

Abstract
Human β-tryptase, a tetrameric trypsin-like serine protease, is an important mediator of the allergic inflammatory responses in asthma. During acute hypersensitivity reactions, mast cells degranulate, releasing active tetramer as a complex with proteoglycans. Extensive efforts have focused on developing therapeutic β-tryptase inhibitors, but its unique activation mechanism is less well-explored. Tryptase is active only after proteolytic removal of the pro-domain followed by tetramer formation via two distinct symmetry-related interfaces. We show that the cleaved I16G mutant cannot tetramerize, likely due to impaired insertion of its N terminus into its “activation pocket,” indicating allosteric linkage at multiple sites on each protomer. We engineered cysteines into each of the two distinct interfaces (Y75C for small or I99C for large) to assess the activity of each tetramer and disulfide-locked dimer. Using size-exclusion chromatography and enzymatic assays, we demonstrate that the two large tetramer interfaces regulate enzymatic activity, elucidating the importance of this protein–protein interaction for allosteric regulation. Notably, the I99C large interface dimer is active, even in the absence of heparin. We show that a monomeric β-tryptase mutant (I99C*/Y75A/Y37bA, where C* is cysteinylated Cys-99) cannot form a dimer or tetramer, yet it is active but only in the presence of heparin. Thus heparin both stabilizes the tetramer and allosterically conditions the active site. We hypothesize that each β-tryptase protomer in the tetramer has two distinct roles, acting both as a protease and as a cofactor for its neighboring protomer, to allosterically regulate enzymatic activity, providing a rationale for direct correlation of tetramer stability with proteolytic activity.
Keywords: allosteric regulation, enzyme mechanism, heparin-binding protein, protein engineering, protein–protein interaction, serine protease, tryptase, tetramerization
Introduction
Human tryptases are trypsin-like serine proteases that are predominantly found in the secretory granules of mast cells, which likely play a critical role in inflammation and host defense (1–4). However, mast cell activation through IgE receptor cross-linking and subsequent secretion of mediators, including tryptase, has also been linked to a number of diseases such as anaphylaxis and asthma as well as nonallergic disorders such as arthritis, inflammatory bowel disease, and bacterial infections (1, 5). In humans, there are four groups of tryptases: α (subtypes I and II), β (subtypes I, II, and III), the membrane-anchored γ (subtypes I and II), and δ (subtypes I and II) (2, 6, 7), although only the α and β groups are preferentially and abundantly expressed in mast cells and stored in their secretory granules. The βI and βII isoforms are extremely similar in sequence, substrate preference, and enzymatic activity (8).
Active β-tryptase is unique in its architecture. It is a noncovalent homotetramer and only fully active as such compared with monomeric trypsin or most other serine proteases (9, 10). The activation of β-tryptase requires the canonical pro-domain removal and subsequent tetramerization, which happens inside the mast cell at pH 5–6 in the presence of serglycin proteoglycan carrying heparin glycosaminoglycan, whereas glycosaminoglycans of lesser negative charge densities (chondroitin sulfates A, B, C, and E) do not support the maturation of β-protryptase to active β-tryptase tetramers (11–14). The crystal structure of human β-tryptase revealed a frame-like, toroidal quaternary structure with a quasi 2-fold symmetry among the four individual protomers, ABCD; there are also positively charged surfaces implicated in heparin binding that span across the two protomer pairs with small interfaces (A:B and C:D), whereas the large interfaces between B:C and D:A lack such a spanning heparin-binding groove (15). The active sites are inside the tetramer and face toward the center of the pore.
Because of the dimensions of the pore entrance (15 × 40 Å), access is limited to small peptides and loops from larger proteins that are flexible enough to enter the pore for cleavage. Substrates are diverse and include VIP, PAR2, pro-stromelysin, pro-urokinase, fibrinogen, cathelicidin, and kininogen, which lead to biological pathway activation in some cases and inhibition in others (16–22). Interestingly, the pore diameter also excludes any circulating endogenous protease inhibitors such as aprotinin, α1-antitrypsin, and α2-macroglobulin, which are too large to access the active sites (23). To date, the only known protein inhibitors of tetrameric β-tryptase are from ticks and leeches (24–26).
Substrate specificity as well as regulation of enzymatic activity are quite different for tetrameric β-tryptase compared with other monomeric serine proteases. This gives tetrameric β-tryptase the ability to cleave a selected set of substrates based on size and P1 selectivity (Arg and Lys) (8) in an environment where endogenous protease inhibitors are present. Dissociation of the tetramer into individual monomers at neutral pH (pH 7.4), facilitated by natural heparin-binding proteins such as antithrombin III (23, 27), and the low concentrations of heparin found in the extracellular environment lead to a dramatic loss in protease activity (27–29). However, monomeric β-tryptase from dissociated tetramer has also been reported to have heparin-dependent proteolytic activity, which can be inhibited by aprotinin, soybean trypsin inhibitor, antithrombin III, and α2-macroglobulin (30–35). A major liability of the published monomeric tryptase data has been the inability to totally exclude the possibility of re-tetramerization, even if only a small fraction. β-Tryptase has also been reported to remain monomeric when bound to anti-tryptase B12 antibody even in the presence of heparin, where enzymatic activity was detected at acidic but not neutral pH (33).
Heparin stabilizes the tetramer complex, affecting both its half-life and enzymatic activity (27, 36). Specific binding sites have been suggested to reside on a continuous positively charged surface stretching along two sides of the tetramer while bridging the small interfaces (37), and several key histidines in this region are critical for pH-dependent tryptase activation (38). Structural superposition of a single protomer from the tetramer complex structure with monomeric active trypsin shows that the overall fold of the two proteases is very similar, except for six surface loops (147s, 70–80s, 37s, 60s, 97s, and 173s loops) arranged around the active site that differ in length and conformation (37); chymotrypsinogen numbering is used throughout (42). Other than their role in forming the active-site cleft, these loops establish the protein–protein contact surfaces between neighboring protomers in the tetramer complex. However, these structural differences provide no obvious explanation why β-tryptase, despite its structural similarity to trypsin, is inactive as a monomer after pro-domain removal and insertion of the N terminus into the canonical “activation pocket” (39–41).
The allosteric connections between full enzymatic activity and the tetrameric state are not fully understood on a molecular level. The two types of protein interfaces involved in tetramer formation, designated the small (500 Å2) and the large (1100 Å2) interfaces, include surface loops that are part of the so-called protease “activation domain” (15, 39). These loops participate in a specific structural rearrangement when transitioning from zymogen to active protease. Here, we engineered stable β-tryptase monomers and dimers that are unable to form tetramers and determined their activities in the presence or absence of heparin, elucidating an additional role for heparin as an allosteric activator. We determined the individual contributions of the small and large interfaces in the tetramer with respect to enzyme activation to understand the complex allosteric relationships between the individual protomers in the tetramer complex.
Results
Individual protomers in the β-tryptase tetramer are active
We wanted to explore the noncatalytic roles of neighboring protomers as well as determine the specific activity of an individual protomer within a tetramer. We expressed and purified monomeric versions of human wildtype (WT) and S195A (chymotrypsinogen numbering is used throughout (42)) zymogens from baculovirus-infected Trichoplusia ni insect cells using Ni-NTA2 chromatography and size-exclusion chromatography (SEC); the S195A mutant is catalytically incompetent in its protease form. We then mixed zymogens of WT and S195A at four respective molar ratios of 4:0, 3:1, 2:2, and 1:3. After activation cleavage with enterokinase (EK) to remove the pro-domain in the presence of heparin, we isolated tetramers of these individual mixtures by SEC, all having identical elution profiles. Because the only difference between the protomers in the tetramer is Ser-195 or Ala-195 at the active site, no bias is expected in the tetramer formation process because the binding interfaces of WT and S195A are identical.
On average, these active tetramers respectively contain 4, 3, 2, or 1 WT and 0, 1, 2, or 3 S195A mutant protomers per tetramer (Fig. 1A). We measured the enzymatic activity and found that tetramers with WT to S195A zymogen mixing ratios (RatioZM, the zymogen mixing ratio) of 3:1, 2:2, and 1:3 had Vmax values relative to the WT tetramer of 72, 52, and 32%, respectively (Fig. 1, B and C). It is important to note that each of the heterotetramers is actually composed of a mixture of five types, with WT to S195A protomer ratios of 4:0, 3:1, 2:2, and 1:3 and 0:4, that will have fractional proportions based on a binomial distribution of protomers (Table 1). For the tryptase heterotetramer that comprises two WT and two S195A mutant protomers, there are actually three geometrically distinct subtypes due to the 2-fold symmetry of the tetramer. Using an alternative “binomial” linear regression analysis method (see supporting note), estimates of Vmax for purely homogeneous populations for each of the individual heterotetramer types were generated, where similar trends were in general agreement with the data in Fig. 1C (see Table S1 and Fig. S1).
Figure 1.

Activity of tetrameric β-tryptase with different WT to S195A protomer ratios. A, cartoon depicting the generation of β-tryptase tetramers following enterokinase cleavage of WT and S195A zymogens at four different zymogen mixing ratios (RatioZM). The heterotetramers are actually a mixture of individual tetramer species weighted according to their binomial distribution (Table 1). B, comparison of the four β-tryptase tetramers with different protomer ratios at 1 nm measured with the chromogenic substrate S-2288. Data were collected in triplicate and fit to the Michaelis-Menten equation; errors are shown as S.D. C, comparison of Vmax and Km values of the different β-tryptase tetramer mixtures with WT; Vmax and Km were normalized to 100% for WT; errors are shown as S.D.
Table 1.
