cells. Furthermore, since ZO-1 localization in cell junctions is regulated by phosphorylation [42-44], we also determined whether stimulation of VCAM-1 induced ZO-1 phosphorylation. Primary cultures of human microvascular endothelial cells from the lung (HMEC-L cells) were treated overnight with TNFα to induce adhesion molecule expression and washed [24, 25]. Then, VCAM-1 was stimulated by receptor crosslinking with anti-VCAM-1 and a secondary antibody as we previously reported [24, 25]. ZO-1 was immunoprecipitated and ZO-1 phosphorylation was examined by western blot. The molecular weight of ZO-1 is 220 kD. Total ZO-1 in the cells was not altered by VCAM-1 stimulation (Figure 2). Stimulation of VCAM-1 induced serine phosphorylation (Figure 2A,B,C,E) but not tyrosine phosphorylation (Figure 2D) of ZO-1 at 15 minutes, the optimal time point for ZO-1 phosphorylation (Figure 2A and Supplement Figure 1A). This is consistent with optimal VCAM-1 stimulation of PKCα activation at 10 minutes, VCAM-1 stimulation of PTP1B activation at 15 minutes, and leukocyte transendothelial migration across monolayers of endothelial cells at 15 minutes [24, 25].
Figure 2. VCAM-1 signals induce serine phosphorylation of ZO-1 in HMEC-L cells.
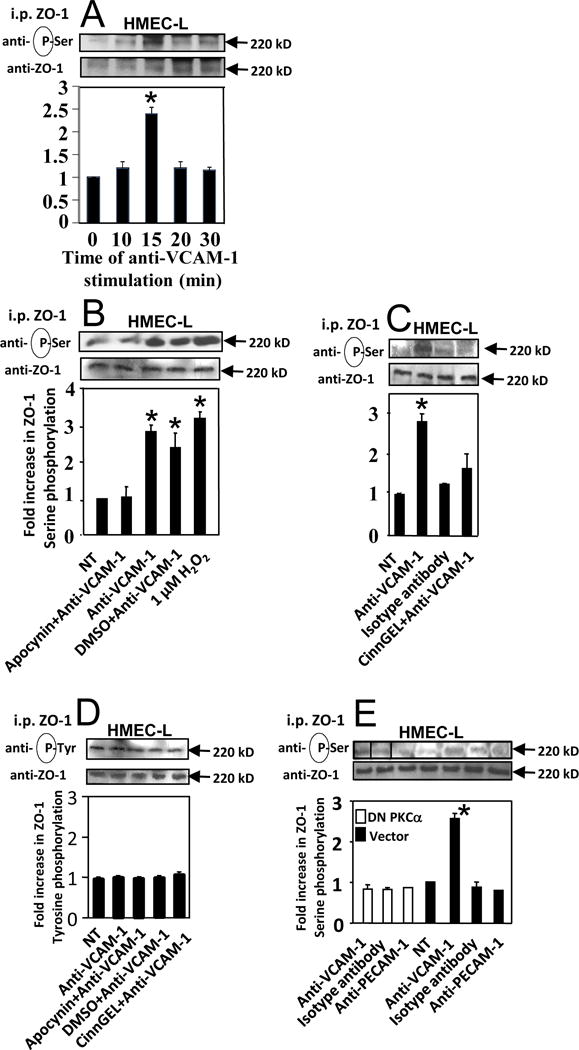
HMEC-L cells (passage 1-4) were stimulated overnight with TNFα. Then the cells were nontreated (NT) or pretreated for 30 minutes with 4mM apocynin (panel B,D), 10μM CinnGEL-2ME (panel C,D),or the vehicle control DMSO (0.1%). Alternatively, HMEC-L cells were transfected with dominant negative PKCα or control vector (panel E). Then, the confluent HMEC-L cells were stimulated with 1μM H2O2 or stimulated with 27 μg/ml anti-VCAM-1 antibody, a control anti-PECAM-1 antibody, or an isotype-matched antibody plus 15 μg/ml a secondary antibody. The optimal time for anti-VCAM-1 stimulation was 15 minutes (panel A and Supplement Figure 1A). ZO-1 was immunoprecipitated (i.p.) and examined by western blot with anti-phosphoserine, anti-phosphotyrosine or anti-ZO-1. The lines in the blots indicate where spaces between lanes were removed; the original micrograph of the blot is in Supplement Figure 1B. All lanes were from the same blot. Data are from 3 experiments and presented as mean ±SEM. *, p<0.05 compared to non-treated or DMSO controls.
It was determined whether inhibition of VCAM-1 signals through NOX2, PKCα and PTP1B blocked VCAM-1-stimulated serine phosphorylation of ZO-1. HMEC-L cells were nontreated or treated with dominant negative PKCα and then VCAM-1 expression was induced by TNFα [24, 25]; as we have reported [24, 25], transfection with dominant negative PKCα increases total PKCα and does not alter VCAM-1 expression. To inhibit PTP1B or NADPH oxidase, TNFα-treated HMEC-L cells were pre-incubated for 30 minutes with the PTP1B specific inhibitor CinnGEL (10μM) [56], the NADPH oxidase inhibitor apocynin (4 mM) or the vehicle control DMSO (0.1%) as we previously described [1, 24, 25]. Apocynin is an inhibitor of NADPH oxidase and xanthine oxidase but we have reported that VCAM-1 does not activate xanthine oxidase during leukocyte transendothelial migration [1]. The transfections and inhibitors did not affect cell viability in these studies, consistent with our previous reports [1, 24, 25]. Importantly, apocynin, CinnGel and dominant negative PKCα blocked VCAM-1-stimulated serine phosphorylation of ZO-1 in HMEC-L cells (Figure 2B,C,E and Supplement Figure 1B). As controls, antibody crosslinking of PECAM-1 on the HMEC-L cells or incubation with isotype control antibodies plus a secondary antibody did not activate serine phosphorylation of ZO-1 (Figure 2C,E), demonstrating specificity of the antibody crosslinking of VCAM-1 for induction of ZO-1 phosphorylation.
We determined whether bypassing the VCAM-1 receptor by exogenous addition of the VCAM-1 signaling intermediate, 1 μM H2O2 [11, 24, 25], stimulated serine phosphorylation of ZO-1. The 1 μM H2O2 did stimulate HMEC-L cell ZO-1 serine phosphorylation at 15 minutes (Figure 2A,B).
VCAM-1 signals induce serine phosphorylation of ZO-1 in constitutively activated endothelial cell lines
The endothelial cell line mHEV was used in these studies because they are constitutively activate with VCAM-1 expression and provide a system to examine the functional outcome of VCAM-1 signaling during leukocyte transendothelial migration [3, 23-25]. Before examining migration, we first examined regulation of ZO-1 by VCAM-1 signals in these cells. We have reported that VCAM-1 stimulation of NOX2, PKCα and PTP1B in the these cells [1, 51, 57] occurs with the same magnitude and time course as TNFα-activated primary cultures of HMEC-L cells. In Figure 3A and Supplement Figure 2A, stimulation of VCAM-1 with anti-VCAM-1 and a secondary antibody induced serine phosphorylation of ZO-1 at 15 minutes, the same time course as for HMEC-L cells (Figure 2). This ZO-1 phosphorylation was blocked by transfection with dominant negative PKCα or treatment with the PTP1B inhibitor CinnGel but not the vector control (Figure 3A and Supplement Figure 2A). Anti-VCAM-1 stimulation did not increase the mHEV cell monolayer permeability (Figure 3D). Exogenous 1 μM H2O2, also induced serine phosphorylation of ZO-1 in monolayers of mHEV cells at 15 minutes (Figure 3B,C and Supplement Figure 2B) and this was blocked by pretreatment of the cells with CinnGEL but not the DMSO solvent control (Figure 3C). In summary, VCAM-1 signals induce serine phosphorylation of ZO-1 in mouse endothelial cells lines and HMEC-L cells.
