

Abstract
Elevation of intraocular pressure (IOP) is a serious adverse effect of glucocorticoid (GC) therapy. Increased extracellular matrix (ECM) accumulation and endoplasmic reticulum (ER) stress in the trabecular meshwork (TM) is associated with GC-induced IOP elevation. However, the molecular mechanisms by which GCs induce ECM accumulation and ER stress in the TM have not been determined. Here, we show that a potent GC, dexamethasone (Dex), activates transforming growth factor β (TGFβ) signaling, leading to GC-induced ECM accumulation, ER stress, and IOP elevation. Dex increased both the precursor and bioactive forms of TGFβ2 in conditioned medium and activated TGFβ-induced SMAD signaling in primary human TM cells. Dex also activated TGFβ2 in the aqueous humor and TM of a mouse model of Dex-induced ocular hypertension. We further show that Smad3−/− mice are protected from Dex-induced ocular hypertension, ER stress, and ECM accumulation. Moreover, treating WT mice with a selective TGFβ receptor kinase I inhibitor, LY364947, significantly decreased Dex-induced ocular hypertension. Of note, knockdown of the ER stress–induced activating transcription factor 4 (ATF4), or C/EBP homologous protein (CHOP), completely prevented Dex-induced TGFβ2 activation and ECM accumulation in TM cells. These observations suggested that chronic ER stress promotes Dex-induced ocular hypertension via TGFβ signaling. Our results indicate that TGFβ2 signaling plays a central role in GC-induced ocular hypertension and provides therapeutic targets for GC-induced ocular hypertension.
Keywords: endoplasmic reticulum stress (ER stress), glucocorticoid, growth factor, SMAD transcription factor, extracellular matrix (ECM) protein, glucocorticoids, ocular hypertension, steroid-induced glaucoma, trabecular meshwork, transforming growth factor β2 (TGFβ2)
Introduction
Glaucoma is the second leading cause of irreversible blindness worldwide. In 2013, an estimated 64.3 million people were affected by glaucoma globally, a number projected to increase to 76 million by 2020 and to 111.8 million by 2040 (1). Primary open-angle glaucoma (POAG)2 is the most common type of glaucoma, accounting for 74% of all glaucoma cases (2). Elevated intraocular pressure (IOP) is an important and modifiable risk factor for the development and progression of POAG (3). Glucocorticoids (GC) have proven to be vital agents for the treatment of a wide range of disorders including various ocular diseases involving inflammation. Although administration of GC has several benefits, topical or systemic GC can lead to ocular hypertension in about 30–50% of patients depending on the route of administration, and sustained GC treatment can lead to secondary iatrogenic open-angle glaucoma if not withdrawn (4–6). Although GC-induced glaucoma is a form of secondary iatrogenic open-angle glaucoma, its clinical presentations are similar in many ways to POAG (5). Steroid responders are at high risk of developing POAG (6), and almost all POAG patients are considered to be steroid responders (7).
It is known that GC-induced ocular hypertension is caused by increased resistance to aqueous humor outflow at the trabecular meshwork (TM) tissue (5, 7–9). However, the molecular pathological mechanisms of how GC treatment leads to increased outflow resistance at the TM are still unclear. Several studies have shown that GC-induced ocular hypertension is associated with increased accumulation of ECM in the TM, particularly in the juxtacanalicular connective tissue region and the inner wall endothelium of Schlemm's canal (10, 11). In addition, GC-induced ocular hypertension is associated with other morphological changes including decreased intratrabecular spaces (7), fingerprint-like depositions in the uveal meshwork (12, 13), fibrillar material in the juxtacanalicular connective tissue region (14), and the presence of α-smooth muscle actin positive myofibroblasts in the Schlemm's cannal region (8). GCs modulate the expression and secretion of various proteins including myocilin (15), fibronectin (16, 17), collagen (17), laminin (18), and elastin (19) in TM cells treated with a potent GC, dexamethasone (Dex). Dex also alters the expression of proteolytic enzymes, which regulate ECM turnover in the TM (20–22). Apart from humans, GC-induced ocular hypertension has been reported in eight other species (23). This phenomenon has also been observed in ex vivo human and bovine anterior perfusion culture (7, 24). Previously, we developed a mouse model of GC-induced ocular hypertension by administering topical Dex eye drops three times daily for several weeks (25). We also recently developed a rapid and sustained GC-induced ocular hypertension mouse model by administrating periocular injections of Dex acetate once a week (9). Using these mouse models and primary human TM cells, we have demonstrated the role of ER stress and increased ECM accumulation in GC-induced ocular hypertension. However, it is not understood how Dex leads to ER stress in the TM.
Transforming growth factor β2 (TGFβ2) is a major regulator of the ECM in the TM. TGFβ2 is known to be involved in the pathogenesis of POAG (26). TGFβ2 levels have been shown to be elevated in the aqueous humor of eyes from POAG patients (27, 28). Treatment of primary human TM cells with recombinant TGFβ2 has been shown to increase the synthesis and deposition of ECM (29, 30) and induce ECM cross-linking enzymes in TM cells (31–33). The binding of active TGFβ2 to its receptor leads to phosphorylation of SMAD2/3 and activation of the SMAD signaling pathway (34). Adenoviral injections of bioactive TGFβ2 elevate IOP in a SMAD3-dependent manner and also induce ECM deposition in mouse TM tissues (35, 36). In addition, exogenous TGFβ2 elevates IOP ex vivo in the human anterior segment perfusion system (29). Although the profibrotic effects of TGFβ2 and GC in TM are well-established, the link between GC and TGFβ2 signaling in the TM has not yet been studied. Several studies have shown inhibitory cross-talk between GC and TGFβ2 signaling in some but not all cell types (37–40). GCs inhibit TGFβ signaling in multiple cell types either by reducing the bioavailability of TGFβ or by regulating the SMAD signaling pathway (37, 38, 40). A synergistic effect of GC and TGFβ signaling was observed in ovarian cancer cells (39). A recent study showed similarities in proteomic changes between GC- and TGFβ2-treated TM cells, suggesting that a similar regulation is employed by both of these pathways (41). We hypothesize that cross-talk between GC and TGFβ2 signaling in the TM plays an important role in inducing ECM accumulation and ER stress, elevating IOP. In the present study, we explored this cross-talk between GC and TGFβ2 signaling and its role in the regulation of ECM, ER stress, and Dex-induced IOP elevation.
Results
Dex increases TGFβ2 in primary human TM cells
We hypothesized that Dex-induced ECM accumulation and ER stress are regulated by TGFβ2 signaling. Inactive TGFβ2 (∼54 kDa) is secreted as a precursor form that associates with latency-associated peptide. The dimeric, mature, and biologically active forms of TGFβ2 (25 kDa) are created with cleavage of the latency-associated peptide (42). We first examined whether treatment of primary human TM cells with Dex alters TGFβ2 and ECM proteins (Fig. 1). Primary human TM cells were treated with either vehicle (Veh) or Dex (100 nm). TGFβ2 and ECM proteins were examined in the conditioned medium and in cellular lysates. Western blot analysis of the conditioned medium demonstrates that Dex increases both pro and active forms of TGFβ2 along with fibronectin (Fig. 1A). Densitometric analysis demonstrates that Dex increases both the active form of TGFβ2 and fibronectin by more than 3-fold over vehicle-treated cells (Fig. 1B). ELISA revealed that Dex significantly increases both the total and active forms of TGFβ2 in the conditioned medium (Fig. 1, C and D). Dex also increases fibronectin, collagen IV, and the active form of TGFβ2 in the cellular lysates (Fig. 1E). Interestingly, Dex induction of ECM and TGFβ2 is also associated with increased ER stress as determined by ATF4 and CHOP analysis in primary human TM cells (Fig. 1E). Dex treatment caused a 1.3-fold increase in TGFβ2 mRNA levels and a 2-fold increase in fibronectin mRNA levels (Fig. 1F). Although Dex induction of fibronectin mRNA expression was not statistically significant, Dex-induced fibronectin protein is evident in Fig. 1, A and E, indicating that primary TM cells responded well to Dex treatment. Together, these data indicate that Dex increases secretion and activation of TGFβ2, which correlates with Dex-induced ECM and ER stress induction in human TM cells.
Figure 1.

Dex increases TGFβ2 levels and ECM proteins and induces ER stress in human TM cells. Primary human TM cells (n = 3 strains) were treated with Veh or Dex (100 nm) for 72 h, and conditioned medium (A–D) and cell lysates (E) were examined for TGFβ2, ECM proteins, and ER stress markers. A, Western blot analysis of conditioned medium demonstrated that Dex treatment increased both the precursor and active forms of secreted TGFβ2 in primary human TM cells. Coomassie staining of gel was used as the loading control (n = 3 cell strains). B, densitometric analysis of the Western blotting was normalized to Coomassie staining, and the -fold change was calculated by dividing densitometric values of Dex-treated samples over Veh-treated samples. Dex increased both TGFβ2 and fibronectin (FN) in human primary TM cells (n = 4). C and D, ELISA for total (C) and active (D) TGFβ2 in the conditioned medium of GTM3 cells showing that Dex significantly increased both the total and active forms of TGFβ2 (n = 3 replicates). E, Western blot analysis of cellular lysates demonstrates that Dex-induced TGFβ2 and ECM is associated with induction of ER stress markers including ATF4 and CHOP (n = 3 cell strains). F, TGFβ2 and fibronectin mRNA expression levels were examined by real-time PCR in primary human TM cells treated with Veh or Dex for 48 h (not statistically significant).