Binomial distribution of specific types of β-tryptase tetramers with WT and/or S195A protomers at different zymogen mixing ratios
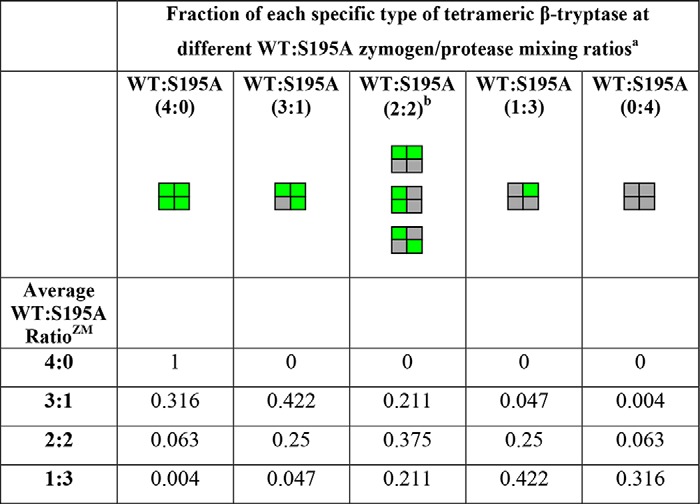
a WT protomers are shown in green and S195A protomers are shown in gray.
b There are three distinct possible geometric subtypes in tetramers that contain two WT protomers and two S195A protomers.
The linear loss in Vmax activity upon increasing S195A protomers indicates that each WT protomer is active even when bound to inactive protomers in the heterotetrameric complexes. In contrast to the Vmax activity, the normalized Km values showed relatively minor reductions to 94, 88, and 71% of WT tetramer for the respective heterotetrameric β-tryptase complexes with RatioZM values of WT to S195A of 3:1, 2:2, and 1:3 (Fig. 1C). Thus, the active-site conformation was minimally affected by the number of inactive mutant protomers in the tetramer.
N-terminal insertion into the activation pocket is required for tetramer formation
Because conversion of serine protease zymogens to active proteases requires cleavage of the pro-domain and subsequent insertion of the newly formed N terminus into the so-called activation pocket (39–41, 43), we wanted to see whether this was important for the tryptase tetramer formation as well. We expressed and purified zymogen β-tryptase mutant I16G as described above. We then subjected it to EK cleavage in the presence of heparin at pH 6.8 to remove the pro-domain, which promotes the formation of β-tryptase tetramers. The removal of the pro-domain and the correct newly formed N terminus (GVGG) was confirmed by Edman degradation. As shown in Fig. 2, WT β-tryptase migrates as a tetramer. However, the I16G N-terminal insertion mutant does not, instead migrating as a monomer with a comparable retention time to the β-tryptase zymogen. Consistent with this finding, truncation of Ile-16 after the pro-domain removal also abrogates tetramer formation under the same conditions (data not shown). We also carried out steered molecular dynamic simulations of the WT protomer and the I16G protomer to investigate dissociation of the N terminus from the activation pocket. Under conditions where an external force was applied to the N terminus to “pull it out” of the pocket, preliminary data show that the difference in energy required was significantly less for the I16G protomer.3 Taken together, this supports the notion that N-terminal insertion and tetramer formation are allosterically linked.
Figure 2.
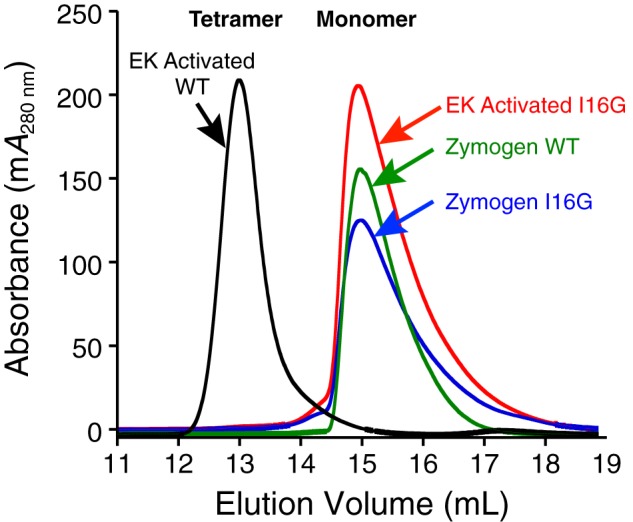
Size-exclusion chromatography of WT and I16G β-tryptase zymogens and proteases. WT β-tryptase forms tetramers after activation and pro-domain removal by EK in the presence of 0.5 mg/ml heparin. WT tetramer had an Ve of 13.0 ml on an S200GL column in SEC buffer. Following identical pro-domain removal by EK with heparin, I16G β-tryptase has a Ve of 15.0 ml in SEC buffer, which is essentially identical to that observed for zymogens of WT and I16G β-tryptase.
Engineered disulfide-linked β-tryptase dimers at the small and large interfaces are active
We next wanted to study the role of neighboring protomers in greater detail to determine how they may affect activity. The small interface (500 Å2) of the tetramer comprises hydrophobic interactions mediated largely by prolines and tyrosines, whereas the large interface (1100 Å2) of the tetramer contains both hydrophobic and ionic interactions. To determine which of the two interfaces is more important for driving coactivation of neighboring β-tryptase protomers, we engineered two β-tryptase mutants that covalently cross-link two protomers in a tetramer between either the small or the large interface. In the small interfaces, Tyr-75 in protomers A and D are proximal to Tyr-75 in protomers B and C, respectively (Fig. 3) (15, 37). In the large interfaces, Ile-99 in protomers A and B are proximal to Ile-99 in protomers C and D, respectively (Fig. 3). Models of Y75C and I99C tryptase mutants show respective distances of 2.4 and 3.2 Å between the thiols of each opposing cysteine (Fig. 3), which are somewhat greater than a typical disulfide bond length of 2.05 Å.
Figure 3.

Disulfide engineering of β-tryptase dimers and tetramers. Tetrameric β-tryptase was engineered such that two of the four protomer interfaces were covalently linked by a disulfide bond. PyMOL was used to measure the distances between the two proposed thiols of Y75C in the small protomer interfaces (A:B and C:D) or I99C in the large protomer interfaces (A:D and B:C). Tetrameric β-tryptase having a mutation at either Y75C or I99C could only dissociate into two distinct disulfide-linked dimers as indicated in the cartoon. The red spheres are the oxygen atoms of Tyr-75. Protomer nomenclature corresponds to that described by Pereira et al. (15).
The Y75C and I99C β-tryptase mutants were individually expressed and purified as zymogens. During the purification procedure, two forms for each mutant were detected by SEC corresponding to a dimeric and a monomeric form (data not shown). Following pro-domain removal of the dimeric form with EK in the presence of heparin and subsequent purification by SEC, we isolated the tetrameric forms, which all eluted at 13.0 ml (Fig. 4A). Analysis of tetramers by SDS-PAGE under nonreducing conditions showed WT migrating as a monomer, whereas the Y75C and I99C mutants migrated as dimers (Fig. 4B). These dimers collapsed to the monomers when analyzed by SDS-PAGE under reducing conditions, consistent with an intermolecular disulfide bond between the two monomers.
Figure 4.

Characterization of WT, Y75C, and I99C β-tryptases. A, size-exclusion chromatography of tetrameric WT, Y75C, and I99C β-tryptases was carried out as in Fig. 2 on an S200GL column in SEC buffer; the dimeric zymogens of Y75C and I99C β-tryptases were activated and then run on SEC. Elution volumes for the tetrameric peaks were 13.0 ml, which were pooled separately. B, SDS-PAGE of pooled tetrameric WT, Y75C, and I99C β-tryptases under nonreducing (NR) and reducing (R) conditions. C, enzymatic activity of tetrameric WT, Y75C, and I99C β-tryptases at 1 nm concentration using S-2288 as a substrate. Data were collected in triplicate and fit to the Michaelis-Menten equation; errors are shown as S.D.
Both of the tetrameric Y75C and I99C mutants were enzymatically active. The respective catalytic efficiencies (kcat/Km) for the Y75C and I99C tetramers were 63 and 33% of that of tetrameric WT β-tryptase (Fig. 4C and Table 2).
Table 2.
Kinetic parameters for WT, Y75C, and I99C β-tryptase tetramers, I99C/Y75A/Y37bA dimer, and I99C*/Y75A/Y37bA monomer
Assays were run as described under “Experimental procedures” in the presence of 100 μg/ml heparin. Data were analyzed as described under “Experimental procedures.” Kinetic constants are mean ± S.E.
| β-Tryptase | Statea | Km | kcatb | kcat/Kmb | Mutant/WT catalytic efficiencyb,c |
|---|---|---|---|---|---|
| μm | s−1 | m−1 s−1 | |||
| WTd | T | 470 ± 60 | 87 ± 10 | 1.8 ± 0.1 × 105 | 1 |
| Y75Cd | T | 320 ± 50 | 37 ± 10 | 1.2 ± 0.2 × 105 | 0.63 |
| I99Cd | T | 820 ± 30 | 50 ± 10 | 0.61 ± 0.1 × 105 | 0.33 |
| I99C/Y75A/Y37bAe | D | 1000 ± 80 | 5.8 ± 0.3 | 0.06 ± 0.04 × 105 | 0.029 |
| I99C*/Y75A/Y37bAf | M | 1600 ± 300 | 6.7 ± 0.7 | 0.04 ± 0.02 × 105 | 0.021 |
a Data refer to oligomeric state when assayed; T is tetramer; D is dimer; M is monomer.
b Data were normalized to monomer (protomer).
c Refers to kcat/Km (mutant)/kcat/Km (WT).
d Concentrations of these tetrameric tryptases were 1 nm; protomer concentrations were 4 nm.
e Concentrations of dimeric tryptase were 100 nm; protomer concentrations were 200 nm.
f Concentrations of monomeric tryptase were 400 nm.
Determination of inter-protomer disulfides in the Y75C and I99C disulfide-linked dimers
To determine that the desired engineered disulfides had indeed formed, we used MS and X-ray crystallography. The correct formation of the inter-protomer disulfide bond in the Y75C mutant, in addition to the canonical WT intramolecular disulfide bonds, was confirmed by MS after tryptic digest of the dimer under nonreducing conditions. Nonreduced MALDI showed a peak at 3655 Da (mass of two EQHLYCQDQLLPVSR, 2 Da for disulfide formation) (data not shown). Upon reduction, the 3655-Da band was not present, and the presence of the single EQHLYCQDQLLPVSR peptide appears, consistent with the formation of a Cys-75–Cys-75 dimer. Mass spectrometric analysis for the intermolecular disulfide for the I99C dimer was considerably more complex, and we pursued an alternative strategy.