Figure 3. VCAM-1 signals induce serine phosphorylation of ZO-1 in endothelial cell lines and did not alter endothelial permeability.
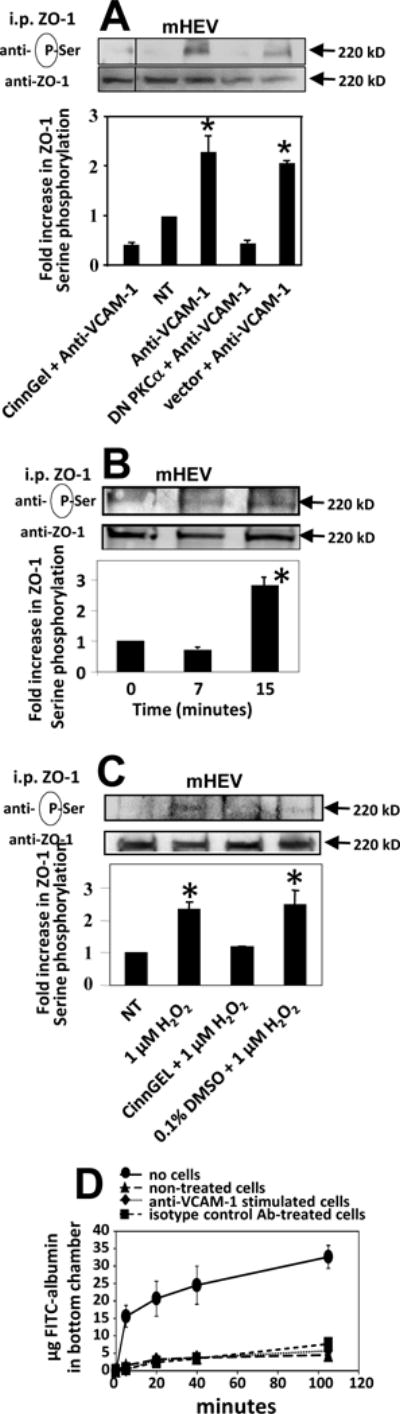
mHEV cells were nontreated (NT), pretreated with inhibitors as in Figure 2 and then stimulated with anti-VCAM-1 plus a secondary antibody for 15 minutes (panels A) as in Figure 2 or with 1μM H2O2 (panels B,C and Supplement Figure 2B). ZO-1 was immunoprecipitated and examined by western blot. The lines in the blots indicate where intervening lanes were removed; the original micrograph is in Supplement Figure 2A. All lanes were from the same blot. In panel D, confluent monolayers of mHEV cells on transwells were stimulated with anti-VCAM-1 or isotype antibody control plus a secondary antibody. FITC-albumin was added to the upper chamber of transwells with mHEV cells or transwells without cells. Medium (25 μl) was collected from the bottom chamber at the indicated time points and the fluorescence measured with a fluorescent plate reader. Data are from 3 experiments and presented as mean ±SEM. *, p<0.05 compared to non-treated, DMSO or vector controls.
Localization of ZO-1 in junctions in endothelial cells is reduced in vitro by VCAM-1 activation of PKCα or the VCAM-1 signaling intermediate 1 μM H2O2
Because allergic lung inflammation induced VCAM-1, angiomotin, and N-Cadherin and decreased ZO-1 in endothelial cells in vivo, it was determined whether stimulation with the VCAM-1 signaling intermediate 1 μM H2O2 [11] alters localization of N-Cadherin, angiomotin, VCAM-1 or ZO-1 in endothelial cell junctions. The mHEV cells expressed ZO-1, VCAM-1, N-Cadherin and angiomotin as compared to the isotype control antibody labeled cells (Figure 4D-G). H2O2 (1 μM) reduced ZO-1 in junctions (Figure 4). Importantly, there was no change in total ZO-1 as determined by western blot (Figure 3B), indicating a change in localization of ZO-1 in the cells rather than loss of ZO-1 in the cells. Briefly, when concentrated junctional ZO-1 dissociates from the junctions and is diluted as it enters the large volume of the cytoplasm, there is a reduction in fluorescent signal from ZO-1 immunolabeling/area. In contrast to effects on ZO-1, H2O2 (1 μM) did not alter localization of VCAM-1, N-Cadherin or angiomotin in endothelial cells (Figure 4).
Figure 4. The VCAM-1-signaling intermediate H2O2 stimulates dissociation of ZO-1 from endothelial cell junctions without altering expression of VCAM-1, N-Cadherin or angiomotin.
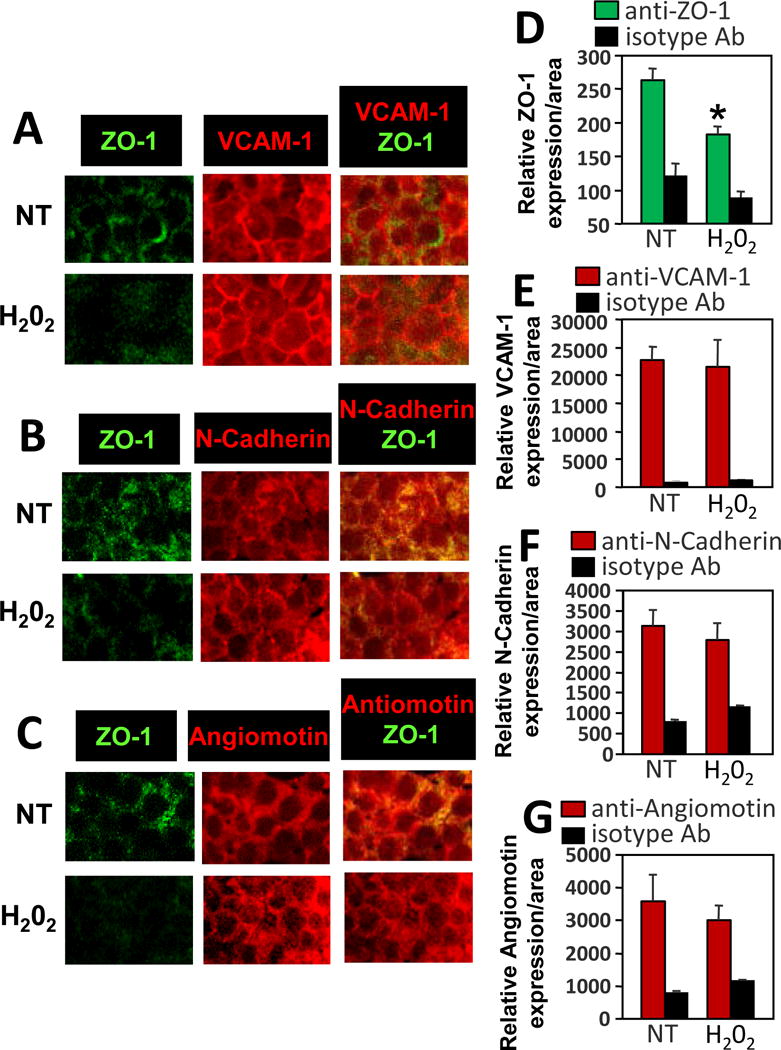
mHEV cells were nontreated (NT) or stimulated with 1μM H2O2 for 15 minutes, fixed and immunolabled for VCAM-1, N-Cadherin or angiomotin or incubated with isotype control antibodies and secondary Texas red-conjugated secondary antibodies. Then the cells were immunolabeled with FITC-conjugated anti-ZO-1 as in Figure 2. Representative wide-field fluorescence micrographs of 3 experiments are shown. ZO-1 is located in cell junctions and VCAM-1 is expressed in cell junctions and on the apical cell surface. DAPI is not included here so that VCAM apical surface expression is not overwhelmed by DAPI. DAPI is included in micrographs in Figure 5. Relative sum of fluorescence pixel intensities per area of the monolayer of endothelial cells were quantified for VCAM-1 or ZO-1 and are shown in the graphs. Data are from 3 experiments and are presented as mean ±SEM. *, p<0.05 compared to NT.