Dex activates the SMAD signaling pathway in human TM cells
Previous studies have shown that TGFβ2 activates the canonical SMAD signaling pathway, which regulates ECM in TM cells (43). Because Dex increases the active form of TGFβ2, we next examined whether Dex also leads to activation of the SMAD signaling pathway in human TM cells. GTM3 cells were treated at various time points with vehicle or Dex (100 nm) under serum-free conditions. Western blot analysis of cellular lysates revealed that Dex treatment increases phosphorylation of SMAD3, starting from 60 min of treatment, compared with vehicle treatment, which correlated well with the timing of induction of TGFβ2 and the ER stress marker GRP78 (Fig. 2A). Cytoplasmic to nuclear translocation of the phosphorylated SMAD complex is a critical step in TGFβ-mediated signal transduction. Therefore, we examined whether Dex increases the nuclear translocation of SMAD3. As shown in Fig. 2B, Western blot analysis demonstrated a prominent increase in pSMAD3 and SMAD3 in the nuclear fractions. GTM3 cells treated with recombinant TGFβ2 (5 ng/ml) alone for 60 min were used as a positive control. The presence of lamin A/C and the absence of GAPDH in nuclear fractions indicated the relative purity of the nuclear fractions. Immunostaining for SMAD4 revealed that Dex dramatically increases nuclear levels of SMAD4 over control (Fig. 2C). The SMAD reporter assay further demonstrated a more than 2-fold increase in SMAD-luciferase activity after 16 h of Dex treatment (Fig. 2D). These data indicate that Dex increases the active form of TGFβ2 and activates the downstream SMAD signaling pathway.
Figure 2.

Dex treatment activates SMAD signaling. A, GTM3 cells treated with Veh or Dex (100 nm) for 0.5, 1, 12 and 24 h under serum-free conditions. Cellular lysates were examined for the timing of TGFβ-SMAD signaling activation. Increases in SMAD3 phosphorylation and TGFβ2 was observed starting at 1 h after Dex treatment. Increased GRP78 and fibronectin was observed after 12 h of Dex treatment (n = 2 replicates). B, GTM3 cells were treated with Veh or Dex for 1, 2, and 12 h under serum-free conditions. Cytoplasmic and nuclear fractions were subjected to Western blot analyses of SMAD-signaling proteins. GTM3 cells treated with recombinant TGFβ2 (5 ng/ml) was used as a positive control for the activation of SMAD signaling. GAPDH and lamin A/C were used to demonstrate the relative purity of cytoplasmic and nuclear fractions, respectively (n = 2 replicates). C, SMAD4 localization was examined by immunostaining in GTM3 cells treated with Veh or Dex for 60 min. Increased nuclear localization of SMAD4 in Dex-treated cells indicates activation of SMAD signaling. n = 2 replicates. D, GTM3 cells transfected with SMAD-luciferase reporter construct for 24 h and treated with Veh or Dex for 16 h under serum-free conditions. “Relative luciferase units” represents firefly luciferase intensities normalized to Renilla activity. A more than 2-fold increase in SMAD reporter activity was observed in Dex-treated GTM3 cells compared with Veh-treated cells (unpaired t test, n = 3).
Inhibition of TGFβ signaling reduces Dex-induced ECM accumulation and ER stress in human TM cells
We next investigated whether inhibition of TGFβ signaling via SMAD3 knockdown abrogates Dex-induced ECM changes and ER stress. SMAD3 knockdown was achieved by various approaches: with CRISPR (CR)-Cas9 targeting of SMAD3 or using a selective chemical inhibitor of SMAD3 phosphorylation (SIS3) or a selective TGFβ receptor kinase I inhibitor (LY364947). Primary human TM cells (n = 3 cell strains) were treated with Dex for 3 days, and immunostaining was performed in nonpermeabilized (no Triton added) cells to stain for extracellular fibronectin. Dex increased extracellular fibronectin staining (Fig. 3A), which was completely blocked by SIS3 and LY364947. Western blot analysis further demonstrated that Dex-induced fibronectin and ER stress markers, including GRP78, GRP94, and CHOP, are reduced when GTM3 cells are treated with SIS3, LY364947, or CR-SMAD3. Reducing the phosphorylation of SMAD3 in GTM3 cells treated with SIS3, LY364947, or CR-SMAD3 further supported inhibition of TGFβ signaling (Fig. 3B). Densitometric analysis of Western blots for fibronectin demonstrated a significant increase in fibronectin after Dex treatment, which was completely blocked by co-treatment with SIS3, LY364947, or SMAD3 knockdown in GTM3 cells (Fig. S1) and in primary human TM cells (Fig. 3C). Inhibition of TGFβ-induced SMAD signaling was further examined using a SMAD reporter assay. GTM3 cells were transfected with SMAD-luciferase constructs and treated with vehicle or Dex with or without TGFβ-signaling inhibitors (LY364947, SIS3, or CR-SMAD3). As shown in Fig. 3D, Dex significantly increased SMAD-luciferase activity, which was significantly reduced in the presence of TGFβ-signaling inhibitors. These results clearly demonstrate that inhibition of TGFβ signaling prevents Dex-induced ECM accumulation and ER stress in TM cells.
Figure 3.

Inhibition of TGFβ signaling attenuates Dex-induced elevation of ECM and ER stress. A, primary human TM cells treated with Dex with or without TGFβ-signaling inhibitors (LY364947 is a TGFβ receptor I kinase inhibitor, and SIS3 is a selective inhibitor of SMAD3 phosphorylation) for 72 h and stained for fibronectin without Triton permeabilization. Dex increased extracellular fibronectin, which was blocked by TGFβ-signaling inhibitors (n = 3 cell strains). Scale bar = 50 μm. B, Western blot analysis of fibronectin, ER stress markers (GRP78, GRP94, and CHOP), and TGFβ-signaling proteins (pSMAD3 and total SMAD3) in GTM3 cells treated with Veh or Dex in the presence or absence of LY364947, SIS3, and CR-SMAD3 (CRISPR-Cas9 targeting SMAD3). C, densitometric analysis of fibronectin Western blotting in human primary TM cells treated with Dex and TGFβ2-signaling inhibitors (n = 3, one-way ANOVA; *, p < 0.05; **, p < 0.01). D, GTM3 cells were transfected with SMAD-luciferase constructs and treated with Veh or Dex with or without TGFβ-signaling inhibitors (LY364947, SIS3, and CR-SMAD3). Dex-induced SMAD reporter activity was significantly reduced in GTM3 cells treated with inhibitors of TGFβ signaling (n = 3 replicates, one-way ANOVA; ***, p < 0.001).
We recently demonstrated that Dex-induced intracellular fibronectin and type I collagen interact and co-localize with KDEL antibody, which recognizes the ER stress markers GRP78 and GRP94 (44). Here, we examined whether inhibition of TGFβ signaling prevents Dex-induced co-localization of fibronectin and type I collagen with KDEL in primary human TM cells (Fig. 4). Human primary TM cells were stained with fibronectin and KDEL antibody (Triton-permeabilized). Co-localization of fibronectin (Fig. 4) or type I collagen ((Fig. S2) with KDEL antibody demonstrated that Dex increases the co-localization of fibronectin or type I collagen with KDEL, which was completely blocked by inhibition of TGFβ signaling using either SIS3 or LY364947 in human primary TM cells. Together, these data indicate that inhibition of TGFβ signaling reduces Dex-induced ECM accumulation and ER stress in human primary TM cells.
Figure 4.
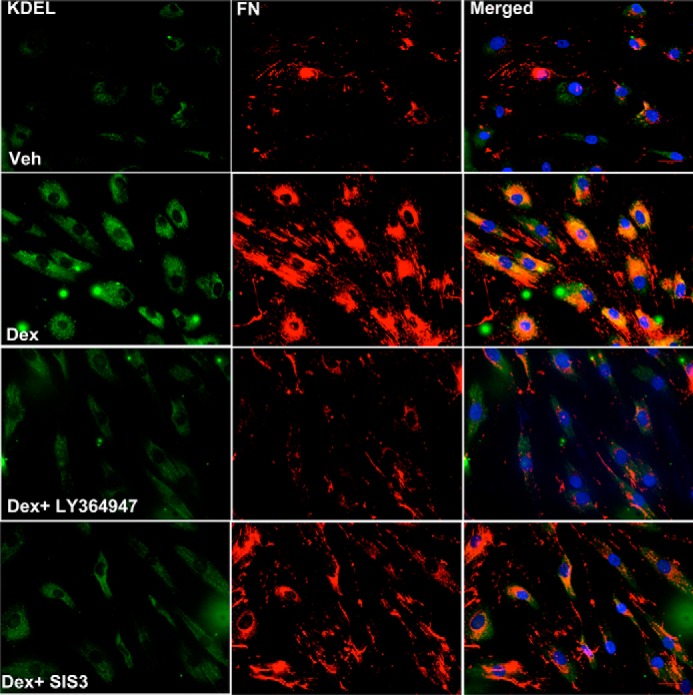
Inhibition of TGFβ signaling blocks Dex-induced intracellular fibronectin co-localization with ER stress markers in primary human TM cells. Primary human TM cells (n = 3 cell strains) were treated with Veh or Dex in the presence or absence of TGFβ-signaling inhibitors (LY364947 and SIS3) for 72 h, and cells were stained for fibronectin and KDEL (Triton-permeabilized cells). The Dex-induced intracellular fibronectin load and its co-localization with KDEL were completely blocked by TGFβ-signaling inhibitors. Scale bar = 50 micron.
Increased TGFβ2 in the aqueous humor of Dex-treated mice
We next explored whether Dex regulates TGFβ2 signaling in vivo. C57 mice were treated with topical vehicle or Dex eye drops daily for several weeks, and TGFβ2 levels were examined in the aqueous humor samples before and after IOP elevation (Fig. 5). Western blotting (Fig. 5, A, C, and E) and densitometric analyses (Fig. 5, B, D, and F) of aqueous humor samples demonstrated a significant induction of the active form of TGFβ2 starting from 3 weeks of Dex treatment. Interestingly, the active form of both TGFβ2 (Fig. 5, A and B) and fibronectin (Fig. S3A) showed little change prior to IOP elevation at 1 week of treatment. However, active TGFβ2 and fibronectin levels were significantly elevated upon 3 and 8 weeks of Dex treatment (Figs. 5, C–F, and S3, B and C). We further examined TGFβ2 levels in the aqueous humor by ELISA. C57 mice were treated with weekly periocular injections of vehicle or Dex for 2 weeks, and aqueous humor samples were subjected to ELISA for TGFβ2. Dex-treated mice demonstrate significant increases in both total and active TGFβ2 compared with vehicle-treated mice (Fig. 6, A and B). Western blot analysis of anterior segment tissues from 3-week vehicle and Dex-treated mice demonstrated that Dex treatment dramatically increases the active form of TGFβ2 in anterior segment tissues (Fig. 6C). Furthermore, immunostaining for TGFβ2 showed little staining in vehicle-treated mice, whereas TGFβ2 appeared to increase in the TM and ciliary body of Dex-treated mice (Fig. 6D). We also determined whether Dex induces Smad signaling in vivo by Western blot analysis of anterior segment tissue lysates. Dex-treated mice had increased pSmad3 compared with vehicle-treated mice, further indicating activation of TGFβ signaling in TM (Fig. 6E).