The tetrameric I99C β-tryptase mutant was crystallized, and its structure was determined at 2.7 Å resolution by X-ray crystallography to assess the disulfides as well as any changes in structure that might explain activity differences with WT. The structure showed that the intermolecular disulfide bond between the Cys-99 residues in each dimer was present (Fig. 5 and Table 3); all the other intramolecular disulfides were indistinguishable from WT. The I99C β-tryptase crystallized in a unit cell nonisomorphous with any of the 23 most homologous entries in the PDB, all of which are β- and α-tryptase structures. Twenty nine residues of each tetramer have contact with neighboring tetramers within 3.5 Å. Pairwise protomer superpositions with WT human β-tryptase (PDB 1A0L) show r.m.s.d. for all Cα atoms of ∼0.45 Å. Nonetheless, there are small changes in the arrangement of protomers within the tetramer, as seen by the slightly larger r.m.s.d. of 0.64 Å upon superposition of the tetramers. Although we cannot definitively explain the lower activity of the I99C mutant compared with WT from the structure of the tetramer, it is possible this could result from restricting dynamic changes in subunit conformations or interactions.
Figure 5.

Crystal structure of tetrameric I99C β-tryptase mutant at 2.7 Å resolution. A, I99C β-tryptase mutant is shown in green and blue, with internal disulfides in yellow and the I99C disulfide in orange. Cysteine residues are shown as spheres for the disulfides; sulfur atoms are yellow for internal disulfides and orange for I99C disulfides. B, tetramer was rotated ∼90°; only one of the disulfide-locked dimers is shown. C, I99C protomer is superposed with one from the WT structure (1A0L), which is shown in beige. The catalytic triad residues are shown as sticks.
Table 3.
Crystallography data collection and refinement statistics for I99C βI-tryptase
| I99C βI-Tryptase | |
|---|---|
| Data | |
| X-ray source | APS 22-ID |
| Wavelength (Å) | 1.0000 |
| Resolution range (Å) | 50.01–2.72 (2.73–2.72) |
| Space group | P3221 |
| Unit cell a, b, c (Å) | 96.743 96.743 238.553 |
| Unit cell α, β, γ (°) | 90 90 120 |
| Total reflections | 387721 |
| Unique reflections | 35711 |
| Multiplicity | 10.9 (11.2) |
| Completeness (%) | 100 (100) |
| Mean I/σ(I) | 19.2 (2.2) |
| Wilson B-factor (Å2) | 60.03 |
| R-symm | 0.11 (1.25) |
| CC(1/2) | 0.999 (0.793) |
| Refinement | |
| Reflections for R-free | 1035 |
| Rwork | 0.193 |
| Rfree | 0.227 |
| No. non-H atoms | 7834 |
| Macromolecules | 7673 |
| Ligands | 100 |
| Ions | 35 |
| Water | 25 |
| Protein residues | 972 |
| r.m.s.d. (bonds) (Å) | 0.009 |
| r.m.s.d. (angles) (°) | 1.09 |
| Ramachandran favorable (%) | 95 |
| Average B-factor (Å2) | 59.2 |
| Macromolecules | 59.6 |
| Ligands | 70.8 |
| Ions | 123.8 |
| Water | 53.5 |
| PDB accession code | 5WI6 |
Dissociation and activity of Y75C and I99C β-tryptases and their disulfide-linked dimeric forms
We next investigated whether the disulfide-linked tetrameric Y75C and I99C mutants could be dissociated into dimers and, if so, whether they are still catalytically active. We used the Fab fragment of murine anti-tryptase antibody B12 (44), which has been reported to dissociate tetrameric β-tryptase into monomers upon binding at neutral pH (33). When tetrameric Y75C and I99C β-tryptase mutants were incubated with excess B12 Fab and the mixture was analyzed by SEC and subsequent SDS-PAGE of eluted protein peaks, we found that both mutants formed complexes with B12 (1 dimer and 2 Fab) and had the same elution volume (Fig. 6). For comparison, tetrameric WT β-tryptase in complex with excess B12 Fab had a larger elution volume, indicative of a smaller complex (1 monomer and 1 Fab). Tetrameric WT β-tryptase alone (four protomers) was also run as a control. In addition, we performed multiangle light–scattering experiments on the eluted protein complexes from the size-exclusion chromatography (SEC-multiangle light scattering) and found that both disulfide-linked β-tryptase mutants in complex with murine B12 Fab had a combined molecular mass of 147 kDa, consistent with a complex of one β-tryptase dimer bound to two Fabs. We concluded from these experiments that murine B12 Fab dissociates both tetrameric β-tryptase mutants into dimers in solution while remaining bound.
Figure 6.

Characterization of WT, Y75C, and I99C β-tryptases in complex with murine B12 Fab. A, size-exclusion chromatography of B12 Fab in complex with WT, Y75C, and I99C β-tryptases was carried out as in Fig. 2 on an S200GL column in SEC buffer. Chromatograms are colored as follows: WT tryptase tetramer is colored black (Peak 3; Ve = 13.0 ml); complexes of B12 Fab with WT are green (Peak 2; Ve = 14.3 ml), Y75C is blue (Peak 4; Ve = 12.1 ml), and I99C is red (Peak 5; Ve = 12.1 ml). Excess free B12 Fab in Peaks 1a,b,c all have a similar Ve = 15.7 ml. B, fractions of main peaks were analyzed by SDS-PAGE under nonreducing conditions.
We then determined and compared the enzymatic activity of Y75C and I99C β-tryptase mutants in the presence or absence of saturating B12 Fab as well as in the presence or absence of a fixed heparin concentration of 100 μg/ml using the chromogenic substrate S-2288 (Fig. 7). We found that tetrameric WT β-tryptase was almost completely inactive at saturating concentrations of B12 Fab independent of the presence or absence of heparin, in accord with previously described results (33). The Y75C β-tryptase mutant was also almost completely inactive in the presence of B12 but only in the absence of heparin. However, in the presence of heparin, the inhibition was incomplete, with 36% residual activity remaining at saturating B12 Fab concentrations (Fig. 7). The activity of the I99C β-tryptase mutant was only mildly affected by B12 Fab at saturating concentrations, with the dimer having 75% of the tetramer activity in the presence of heparin. However, it was quite striking that the I99C dimer retained 77% of its activity with B12 Fab in the absence of heparin, which is essential for both WT and the Y75C dimer. B12 Fab binding may have additional allosteric effects on activity due to its interaction with the dimer, distinct from its ability to dissociate the tetramer. In addition, the difference in the preservation of amidolytic activity by heparin for the Y75C β-tryptase mutant (35%) was also much greater compared with that for the I99C mutant (2%), which is essentially heparin-independent (Fig. 7). This is consistent with heparin having a much greater activation role on the small interface, even when it is locked with a disulfide bond. Overall, however, dimerization at the large interface is more important for activity enhancement than at the small interface.
Figure 7.
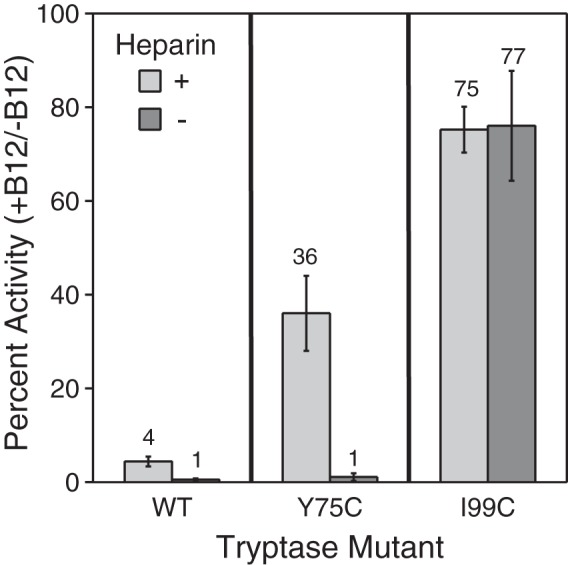
Activity of tryptase WT and disulfide-locked dimer mutants after dissociation with B12 Fab. The percent activity remaining of WT, Y75C, and I99C tryptase mutants (1 nm) in the presence of B12 Fab (125 nm) compared with its absence was determined in the presence (light gray) and absence (dark gray) of 0.1 mg/ml heparin. Tryptase variants were incubated with B12 Fab for 15 min at room temperature prior to activity measurements. The percent activity for each tryptase variant in the absence of B12 Fab and the presence and absence of 0.1 mg/ml heparin is defined as 100%. Data were collected in triplicate for two independent determinations; errors are shown as S.D.
Disulfide-linked β-tryptase dimers and a monomer that cannot tetramerize show an allosteric activation role for heparin
To eliminate any effects from the addition of B12 Fab while studying the effects of protomers on one another, we generated the β-tryptase triple mutant (I99C/Y75A/Y37bA), which keeps the disulfide-linked I99C dimer at the large interface but prevents subsequent tetramer formation via the small interface due to alanine mutation of two tyrosines in the small interface. As described above, we isolated β-tryptase dimer and monomer zymogens of the same mutant during protein purification, where the dimer is linked by the intermolecular disulfide bond. Serendipitously, we found monomeric fractions that were cysteinylated at I99C (I99C*), which prevents dimerization with another β-tryptase monomer via the large interface, whereas the mutations Y75A and Y37bA abrogate interactions via the small interface, thus preventing tetramer formation. β-Tryptase monomer and dimer were treated with n-isopropylacrylamide (NIPIA)–alkylating agent to cap the free cysteines under both reducing and nonreducing conditions prior to enzymatic peptide digestion. Bottom-up MS analyses of the chymotryptic peptide containing I99C (TAQCGADIALL) were searched with variable modifications for NIPIA (+99 Da) and cysteinylation (+119 Da) on cysteine residues. The β-tryptase monomer shows a high abundance of cysteinylation modifications under nonreducing conditions. In contrast, the dimer shows a much lower degree of cysteinylation, consistent with cysteinylation of the monomer preventing dimerization.