VCAM-1 induction of 1μM H2O2 activates PKCα. Therefore the mHEV cells were stimulated with 1μM H2O2 in the presence and absence of the PKCα inhibitor Gö-6976. The Gö-6976 blocked the H2O2-induced loss of ZO-1 in endothelial cell junctions (Figure 5). This is consistent with the PKCα-dependence of VCAM-1-stimulated ZO-1 phosphorylation shown in Figures 2 and 3.
Figure 5. The VCAM-1-signaling intermediate H2O2 stimulates dissociation of ZO-1 from endothelial cell junctions and this is blocked by inhibition of PKCα.
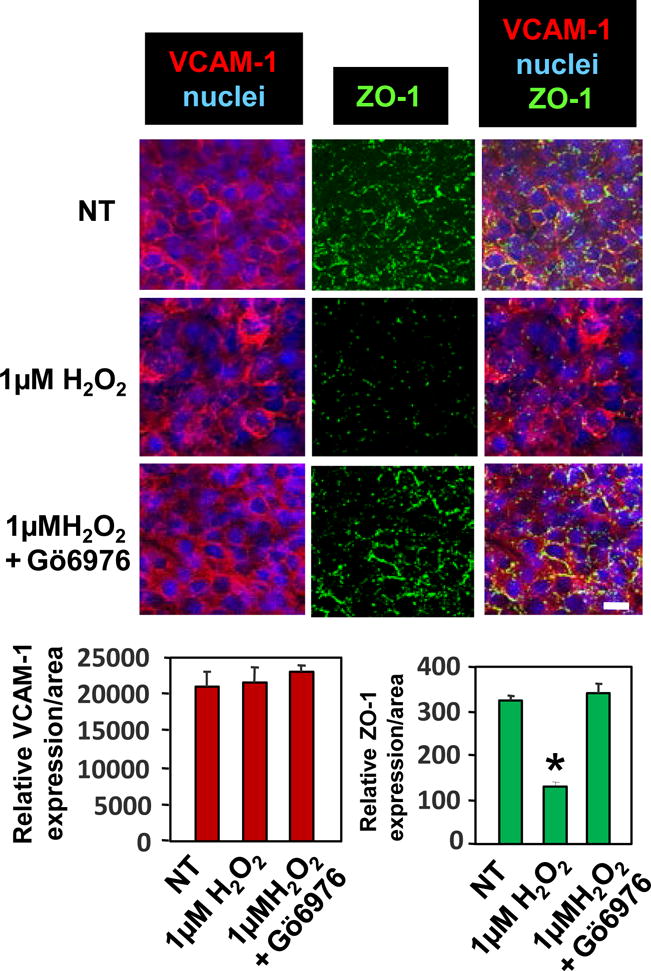
mHEV cells were nontreated (NT) or pretreated with the PKCα inhibitor Gö6976 (2.3 nM) for 30 minutes and washed. The cells were stimulated with 1μM H2O2 for 15 minutes, fixed and immunolabled for ZO-1 and VCAM-1, and then nuclei were labeled with 1 μg/ml DAPI. Representative wide-field fluorescence micrographs of 3 separate experiments are shown. ZO-1 is located in cell junctions and VCAM-1 is expressed in cell junctions and on the apical cell surface. Bar, 10 μm. Relative sum of fluorescence pixel intensities for immunolabeling for VCAM-1 or ZO-1 were quantified and shown in the graphs. Data are presented as mean ±SEM. *, p<0.05 compared to NT.
Lymphocyte Binding stimulates dissociation of ZO-1 from endothelial cell junctions
It was determined whether VCAM-1-dependent lymphocyte migration [1, 27, 58] stimulated the dissociation of ZO-1 from endothelial cell junctions. We have reported that lymphocyte migration across these endothelial cell lines requires adhesion to VCAM-1 and not other adhesion molecules [1, 51, 52, 57]. These endothelial cells also constitutively produce the chemokine MCP-1 [54] which induces a high affinity state in the lymphocyte integrins that then bind to VCAM-1. mHEV cells were either non-treated or pretreated with irreversible inhibitors of NADPH oxidase (4 mM apocynin), PKCα (2.3 nM Gö-6976), or PTP1B (10 μM CinnGEL-2ME) and then washed as we previously described [1, 24, 25]. The cells were placed into a parallel plate flow chamber and lymphocytes were added for 15 minutes under laminar flow at 2 dynes/cm2 because this is the flow rate in postcapillary venules in vivo where leukocytes migrate into inflammatory sites [24, 25]. After 15 minutes, the cells were fixed and immunolabeled for ZO-1 and lymphocyte CD45 and then examined by confocal microscopy. The diagram in Figure 6B indicates the location of the three-dimensional imaging at the apical surface of the endothelial cells. In the paths where the bound lymphocytes had rolled (Figure 6A column a, dashed boxes in the XY on-face view of the three dimensional image, and Figure 6C, arrow in diagram of area analyzed), there was a significant loss of ZO-1 from untreated or DMSO solvent-treated endothelial cells (Figure 6D). The loss of ZO-1 is in the direction of leukocyte rolling as indicated with an arrow (Figure 6A lower left micrograph). In the representative Z-slice vertically through the cells in Figure 6A column a, the lymphocyte is located between the DAPI (blue)-labeled nuclei of the endothelial cells, indicating that the lymphocyte is between the endothelial cells in the process of migration. The other cells from this XY image and in 4 experiments were also migrating between the endothelial cells (data not shown). In contrast, in the paths where the bound lymphocytes had rolled on apocynin-pretreated, CinnGEL-pretreated or Gö-6976 -pretreated endothelial cells (Figure 6A columns b,c,d of the XY on-face view of the three dimensional images), there was no loss of ZO-1 from the endothelial junctions and the lymphocytes had not migrated into the junctions (Figure 6D). Briefly, in the Z-slices in columns b,c,d of Figure 6A, the lymphocytes are located above ZO-1 (green) and the nuclei (blue) of the inhibitor-pretreated endothelial cells, suggesting that the lymphocytes bound to the endothelial cell but did not migrate between the endothelial cells. This is consistent for 2-4 lymphocytes/image and 4 experiments for each treatment. The total amount of ZO-1 in Figure 6A column a is less than Figure 6A columns b-d in all experiments; this may be because lymphocytes that had rolled across VCAM-1 on the endothelial cells and then rolled off the monolayer during the 15 minutes would have induced some VCAM-1 signals for changes in ZO-1 localization despite rolling off the monolayer. In summary, lymphocyte binding to VCAM-1 [1, 27, 58] stimulates VCAM-1 signals that induce ZO-1 phosphorylation and dissociation of ZO-1 from junctions. Thus, inhibition of these VCAM-1 signals blocked ZO-1 changes and blocked leukocyte transendothelial migration (Figure 6A).
Figure 6. Lymphocyte migration stimulates dissociation of ZO-1 from endothelial cell junctions.