Figure 5.

Increased active form of TGFβ2 in the aqueous humor of Dex-treated mice. BL6 mice were treated with Veh (V) or Dex (D) eye drops for 8 weeks, and aqueous humor samples were collected prior to IOP elevation (A and B, 1 week) and after significant IOP elevation (C and D, 3 weeks; E and F, 8 weeks) and subjected to Western blotting analyses of TGFβ2. The top panel shows Western blotting for TGFβ2, and the bottom panel shows Coomassie staining of gel for total protein loading. Western blotting and densitometric analysis demonstrated no significant change in TGFβ2 prior to IOP elevation at 1 week of Dex treatment (B). Dex treatment significantly elevated TGFβ2 at 3 and 8 weeks (D and F) of treatment, indicating a strong association of TGFβ2 with IOP elevation (n = 6, each group at 1 week; n = 4, each group at 3 weeks; and n = 5 (Veh) and 6 (Dex) at 8 weeks of treatment; unpaired t test).
Figure 6.

Dex increases TGFβ2 in the aqueous humor and in the TM tissues. A and B, total (A) and active (B) forms of TGFβ2 were analyzed by ELISA in aqueous humor from Veh (V)- and Dex (D)-treated mice at 2 weeks. Dex treatment significantly increased both the total and active forms of TGFβ2 in the aqueous humor. Unpaired t test; n = 4 mice. C, anterior segment tissue lysates from Veh- or Dex-treated mice at 3 weeks were subjected to Western blot analysis of TGFβ2. Dex treatment induced the active form of TGFβ2 in the anterior segment tissue lysates (n = 3 in each group). D, immunostaining for TGFβ2 in the anterior segment tissues revealed increased immunostaining for total TGFβ2 in the TM of Dex-treated mice (n = 3, Veh- and Dex-treated mice). Arrows indicate TM tissue. C, cornea; CB, ciliary body. Scale bar = 50 μm. E, Western blot analyses of anterior segment tissues from 5-week-treated mice demonstrate that Dex increased phosphorylation of Smad3. GAPDH was utilized as a loading control.
Smad3−/− mice are protected from Dex-induced ocular hypertension, ER stress, and abnormal ECM accumulation
To further test whether TGFβ signaling regulates Dex-induced ocular hypertension, we utilized Smad3−/− mice. A previous study shows that TGFβ2-induced ECM remodeling in the TM and IOP elevation depends on SMAD3 (36). Weekly bilateral periocular injections of vehicle or Dex acetate suspension were given to 3-month-old Smad3−/− and WT littermates. Conscious IOPs were measured as described previously (9). A significant increase in IOP was observed in WT mice after 1, 2, and 3 weeks of Dex treatment compared with vehicle-treated WT mice (Fig. 7A). Notably, Dex did not significantly increase IOP in Smad3−/− mice compared with WT mice treated with Dex. Interestingly, IOPs in Smad3−/− mice were similar to WT mice treated with vehicle, suggesting complete protection from Dex-induced ocular hypertension. Smad3+/− (heterozygous) mice responded with elevated IOP measurements similar to WT mice with Dex treatment (data not shown). We further examined whether Smad3−/− mice were also protected from Dex-induced ECM accumulation and ER stress in the anterior segment lysates (Fig. 7, B and C). Western blotting (Fig. 7B) and its densitometric analysis (Fig. 7C) clearly demonstrated that Dex significantly increases fibronectin and ER stress markers including GRP78, GRP94, ATF4, and CHOP, which are significantly reduced in Smad3−/− mice. These data demonstrate that inhibition of TGFβ2 signaling via the genetic loss of Smad3 prevents Dex-induced abnormal ECM, ER stress, and ocular hypertension in mice.
Figure 7.

Smad3−/− mice are protected from Dex-induced ocular hypertension, ER stress, and abnormal ECM accumulation. Three-month-old WT (Smad3+/+) and Smad3−/− littermates were given periocular injections of Veh or Dex suspension for 3 weeks. A, Dex treatment significantly elevated IOP in WT mice, whereas IOP in Dex-treated Smad3−/− mice was similar to Veh-treated WT mice, indicating that Smad3−/− mice are protected from Dex-induced ocular hypertension (n = 8–10 in each group, one-way ANOVA). Western blotting (B) and densitometric analysis (C) of anterior segment lysates of above described mice revealed that Dex induced ECM (fibronectin) and ER stress (GRP78, GRP94, CHOP and ATF4) in WT mice, which were significantly decreased in Smad3−/− mice treated with Dex (n = 4 in each group, two-way ANOVA). *, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus Veh-treated Smad3+/+ mice; #, p < 0.05; ##, p < 0.01; and ###, p < 0.001 versus Dex-treated Smad3+/+ mice).
Inhibition of TGFβ signaling via treatment with LY364947 reduces Dex-induced ocular hypertension in mice
We further examined whether inhibition of TGFβ2 signaling blocks Dex-induced IOP elevation via treating mice with topical ocular eye drops of LY364947 (selective TGFβ receptor kinase I inhibitor). C57BL/6J mice were treated with weekly bilateral periocular injections of vehicle or Dex acetate suspension as described previously (9). After 1 week of Dex injection, conscious IOP revealed that Dex treatment elevated IOP significantly compared with vehicle-injected mice (data not shown). Topical ocular LY364947 eye drops (1%) were given to the right eyes, and the contralateral left eyes were given vehicle eye drops twice daily after 1 week of periocular injections (Fig. 8). The left eyes, treated with periocular Dex and vehicle control eye drops, showed significantly elevated IOP after 2 and 3 weeks of treatment compared with periocular vehicle injected mice. The right eyes, treated with periocular Dex acetate suspension and LY364947 eye drops, demonstrated a significant reduction in IOP compared with the contralateral left eyes treated with periocular Dex acetate suspension and vehicle eyes drops. We did not observe any significant change in IOP in mice treated with periocular vehicle suspension and LY364947 eye drops compared with mice treated with periocular vehicle suspension and vehicle control eye drops. These data indicate that inhibition of TGFβ2 signaling blocks Dex-induced IOP elevation.
Figure 8.
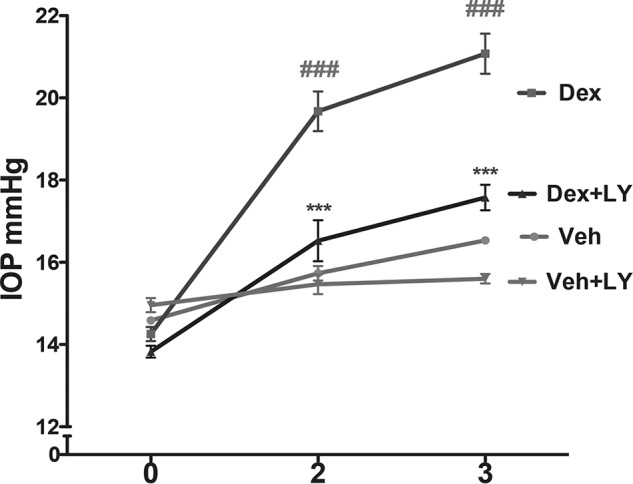
Inhibition of TGFβ signaling via treatment with LY364947 reduces Dex-induced ocular hypertension in mice. BL6 mice were treated with weekly periocular injections of Veh or Dex acetate suspension; topical ocular LY364947 (LY) eye drops were given to the right eyes, and the contralateral left eyes were given vehicle control eye drops twice daily after 1 week of periocular injections. The left eyes, treated with periocular Dex and vehicle eye drops, developed significantly elevated IOP after 2 and 3 weeks of treatment compared with periocular Veh-injected mice. The right eyes, treated with periocular Dex acetate suspension and LY364947 eye drops, demonstrated significant reduction in IOP compared with the contralateral left eyes treated with periocular Dex acetate suspension and vehicle eyes drops (n = 4 in periocular Veh-treated group and n = 8 in periocular Dex-treated group; two-way ANOVA). ###, p < 0.001 versus periocular Veh + vehicle control eye drops; ***, p < 0.001versus periocular Dex + vehicle eye drops.
ATF4 and CHOP is involved in Dex-induced TGFβ2 activation and ECM accumulation in TM cells
As shown previously (25) and in Fig. 1, Dex-induced TGFβ2 activation is associated with increased ATF4 and CHOP in TM cells. Moreover, Chop knockout mice are protected from Dex-induced ocular hypertension (25). Therefore, we examined whether Dex-induced TGFβ2 activation is regulated by ATF4 and CHOP in human TM cells. First, we examined whether loss of ATF4 or CHOP inhibits TGFβ2 signaling, thus preventing an abnormal ECM accumulation and induction of ER stress (Fig. 9A). GTM3 cells were transfected with CRISPR-Cas9 targeting CHOP or ATF4 for 24 h and then treated with vehicle or Dex for an additional 48 h. Western blot analysis demonstrated that Dex increases precursor and active TGFβ2, fibronectin, and ER stress markers including GRP78, ATF4, and CHOP, which were reduced in cells pretreated with CR-ATF4 or CR-CHOP resulting in knockdown of these proteins. Western blotting data confirmed that CR-ATF4 or CR-CHOP treatment reduces protein levels of ATF4 and CHOP in TM cells transfected with these plasmids and treated with Dex. We further confirmed that CRISPR-Cas9 targeting ATF4 reduces ATF4 levels in control GTM cells, although control Cas9 plasmids did not alter ATF4 levels (Fig. S4). We next examined whether ATF4 knockdown prevents Dex-induced TGFβ2 in the conditioned medium. GTM3 cells were transduced with adenovirus 5 expressing CRISPR-Cas9 targeting ATF4 or a dominant-negative inhibitor of ATF4 (45) followed by treatment with vehicle or Dex for 48 h. Western blot analysis of the conditioned medium demonstrated that ATF4 knockdown decreases Dex-induced expression of pro and active TGFβ2 levels (Fig. 9B). Coomassie staining demonstrated relatively similar protein loading of the conditioned medium (Fig. S5). We also observed that knockdown of ATF4 or CHOP reduced both active and total TGFβ2 levels in the conditioned medium of control GTM cells (Fig. S6).