After pro-domain removal in the presence of heparin, both the dimeric and monomeric β-tryptase triple mutants were analyzed by SEC in the presence or absence of heparin (Fig. 8). In the absence of heparin in SEC buffer (10 mm MOPS, pH 6.8, 2 m NaCl), the elution volumes of the dimeric triple mutant and its cysteinylated I99C* monomeric form were larger than WT β-tryptase tetramer, consistent with their inability to form tetramers after pro-domain removal and with heparin present. In the presence of heparin in TNH buffer (50 mm Tris, pH 8, 150 mm NaCl, 0.1 mg/ml heparin, 0.02% NaN3), the triple mutant β-tryptase dimer and monomer had slightly smaller elution volumes compared to when heparin was absent. The heparin used in this experiment had an average molecular mass of 18 kDa, which explains the change in elution volumes of β-tryptase due to forming a complex with heparin. Both the dimer and the monomer had elution volumes larger than those of a tetramer bound to heparin. We conclude that the Y75A and Y37bA mutations prevent β-tryptase from interacting with another tryptase protomer via the small interface and that the β-tryptase dimer and cysteinylated I99C* monomer mutants remain homogeneously dimeric or monomeric, respectively, in solution, even in the presence of heparin.
Figure 8.
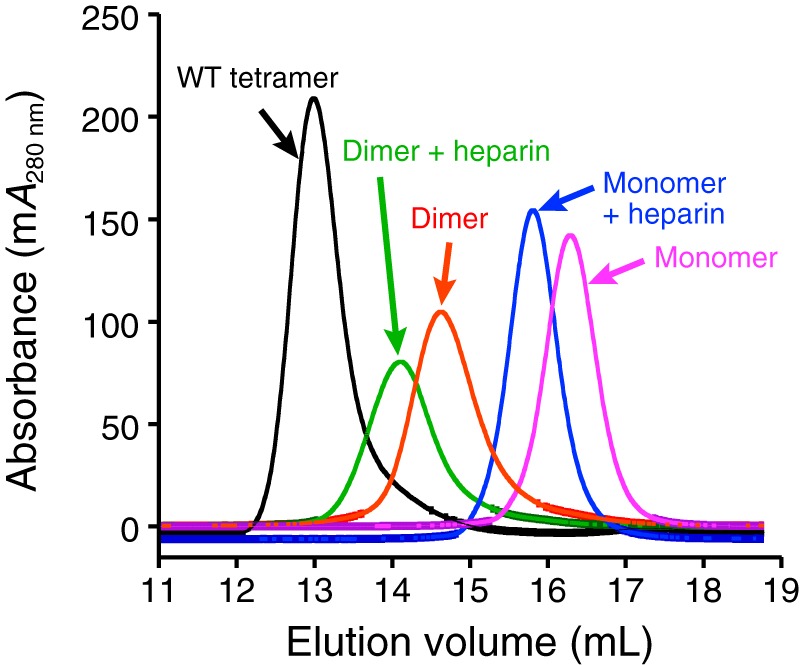
Size-exclusion chromatography of tetrameric WT, dimeric I99C/Y75A/Y37bA, and monomeric I99C*/Y75A/Y37bA β-tryptases in the presence or absence of heparin. After pro-domain removal by EK, proteins were analyzed on an S200GL column in either SEC buffer (without heparin) or in TNH buffer (with heparin). Elution volumes for WT tetramer without heparin (13.0 ml), I99C/Y75A/Y37bA dimer with heparin (14.1 ml), I99C/Y75A/Y37bA dimer without heparin (14.6 ml), I99C*/Y75A/Y37bA monomer with heparin (15.8 ml), and I99C*/Y75A/Y37bA monomer without heparin (16.3 ml) are as indicated in parentheses.
We determined the enzymatic activity of the activated dimeric β-tryptase triple mutant at different heparin and substrate concentrations, ranging from 0 to 100 μg/ml and 23.4 to 3000 μm, respectively. We found that the rate data for the engineered β-tryptase dimer mutant fit well to an allosteric sigmoidal equation for enzyme kinetics, where v = Vmax·[S]H/(K′ + [S]H) and H is the Hill coefficient (45), which differs from the hyperbolic response curves observed for the WT tetramer (Fig. 9A; Table 2). Sigmoidal enzyme kinetics are consistent with a mechanism of allosteric regulation and can be suitably described by the Monod-Wyman-Changeux model, where two identical subunits of a homodimeric protein complex allosterically activate one another in the presence of increasing amounts of substrate (46–48). In the case of β-tryptase, this positive allosteric effect seems to be transmitted through the interaction of the large interface. This effect can now be detected in the β-tryptase dimer mutant, because that area is covalently held together and the two protomers cannot dissociate. The average Hill slopes for all the curves in Fig. 9A are 1.54 ± 0.07 (± S.D.). A Hill coefficient greater than 1 suggests that the β-tryptase dimer has more than one substrate-binding site with positive cooperativity.
Figure 9.

Dependence of velocity on substrate and heparin for activated dimeric I99C/Y75A/Y37bA and monomeric I99C*/Y75A/Y37bA β-tryptase mutants. A, activated I99C/Y75A/Y37bA dimer data for the dependence of velocity with 2 mm S-2288 substrate at different heparin concentrations are shown. Data were collected in triplicate and fit to a sigmoidal equation. B, activated I99C/Y75A/Y37bA dimer data for the dependence of velocity on heparin at different S-2288 substrate concentrations are shown. C, activated I99C*/Y75A/Y37bA monomer data for the dependence of velocity on S-2288 substrate at different heparin concentrations are shown. D, activated I99C*:Y75A:Y37bA monomer data for the dependence of velocity on heparin at different S-2288 substrate concentrations are shown. Data for B, C, and D were each collected in triplicate and fit to hyperbolic binding equations and the 1:1 binding isotherm for B and D or Michaelis-Menten equation for C. Errors in all data are shown as S.D. Tryptase dimer and monomer concentrations were 100 and 400 nm, respectively.
In addition, the overall reaction velocity was enhanced with increasing amounts of heparin. This indicates that heparin must also have a positive allosteric effect on the active site on each protomer of the dimer, because tetramerization is eliminated due to the mutations in the small interface. When plotting the same data as a function of heparin concentration versus reaction velocity at different substrate concentrations, we found that the data best fit a hyperbolic response curve (Fig. 9B). This suggests that heparin may positively affect each protomer in the β-tryptase dimer individually in an allosteric fashion.
To further test this hypothesis, we used the activated cysteinylated monomer of this β-tryptase triple mutant, which can neither dimerize nor tetramerize in the presence of heparin, and we tested its enzymatic activity at different heparin and substrate concentrations, ranging from 0 to 100 μg/ml and 23.4 to 3000 μm, respectively (Fig. 9, C and D). When we plotted the reaction velocity versus the substrate concentration (Fig. 9C; Table 2), we detected hyperbolic response curves of the enzyme kinetics, unlike the sigmoidal response in the dimer (Fig. 9A). Furthermore, the reaction velocity of this β-tryptase monomer mutant increased proportionally with increasing concentrations of heparin, indicating that heparin is a positive allosteric activator of a β-tryptase monomer. The Km value for each hyperbolic activity curve was determined, which showed a trend of decreasing Km values at increasing heparin concentrations. When we plotted the same reaction velocities as a function of heparin concentration at different fixed substrate concentrations (Fig. 9D), hyperbolic response curves were observed. To ensure that the active monomeric β-tryptase triple mutant remained monomeric under these assay conditions, we tested its activity in the presence or absence of aprotinin and found that it can be completely inhibited at saturating concentrations of aprotinin. For comparison, the chromogenic activity of WT tetrameric β-tryptase cannot be inhibited by aprotinin (Fig. 10). Because of its size, aprotinin cannot enter the pore of the tetramer and access the active sites, but it can bind to the active sites of monomeric tryptase (33). In summary, we conclude that heparin allosterically conditions the active site for better substrate binding. As discussed below, it is important to note that the allosteric kinetics observed herein can also be described by the general modifier mechanism, first described by Botts and Morales (49) and significantly expanded upon by Baici (50).
Figure 10.
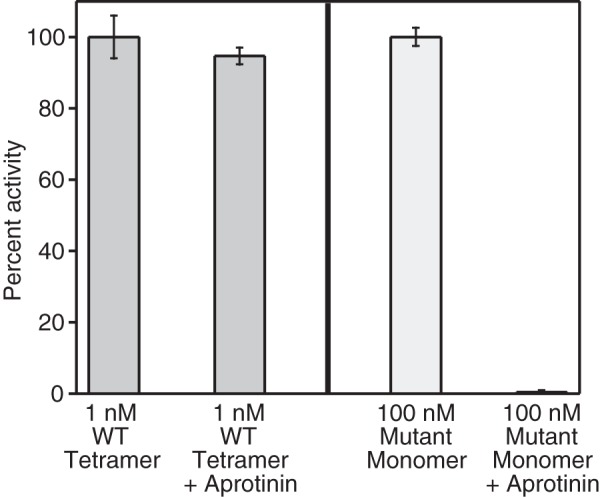
Aprotinin only inhibits monomeric β-tryptase. Relative enzymatic activity of tetrameric WT and monomeric mutant (I99C*/Y75A/Y37bA) β-tryptases in the presence or absence of 10 μm aprotinin is shown. Activities were determined in TNH buffer with 1 mm S-2288. Tetramer activity was assayed at 1 nm and normalized to 1 nm WT; monomer activity was assayed at 100 nm and normalized to 100 nm mutant monomer. Data were collected in triplicate; errors are shown as S.D.