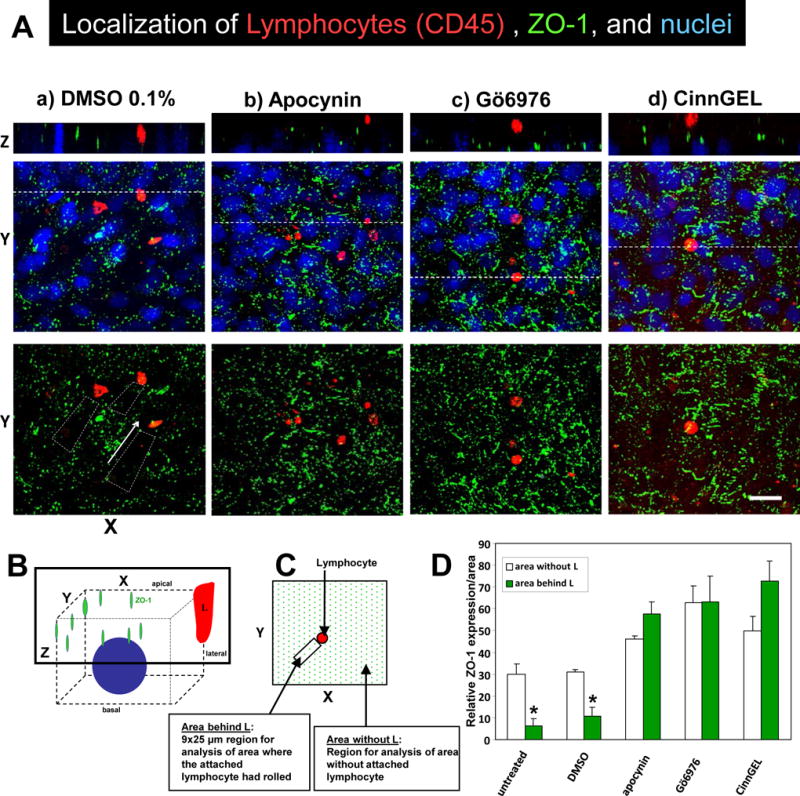
A) Confluent monolayers of mHEV cells were non-treated or pretreated for 30 minutes with 0.1% DMSO vehicle control, 4 mM apocynin, 2.3 nM Gö6976 or 10μM CinnGEL-2ME. The monolayers were washed 5 times, placed in a parallel plate flow chamber and then lymphocytes were added for 15 minutes under 2 dynes/cm2 laminar flow. The monolayers were fixed and immunolabeled for ZO-1 (green) and CD45 (red). The nuclei (blue) are labeled with 1 μg/ml DAPI. The fluorescence was examined by confocal microscopy and representative micrographs of 3 experiments are shown. Shown are XY on-face of 3D images or a Z-stack of the images at the location of the dotted line in the panel below it. Arrows indicate the direction of laminar flow. Bar, 20 μm. B) Diagram of 3D confocal analysis. C) Diagram of 9×25 μm XY regions of the Z stack behind the lymphocyte that was analyzed for intensity of labeling of ZO-1 in the Z stack. D) Relative intensity of ZO-1 labeling/area in the 9×25 μm XY regions of a Z stack as described in panel C. Data are from 3 experiments and presented as mean ±SEM. *, p<0.05.
Overexpression of ZO-1 increases ZO-1 in cell junctions and blocks lymphocyte migration
Inhibition of VCAM-1 signals blocked ZO-1 phosphorylation and ZO-1 dissociation from cell junctions (Figures 2,3,5,6) and inhibition of VCAM-1 signals blocks VCAM-1-dependent leukocyte transendothelial migration (Figure 6A) [1, 24, 25], suggesting that ZO-1 phosphorylation and dissociation from junctions is critical for leukocyte transendothelial migration. So, we determined whether overexpression of ZO-1 blocked VCAM-1-dependent lymphocyte transendothelial migration across monolayers of mHEV cells under physiological laminar flow at 2 dynes/cm2, the rate of flow at postcapillary venules. Stable transfection of ZO-1-EGFP by the endothelial cells increased mHEV cell expression of ZO-1 as determined by western blot (Figure 7A), flow cytometry for EGFP (Figure 7B), and fluorescence microscopy (Figure 7C). ZO-1 overexpression did not alter VCAM-1 expression (Figure 7B). The overexpression of ZO-1 significantly blocked VCAM-1-dependent lymphocyte transendothelial migration (Figure 7D). The vector did not affect VCAM-1-dependent migration (Figure 7D).
Figure 7. Overexpression of ZO-1 blocks VCAM-1-dependent leukocyte transendothelial migration.
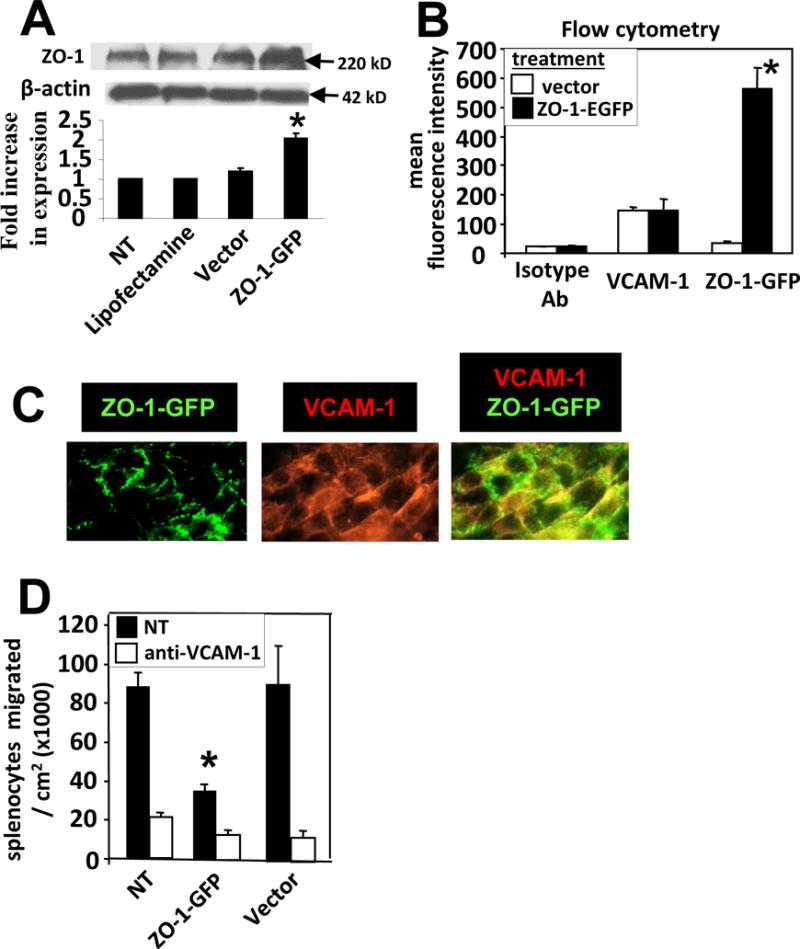
mHEV cells were stably transfected with ZO-1-EGFP. A) ZO-1 examined by western blot for non-treated cells (NT), lipofectamine-treated cells, vector-treated cells and ZO-1-EGFP transfected cells. ZO-1 is 220 kD and ZO-1-EGFP is about 247 kD. Therefore, this results in a wider band in the western blot for the cells expressing ZO-1-EGFP. B) mHEVa cells, transfected with vector or ZO-1-EGFP, were Immunolabeled with anti-VCAM-1 or isotype control antibodies (Ab), or examined for ZO-1-EGFP expression by flow cytometry. C) Representative micrographs of ZO-1-EGFP transfected mHEV cells and co-localization (yellow) in junctions labeled with rat anti-mouse VCAM-1 and a secondary donkey anti-rat IgG Texas Red. N=3 experiments. D) Spleen leukocyte transendothelial migration at 15 minutes in a parallel plate flow chamber at 2 dynes/cm2. *, p<0.05 compared to controls. Data are from 3 experiments and are presented as mean ±SEM.