Figure 9.

Chronic ER stress–induced ATF4 and CHOP are involved in Dex-induced TGFβ2. A, GTM3 cells transiently transfected with CRISPR-Cas9-CHOP or CRISPR-Cas9-ATF4 and treated with Veh or Dex for 48 h. Cell lysates were analyzed for expression levels of fibronectin (FN), TGFβ2, and ER stress markers by Western blotting. Knockdown of ATF4 or CHOP inhibits TGFβ2 signaling and prevents abnormal Dex-induced ECM accumulation and ER stress in TM cells. n = 3 replicates. B, GTM3 cells were transfected with CRISPR-Cas9-ATF4 (Cr ATF4) or a dominant-negative inhibitor of ATF4 (Mut ATF4) and treated with Dex for 48 h. The conditioned medium was examined for TGFβ2 by Western blot analysis. n = 3 replicates. C, GTM3 cells, transfected with SMAD-luciferase and CRISPR-Cas9 targeting SMAD3 or CHOP or ATF4 constructs, were treated with Veh or Dex for 24 h. Knockdown of ATF4 or CHOP in Dex-treated TM cells prevented SMAD activation in a SMAD-luciferase assay (n = 3, one-way ANOVA). ***, p < 0.001). D, GTM3 cells were transduced with adenovirus 5–expressing ATF4 for 48 h. The conditioned medium was examined for TGFβ2 by Western blot analysis (n = 3 replicates). E = control adenovirus. E, GTM3 cells were transfected with SMAD-luciferase constructs and treated with Veh, Dex, or adenovirus 5–expressing ATF4 (Ad5-ATF4). Treatment with recombinant TGFβ2 was utilized as a positive control for activation of SMAD signaling. Both Dex and overexpression of ATF4 alone were able to increase SMAD-luciferase activity significantly. A combination of Dex with ATF4 overexpression further increased SMAD-luciferase activity compared with ATF4 alone. n = 3, one-way ANOVA; *, p < 0.05; ***, p < 0.001.
We assessed whether knockdown of ATF4 and CHOP would reduce the Dex-induced SMAD signaling pathway using a SMAD-luciferase assay (Fig. 9C). GTM3 cells were co-transfected with SMAD-luciferase constructs and plasmids expressing CRISPR-Cas9 targeting ATF4 or CHOP for 24 h followed by treatment with vehicle or Dex for an additional 24 h. A SMAD reporter assay revealed that Dex significantly increased SMAD-luciferase activity, which was significantly reduced by genetic knockdown of ATF4 or CHOP. We also observed that control Cas9 plasmids did not alter SMAD-luciferase activity significantly in control GTM3 cells compared with cells treated with Lipofectamine alone (Fig. S7). In addition, CRISPR-Cas9 targeting ATF4 or CHOP did not reduce SMAD-luciferase activity significantly in control GTM3 cells compared with control Cas9 treatment (Fig. S7). We next examined whether overexpression of ATF4 alone was sufficient to induce TGFβ2 in TM cells without Dex treatment (Fig. 9D). GTM3 cells were transduced with adenovirus-expressing ATF4 (45), and the conditioned medium was examined for TGFβ2. Western blot analysis demonstrated that overexpression of ATF4 alone is sufficient to increase the precursor and active forms of TGFβ2 in TM cells. Western blot analysis of cell lysates confirmed that GTM3 cells transduced with Ad5-ATF4 overexpressed ATF4 compared with Ad5-empty–treated cells (Fig. S8). Consistent with this finding, the overexpression of ATF4 increased SMAD-luciferase activity in a manner similar to Dex treatment. Interestingly, co-treatment with Dex and ATF4 further enhanced SMAD activity, suggesting synergistic interactions (Fig. 9E). These data support the idea that ATF4 and CHOP are involved in the activation of TGFβ2 signaling in Dex-induced ocular hypertension.
Discussion
GC-induced IOP elevation is associated with increased ECM accumulation and ER stress in the TM. However, it is not understood how GC induces increased ECM accumulation and ER stress in the TM. Here, we have demonstrated that Dex-induced IOP elevation, ECM accumulation, and ER stress are mediated by TGFβ2 signaling in the TM. Dex induces the secretion and activation of TGFβ2, which triggers canonical SMAD signaling to increase ECM synthesis and deposition, inducing ER stress in primary human TM cells and in mouse TM and resulting in elevated IOP. The inhibition of TGFβ signaling prevents Dex-induced ECM deposition and ER stress and rescues Dex-induced IOP elevation. These findings indicate that the activation of TGFβ2 signaling is responsible for ECM remodeling, ER stress, and IOP elevation in GC-induced glaucoma.
TGFβ2 is secreted in the latent form and remains inactive with latent TGFβ-binding proteins and latency-associated peptide, and the active form of TGFβ2 is required for downstream signal transduction (46). The active form of TGFβ2 is increased in the aqueous humor of POAG patients (47). The overexpression of WT TGFβ2 (biologically inactive) alone is not enough to elevate IOP in mice, but expression of a bioactivated form of TGFβ2 in the TM is required to elevate IOP in mice (35). Consistent with this, we observed that Dex increased the active form of TGFβ2 levels in conditioned medium and aqueous humor collected from Dex-treated mice, suggesting that Dex induces extracellular activation of TGFβ2, which is important for the activation of downstream TGFβ2 signaling. We also observed that the activation of SMAD signaling and the timing of activation of TGFβ2 in the aqueous humor in Dex-treated mice correlated well with IOP elevation, indicating that the activation of TGFβ2 in the aqueous humor is associated with IOP elevation. Importantly, inhibiting TGFβ signaling completely protects Dex-induced ECM accumulation, ER stress, and IOP elevation.
Dex increased TGFβ2 levels as well as downstream SMAD proteins within 60 min of treatment in cultured TM cells, suggesting that the effects of Dex on TGFβ2 are post-translational. In addition, the timing of TGFβ2 induction by Dex correlates well with the timing of fibronectin and ER stress induction. Therefore, Dex-induced fibrosis and ER stress are most likely regulated by the TGFβ2 signaling pathway. In support of this hypothesis, we observed that inhibition of TGFβ signaling at the TGFβ receptor level or SMAD3 level prevents Dex-induced fibrosis and ER stress in primary human TM cells. More importantly, the loss of SMAD3, which is required for TGFβ2-induced IOP elevation, protects from Dex-induced IOP elevation, ECM accumulation, and induction of ER stress. Inhibition of TGFβ signaling via LY364947 significantly reduces Dex-induced ocular hypertension. The TGFβ-signaling inhibitors utilized in this study are not exclusively selective for TGFβ2 signal transduction, as these inhibitors can also block TGFβ1 signaling. Because TGFβ2 is a major isoform in the TM and the fact that Dex activates the secretion of TGFβ2, it is likely that the effects observed in this study are attributable to TGFβ2 rather than TGFβ1. Our analysis of SMAD signaling using a SMAD-luciferase reporter assay, which measures several SMADs, demonstrates that Dex increases total SMAD transcriptional activity. Therefore, it is likely that Dex may increase other SMADs rather than just SMAD3 alone. However, SMAD3 has been shown to be required for TGFβ2-induced ocular hypertension (36). Consistent with this understanding, we observed that inhibition of SMAD3 was able to prevent Dex-induced ocular hypertension. These studies indicate an essential role for activated TGFβ2 signaling in Dex-induced ocular hypertension. Because GCs are known to influence a variety of pathways, the influence of these other pathways on Dex-induced fibrosis cannot be eliminated.
Although it is known that ER stress plays a role in fibrosis (48, 49), it is not understood how ER stress can lead to fibrosis. Based on the findings of our study, it is plausible that chronic ER stress can modify TGFβ signaling, which may worsen already existing fibrosis. Chronic ER stress–induced transcriptional factors such as ATF4 and CHOP are known to be involved in cell dysfunction and death (45, 50, 51). However, it is not understood whether these chronic ER stress factors play any role in fibrosis. In our previous (25) and current studies, we observed that Dex-induced ATF4 and CHOP is associated with Dex-induced abnormal ECM accumulation and TGFβ2 activation. Genetic knockdown of ATF4 or CHOP prevents Dex-induced TGFβ2 activation and ECM accumulation in TM cells. Consistent with this, the overexpression of ATF4 alone is sufficient to induce TGFβ2 activation without Dex treatment. Interestingly, we have demonstrated previously that Chop knockout mice are protected from Dex-induced ocular hypertension. These findings suggest that chronic ER stress leads to TGFβ2 activation, which in turn leads to fibrosis in the TM. It is interesting to note that both ATF4 and CHOP are significantly elevated in glaucomatous TM tissues, and chronic ER stress is associated with abnormal ECM accumulation in glaucomatous TM tissues (44, 52). Considering the known role of TGFβ2 in glaucoma, it is possible that chronic ER stress in the TM may activate TGFβ2, which can further exacerbate ER stress via induction of abnormal ECM accumulation. Alternatively, elevated TGFβ2 levels in glaucoma may induce ER stress via abnormal ECM accumulation in the TM. In addition, TGFβ2 may directly induce chronic ER stress factors including ATF4 and CHOP, which can further potentiate TGFβ signaling, worsening abnormal ECM accumulation in TM. Future studies will be aimed at understanding whether TGFβ2 can directly induce ER stress in the TM.