Allosteric linkage between protomers at the large interface
To further identify and investigate key interactions of the large interface important for transducing allosteric activation, we chose to focus on the 60s and 170s loops, both of which have the largest number of residue insertions (5 and 9, respectively), when ranking loop differences between tryptase and trypsin (37). We generated a tetrameric Y173dA β-tryptase mutant, because Y173d resides at the outer tip of the 170s loop and interacts with the extended 60s loop of the neighboring protomer, placing the side chain in a hydrophobic pocket created by Val-60c, Val-90, Ile-88, Val-59, and Gly-60 (Fig. 11A). In close proximity to the 60s loop is the essential catalytic His-57. We produced the Y173dA mutant, which could form tetramers, but tetramer enzymatic activity was reduced by ∼40% when compared with WT β-tryptase under identical conditions (Fig. 11B). The Y173dA tetramer shows a 43% reduction in Vmax when compared with that for WT. Overall, the catalytic efficiency of the Y173dA mutant was reduced by 27%. The relative activity of mutant to WT (mutant/WT = 0.6 ± 0.1) remained constant over β-tryptase tetramer concentration ranging from 2 to 0.1 nm, thus eliminating the possibility that reduced activity of Y173dA stems from a significantly less stable tetramer complex (data not shown). Overall, these results show an allosteric effect of one protomer on its neighboring protomer that results in regulation of its proteolytic activity.
Figure 11.

Characterization of the Y173dA tetramer mutant. A, Y173d from one protomer in the large interface lies in a hydrophobic pocket of its neighboring protomer. It is sandwiched between Val-90 and Val-60c with surrounding Val-59, Gly-60, and Ile-88 hydrophobic residues; the 60s loop is relatively close to the catalytic His-57 (red). B, dependence of reaction velocity on S-2288 substrate for tetrameric WT β-tryptase and the Y173dA mutant. Data were collected in triplicate and fit to the Michaelis-Menten equation; errors are shown as S.D.
Discussion
We first sought to address the role of each protomer within the β-tryptase tetramer to understand the requirement for tetramers as the only active forms. By incorporating individual WT and inactive S195A mutant protomers into the tetramer at different ratios, we clearly see a strong dependence of activity that directly correlates with the molar ratio of WT protomers (Fig. 1). Under those experimental conditions, each WT protomer contributes ∼25% of the total amidolytic activity of the WT tetramer, whereas in striking contrast, the WT monomer itself has no amidolytic activity. From our combined data, we conclude that each protomer, whether WT or S195A mutant, can act as a coactivator or cofactor for their neighboring protomers in the tetrameric complex. The specific activity of a protomer in the tetrameric state is dramatically different when compared with its monomeric state (i.e. monomeric β-tryptase), which has essentially no activity at neutral pH in the absence of heparin (see below) and as reported previously (27, 28, 36, 51).
Insertion of the N-terminal residue 16 into the activation pocket after pro-domain removal, driven by a salt bridge between the positively charged N terminus and Asp-194 and hydrophobic interactions between the side chain of residue 16 (typically Ile or Val) and nonpolar residues found in the pocket, is a hallmark of trypsin-like serine protease activation (39–41, 43). It is coupled to conformational changes in the activation domain loops and the opening of the P1 specificity pocket to accept substrates. Mutation of the N-terminal WT residue to other residues or truncation preventing productive interactions with the activation domain pocket results in inactive serine proteases, despite pro-domain removal (43). The same requirement for N-terminal insertion is found for transactivation, for example in the streptokinase-dependent activation of plasminogen (52–54). Here, we also show that mutation of the corresponding residue in β-tryptase (I16G) directly abrogates the ability of monomers to form tetramers after pro-domain removal, rendering the β-tryptase inactive. Because many parts of the flexible and surface-exposed activation domain loops in β-tryptase are involved in making specific protein–protein interactions with other protomers to establish the quaternary tetramer structure, these data suggest that N-terminal insertion of Ile-16 into the activation domain pocket promotes and stabilizes a conformation of these activation domain loops that allows for successful tetramer formation. Our results are in accord with previous data suggesting that monomeric β-tryptase exists in a zymogen-like conformation (55). We propose that there is a powerful allosteric link between correct insertion of the N-terminal Ile-16 into the activation domain pocket and tetramerization of β-tryptase monomers.
Engineering disulfide-locked dimers at the small (Y75C) and large (I99C) interfaces led to several important results. First, we show that tetrameric versions of these locked dimers are still active, albeit somewhat less so than WT. It was somewhat surprising that the dimeric versions migrated with small, but discernable, changes in apparent sizes by SDS-PAGE analysis. The observation that the Y75C dimer runs as a slightly lower molecular weight form under nonreducing and denaturing conditions compared with the I99C dimer (Fig. 4B) suggests that the I99C dimer is less compact, consistent with I99C residing in a flexible loop, and likely resulting in a more elongated shape compared with the Y75C dimer. The collapse of the Y75C and I99C dimers to bands that migrate identically when reduced with DTT confirms they are both disulfide-linked dimers.
The lack of isomorphism between crystals of I99C and WT β-tryptase leads to intermolecular environments differing in a nontrivial way. This observation, coupled with the resolutions of the two structures (2.72 and 3.0 Å, respectively), leads us to conclude that there is no observable change caused by the I99C mutation. The Glu-Gly-Arg chloromethyl ketone inhibitor (ligand 0GJ in PDB nomenclature) is seen in six other PDB entries, all of them trypsin-like serine proteases. There is close similarity in how these inhibitors bind their protease-active sites, except among the distal glutamic acid components, which vary according to intermolecular contacts or details of the substrate-binding clefts. Despite the absence of significant structural alterations in I99C, the enzymatic activity of this mutant was lower than that of WT β-tryptase, which may be caused by rigidification of the complex due to the intermolecular disulfide link as discussed above.
We then analyzed the effects of B12 Fab on both the stability and activity of the tetramers. B12 Fab dissociates tetrameric WT β-tryptase into monomers upon binding at neutral pH (33). B12 Fab clearly dissociates Y75C and I99C tetramers into their respective disulfide-locked dimers (Fig. 6). More importantly, we found that these dimers were still active as dimers in the presence of heparin (Fig. 7A), unlike WT monomers, which were almost completely inactive under these conditions, as described previously (33). Notably, the I99C large interface dimer even maintained substantial activity in the absence of heparin (Fig. 7B), showing the predominant role this interface plays in generating an active tryptase conformation.
To avoid any effects arising from B12 binding to dimers, we engineered a dimer at the large interface (I99C/Y75A/Y37bA) unable to form a tetramer based on SEC (Fig. 8) due to replacement of the two key tyrosines with alanines in the small interface. In the process, we also fortuitously generated a monomer (I99C*/Y75A/Y37bA) that was also unable to tetramerize or dimerize (Fig. 8). This was due to cysteinylation of the introduced Cys residue at position 99 during expression of the zymogen to form a disulfide bond, which sterically prevents association with another monomer. To our knowledge, this is the first time a free β-tryptase monomer that cannot tetramerize has been described (see below). There are many previous studies assessing the activity of β-tryptase monomers, some having contradictory conclusions, for example, are monomers active or inactive? These reports must be interpreted carefully as one cannot exclude the possibility that β-tryptase monomers could re-tetramerize, at least to some extent (56). Furthermore, what level of activity constitutes being active can confound interpretations. The monomer we used has no such liabilities, as it cannot tetramerize. Overall, our data support that monomers can be active at neutral pH, but only in the presence of heparin in accord with previous data and models (31, 32, 35), and with much lower specific activities than WT (Table 2). The relative specific activity of a monomer is 2.1% of WT, when compared using a per protomer basis. The fact that a protomer in the tetrameric state is 48-fold more active than a bona fide monomer points to the benefit of tetramerization and the complex allostery involved to maximize catalytic efficiency. The same can be said for the dimer state. Although it is fascinating that the large interface dimer is active, even in the absence of heparin, it is much less active than the tetramer, which is the physiologically relevant state.
The dependence of activity on substrate and heparin for both the I99C/Y75A/Y37bA locked dimer (Fig. 9, A and B) and the cysteinylated I99C*/Y75A/Y37bA monomer (Fig. 9, C and D) was interesting. Notably, we saw that the dimer activity had a sigmoidal dependence on substrate with positive cooperativity at all heparin concentrations, whereas the dependence on heparin at different substrate concentrations was hyperbolic. The positive cooperativity for the dimer suggests that substrate binding at one protomer increases the binding and subsequent turnover at the other protomer. The most likely allosteric pathway involves transmission from the first active site to its 60s loop, which is in contact with the 170s loop from its neighbor, which has a path to the second active site. The activity of the homodimer, with its two active sites and substrate-dependent sigmoidal kinetics, is consistent with the Monod-Wyman-Changeux model of allostery (46, 47). Although the activity of the dimer is boosted by heparin, the sigmoidal kinetics still remain.
This was not the case with the monomer, which showed hyperbolic kinetics versus either substrate or heparin. The decrease in Km with increasing heparin is consistent with a role in conditioning the active site and acting as a positive allosteric activator. Regulation of proteases has been previously observed by diverse allosteric effectors such as cations, small molecules, proteins, and oligomers such as heparin (57). The hyperbolic behavior by heparin on monomeric tryptase can be described by the general allosteric modifier mechanism of Botts and Morales (49) and Baici (50), which has also been applied to proteases, including serine proteases (50, 58).
Previous reports, which describe the enzymatic activity of monomeric β-tryptase isolated from activated tetramer, have shown active monomer, but only in the presence of heparin and generally at more acidic pH values (30–35). Our findings also have some similarities with a previously proposed allosteric role for heparin that was determined by molecular dynamics simulations on human βII-tryptase, which determined an energetically more favorable binding of an active site small–molecule inhibitor to the P1 pocket in the presence of a 4-mer heparin molecule (59).
Part of the large interface involves the 170s loop of tryptase, which is nine residues longer than the same region in trypsin. The potential importance of the 60s and 170s loops in the large interface has been noted in prior papers discussing the tetramer structure (15, 37). We now provide experimental evidence to support this. The Y173dA β-tryptase mutant was still able to form a tetramer, but clearly it has a detrimental effect on the activity (Fig. 11B). We think this is due to an allosteric effect that is likely transmitted to His-57, as they are in reasonable proximity (Fig. 11A). This also supports our hypothesis that each β-tryptase protomer in the tetramer acts as a cofactor for the neighboring protomers, with the large interface being the dominant site for transducing allosteric activation. The fact that removal of the single aromatic side chain sitting in a hydrophobic pocket at the large interface has an inhibitory effect on activity suggests that it may be possible to develop small molecules that could bind at these exosites as an alternative strategy to develop tryptase inhibitors.