DISCUSSION
We demonstrated that lungs with allergic inflammation had increased endothelial cell expression of VCAM-1, N-Cadherin and angiomotin in endothelial cells and had decreased detection of ZO-1 in endothelial cell junctions. VCAM-1 activated serine phosphorylation of ZO-1 and induced a reduction of ZO-1 at endothelial cell junctions but not alter total levels of ZO-1 in the cells. This VCAM-1 activation of ZO-1 phosphorylation and reduction in ZO-1 localization at cell junctions was induced through VCAM-1 intracellular signals, including NOX2, PKCα, PTP1B, and 1 μM H2O2. Overexpression of ZO-1 increased ZO-1 in cell junctions and blocked leukocyte transendothelial migration. Thus, VCAM-1 signaling in endothelial cells during recruitment of leukocytes induces changes in endothelial cell junctions for leukocyte transendothelial migration.
These studies are the first to report that with allergic inflammation, there is increased N-Cadherin and angiomotin and decreased ZO-1. ZO-1 was not contiguous in the endothelial cell junctions which is consistent with previous reports that ZO-1 is not contiguous in junctions of endothelial cells [59, 60]. An increase in expression of N-Cadherin in lung endothelial cells has also been reported to increase with neutophilic inflammation induced by administration of endotoxin [61, 62], but the mechanism was not defined. The increase in angiomotin in mouse lungs with allergic inflammation is consistent with studies in humans which demonstrated that angiomotin is expressed by lung endothelial cells in patients with asthma [48]. Moreover, angiomotin has been reported to localize with ZO-1, recruits ZO-1 to junctions in CHO cells and can be localized with cadherins in cell junctions [45-47]. Whether angiomotin mediates recovery of de-phosporylated ZO-1 back to junctions after leukocyte transendothelial migration will be addressed in future studies.
ZO-1 associates with multiple junction proteins in adherent cells [33-42, 45] and is regulated by ZO-1 phosphorylation [31, 38, 63]. In rabbit nasal epithelium, PKC-dependent serine phosphorylation of ZO-1 is induced by poly-L-arginine [42]. In okadaic acid-treated epithelial cells, serine or threonine phosphorylation but not tyrosine phosphorylation of ZO-1 is important for the release of ZO-1 from cell junctions [64, 65]. We demonstrated here that VCAM-1 signaling stimulated serine phosphorylation but not tyrosine phosphorylation of ZO-1 in endothelial cells and reduced ZO-1 in cell junctions.
Previous reports on H2O2 modulation of junctions focus on high levels of H2O2 (20-1000 μM), whereas VCAM-1 stimulates production of only 1 μM H2O2. High toxic concentrations of H2O2 (>200 μM H2O2) induce an increase in phosphorylation of ZO-1 and loss of ZO-1 from junctions [43], but it was not known whether ZO-1 is modulated during VCAM-1 signaling. Moreover, since we have reported that >20 μM H2O2 and 1 μM H2O2 have opposing effects on the cell signals PKCα, PTP1B and MMPs [23-25, 28, 29], the reports on mechanisms for the modulation of cells junctions by >20 μM H2O2 would not be consistent with what occurs for VCAM-1 signaling during transendothelial migration. We demonstrate that VCAM-1 signals through 1 μM H2O2 induce phosphorylation of ZO-1. Consistent with VCAM-1 induction of ZO-1 serine phosphorylation, exogenous addition of 1 μM H2O2, which corresponds to the levels of H2O2 produced from NOX2-generated superoxide during VCAM-1 signaling [1, 27, 51], was sufficient to induce serine phosphorylation of ZO-1 and dissociation of ZO-1 from endothelial cell junctions. The time course for induction of ZO-1 serine phosphorylation was at 15 minutes for activation by either VCAM-1 or 1 μM H2O2.
There are differences in ZO-1 regulation by cell type and stimulant. In addition to serine phosphorylation of ZO-1, ZO-1 can be tyrosine phosphorylated in epithelial cells. Tyrosine phosphorylation of ZO-1 correlates with a decrease in transmembrane electrical resistance (TER) in hepatocyte growth factor-stimulated epithelial cells [66], correlates with a decrease in TER in oxidatively stressed Caco-2 cells [67], and correlates with increased permeability after intra-ocular injection of VEGF [44]. Conversely, after ZO-1 associates with the epidermal growth factor receptor, there is an increase in ZO-1 tyrosine phosphorylation that induces organization of junctions in A431 cells and primary colorectal cancer cells [68, 69]. We have shown that VCAM-1 signaling increases serine phosphorylation of PTP1B but does not alter tyrosine phosphorylation of ZO-1.
A model for VCAM-1 signals that function during leukocyte transendothelial migration is as follows (Figure 8): VCAM-1 activates a calcium release, calcium channels, and Rac-1 [2, 22]. VCAM-1 activation of Rac-1 induces increases in endothelial cell intercellular gaps and leukocyte transendothelial migration [2, 22]. This activation of Rac-1 stimulates NOX2 that generates low levels of superoxide, resulting in 1 μM H2O2 [1, 22]. This H2O2 oxidizes a cysteine in the pro-domain of endothelial cell-associated MMPs which induces release of the pro-domain and activation of the MMPs [23]. The H2O2 also diffuses through membranes at 100 μm/sec [70] and oxidizes cysteines in the regulatory domain of PKCα [71] but does not oxidize the cysteines in the catalytic site of PTP1B [25], indicating compartmentalization of intracellular targets for oxidation. The oxidized PKCα undergoes autophosphorylation and activation. PKCα then stimulates serine phosphorylation and activation of PTP1B [25]. Thus, MMPs and PKCα are two independent targets of oxidation during VCAM-1 signaling. In this report, the inhibition of PKCα or PTP1B during VCAM-1 signaling blocked VCAM-1-stimulated ZO-1 phosphorylation, suggesting that the intracellular ZO-1 phosphorylation is dependent on intracellular PKCα and PTP1B and that MMP activity is not necessary for the regulation of ZO-1 phosphorylation. We also report here that these signals through PTP1B indirectly induce serine but not tyrosine phosphorylation of ZO-1. Kinases for this serine phosphorylation of ZO-1 are under investigation. Thus, VCAM-1-stimulated serine phosphorylation of ZO-1 induces ZO-1 dissociation from endothelial cell junctions without altering localization of VCAM-1, N-Cadherin or angiomotin. Phosphorylation of ZO-1 results in loss of junction affinity [32, 72] and allows the passage of leukocytes during transendothelial migration.
Figure 8. Model of VCAM-1 signaling.