We have shown previously that the knockout of Chop in mice protects against Dex-induced ocular hypertension. However, the exact mechanism was not clear. Based on our current findings, it is likely that the knockout of Chop in mice protects against Dex-induced ocular hypertension via inhibiting Dex-induced TGFβ2 signaling, thus preventing abnormal ECM accumulation in the TM. Consistent with this understanding, we demonstrated previously that Chop knockout mice prevent Dex-induced ECM accumulation (25). Although it is not clear from this study how ATF4 and CHOP activate TGFβ2, it is possible that ATF4 and CHOP may interact with GC-induced response elements to modulate TGFβ2 activation and signaling. A previous study demonstrates that a major ER chaperone, calreticulin, is required for TGFβ-induced fibrosis (53). Because TGFβ2 and ECM proteins are synthesized and processed in the ER, it is possible that ER stress plays an important role in TGFβ-mediated ECM remodeling.
Although it is clear that ATF4 and CHOP are involved in Dex-induced TGFβ2 activation, there are several other factors including integrins, proteases, and TSP1 that may be involved in Dex-induced TGFβ2 activation in the TM (46, 54). It is possible that Dex increases these extracellular factors involved in the activation of TGFβ2. For example, TSP1 has been shown to induce the activation of TGFβ2 (55). Interestingly, TSP1 levels were found to be increased in one-third of POAG patients, and TSP1 is induced by Dex in vitro (56). Furthermore, TSP1-null mice have a lower IOP when compared with their WT littermates (57). Because TGFβ2 lacks an RDG motif (46), it is unlikely that integrins are involved in Dex-induced TGFβ2 activation. Both MMP2 and MMP9 have been shown to activate TGFβ (54). It is likely that both MMP and TSP1 play a role in modulating Dex-induced TGFβ activation in the TM.
In non-ocular cell types, the cross-talk between GC and TGFβ2 pathways has different effects depending on the cell type. Some studies have demonstrated synergistic effects when cells are treated with both Dex and TGFβ2. Wickert et al. (58) report a strong induction of TGFβ-mediated PAI-1 and connective tissue growth factor (CTGF) expression in rat hepatocytes upon simultaneous stimulation with TGFβ and Dex compared with that of TGFβ alone. Similarly, Kimura et al. (59) report that GC enhance TGFβ-induced PAI-1 expression in human renal proximal tubular cells. In contrast, other studies have described opposing interactions between GC and TGFβ in lung fibroblasts (60) and hepatic stellate cells (40). It appears that these interactions are cell type–dependent. Differential expression of GC receptors may play a major role in determining these contrasting effects of GC on TGFβ signaling. GRα is responsible for the physiological and pharmacological effects of GC, whereas GRβ acts as a negative regulator of GR activity (61). The lower expression of GRβ in TM cells (62) may contribute to these differential interactions.
In summary, this study demonstrates that cross-talk between GC and TGFβ2 signaling regulates GC-induced ECM remodeling in the TM. Similar cross-talk between these pathways may also play a major role in the pathophysiology of POAG. Furthermore, our studies demonstrate that the manipulation of these interactions can provide novel interventions for steroid-induced glaucoma.
Materials and methods
Antibodies
Antibodies were purchased from the following sources: TGFβ2 (catalogue No. SC-90, Santa Cruz Biotechnology, Dallas, TX), fibronectin (catalogue No. Ab2413, Abcam, Cambridge, MA), KDEL (catalogue No. Ab12223, Abcam), collagen type IV (catalogue No. SAB4500369, Sigma-Aldrich), collagen I (catalogue No. NB600-408, Novus Biologicals, Littleton, CO), ATF4 (catalogue No. SC-200, Santa Cruz Biotechnology), CHOP (catalogue No. 13172, Novus Biologicals), GRP78 (N-20, catalogue No. SC-1050, Santa Cruz Biotechnology), GRP94 (H-212, catalogue No. SC-11402, Santa Cruz Biotechnology), SMAD3 (catalogue No. C67H9, Cell Signaling Technology, Danvers, MA), phospho-SMAD3 (catalogue No. S423/425, Cell Signaling Technology), and GAPDH (catalogue No. 3683, Cell Signaling Technology).
Experimental animals
C57BL/6J (BL6) mice and Smad3 knockout (Smad3−/−) mice on pure 129 background were obtained from The Jackson Laboratory. Smad3−/− mice were bred and genotyped to ensure that each mouse had the genotype as reported previously (36, 63, 64). Animals were fed standard chow ad libitum and housed in 12-h light/12-h dark conditions. All experimental procedures were approved by the University of North Texas Health Science Center Institutional Animal Care and Use Committee review board.
Dex treatment of mice
Topical 0.1% Dex phosphate (Bausch & Lomb Inc.) or vehicle eye drops containing sterile phosphate-buffered saline (PBS) were applied to 3-month-old C57BL/6J mice three times daily for 8 weeks (Figs. 5 and 6) as described previously (25). At the end of the treatment, aqueous humor or anterior segment tissues were collected for Western blot analysis, whereas whole eyes were fixed and sectioned for immunostaining. We also utilized our recently developed mouse model of Dex-induced ocular hypertension (9). BL6 mice were obtained from The Jackson Laboratory, and baseline IOP were measured to ensure that each mouse had normal IOP before treatment. Bilateral periocular vehicle or Dex acetate injections were performed every week as described previously (9). To evaluate the effects of genetic knockdown of SMAD3, WT or Smad3−/− littermates were given weekly periocular injections of vehicle or Dex acetate for 3 weeks. Conscious IOPs (IOPs measured without keeping mice under any anesthesia) were measured weekly. To determine the effects of LY364947 on Dex-induced ocular hypertension, BL6 mice were given weekly bilateral periocular injections of Veh or Dex acetate suspension. One week after periocular injections, the right eyes were treated with LY364947 eye drops (1%), whereas the contralateral left eyes were treated with vehicle control (1% DMSO in water) for 2 weeks. IOP was monitored weekly.
Dex treatment of human TM cells
Four different primary human TM cell strains and a transformed GTM3 cell line (65) were cultured in DMEM (Sigma) supplemented with 10% fetal bovine serum (Atlas Biologicals, Fort Collins, CO), l-glutamine (Gibco, Life Technologies), and penicillin-streptomycin (Gibco, Life Technologies). For the characterization of primary human TM cells, cells were examined for the expression of fibronectin, collagen, and laminin as well as for Dex induction of cross-linked actin networks and myocilin as described previously (15, 66, 67). The human primary TM cells (n = 4) or GTM3 cells were treated with either vehicle (0.1% ethanol) or Dex (100 nm) (Sigma-Aldrich) in serum-free conditions at various time points, and the conditioned medium and lysates were collected for Western blot analysis. The proteins in the media were concentrated using previously described StrataClean resin (Agilent Technologies) (68). For immunostaining analysis, the cells were fixed in 4% paraformaldehyde and stained with the appropriate primary and secondary antibodies.
IOP measurements
Conscious IOP measurements were carried out using rebound tonometry as described previously (69). Mice were restrained in plastic cones and secured in a custom-made restrainer. After a few minutes of acclimatization, IOPs were recorded in a masked manner. All IOP measurements were recorded between 10 a.m. and 1 p.m.
Immunostaining
Eyes from vehicle- or Dex-treated mice were enucleated, fixed in 4% paraformaldehyde, dehydrated, and embedded in paraffin. 10-μm thick sections were made using rotatory microtome. The deparaffinized and rehydrated sections were subjected to antigen retrieval in a sodium citrate buffer (pH 6). The slides were blocked in 10% goat serum containing 0.5% Triton X-100 for 2 h and incubated overnight with the appropriate primary antibody (1:50) in 10% goat serum followed by a 1.5-h incubation with the appropriate Alexa Fluor secondary antibodies (1:200, Life Technologies). The slides were mounted in DAPI mounting solution, and images were taken using a Keyence microscope (Itasca, IL). Slides incubated without primary antibody served as a negative control (data not shown). The primary human TM or transformed GTM3 cells cultured in 8-chamber slides were fixed in 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 for 10 min followed by 1 h of blocking with 10% goat serum. The slides were incubated overnight with the appropriate primary antibodies (1:500 for fibronectin and type I collagen). Next day, the slides were washed three times with 1× PBS and incubated with the appropriate Alexa Fluor secondary antibodies for 1.5 h. The slides were washed three times with 1× PBS before being mounted in DAPI mounting solution. To examine extracellular fibronectin in primary human TM cells, TM cells were fixed and stained with fibronectin antibody without Triton permeabilization. To examine intracellular fibronectin and its co-localization with KDEL, TM cells were fixed, permeabilized with 0.5% Triton for 15 min, and stained with fibronectin and KDEL.
Western blot analysis
TM cells or mouse anterior segments dissected from enucleated eyes were lysed in lysis buffer as described previously (68). Equal protein concentrations of lysates were loaded and run in 4–12% bis-Tris gels (NuPAGE bis-Tris gels, Life Technologies) and transferred onto polyvinylidene difluoride membrane. The blots were blocked in 10% nonfat dried milk prepared in 1× PBST (PBS with Tween 20) for 2 h and then incubated with the appropriate primary antibodies (1:1000) overnight at 4 °C. The blots were washed three times with 1× PBST followed by a secondary antibody (horseradish peroxidase–conjugated) incubation for 1.5 h. The blots were developed using ECL detection reagents (SuperSignal West Femto maximum sensitivity substrate, Life Technologies). For phosphorylated Smad3, SuperBlock (PBS) blocking buffer (Life Technologies) was used instead of 10% nonfat dried milk for blocking and antibody incubation.