The 170s loop has previously been shown be involved in allosteric activation of blood coagulation factor VII (FVII), where binding to its cofactor tissue factor stabilizes a specific conformation of the helix preceding the long 170s loop (60). The binding of activated FVII (FVIIa) to tissue factor in the presence of Ca2+ ions results in maximal enzymatic activity; FVIIa alone shows very little proteolytic activity despite pro-domain removal (61–63).
In conclusion, β-tryptase reaches its full enzymatic activity after canonical pro-domain removal and stable tetramer formation in the presence of heparin. Complex formation via the large interface (I99C) in the tetramer is essential for allosteric enzyme activation, and we surmise that the interaction via the small interface (Y75C) that is facilitated by heparin stabilizes two β-tryptase dimers as a tetramer to establish full enzymatic activity. The interaction via the large interface results in positive allosteric cooperativity between the two protomers in the dimer. β-Tryptase could be viewed as an active dimer that is further stabilized by interacting with another dimer (a dimer of dimers), hence becoming a tetramer. Each protomer in the tetramer has two distinct roles, one as a protease and a second one as a cofactor for its neighboring protomer in the large interface. Heparin also appears to have two roles, one in stabilizing the tetramer and a separate one in allosterically conditioning the active site.
Experimental procedures
Recombinant expression of β-tryptase in insect cells and mammalian cells
The sequence encoding mature wildtype (WT) human βI-tryptase (Uniprot Q15661) from Ile-31–Pro-275 (Ile-16–Pro-246 chymotrypsinogen numbering) was cloned into a modified pAcGP67A vector behind the polyhedron promoter and the gp67 secretion signal sequence. Unless noted otherwise, tryptase or β-tryptase refers to βI-tryptase, and chymotrypsinogen numbering is used throughout; we also provide full-length numbering for tryptase (Fig. S2). Expression constructs contain an N-terminal His6-tag directly followed by an EK cleavage site directly fused to the mature N-terminal Ile-16 of β-tryptase. Site-directed mutagenesis was performed using standard QuikChange protocols (Stratagene) to generate β-tryptase mutants. All constructs were confirmed by DNA sequencing. Recombinant baculovirus were generated using the BaculoGold system (BD Biosciences) in Sf9 cells following standard protocols. T. ni cells were infected for large-scale protein production and harvested 48 h post-infection. The harvested media were supplemented with 1 mm NiCl2, 5 mm CaCl2, and 20 mm Tris, pH 8, shaken for 30 min and then centrifuged for 20 min at 8500 × g to remove the cells and precipitate from media. The supernatant media was filtered through a 0.22-μm PES filter prior to loading onto an Ni-NTA affinity column.
Purification of tetrameric WT and mutant β-tryptases
Insect cell media containing secreted His6-tagged zymogen β-tryptase (WT or mutant) was loaded onto a 10-ml Ni-NTA Superflow column (Qiagen) at a volumetric flow rate of 170 cm/h. The column was washed with 10 column volumes of wash buffer (20 mm Tris, pH 8, 10 mm imidazole, 300 mm NaCl) and eluted with 8 column volumes of elution buffer (20 mm Tris, pH 8, 300 mm imidazole, 300 mm NaCl). Fractions assayed by SDS-PAGE containing β-tryptase were pooled, concentrated, and loaded onto an S200 column (GE Healthcare) for further purification by SEC using SEC buffer (10 mm MOPS pH 6.8, 2 m NaCl) at flow rates recommended by the manufacturer. Fractions containing zymogen β-tryptase (monomeric) were pooled and concentrated. Zymogen β-tryptase was then cleaved overnight at room temperature at a concentration of 2 mg/ml in 10 mm MOPS, pH 6.8, 0.2 m NaCl containing 0.5 mg/ml heparin (H3393; average mass ∼18 kDa; Sigma) and 0.1 mg/ml EK (New England Biolabs, Ipswich, MA). This step removes the N-terminal His6-tag and results in tetramerization and proteolytically active β-tryptase, which has IVGG as the newly formed N-terminal sequence starting at residue 16. Tetrameric β-tryptase was then subjected to SEC using an S200 column (GE Healthcare) in SEC buffer to purify tetrameric β-tryptase by removing EK and any uncleaved zymogen β-tryptase. The same protocol was used to purify and activate β-tryptase mutant I16G, which failed to form tetramer as determined by SEC.
β-Tryptase mutants Y75C and I99C as well as the catalytically inactive mutant S195A were purified by nickel-affinity chromatography as described above. Disulfide-linked β-tryptase dimer mutants were then separated from nondisulfide-linked β-tryptase monomer mutants by SEC on an S200 column as above. Disulfide-linked dimer and monomer mutants were further processed by EK cleavage as described above for WT β-tryptase to form active tetramers (mutants Y75C and I99C), active dimers (mutant I99C/Y75A/Y37bA), as well as active monomers (mutant I99C*/Y75A/Y37bA (where C* is cysteinylated Cys). After EK cleavage of the β-tryptase monomer mutant to remove the pro-domain, the I99C*/Y75A/Y37bA β-tryptase mutant was purified by an orthogonal Ni-NTA column to remove the His-tagged EK and any uncleaved β-tryptase. The flow-through was concentrated, mixed with equimolar amounts of anti-penta-His antibody (Qiagen, Germantown, MD), and subjected to SEC on an S200 column in SEC buffer to remove any residual EK and uncleaved β-tryptase from the activated β-tryptase monomer mutant.
Molecular weight analysis of WT and β-tryptase mutants alone or in complex with B12 Fab
Size-exclusion chromatography was employed to analyze WT and mutant β-tryptase variants alone or bound to murine B12 Fab (2-fold molar excess) using an S200GL column (25-ml bed volume; GE Healthcare) in either SEC buffer (10 mm MOPS, pH 6.8, 2 m NaCl) or TNH buffer (50 mm Tris, pH 8, 150 mm NaCl, 0.1 mg/ml heparin, 0.02% NaN3) at 0.5 ml/min flow rate. All fractions containing protein as indicated by absorbance at A280 nm were analyzed by SDS-PAGE and Coomassie Blue staining to identify the protein components in these fractions. The elution volumes (Ve) of β-tryptase alone or in complex with Fab were compared, and the protein peaks eluted from the column were analyzed by multiangle static light scattering (MALS) using a DAWN HELIOS II machine (Wyatt Technology, Santa Barbara, CA).
β-Tryptase enzymatic activity assay
The enzymatic activities of 1 nm tetrameric β-tryptase and 100 nm dimeric or 100 nm monomeric β-tryptase triple mutants were measured at room temperature after 10 min of incubation in TNH buffer and initiating with 2 mm S-2288 chromogenic substrate (DiaPharma Group, Inc., West Chester, OH); the final volume was 200 μl. The reaction velocity was determined by the rate at which p-nitroaniline (pNA) is released, which was measured spectrophotometrically at 405 nm using a SpectraMax M5e plate reader (Molecular Devices, Sunnyvale, CA). The conversion factor was 1 mA405 nm = 6.59 μm pNA. The activities of either WT or mutant β-tryptase variants were measured as above in the presence or absence of heparin, at different concentrations of murine B12 Fab or aprotinin as indicated. Using Kaleidagraph (Synergy Software, Reading, PA), data were fit to either the hyperbolic Michaelis-Menten equation or to the allosteric sigmoidal equation (v = Vmax·[S]H/(K′ + [S]H), where H is the Hill coefficient (see Equation 5.47 or 12.7 in Copeland (45)). Experiments were performed in triplicate, and kinetic constants from the fits are reported as mean ± S.E. Data points on activity plots at specific concentrations are mean ± S.D.
Disulfide mapping by peptide mass fingerprinting
Purified Y75C β-tryptase protein was digested by trypsin (1:100, enzyme/substrate) (Promega, Madison, WI) for 18 h at 37 °C with and without 100 mm dithiothreitol (DTT) for 60 min at 60 °C. Digests were acidified to a final concentration of 0.1% TFA. 1 μl of each digest was spotted on a MALDI target plate with an equal volume of α-cyano-4-hydroxycinnamic acid (Agilent, Santa Clara, CA). Samples were analyzed on an ABI 4800 Plus MALDI-TOF/TOF mass spectrometer (AB Sciex, Redwood City, CA), and peptide mass spectra were subjected to manual interpretation by peptide-mass-fingerprinting (64) analysis for disulfide linkages and compared with the theoretical tryptic peptide mass list, including all possible disulfide-linked peptides generated by an in-house program.
Cysteinylation mapping by LC/MS/MS
Purified I99C/Y75A/Y37bA β-tryptase protein was digested by chymotrypsin (1:50, enzyme/substrate) (Millipore-Sigma) for 18 h at 37 °C with and without 10 mm DTT for 60 min at 60 °C and alkylated with 20 mm NIPIA for 20 min at 25 °C. Digests were acidified to a final concentration of 0.1% TFA. Samples were injected via an auto-sampler onto a 75-μm × 100-mm column (BEH, 1.7 μm, Waters Corp.) at a flow rate of 1 μl/min using a NanoAcquity UPLC (Waters Corp). A gradient from 98% solvent A (water + 0.1% formic acid) to 80% solvent B (acetonitrile + 0.08% formic acid) was applied over 40 min. Samples were analyzed on-line via nanospray ionization into a hybrid LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific). Data were collected in data-dependent mode with the parent ion being analyzed in the orbitrap and the top eight most abundant ions being selected for fragmentation and analysis in the LTQ. Tandem mass spectrometric data were analyzed using the Mascot search algorithm (Matrix Sciences, Boston, MA) against I99C/Y75A/Y37bA β-tryptase protein sequence with variable modifications NIPIA (+99 Da) and cysteinylation (+119 Da) on cysteine residues. Extracted ion chromatograms of the MS1 precursor mass containing I99C (TAQCGADIALL) peptide with cysteinylation were extracted to calculate the relative abundance of modification.