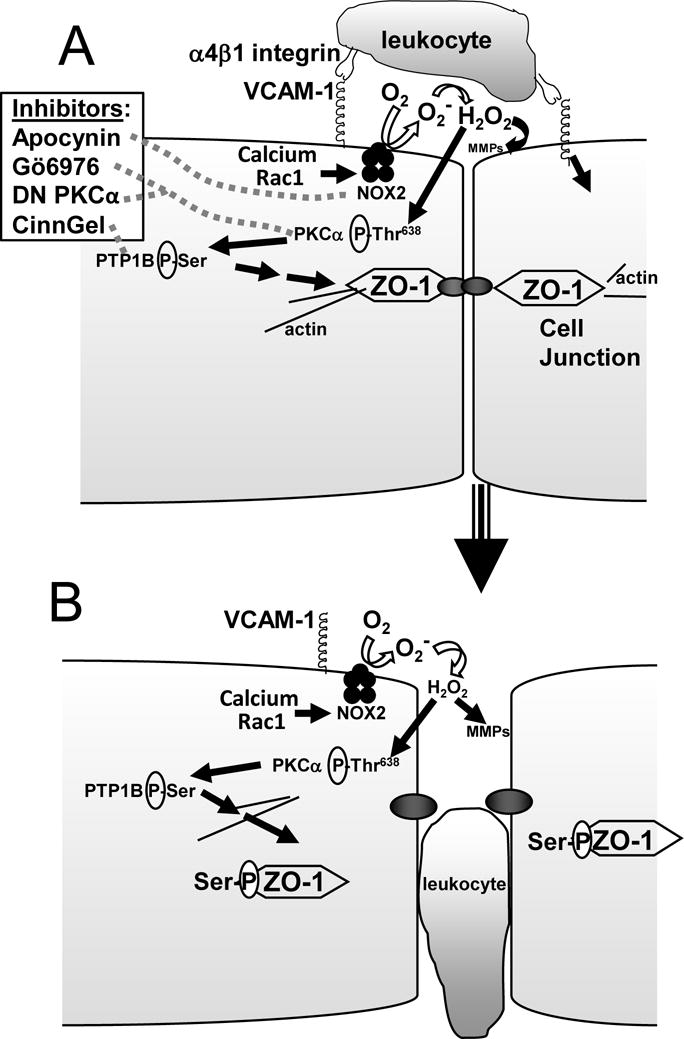
A) Leukocyte binding to VCAM-1 activates intracellular signaling in endothelial cells. The inhibitors of Nox2, PKCα, and PTP1B that are used in Figures 1-4 are indicated with dotted grey lines. B) Model for VCAM-1-induced changes in endothelial cell junction proteins for the passage of leukocytes.
In summary, we demonstrate that during allergic inflammation, lung endothelial cells alter ZO-1 localization and increase expression of N-Cadherin, angiomotin, and VCAM-1. Furthermore, VCAM-1 signal transduction induces serine phosphorylation of ZO-1 and reduces ZO-1 localization at the site of leukocyte-endothelial cell interaction.
Supplementary Material
Acknowledgments
NIH RO1 HL69428, NIH R01 AT004837, NIH R01HL111624, R01AI127695, and American Heart Association 09GRNT2260905 (J.M.C-M) supported this study.
Footnotes
Authorship Contributions.
Joan M. Cook-Mills conceived of the study design and participated in performing experiments, statistical analysis, interpretations, and manuscript preparation. Hiam Abdala-Valencia, Timothy S. Kountz and Michelle E. Marchese participated in performing and interpreting experiments.
Conflict of Interest Disclosures.
The authors have no conflicts of interest.
References
- 1.Matheny HE, Deem TL, Cook-Mills JM. Lymphocyte Migration through Monolayers of Endothelial Cell Lines involves VCAM-1 Signaling via Endothelial Cell NADPH Oxidase. J Immunol. 2000;164:6550–6559. doi: 10.4049/jimmunol.164.12.6550. [DOI] [PubMed] [Google Scholar]
- 2.van Wetering S, van den Berk N, van Buul JD, Mul FPJ, Lommerse I, Mous R, ten Klooster JP, Zwaginga JJ, Hordijk PL. VCAM-1-mediated Rac signaling controls endothelial cell-cell contacts and leukocyte transmigration. American journal of physiology Cell Physiol. 2003;285:C343–C352. doi: 10.1152/ajpcell.00048.2003. [DOI] [PubMed] [Google Scholar]
- 3.Chin JE, Hatfield CA, Winterrowd GE, Brashler JR, Vonderfecht SL, Fidler SF, Griffin RL, Kolbasa KP, Krzesicki RF, Sly LM, Staite ND, Richards IM. Airway recruitment of leukocytes in mice is dependent on alpha4-integrins and vascular cell adhesion molecule-1. Am J Physiol. 1997;272:L219–L229. doi: 10.1152/ajplung.1997.272.2.L219. [DOI] [PubMed] [Google Scholar]
- 4.Hakugawa J, Bae SJ, Tanaka Y, Katayama I. The inhibitory effect of anti-adhesion molecule antibodies on eosinophil infiltration in cutaneous late phase response in Balb/c mice sensitized with ovalbumin (OVA) J Derm. 1997;24:73–79. doi: 10.1111/j.1346-8138.1997.tb02747.x. [DOI] [PubMed] [Google Scholar]
- 5.Sagara H, Matsuda H, Wada N, Yagita H, Fukuda T, Okumura K, Makino S, Ra C. A monoclonal antibody against very late activation antigen-4 inhibits eosinophil accumulation and late asthmatic response in a guinea pig model of asthma. International Archives Allergy Immunol. 1997;112:287–294. doi: 10.1159/000237468. [DOI] [PubMed] [Google Scholar]
- 6.Boyce JA, Mellor EA, Perkins B, Lim YC, Luscinskas FW. Human mast cell progenitors use alpha4-integrin, VCAM-1, and PSGL-1 E-selectin for adhesive interactions with human vascular endothelium under flow conditions. Blood. 2002;99:2890–6. doi: 10.1182/blood.v99.8.2890. [DOI] [PubMed] [Google Scholar]
- 7.Abonia JP, Austen KF, Rollins BJ, Joshi SK, Flavell RA, Kuziel WA, Koni PA, Gurish MF. Constitutive homing of mast cell progenitors to the intestine depends on autologous expression of the chemokine receptor CXCR2. Blood. 2005;105:4308–13. doi: 10.1182/blood-2004-09-3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abonia JP, Hallgren J, Jones T, Shi T, Xu Y, Koni P, Flavell RA, Boyce JA, Austen KF, Gurish MF. Alpha-4 integrins and VCAM-1, but not MAdCAM-1, are essential for recruitment of mast cell progenitors to the inflamed lung. Blood. 2006;108:1588–94. doi: 10.1182/blood-2005-12-012781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hallgren J, Jones TG, Abonia JP, Xing W, Humbles A, Austen KF, Gurish MF. Pulmonary CXCR2 regulates VCAM-1 and antigen-induced recruitment of mast cell progenitors. Proc Natl Acad Sci U S A. 2007;104:20478–83. doi: 10.1073/pnas.0709651104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alcaide P, Jones TG, Lord GM, Glimcher LH, Hallgren J, Arinobu Y, Akashi K, Paterson AM, Gurish MA, Luscinskas FW. Dendritic cell expression of the transcription factor T-bet regulates mast cell progenitor homing to mucosal tissue. J Exp Med. 2007;204:431–9. doi: 10.1084/jem.20060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook-Mills JM, Marchese ME, Abdala-Valencia H. Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants. Antioxid Redox Signal. 2011;15:1607–1638. doi: 10.1089/ars.2010.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Lin SX, Amin S, Overbergh L, Maggiolino G, Chan LS. VCAM-1 blockade delays disease onset, reduces disease severity and inflammatory cells in an atopic dermatitis model. Immunol Cell Biol. 2010;88:334–42. doi: 10.1038/icb.2009.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soriano A, Salas A, Salas A, Sans M, Gironella M, Elena M, Anderson DC, Pique JM, Panes J. VCAM-1, but not ICAM-1 or MAdCAM-1, immunoblockade ameliorates DSS-induced colitis in mice. Lab Invest. 2000;80:1541–1551. doi: 10.1038/labinvest.3780164. [DOI] [PubMed] [Google Scholar]
- 14.Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res. 2000;87:153–9. doi: 10.1161/01.res.87.2.153. [DOI] [PubMed] [Google Scholar]
- 15.Ou R, Zhang M, Huang L, Flavell RA, Koni PA, Moskophidis D. Regulation of immune response and inflammatory reactions against viral infection by VCAM-1. J Virol. 2008;82:2952–65. doi: 10.1128/JVI.02191-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scherbarth S, Orr FW. Intravital videomicroscopic evidence for regulation of metastasis by the hepatic microvasculature: effects of interleukin-1alpha on metastasis and the location of B16F1 melanoma cell arrest. Cancer Res. 1997;57:4105–4110. [PubMed] [Google Scholar]
- 17.Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, Fuentes AM, Anasagasti MJ, Martin J, Carrascal T, Walsh P, Reznikov LL, Kim SH, Novick D, Rubinstein M, Dinarello CA. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci USA. 2000;97:734–739. doi: 10.1073/pnas.97.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kavanagh DP, Durant LE, Crosby HA, Lalor PF, Frampton J, Adams DH, Kalia N. Haematopoietic stem cell recruitment to injured murine liver sinusoids depends on (alpha)4(beta)1 integrin/VCAM-1 interactions. Gut. 2010;59:79–87. doi: 10.1136/gut.2008.168054. [DOI] [PubMed] [Google Scholar]