SMAD reporter assay
TGFβ2-induced signal transduction was assessed using the Cignal SMAD reporter assay kit (Qiagen, Germantown, MD) in GTM3 cells. The SMAD reporter is a mixture of an inducible SMAD-luciferase construct (encodes firefly luciferase reporter gene under the control of a minimal cytomegalovirus promoter and tandem repeats of SMAD-binding element) and a constitutively expressing Renilla luciferase construct (40:1). GTM3 cells were plated into 96-well plates and transfected with negative, positive, and SMAD reporter constructs using Lipofectamine 3000 (Life Technologies). To determine the effects of inhibition of TGFβ signaling on Dex-mediated SMAD activity, GTM3 cells were treated with or without chemical inhibitors of TGFβ signaling (SIS3 (10 μm) and LY364947 (5 μm)) along with Dex in serum-free medium for 16 h. To determine the effects of ATF4 or CHOP on Dex-induced activation of TGFβ signaling, GTM3 cells were treated with CR-Cas9 control, CR-SMAD3, CR-CHOP, and CR-ATF4 plasmid constructs with SMAD reporter constructs. After 24 h of transfection, cells were treated with vehicle, Dex (100 ng/ml), or TGFβ2 (5 ng/ml) for 16 h. Luciferase assays were carried out using the Dual-luciferase reporter assay system (Promega, Madison, WI) according to the manufacturer's protocol.
ELISA for mouse TGFβ2
A mouse TGFβ2 DuoSet ELISA kit (catalogue No. DY7346-05, R&D Systems) was utilized to quantify total and active TGFβ2 levels in the aqueous humor samples from mice. Nine-month-old C57 mice were treated weekly with periocular injections of vehicle (left eye) and Dex suspension (right eye). After 2 weeks of injections, ∼5 μl of aqueous humor was obtained from each eye. 3 μl of aqueous humor was utilized for the analysis of active TGFβ2, and the remaining 2 μl of aqueous humor was used to quantify the total TGFβ2. As per the manufacturer's instructions, samples were processed by acid activation and neutralization (1 N HCl and 1.2 N NaOH/0.5 m HEPES) prior to quantifying the total TGFβ2; the results obtained were multiplied by a dilution factor of 1.4, whereas active TGFβ2 levels were determined in samples without processing. Briefly, a 96-well microplate was coated with 100 μl/well of a working concentration of capture antibody and incubated overnight at room temperature. Next day, the plate was washed three times with 1× wash buffer (400 μl/well), blocked with 300 μl/well reagent diluent, and incubated at room temperature for 1 h. The plate was washed three times with 1× wash buffer and incubated with 100 μl/well sample, control, or standards in reagent diluent for 2 h at room temperature. Following the three washes, 100 μl of the detection antibody diluted in reagent diluent was added to each well, and the mixture was incubated for 2 h at room temperature. The plate was washed three times with 1× wash buffer, a 100 μl of the working dilution of streptavidin–horseradish peroxidase was added to each well, and the mixture was incubated for 20 min at room temperature. Following the washing step, 100 μl of substrate solution was added to each well, and the mixture was further incubated for 20 min at room temperature. Finally, 50 μl of stop solution was added to each well, and the optical density of the plate was determined immediately using a microplate reader at a 450-nm wavelength with a 570-nm wavelength correction.
ELISA for human TGFβ2
A human TGFβ2 Quantikine ELISA kit (catalogue No. DB250, R&D Systems) was used to quantify total and active TGFβ2 levels in the conditioned medium from GTM3 cells treated with vehicle and Dex for 72 h in serum-free media. As per the manufacturer's instructions, samples were processed by acid activation and neutralization (1 N HCl and 1.2 N NaOH/0.5 m HEPES) prior to quantifying the total TGFβ2; the results obtained were multiplied by a dilution factor of 7.8. Active TGFβ2 levels were determined in samples without processing. Briefly, 100 μl/well assay diluent RD1-17 was added to an ELISA plate precoated with mAb specific for TGFβ2. This was followed by the addition of 100 μl/well standards, control, and both activated and nonactivated test samples (in duplicates). After 2 h of incubation at room temperature, the ELISA plate was washed with 1× wash buffer (400 μl/well) at least three times, and 200 μl/well TGFβ2 conjugate was added. The ELISA plate was further incubated for 2 h at room temperature and then washed three times. 200 μl/well substrate solution was added, and the mixture was incubated for 20 min at room temperature. Finally, 50 μl/well stop solution was added to the plate, and the absorbance was read using a microplate reader at 450 nm with a wavelength correction of 570 nm.
Real-time PCR analysis
Quantitative real-time PCR analysis was carried out in primary human TM cells (n = 3 cell strains) treated with vehicle or Dex for 48 h in serum-free conditions. Total RNA was isolated using a Mini Total RNA kit (IBI Scientific) according to the manufacturer's instructions. The purity and concentration of the isolated RNA was examined by NanoDrop 2000 (Thermo Fisher Scientific). An equal amount of RNA was used for cDNA synthesis using a SuperScript VILO cDNA synthesis kit (Thermo Fisher Scientific). Quantitative PCR was performed using 2× SsoAdvanced SYBR Green supermix (Bio-Rad) in a CFX96 thermocycler (Bio-Rad). The PCR thermal profiles consisted of an initial incubation at 95 °C for 60 s and 40 cycles at 95 °C for 60 s, 60 °C for 45 s, and 72 °C for 45 s followed by a final dissociation curve step. The primer pairs used for PCR included: fibronectin, 5′-AGCGGACCTACCTAGGCAAT-3′ and 5′-GGTTTGCGATGGTACAGCTT-3′; TGFβ2, 5′-ATCCCGCCCACTTTCTACAG-3′ and 5′-GCCATTCATGAACAGCATCA-3′; and GAPDH, 5′-GGATGATGTTCTGGAGAGCC-3′ and 5′-CATCACCATCTTCCAGGAGC-3′). The PCR cycle threshold (Ct) values were obtained from the CFX96 thermocycler (Bio-Rad). Dex-induced mRNA expression of fibronectin and TGFβ2 compared with vehicle was calculated by the ΔΔCt method using GAPDH as an internal control as reported previously (68, 70).
Nuclear and cytoplasmic extraction
GTM3 cells treated with vehicle or Dex at various time points were used to extract nuclear and cytoplasmic fractions with NE-PER nuclear and cytoplasmic extraction reagent kits (catalogue No. 78835, Thermo Fisher Scientific) according to the manufacturer's instructions. Briefly, the treated GTM3 cells were harvested by trypsin-EDTA and centrifuged at 500 × g for 5 min. The cell pellet was suspended in ice-cold cytoplasmic extraction reagent I (CER I) with vigorous vortexing. The suspension was incubated for 10 min on ice followed by the addition of ice-cold cytoplasmic extraction reagent II (CER II), vortexing, and incubating for 1 min on ice. The tube was centrifuged at high speed (16,000 × g) for 5 min. The supernatant, which contained the cytosolic fraction, was collected in a different prechilled tube. The pellet, which contained the nuclei, was suspended in ice-cold nuclear extraction reagent by repeated cycles of vigorous vortexing plus 10 min of incubation on ice for four times. The tube was centrifuged at high speed (16,000 × g) for 10 min, and the supernatant (nuclear fraction) was collected in a different prechilled tube for subsequent Western blot analysis.
ATF4 or CHOP knockdown/overexpression
GTM3 cells were transfected with CRISPR-Cas9-ATF4 or CRISPR-Cas9-CHOP plasmid constructs using Lipofectamine 3000 and then treated with vehicle or Dex for an additional 48 h (Fig. 9A). We also knocked down ATF4 using adenovirus 5–expressing CRISPR-Cas9–targeting ATF4 or dominant-negative ATF4ΔRK (which inhibits endogenous ATF4 activity) as described previously (45) at multiplicity of infection = 100 for 48 h. Cells were treated with vehicle or Dex for another 48 h (Fig. 9B). For overexpression of ATF4, GTM3 cells were transduced with Ad5-empty or ATF4 at multiplicity of infection = 100 for 24 h (Fig. 9D). The conditioned medium was subjected to Western blot analysis.
Statistical analysis
All data are presented as mean ± S.E. Statistical significance between two groups was analyzed using the unpaired 2-tailed Student's t test. For data between multiple groups, one-way ANOVA or two-way ANOVA was used. p ≤ 0.05 was considered statistically significant.
Author contributions
R. B. K., C. S., V. C. S., and G. S. Z. resources; R. B. K., P. M., P. P., C. S., and G. S. Z. data curation; R. B. K., P. M., P. P., and G. S. Z. formal analysis; R. B. K. and G. S. Z. investigation; R. B. K. and G. S. Z. methodology; R. B. K., V. C. S., and G. S. Z. writing-review and editing; G. S. Z. conceptualization; G. S. Z. funding acquisition; G. S. Z. writing-original draft; G. S. Z. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Abbot Clark, Sherri Harris, and Sandra Maansson for sharing mice, animal colony maintenance, and paraffin sectioning, respectively. We also thank Dr. John Hulleman for providing critical feedback on this manuscript.
This work was supported by National Institutes of Health Grants EY022077, EY026177, and EY024259 from the NEI and by funding from the North Texas Eye Research Institute in Fort Worth, TX. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S8.
- POAG
- primary open-angle glaucoma
- IOP
- intraocular pressure
- GC
- glucocorticoid
- TM
- trabecular meshwork
- ECM
- extracellular matrix
- Dex
- dexamethasone
- ER
- endoplasmic reticulum
- TGFβ
- transforming growth factor β
- Veh
- vehicle
- CHOP
- CCAT/enhancer-binding protein (C/EBP) homologous protein
- GADH
- glyceraldehyde-3-phosphate dehydrogenase
- DAPI
- 4′,6-diamidino-2-phenylindole
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ANOVA
- analysis of variance
- Ad5
- adenovirus 5
- CR
- CRISPR.