Production of murine B12 Fab
Murine B12 Fab was generated as described previously by Fukuoka and Schwartz (33).
Crystallization and structural determination of the I99C tetramer
The tetrameric I99C β-tryptase mutant was incubated with a 10-fold molar excess of Glu-Gly-Arg chloromethyl ketone protease inhibitor (ENZO Life Sciences Inc., Farmingdale, NY) and then purified by SEC on an S200GL (GE Healthcare) in 10 mm MOPS, pH 6.1, 2 m NaCl. The β-tryptase mutant was then crystallized essentially as described previously (65). The crystals were dipped in artificial mother liquor containing 15% ethylene glycol and preserved for data collection by sudden immersion in liquid nitrogen. Diffraction data extending to 2.72 Å were collected in a trigonal lattice from one crystal at 110 K using 1-Å X-rays at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the APS and reduced using standard methods (66–69) in point group 321. A Matthews coefficient Vm of 3.2 Å3/Da suggested a single tetramer in the asymmetric unit. The structure was solved (70) in space group P3221 using a tetramer search probe of WT β-tryptase from PDB entry 1A0L (15). Limited refinement (71) of a model of I99C (72) provided electron density maps consistent with covalent attachment of a tripeptide chloromethyl ketone inhibitor in all four active sites. Model manipulation reflecting the larger features in an Fo − Fc electron density map and more refinement (73), including automatic addition of water atoms, produced maps consistent with Cys substitution at residue 99. The final refinements applied a noncrystallographic symmetry restraint across the β-tryptase protomers in one span. Data reduction and refinement statistics are presented in Table 3.
Author contributions
H. R. M. designed and carried out most of the experiments, analyzed the results, and wrote the manuscript. P. S. L. carried out mass spectrometry experiments, analyzed data, and helped write the manuscript. Y. F. designed all the expression constructs. C. E. guided the crystallography, determined the structure, and helped write the manuscript. W. F. F. carried out “binomial” linear regression analysis to calculate Vmax values for heterotetramer subtypes and helped to write the manuscript. L. B. S. supplied B12 antibody and helped to analyze data and write the manuscript. R. A. L. helped to design experiments, analyzed the results, and wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Christine Tam for cloning expression constructs and Kyle Mortara and the Biomolecular Resources Department for expression of β-tryptase in insect cells. We thank Daniel Kirchhofer and Terry Lipari for very helpful discussions and reagents. We thank Wendy Sandoval, Lawren Wu, and Tangsheng Yi for their support. We thank Jeff Holden for steered molecular dynamic simulations. We acknowledge the Antibody Production Group and Mark Ultsch for producing B12 IgG and Fab proteins. We thank Ping Wu for harvesting protein crystals and John J. Chrzas for data collection at the Advanced Photon Source. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, and Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline.
The authors were all employees of Genentech, Inc., when the work was carried out except for L. B. S. L. B. S. is a paid consultant for Genentech, Inc.
This article contains Figs. S1–S2, Table S1, supporting note, and supporting Refs. 1 and 2.
The atomic coordinates and structure factors (code 5WI6) have been deposited in the Protein Data Bank (http://wwpdb.org/).
J. K. Holden and R. A. Lazarus, unpublished results.
- Ni-NTA
- nickel-nitrilotriacetic acid
- SEC
- size-exclusion chromatography
- EK
- enterokinase
- NIPIA
- n-isopropylacrylamide
- pNA
- p-nitroaniline
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- fVII
- factor VII.
References
- 1. Caughey G. H. (2016) Mast cell proteases as pharmacological targets. Eur. J. Pharmacol. 778, 44–55 10.1016/j.ejphar.2015.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hallgren J., and Pejler G. (2006) Biology of mast cell tryptase. An inflammatory mediator. FEBS J. 273, 1871–1895 10.1111/j.1742-4658.2006.05211.x [DOI] [PubMed] [Google Scholar]
- 3. McNeil H. P., Adachi R., and Stevens R. L. (2007) Mast cell-restricted tryptases: structure and function in inflammation and pathogen defense. J. Biol. Chem. 282, 20785–20789 10.1074/jbc.R700017200 [DOI] [PubMed] [Google Scholar]
- 4. Wernersson S., and Pejler G. (2014) Mast cell secretory granules: armed for battle. Nat. Rev. Immunol. 14, 478–494 10.1038/nri3690 [DOI] [PubMed] [Google Scholar]
- 5. Rivera J., Fierro N. A., Olivera A., and Suzuki R. (2008) New insights on mast cell activation via the high affinity receptor for IgE. Adv. Immunol. 98, 85–120 10.1016/S0065-2776(08)00403-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Caughey G. H. (2007) Mast cell tryptases and chymases in inflammation and host defense. Immunol. Rev. 217, 141–154 10.1111/j.1600-065X.2007.00509.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reimer J. M., Samollow P. B., and Hellman L. (2010) High degree of conservation of the multigene tryptase locus over the past 150–200 million years of mammalian evolution. Immunogenetics 62, 369–382 10.1007/s00251-010-0443-2 [DOI] [PubMed] [Google Scholar]
- 8. Harris J. L., Niles A., Burdick K., Maffitt M., Backes B. J., Ellman J. A., Kuntz I., Haak-Frendscho M., and Craik C. S. (2001) Definition of the extended substrate specificity determinants for β-tryptases I and II. J. Biol. Chem. 276, 34941–34947 10.1074/jbc.M102997200 [DOI] [PubMed] [Google Scholar]
- 9. Schwartz L. B., Lewis R. A., Seldin D., and Austen K. F. (1981) Acid hydrolases and tryptase from secretory granules of dispersed human lung mast cells. J. Immunol. 126, 1290–1294 [PubMed] [Google Scholar]
- 10. Schwartz L. B., Lewis R. A., and Austen K. F. (1981) Tryptase from human pulmonary mast cells. Purification and characterization. J. Biol. Chem. 256, 11939–11943 [PubMed] [Google Scholar]
- 11. Sakai K., Ren S., and Schwartz L. B. (1996) A novel heparin-dependent processing pathway for human tryptase. Autocatalysis followed by activation with dipeptidyl peptidase I. J. Clin. Invest. 97, 988–995 10.1172/JCI118523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mulloy B., Lever R., and Page C. P. (2017) Mast cell glycosaminoglycans. Glycoconj. J. 34, 351–361 10.1007/s10719-016-9749-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rönnberg E., Melo F. R., and Pejler G. (2012) Mast cell proteoglycans. J. Histochem. Cytochem. 60, 950–962 10.1369/0022155412458927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stevens R. L., and Adachi R. (2007) Protease-proteoglycan complexes of mouse and human mast cells and importance of their β-tryptase-heparin complexes in inflammation and innate immunity. Immunol. Rev. 217, 155–167 10.1111/j.1600-065X.2007.00525.x [DOI] [PubMed] [Google Scholar]
- 15. Pereira P. J., Bergner A., Macedo-Ribeiro S., Huber R., Matschiner G., Fritz H., Sommerhoff C. P., and Bode W. (1998) Human β-tryptase is a ring-like tetramer with active sites facing a central pore. Nature 392, 306–311 10.1038/32703 [DOI] [PubMed] [Google Scholar]
- 16. Corvera C. U., Déry O., McConalogue K., Böhm S. K., Khitin L. M., Caughey G. H., Payan D. G., and Bunnett N. W. (1997) Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor 2. J. Clin. Invest. 100, 1383–1393 10.1172/JCI119658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gruber B. L., Marchese M. J., Suzuki K., Schwartz L. B., Okada Y., Nagase H., and Ramamurthy N. S. (1989) Synovial procollagenase activation by human mast cell tryptase dependence upon matrix metalloproteinase 3 activation. J. Clin. Invest. 84, 1657–1662 10.1172/JCI114344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maier M., Spragg J., and Schwartz L. B. (1983) Inactivation of human high molecular weight kininogen by human mast cell tryptase. J. Immunol. 130, 2352–2356 [PubMed] [Google Scholar]
- 19. Molino M., Barnathan E. S., Numerof R., Clark J., Dreyer M., Cumashi A., Hoxie J. A., Schechter N., Woolkalis M., and Brass L. F. (1997) Interactions of mast cell tryptase with thrombin receptors and PAR-2. J. Biol. Chem. 272, 4043–4049 10.1074/jbc.272.7.4043 [DOI] [PubMed] [Google Scholar]
- 20. Schiemann F., Brandt E., Gross R., Lindner B., Mittelstädt J., Sommerhoff C. P., Schulmistrat J., and Petersen F. (2009) The cathelicidin LL-37 activates human mast cells and is degraded by mast cell tryptase: Counter-regulation by CXCL4. J. Immunol. 183, 2223–2231 10.4049/jimmunol.0803587 [DOI] [PubMed] [Google Scholar]
- 21. Schwartz L. B., Bradford T. R., Littman B. H., and Wintroub B. U. (1985) The fibrinogenolytic activity of purified tryptase from human lung mast cells. J. Immunol. 135, 2762–2767 [PubMed] [Google Scholar]
- 22. Stack M. S., and Johnson D. A. (1994) Human mast cell tryptase activates single-chain urinary-type plasminogen activator (pro-urokinase). J. Biol. Chem. 269, 9416–9419 [PubMed] [Google Scholar]
- 23. Alter S. C., Kramps J. A., Janoff A., and Schwartz L. B. (1990) Interactions of human mast cell tryptase with biological protease inhibitors. Arch. Biochem. Biophys. 276, 26–31 10.1016/0003-9861(90)90005-J [DOI] [PubMed] [Google Scholar]