- 19.Berdnikovs S, Abdala-Valencia H, Cook-Mills JM. Endothelial cell PTP1B regulates leukocyte recruitment during allergic inflammation. Am J Physiol Lung Cell Mol Physiol. 2013;304:L240–9. doi: 10.1152/ajplung.00375.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdala-Valencia H, Earwood J, Bansal S, Jansen M, Babcock G, Garvy B, Wills-Karp M, Cook-Mills JM. Nonhematopoietic NADPH oxidase regulation of lung eosinophilia and airway hyperresponsiveness in experimentally induced asthma. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1111–25. doi: 10.1152/ajplung.00208.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cook-Mills JM, Deem TL. Active Endothelial Cell Function During Inflammation. J Leukocyte Biol. 2005;77:487–495. doi: 10.1189/jlb.0904554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook-Mills JM, Johnson JD, Deem TL, Ochi A, Wang L, Zheng Y. Calcium mobilization and Rac1 activation are required for VCAM-1 (vascular cell adhesion molecule-1) stimulation of NADPH oxidase activity. Biochem J. 2004;378:539–547. doi: 10.1042/BJ20030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deem TL, Cook-Mills JM. Vascular Cell Adhesion Molecule-1 (VCAM-1) Activation of Endothelial Cell Matrix Metalloproteinases: Role of Reactive Oxygen Species. Blood. 2004;104:2385–2393. doi: 10.1182/blood-2004-02-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdala-Valencia H, Cook-Mills JM. VCAM-1 Signals Activate Endothelial Cell Protein Kinase Cα Via Oxidation. J Immunol. 2006;177:6379–6387. doi: 10.4049/jimmunol.177.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deem TL, Abdala-Valencia H, Cook-Mills JM. VCAM-1 Activation of PTP1B in Endothelial Cells. J Immunol. 2007;178:3865–3873. doi: 10.4049/jimmunol.178.6.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keshavan P, Deem TL, Schwemberger SJ, Babcock GF, Cook-Mills JM, Zucker SD. Unconjugated bilirubin inhibits VCAM-1-mediated transendothelial leukocyte migration. J Immunol. 2005;174:3709–3718. doi: 10.4049/jimmunol.174.6.3709. [DOI] [PubMed] [Google Scholar]
- 27.Tudor KSRS, Hess KL, Cook-Mills JM. Cytokines Modulate Endothelial Cell Intracellular Signal Transduction Required for VCAM-1-dependent Lymphocyte Transendothelial Migration. Cytokine. 2001;15:196–211. doi: 10.1006/cyto.2001.0922. [DOI] [PubMed] [Google Scholar]
- 28.Fialkow L, Chan CK, Downey GP. Inhibition of CD45 during neutrophil activation. J Immunol. 1997;158:5409–5417. [PubMed] [Google Scholar]
- 29.Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest. 1996;98:2572–2579. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furuse M, Furuse K, Sasaki H, Tsukita S. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol. 2001;153:263–272. doi: 10.1083/jcb.153.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luscinskas FW, Ma S, Nusrat A, Parkos CA, Shaw SK. The role of endothelial cell lateral junctions during leukocyte trafficking. Immunol Rev. 2002;186:57–67. doi: 10.1034/j.1600-065x.2002.18606.x. [DOI] [PubMed] [Google Scholar]
- 32.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–9. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsukita S, Katsuno T, Yamazaki Y, Umeda K, Tamura A, Tsukita S. Roles of ZO-1 and ZO-2 in establishment of the belt-like adherens and tight junctions with paracellular permselective barrier function. Ann N Y Acad Sci. 2009;1165:44–52. doi: 10.1111/j.1749-6632.2009.04056.x. [DOI] [PubMed] [Google Scholar]
- 34.Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, Schwartz MA, Matter K, Balda MS. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol. 2015;208:821–38. doi: 10.1083/jcb.201404140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zemljic-Harpf AE, Godoy JC, Platoshyn O, Asfaw EK, Busija AR, Domenighetti AA, Ross RS. Vinculin directly binds zonula occludens-1 and is essential for stabilizing connexin-43-containing gap junctions in cardiac myocytes. J Cell Sci. 2014;127:1104–16. doi: 10.1242/jcs.143743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Itoh M, Sasaki H, Furuse M, Ozaki H, Kita T, Tsukita S. Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J Cell Biol. 2001;154:491–497. doi: 10.1083/jcb.200103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ebnet K, Schulz CU, Meyer Zu Brickwedde MK, Pendl GG, Vestweber D. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem. 2000;275:27979–27988. doi: 10.1074/jbc.M002363200. [DOI] [PubMed] [Google Scholar]
- 38.Muller SL, Portwich M, Schmidt A, Utepbergenov DI, Huber O, Blasig IE, Krause G. The tight junction protein occludin and the adherens junction protein alpha-catenin share a common interaction mechanism with ZO-1. J Biol Chem. 2005;280:3747–56. doi: 10.1074/jbc.M411365200. [DOI] [PubMed] [Google Scholar]
- 39.Utepbergenov DI, Fanning AS, Anderson JM. Dimerization of the scaffolding protein ZO-1 through the second PDZ domain. J Biol Chem. 2006;281:24671–7. doi: 10.1074/jbc.M512820200. [DOI] [PubMed] [Google Scholar]
- 40.Nomme J, Fanning AS, Caffrey M, Lye MF, Anderson JM, Lavie A. The Src Homology 3 Domain Is Required for Junctional Adhesion Molecule Binding to the Third PDZ Domain of the Scaffolding Protein ZO-1. J Biol Chem. 2011;286:43352–60. doi: 10.1074/jbc.M111.304089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim E, Lee Y, Kim JS, Song BS, Kim SU, Huh JW, Lee SR, Kim SH, Hong Y, Chang KT. Extracellular domain of V-set and immunoglobulin domain containing 1 (VSIG1) interacts with sertoli cell membrane protein, while its PDZ-binding motif forms a complex with ZO-1. Mol Cells. 2010;30:443–8. doi: 10.1007/s10059-010-0138-4. [DOI] [PubMed] [Google Scholar]
- 42.Ohtake K, Maeno T, Ueda H, Ogihara M, Natsume H, Morimoto Y. Poly-L-arginine enhances paracellular permeability via serine/threonine phosphorylation of ZO-1 and tyrosine dephosphorylation of occludin in rabbit nasal epithelium. Pharmaceut Res. 2003;20:1838–1845. doi: 10.1023/b:pham.0000003383.86238.d1. [DOI] [PubMed] [Google Scholar]
- 43.Kevil CG, Oshima T, Alexander JS. The role of p38 MAP kinase in hydrogen peroxide mediated endothelial solute permeability. Endothelium. 2001;8:107–116. doi: 10.3109/10623320109165320. [DOI] [PubMed] [Google Scholar]
- 44.Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. 1999;274:23463–23467. doi: 10.1074/jbc.274.33.23463. [DOI] [PubMed] [Google Scholar]
- 45.Bratt A, Birot O, Sinha I, Veitonmaki N, Aase K, Ernkvist M, Holmgren L. Angiomotin regulates endothelial cell-cell junctions and cell motility. J Biol Chem. 2005;280:34859–69. doi: 10.1074/jbc.M503915200. [DOI] [PubMed] [Google Scholar]