References
- 1. Tham Y. C., Li X., Wong T. Y., Quigley H. A., Aung T., and Cheng C. Y. (2014) Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 121, 2081–2090 10.1016/j.ophtha.2014.05.013 [DOI] [PubMed] [Google Scholar]
- 2. Quigley H. A., and Broman A. T. (2006) The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol 90, 262–267 10.1136/bjo.2005.081224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosenthal J., and Leske M. C. (1980) Open-angle glaucoma risk factors applied to clinical area. J. Am. Optom. Assoc. 51, 1017–1024 [PubMed] [Google Scholar]
- 4. Jones R. 3rd, and Rhee D. J. (2006) Corticosteroid-induced ocular hypertension and glaucoma: A brief review and update of the literature. Curr. Opin. Ophthalmol. 17, 163–167 [DOI] [PubMed] [Google Scholar]
- 5. Clark A. F., and Wordinger R. J. (2009) The role of steroids in outflow resistance. Exp. Eye Res. 88, 752–759 10.1016/j.exer.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 6. Armaly M. F., and Becker B. (1965) Intraocular pressure response to topical corticosteroids. Fed. Proc. 24, 1274–1278 [PubMed] [Google Scholar]
- 7. Clark A. F., Wilson K., de Kater A. W., Allingham R. R., and McCartney M. D. (1995) Dexamethasone-induced ocular hypertension in perfusion-cultured human eyes. Invest. Ophthalmol. Vis. Sci. 36, 478–489 [PubMed] [Google Scholar]
- 8. Overby D. R., Bertrand J., Tektas O. Y., Boussommier-Calleja A., Schicht M., Ethier C. R., Woodward D. F., Stamer W. D., and Lütjen-Drecoll E. (2014) Ultrastructural changes associated with dexamethasone-induced ocular hypertension in mice. Invest. Ophthalmol. Vis. Sci. 55, 4922–4933 10.1167/iovs.14-14429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel G. C., Phan T. N., Maddineni P., Kasetti R. B., Millar J. C., Clark A. F., and Zode G. S. (2017) Dexamethasone-induced ocular hypertension in mice: Effects of myocilin and route of administration. Am. J. Pathol. 187, 713–723 10.1016/j.ajpath.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lütjen-Drecoll E. (1973) Structural factors influencing outflow facility and its changeability under drugs: A study in Macaca arctoides. Invest. Ophthalmol. 12, 280–294 [PubMed] [Google Scholar]
- 11. Mäepea O., and Bill A. (1992) Pressures in the juxtacanalicular tissue and Schlemm's canal in monkeys. Exp. Eye Res. 54, 879–883 10.1016/0014-4835(92)90151-H [DOI] [PubMed] [Google Scholar]
- 12. Johnson D., Gottanka J., Flügel C., Hoffmann F., Futa R., and Lütjen-Drecoll E. (1997) Ultrastructural changes in the trabecular meshwork of human eyes treated with corticosteroids. Arch. Ophthalmol. 115, 375–383 10.1001/archopht.1997.01100150377011 [DOI] [PubMed] [Google Scholar]
- 13. Rohen J. W., Linnér E., and Witmer R. (1973) Electron microscopic studies on the trabecular meshwork in two cases of corticosteroid-glaucoma. Exp. Eye Res. 17, 19–31 10.1016/0014-4835(73)90164-4 [DOI] [PubMed] [Google Scholar]
- 14. Wordinger R. J., and Clark A. F. (1999) Effects of glucocorticoids on the trabecular meshwork: Towards a better understanding of glaucoma. Prog. Retin. Eye Res. 18, 629–667 10.1016/S1350-9462(98)00035-4 [DOI] [PubMed] [Google Scholar]
- 15. Clark A. F., Steely H. T., Dickerson J. E. Jr., English-Wright S., Stropki K., McCartney M. D., Jacobson N., Shepard A. R., Clark J. I., Matsushima H., Peskind E. R., Leverenz J. B., Wilkinson C. W., Swiderski R. E., Fingert J. H., Sheffield V. C., and Stone E. M. (2001) Glucocorticoid induction of the glaucoma gene MYOC in human and monkey trabecular meshwork cells and tissues. Invest. Ophthalmol. Vis. Sci. 42, 1769–1780 [PubMed] [Google Scholar]
- 16. Steely H. T., Browder S. L., Julian M. B., Miggans S. T., Wilson K. L., and Clark A. F. (1992) The effects of dexamethasone on fibronectin expression in cultured human trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 33, 2242–2250 [PubMed] [Google Scholar]
- 17. Zhou L., Li Y., and Yue B. Y. (1998) Glucocorticoid effects on extracellular matrix proteins and integrins in bovine trabecular meshwork cells in relation to glaucoma. Int. J. Mol. Med. 1, 339–346 [PubMed] [Google Scholar]
- 18. Dickerson J. E. Jr., Steely H. T. Jr., English-Wright S. L., and Clark A. F. (1998) The effect of dexamethasone on integrin and laminin expression in cultured human trabecular meshwork cells. Exp. Eye Res. 66, 731–738 10.1006/exer.1997.0470 [DOI] [PubMed] [Google Scholar]
- 19. Yun A. J., Murphy C. G., Polansky J. R., Newsome D. A., and Alvarado J. A. (1989) Proteins secreted by human trabecular cells. Glucocorticoid and other effects. Invest. Ophthalmol. Vis. Sci. 30, 2012–2022 [PubMed] [Google Scholar]
- 20. Samples J. R., Alexander J. P., and Acott T. S. (1993) Regulation of the levels of human trabecular matrix metalloproteinases and inhibitor by interleukin-1 and dexamethasone. Invest. Ophthalmol. Vis. Sci. 34, 3386–3395 [PubMed] [Google Scholar]
- 21. Snyder R. W., Stamer W. D., Kramer T. R., and Seftor R. E. (1993) Corticosteroid treatment and trabecular meshwork proteases in cell and organ culture supernatants. Exp. Eye Res. 57, 461–468 10.1006/exer.1993.1148 [DOI] [PubMed] [Google Scholar]
- 22. el-Shabrawi Y., Eckhardt M., Berghold A., Faulborn J., Auboeck L., Mangge H., and Ardjomand N. (2000) Synthesis pattern of matrix metalloproteinases (MMPs) and inhibitors (TIMPs) in human explant organ cultures after treatment with latanoprost and dexamethasone. Eye (Lond.) 14, 375–383 [DOI] [PubMed] [Google Scholar]
- 23. Overby D. R., and Clark A. F. (2015) Animal models of glucocorticoid-induced glaucoma. Exp. Eye Res. 141, 15–22 10.1016/j.exer.2015.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mao W., Tovar-Vidales T., Yorio T., Wordinger R. J., and Clark A. F. (2011) Perfusion-cultured bovine anterior segments as an ex vivo model for studying glucocorticoid-induced ocular hypertension and glaucoma. Invest. Ophthalmol. Vis. Sci. 52, 8068–8075 10.1167/iovs.11-8133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zode G. S., Sharma A. B., Lin X., Searby C. C., Bugge K., Kim G. H., Clark A. F., and Sheffield V. C. (2014) Ocular-specific ER stress reduction rescues glaucoma in murine glucocorticoid-induced glaucoma. J. Clin. Invest. 124, 1956–1965 10.1172/JCI69774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lütjen-Drecoll E. (2005) Morphological changes in glaucomatous eyes and the role of TGFβ2 for the pathogenesis of the disease. Exp. Eye Res. 81, 1–4 10.1016/j.exer.2005.02.008 [DOI] [PubMed] [Google Scholar]
- 27. Inatani M., Tanihara H., Katsuta H., Honjo M., Kido N., and Honda Y. (2001) Transforming growth factor-β2 levels in aqueous humor of glaucomatous eyes. Graefes Arch. Clin. Exp. Ophthalmol. 239, 109–113 10.1007/s004170000241 [DOI] [PubMed] [Google Scholar]
- 28. Agarwal P., Daher A. M., and Agarwal R. (2015) Aqueous humor TGF-β2 levels in patients with open-angle glaucoma: A meta-analysis. Mol. Vis. 21, 612–620 [PMC free article] [PubMed] [Google Scholar]
- 29. Fleenor D. L., Shepard A. R., Hellberg P. E., Jacobson N., Pang I. H., and Clark A. F. (2006) TGFβ2-induced changes in human trabecular meshwork: Implications for intraocular pressure. Invest. Ophthalmol. Vis. Sci. 47, 226–234 10.1167/iovs.05-1060 [DOI] [PubMed] [Google Scholar]
- 30. Wordinger R. J., Fleenor D. L., Hellberg P. E., Pang I. H., Tovar T. O., Zode G. S., Fuller J. A., and Clark A. F. (2007) Effects of TGF-β2, BMP-4, and gremlin in the trabecular meshwork: Implications for glaucoma. Invest. Ophthalmol. Vis. Sci. 48, 1191–1200 10.1167/iovs.06-0296 [DOI] [PubMed] [Google Scholar]
- 31. Sethi A., Jain A., Zode G. S., Wordinger R. J., and Clark A. F. (2011) Role of TGFβ/Smad signaling in gremlin induction of human trabecular meshwork extracellular matrix proteins. Invest. Ophthalmol. Vis. Sci. 52, 5251–5259 10.1167/iovs.11-7587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tovar-Vidales T., Fitzgerald A. M., Clark A. F., and Wordinger R. J. (2013) Transforming growth factor-β2 induces expression of biologically active bone morphogenetic protein-1 in human trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 54, 4741–4748 10.1167/iovs.13-12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Welge-Lüssen U., May C. A., and Lütjen-Drecoll E. (2000) Induction of tissue transglutaminase in the trabecular meshwork by TGF-β1 and TGF-β2. Invest. Ophthalmol. Vis. Sci. 41, 2229–2238 [PubMed] [Google Scholar]
- 34. Guo X., and Wang X. F. (2009) Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 19, 71–88 10.1038/cr.2008.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shepard A. R., Millar J. C., Pang I. H., Jacobson N., Wang W. H., and Clark A. F. (2010) Adenoviral gene transfer of active human transforming growth factor-β2 elevates intraocular pressure and reduces outflow facility in rodent eyes. Invest. Ophthalmol. Vis. Sci. 51, 2067–2076 10.1167/iovs.09-4567 [DOI] [PubMed] [Google Scholar]