- 46.Nishimura M, Kakizaki M, Ono Y, Morimoto K, Takeuchi M, Inoue Y, Imai T, Takai Y. JEAP, a novel component of tight junctions in exocrine cells. J Biol Chem. 2002;277:5583–7. doi: 10.1074/jbc.M110154200. [DOI] [PubMed] [Google Scholar]
- 47.Ernkvist M, Birot O, Sinha I, Veitonmaki N, Nystrom S, Aase K, Holmgren L. Differential roles of p80- and p130-angiomotin in the switch between migration and stabilization of endothelial cells. Biochim Biophys Acta. 2008;1783:429–37. doi: 10.1016/j.bbamcr.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 48.Nakajima Y, Nakamura Y, Shigeeda W, Tomoyasu M, Deguchi H, Tanita T, Yamauchi K. The role of tumor necrosis factor-alpha and interferon-gamma in regulating angiomotin-like protein 1 expression in lung microvascular endothelial cells. Allergol Int. 2013;62:309–22. doi: 10.2332/allergolint.12-OA-0528. [DOI] [PubMed] [Google Scholar]
- 49.Jian MY, Liu Y, Li Q, Wolkowicz P, Alexeyev M, Zmijewski J, Creighton J. N-cadherin coordinates AMP kinase-mediated lung vascular repair. Am J Physiol Lung Cell Mol Physiol. 2016;310:L71–85. doi: 10.1152/ajplung.00227.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cook-Mills JM, Gallagher JS, Feldbush TL. Isolation and characterization of high endothelial cell lines derived from mouse lymph nodes. In Vitro Cell Develop Biol. 1996;32:167–177. doi: 10.1007/BF02723682. [DOI] [PubMed] [Google Scholar]
- 52.Tudor KSRS, Deem TL, Cook-Mills JM. Novel alpha 4-integrin ligands on an endothelial cell line. Biochem Cell Biol. 2000;78:99–113. [PubMed] [Google Scholar]
- 53.Chen YH, Lu Q, Goodenough DA, Jeansonne B. Nonreceptor tyrosine kinase c-Yes interacts with occludin during tight junction formation in canine kidney epithelial cells. Mol Biol Cell. 2002;13:1227–1237. doi: 10.1091/mbc.01-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qureshi MH, Cook-Mills J, Doherty DE, Garvy BA. TNF-alpha-Dependent ICAM-1- and VCAM-1-Mediated Inflammatory Responses Are Delayed in Neonatal Mice Infected with Pneumocystis carinii. J Immunol. 2003;171:4700–4707. doi: 10.4049/jimmunol.171.9.4700. [DOI] [PubMed] [Google Scholar]
- 55.Ager A, Mistry S. Interaction between lymphocytes and cultured high endothelial cells: an {Iin vitro} model of lymphocyte migration across high endothelial venule endothelium. Eur J Immunol. 1988;18:1265–1274. doi: 10.1002/eji.1830180818. [DOI] [PubMed] [Google Scholar]
- 56.Moran EJ, Sarshar S, Cargill JF, Shahbaz MM, Lio A, Mjalli AMM, Arstrong RW. Radio Frequency Tag Encoded Combinatorial Library Method for the Discovery of Tripeptide-Subsituted Cinnamic Acid Inhibitors of the Protein Tyrosine Phosphatase PTP1B. J Am Chem Soc. 1995;117:10787–10788. [Google Scholar]
- 57.Cook-Mills JM. Hydrogen Peroxide Activation of Endothelial Cell-Associated MMPs During VCAM-1-dependent Leukocyte Migration. Cell Mol Biol. 2006;52:8–16. [PMC free article] [PubMed] [Google Scholar]
- 58.Berdnikovs S, Abdala-Valencia H, McCary C, Somand M, Cole R, Garcia A, Bryce P, Cook-Mills J. Isoforms of Vitamin E have Opposing Immunoregulatory Functions during Inflammation by Regulating Leukocyte Recruitment. J Immunol. 2009;182:4395–4405. doi: 10.4049/jimmunol.0803659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patibandla PK, Tyagi N, Dean WL, Tyagi SC, Roberts AM, Lominadze D. Fibrinogen induces alterations of endothelial cell tight junction proteins. J Cell Physiol. 2009;221:195–203. doi: 10.1002/jcp.21845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao YY, Zhao LN, Wang P, Miao YS, Liu YH, Wang ZH, Ma J, Li Z, Li ZQ, Xue YX. Overexpression of miR-18a negatively regulates myocyte enhancer factor 2D to increase the permeability of the blood-tumor barrier via Kruppel-like factor 4-mediated downregulation of zonula occluden-1, claudin-5, and occludin. J Neurosci Res. 2015;93:1891–902. doi: 10.1002/jnr.23628. [DOI] [PubMed] [Google Scholar]
- 61.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 62.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–70. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 63.Schneeberger E, Lynch R. The tight junction: a multifunctional complex. American journal of physiology Cell Physiol. 2004;286:C1213–C1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 64.Balda MS, Whitney JA, Flores C, Gonzalez S, Cereijido M, Matter K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134:1031–49. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singer KL, Stevenson BR, Woo PL, Firestone GL. Relationship of serine/threonine phosphorylation/dephosphorylation signaling to glucocorticoid regulation of tight junction permeability and ZO-1 distribution in nontransformed mammary epithelial cells. J Biol Chem. 1994;269:16108–15. [PubMed] [Google Scholar]
- 66.Jin M, Barron E, He S, Ryan SJ, Hinton DR. Regulation of RPE intercellular junction integrity and function by hepatocyte growth factor. Invest Ophthalmol Visual Sci. 2002;43:2782–2790. [PubMed] [Google Scholar]
- 67.Rao RK, Basuroy S, Rao VU, Karnaky KJ, Jr, Gupta A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. 2002;368:471–481. doi: 10.1042/BJ20011804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Itallie CM, Balda MS, Anderson JM. Epidermal growth factor induces tyrosine phosphorylation and reorganization of the tight junction protein ZO-1 in A431 cells. J Cell Sci. 1995;108:1735–1742. doi: 10.1242/jcs.108.4.1735. [DOI] [PubMed] [Google Scholar]
- 69.Kaihara T, Kawamata H, Imura J, Fujii S, Kitajima K, Omotehara F, Maeda N, Nakamura T, Fujimori T. Redifferentiation and ZO-1 reexpression in liver-metastasized colorectal cancer: possible association with epidermal growth factor receptor-induced tyrosine phosphorylation of ZO-1. Cancer Sci. 2003;94:166–172. doi: 10.1111/j.1349-7006.2003.tb01414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mathai JC, Sitaramam V. Stretch sensitivity of transmembrane mobility of hydrogen peroxide through voids in the bilayer. Role of cardiolipin. J Biol Chem. 1994;269:17784–17793. [PubMed] [Google Scholar]
- 71.Gopalakrishna R, Anderson WB. Ca2+- and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc Natl Acad Sci USA. 1989;86:6758–6762. doi: 10.1073/pnas.86.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anderson JM, Van Itallie CM, Fanning AS. Setting up a selective barrier at the apical junction complex. Curr Opin Cell Biol. 2004;16:140–5. doi: 10.1016/j.ceb.2004.01.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.