- 36. McDowell C. M., Tebow H. E., Wordinger R. J., and Clark A. F. (2013) Smad3 is necessary for transforming growth factor-β2 induced ocular hypertension in mice. Exp. Eye Res. 116, 419–423 10.1016/j.exer.2013.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song C. Z., Tian X., and Gelehrter T. D. (1999) Glucocorticoid receptor inhibits transforming growth factor-β signaling by directly targeting the transcriptional activation function of Smad3. Proc. Natl. Acad. Sci. U.S.A. 96, 11776–11781 10.1073/pnas.96.21.11776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schwartze J. T., Becker S., Sakkas E., Wujak Ł. A., Niess G., Usemann J., Reichenberger F., Herold S., Vadász I., Mayer K., Seeger W., and Morty R. E. (2014) Glucocorticoids recruit Tgfbr3 and Smad1 to shift transforming growth factor-β signaling from the Tgfbr1/Smad2/3 axis to the Acvrl1/Smad1 axis in lung fibroblasts. J. Biol. Chem. 289, 3262–3275 10.1074/jbc.M113.541052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pan X. Y., Wang Y., Su J., Huang G. X., Cao D. M., Qu S., and Lu J. (2015) The mechanism and significance of synergistic induction of the expression of plasminogen activator inhibitor-1 by glucocorticoid and transforming growth factor beta in human ovarian cancer cells. Mol. Cell. Endocrinol. 407, 37–45 10.1016/j.mce.2015.03.005 [DOI] [PubMed] [Google Scholar]
- 40. Bolkenius U., Hahn D., Gressner A. M., Breitkopf K., Dooley S., and Wickert L. (2004) Glucocorticoids decrease the bioavailability of TGF-β which leads to a reduced TGF-β signaling in hepatic stellate cells. Biochem. Biophys. Res. Commun. 325, 1264–1270 10.1016/j.bbrc.2004.10.164 [DOI] [PubMed] [Google Scholar]
- 41. Bollinger K. E., Crabb J. S., Yuan X., Putliwala T., Clark A. F., and Crabb J. W. (2012) Proteomic similarities in steroid responsiveness in normal and glaucomatous trabecular meshwork cells. Mol. Vis. 18, 2001–2011 [PMC free article] [PubMed] [Google Scholar]
- 42. Khalil N. (1999) TGF-β: From latent to active. Microbes Infect. 1, 1255–1263 10.1016/S1286-4579(99)00259-2 [DOI] [PubMed] [Google Scholar]
- 43. Tovar-Vidales T., Clark A. F., and Wordinger R. J. (2011) Transforming growth factor-β2 utilizes the canonical Smad-signaling pathway to regulate tissue transglutaminase expression in human trabecular meshwork cells. Exp. Eye Res. 93, 442–451 10.1016/j.exer.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kasetti R. B., Maddineni P., Millar J. C., Clark A. F., and Zode G. S. (2017) Increased synthesis and deposition of extracellular matrix proteins leads to endoplasmic reticulum stress in the trabecular meshwork. Sci. Rep. 7, 14951 10.1038/s41598-017-14938-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lange P. S., Chavez J. C., Pinto J. T., Coppola G., Sun C. W., Townes T. M., Geschwind D. H., and Ratan R. R. (2008) ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 205, 1227–1242 10.1084/jem.20071460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T., and Springer T. A. (2011) Latent TGF-β structure and activation. Nature 474, 343–349 10.1038/nature10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tripathi R. C., Li J., Chan W. F., and Tripathi B. J. (1994) Aqueous humor in glaucomatous eyes contains an increased level of TGF-β 2. Exp. Eye Res. 59, 723–727 10.1006/exer.1994.1158 [DOI] [PubMed] [Google Scholar]
- 48. Lawson W. E., Cheng D. S., Degryse A. L., Tanjore H., Polosukhin V. V., Xu X. C., Newcomb D. C., Jones B. R., Roldan J., Lane K. B., Morrisey E. E., Beers M. F., Yull F. E., and Blackwell T. S. (2011) Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. U.S.A. 108, 10562–10567 10.1073/pnas.1107559108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tanjore H., Lawson W. E., and Blackwell T. S. (2013) Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim. Biophys. Acta 1832, 940–947 10.1016/j.bbadis.2012.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Malhi H., and Kaufman R. J. (2011) Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809 10.1016/j.jhep.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H. P., and Ron D. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 10.1101/gad.1250704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peters J. C., Bhattacharya S., Clark A. F., and Zode G. S. (2015) Increased endoplasmic reticulum stress in human glaucomatous trabecular meshwork cells and tissues. Invest. Ophthalmol. Vis. Sci. 56, 3860–3868 10.1167/iovs.14-16220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zimmerman K. A., Graham L. V., Pallero M. A., and Murphy-Ullrich J. E. (2013) Calreticulin regulates transforming growth factor-β-stimulated extracellular matrix production. J. Biol. Chem. 288, 14584–14598 10.1074/jbc.M112.447243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Annes J. P., Munger J. S., and Rifkin D. B. (2003) Making sense of latent TGFβ activation. J. Cell Sci. 116, 217–224 10.1242/jcs.00229 [DOI] [PubMed] [Google Scholar]
- 55. Murphy-Ullrich J. E., and Poczatek M. (2000) Activation of latent TGF-β by thrombospondin-1: Mechanisms and physiology. Cytokine Growth Factor Rev. 11, 59–69 10.1016/S1359-6101(99)00029-5 [DOI] [PubMed] [Google Scholar]
- 56. Flugel-Koch C., Ohlmann A., Fuchshofer R., Welge-Lussen U., and Tamm E. R. (2004) Thrombospondin-1 in the trabecular meshwork: Localization in normal and glaucomatous eyes, and induction by TGF-β1 and dexamethasone in vitro. Exp. Eye Res. 79, 649–663 10.1016/j.exer.2004.07.005 [DOI] [PubMed] [Google Scholar]
- 57. Haddadin R. I., Oh D. J., Kang M. H., Villarreal G. Jr., Kang J. H., Jin R., Gong H., and Rhee D. J. (2012) Thrombospondin-1 (TSP1)-null and TSP2-null mice exhibit lower intraocular pressures. Invest. Ophthalmol. Vis. Sci. 53, 6708–6717 10.1167/iovs.11-9013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wickert L., Chatain N., Kruschinsky K., and Gressner A. M. (2007) Glucocorticoids activate TGF-β induced PAI-1 and CTGF expression in rat hepatocytes. Comp. Hepatol. 6, 5 10.1186/1476-5926-6-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kimura H., Li X., Torii K., Okada T., Kamiyama K., Mikami D., Kasuno K., Takahashi N., and Yoshida H. (2011) Glucocorticoid enhances hypoxia- and/or transforming growth factor-β-induced plasminogen activator inhibitor-1 production in human proximal renal tubular cells. Clin. Exp. Nephrol. 15, 34–40 10.1007/s10157-010-0351-7 [DOI] [PubMed] [Google Scholar]
- 60. Wen F. Q., Kohyama T., Sköld C. M., Zhu Y. K., Liu X., Romberger D. J., Stoner J., and Rennard S. I. (2003) Glucocorticoids modulate TGF-β production by human fetal lung fibroblasts. Inflammation 27, 9–19 10.1023/A:1022683010976 [DOI] [PubMed] [Google Scholar]
- 61. Zhang X., Clark A. F., and Yorio T. (2005) Regulation of glucocorticoid responsiveness in glaucomatous trabecular meshwork cells by glucocorticoid receptor-β. Invest. Ophthalmol. Vis. Sci. 46, 4607–4616 10.1167/iovs.05-0571 [DOI] [PubMed] [Google Scholar]
- 62. Jain A., Wordinger R. J., Yorio T., and Clark A. F. (2014) Role of the alternatively spliced glucocorticoid receptor isoform GRβ in steroid responsiveness and glaucoma. J. Ocul. Pharmacol. Ther. 30, 121–127 10.1089/jop.2013.0239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang X., Letterio J. J., Lechleider R. J., Chen L., Hayman R., Gu H., Roberts A. B., and Deng C. (1999) Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 18, 1280–1291 10.1093/emboj/18.5.1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhu Y., Richardson J. A., Parada L. F., and Graff J. M. (1998) Smad3 mutant mice develop metastatic colorectal cancer. Cell 94, 703–714 10.1016/S0092-8674(00)81730-4 [DOI] [PubMed] [Google Scholar]
- 65. Pang I. H., Shade D. L., Clark A. F., Steely H. T., and DeSantis L. (1994) Preliminary characterization of a transformed cell strain derived from human trabecular meshwork. Curr. Eye Res. 13, 51–63 10.3109/02713689409042398 [DOI] [PubMed] [Google Scholar]
- 66. Clark A. F., Brotchie D., Read A. T., Hellberg P., English-Wright S., Pang I. H., Ethier C. R., and Grierson I. (2005) Dexamethasone alters F-actin architecture and promotes cross-linked actin network formation in human trabecular meshwork tissue. Cell Motil. Cytoskeleton 60, 83–95 10.1002/cm.20049 [DOI] [PubMed] [Google Scholar]
- 67. Stamer W. D., and Clark A. F. (2017) The many faces of the trabecular meshwork cell. Exp. Eye Res. 158, 112–123 10.1016/j.exer.2016.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kasetti R. B., P T., Millar J. C., and Zode G. S. (2016) Expression of mutant myocilin induces abnormal intracellular accumulation of selected extracellular matrix proteins in the trabecular meshwork. Invest. Ophthalmol. Vis. Sci. 57, 6058–6069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang W. H., Millar J. C., Pang I. H., Wax M. B., and Clark A. F. (2005) Noninvasive measurement of rodent intraocular pressure with a rebound tonometer. Invest. Ophthalmol. Vis. Sci. 46, 4617–4621 10.1167/iovs.05-0781 [DOI] [PubMed] [Google Scholar]
- 70. Tovar-Vidales T., Fitzgerald A. M., and Clark A. F. (2016) Human trabecular meshwork cells express BMP antagonist mRNAs and proteins. Exp. Eye Res. 147, 156–160 10.1016/j.exer.2016.05.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.