- 24. Paesen G. C., Siebold C., Harlos K., Peacey M. F., Nuttall P. A., and Stuart D. I. (2007) A tick protein with a modified Kunitz fold inhibits human tryptase. J. Mol. Biol. 368, 1172–1186 10.1016/j.jmb.2007.03.011 [DOI] [PubMed] [Google Scholar]
- 25. Sommerhoff C. P., Söllner C., Mentele R., Piechottka G. P., Auerswald E. A., and Fritz H. (1994) A Kazal-type inhibitor of human mast cell tryptase: isolation from the medical leech Hirudo medicinalis, characterization, and sequence analysis. Biol. Chem. Hoppe Seyler 375, 685–694 10.1515/bchm3.1994.375.10.685 [DOI] [PubMed] [Google Scholar]
- 26. Pohlig G., Fendrich G., Knecht R., Eder B., Piechottka G., Sommerhoff C. P., and Heim J. (1996) Purification, characterization and biological evaluation of recombinant leech-derived tryptase inhibitor (rLDTI) expressed at high level in the yeast Saccharomyces cerevisiae. Eur. J. Biochem. 241, 619–626 10.1111/j.1432-1033.1996.00619.x [DOI] [PubMed] [Google Scholar]
- 27. Schwartz L. B., and Bradford T. R. (1986) Regulation of tryptase from human lung mast cells by heparin. Stabilization of the active tetramer. J. Biol. Chem. 261, 7372–7379 [PubMed] [Google Scholar]
- 28. Schechter N. M., Eng G. Y., and McCaslin D. R. (1993) Human skin tryptase: kinetic characterization of its spontaneous inactivation. Biochemistry 32, 2617–2625 10.1021/bi00061a020 [DOI] [PubMed] [Google Scholar]
- 29. Schechter N. M., Eng G. Y., Selwood T., and McCaslin D. R. (1995) Structural changes associated with the spontaneous inactivation of the serine proteinase human tryptase. Biochemistry 34, 10628–10638 10.1021/bi00033a038 [DOI] [PubMed] [Google Scholar]
- 30. Addington A. K., and Johnson D. A. (1996) Inactivation of human lung tryptase: evidence for a re-activatable tetrameric intermediate and active monomers. Biochemistry 35, 13511–13518 10.1021/bi960042t [DOI] [PubMed] [Google Scholar]
- 31. Fajardo I., and Pejler G. (2003) Formation of active monomers from tetrameric human β-tryptase. Biochem. J. 369, 603–610 10.1042/bj20021418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fukuoka Y., and Schwartz L. B. (2004) Human β-tryptase: detection and characterization of the active monomer and prevention of tetramer reconstitution by protease inhibitors. Biochemistry 43, 10757–10764 10.1021/bi049486c [DOI] [PubMed] [Google Scholar]
- 33. Fukuoka Y., and Schwartz L. B. (2006) The B12 anti-tryptase monoclonal antibody disrupts the tetrameric structure of heparin-stabilized β-tryptase to form monomers that are inactive at neutral pH and active at acidic pH. J. Immunol. 176, 3165–3172 10.4049/jimmunol.176.5.3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fukuoka Y., and Schwartz L. B. (2007) Active monomers of human β-tryptase have expanded substrate specificities. Int. Immunopharmacol. 7, 1900–1908 10.1016/j.intimp.2007.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schechter N. M., Choi E. J., Selwood T., and McCaslin D. R. (2007) Characterization of three distinct catalytic forms of human tryptase-β: their interrelationships and relevance. Biochemistry 46, 9615–9629 10.1021/bi7004625 [DOI] [PubMed] [Google Scholar]
- 36. Alter S. C., Metcalfe D. D., Bradford T. R., and Schwartz L. B. (1987) Regulation of human mast cell tryptase. Effects of enzyme concentration, ionic strength and the structure and negative charge density of polysaccharides. Biochem. J. 248, 821–827 10.1042/bj2480821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sommerhoff C. P., Bode W., Pereira P. J., Stubbs M. T., Stürzebecher J., Piechottka G. P., Matschiner G., and Bergner A. (1999) The structure of the human βII-tryptase tetramer: Fo (u) r better or worse. Proc. Natl. Acad. Sci. U.S.A. 96, 10984–10991 10.1073/pnas.96.20.10984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hallgren J., Backström S., Estrada S., Thuveson M., and Pejler G. (2004) Histidines are critical for heparin-dependent activation of mast cell tryptase. J. Immunol. 173, 1868–1875 10.4049/jimmunol.173.3.1868 [DOI] [PubMed] [Google Scholar]
- 39. Huber R., and Bode W. (1978) Structural basis of the activation and action of trypsin. Acc. Chem. Res. 11, 114–122 10.1021/ar50123a006 [DOI] [Google Scholar]
- 40. Hedstrom L. (2002) Serine protease mechanism and specificity. Chem. Rev. 102, 4501–4524 10.1021/cr000033x [DOI] [PubMed] [Google Scholar]
- 41. Khan A. R., and James M. N. (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 7, 815–836 10.1002/pro.5560070401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang D., Bode W., and Huber R. (1985) Bovine chymotrypsinogen A X-ray crystal structure analysis and refinement of a new crystal form at 1.8 A resolution. J. Mol. Biol. 185, 595–624 10.1016/0022-2836(85)90074-9 [DOI] [PubMed] [Google Scholar]
- 43. Hedstrom L., Lin T. Y., and Fast W. (1996) Hydrophobic interactions control zymogen activation in the trypsin family of serine proteases. Biochemistry 35, 4515–4523 10.1021/bi951928k [DOI] [PubMed] [Google Scholar]
- 44. Schwartz L. B., Bradford T. R., Rouse C., Irani A. M., Rasp G., Van der Zwan J. K., and Van der Linden P. W. (1994) Development of a new, more sensitive immunoassay for human tryptase: Use in systemic anaphylaxis. J. Clin. Immunol. 14, 190–204 10.1007/BF01533368 [DOI] [PubMed] [Google Scholar]
- 45. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis, 2nd Ed., pp. 137–141 and pp. 373–381, John Wiley & Sons, Inc., New York [Google Scholar]
- 46. Monod J., Wyman J., and Changeux J. P. (1965) On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 12, 88–118 10.1016/S0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- 47. Changeux J. P. (2012) Allostery and the Monod-Wyman-Changeux model after 50 years. Annu. Rev. Biophys. 41, 103–133 10.1146/annurev-biophys-050511-102222 [DOI] [PubMed] [Google Scholar]
- 48. Hilser V. J., Wrabl J. O., and Motlagh H. N. (2012) Structural and energetic basis of allostery. Annu. Rev. Biophys. 41, 585–609 10.1146/annurev-biophys-050511-102319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Botts J., and Morales M. (1953) Analytical description of the effects of modifiers and of enzyme multivalency upon the steady state catalyzed reaction rate. Trans. Faraday Soc. 49, 696–707 10.1039/tf9534900696 [DOI] [Google Scholar]
- 50. Baici A. (2015) Kinetics of Enzyme-Modifier Interactions: Selected Topics in the Theory and Diagnosis of Inhibition and Activation Mechanisms, Springer-Verlag, Wien, Austria [Google Scholar]
- 51. Schwartz L. B., Bradford T. R., Lee D. C., and Chlebowski J. F. (1990) Immunologic and physicochemical evidence for conformational changes occurring on conversion of human mast cell tryptase from active tetramer to inactive monomer. Production of monoclonal antibodies recognizing active tryptase. J. Immunol. 144, 2304–2311 [PubMed] [Google Scholar]
- 52. Wang S., Reed G. L., and Hedstrom L. (1999) Deletion of Ile1 changes the mechanism of streptokinase: Evidence for the molecular sexuality hypothesis. Biochemistry 38, 5232–5240 10.1021/bi981915h [DOI] [PubMed] [Google Scholar]
- 53. Wang S., Reed G. L., and Hedstrom L. (2000) Zymogen activation in the streptokinase-plasminogen complex. Ile1 is required for the formation of a functional active site. Eur. J. Biochem. 267, 3994–4001 10.1046/j.1432-1327.2000.01434.x [DOI] [PubMed] [Google Scholar]
- 54. Boxrud P. D., Verhamme I. M., Fay W. P., and Bock P. E. (2001) Streptokinase triggers conformational activation of plasminogen through specific interactions of the amino-terminal sequence and stabilizes the active zymogen conformation. J. Biol. Chem. 276, 26084–26089 10.1074/jbc.M101966200 [DOI] [PubMed] [Google Scholar]
- 55. Selwood T., McCaslin D. R., and Schechter N. M. (1998) Spontaneous inactivation of human tryptase involves conformational changes consistent with conversion of the active site to a zymogen-like structure. Biochemistry 37, 13174–13183 10.1021/bi980780c [DOI] [PubMed] [Google Scholar]
- 56. Ren S., Sakai K., and Schwartz L. B. (1998) Regulation of human mast cell β-tryptase: Conversion of inactive monomer to active tetramer at acid pH. J. Immunol. 160, 4561–4569 [PubMed] [Google Scholar]
- 57. Merdanovic M., Mönig T., Ehrmann M., and Kaiser M. (2013) Diversity of allosteric regulation in proteases. ACS Chem. Biol. 8, 19–26 10.1021/cb3005935 [DOI] [PubMed] [Google Scholar]
- 58. Di Cera E., Hopfner K. P., and Dang Q. D. (1996) Theory of allosteric effects in serine proteases. Biophys. J. 70, 174–181 10.1016/S0006-3495(96)79558-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang Y., Zheng Q. C., Kong C. P., Tian Y., Zhan J., Zhang J. L., and Zhang H. X. (2015) Heparin makes differences: a molecular dynamics simulation study on the human βII-tryptase monomer. Mol. Biosyst. 11, 252–261 10.1039/C4MB00381K [DOI] [PubMed] [Google Scholar]
- 60. Banner D. W., D'Arcy A., Chène C., Winkler F. K., Guha A., Konigsberg W. H., Nemerson Y., and Kirchhofer D. (1996) The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature 380, 41–46 10.1038/380041a0 [DOI] [PubMed] [Google Scholar]
- 61. Davie E. W. (1995) Biochemical and molecular aspects of the coagulation cascade. Thromb. Haemost. 74, 1–6 [PubMed] [Google Scholar]
- 62. Nemerson Y. (1988) Tissue factor and hemostasis. Blood 71, 1–8 [PubMed] [Google Scholar]
- 63. Rapaport S. I., and Rao L. V. (1995) The tissue factor pathway: How it has become a “prima ballerina”. Thromb. Haemost. 74, 7–17 [PubMed] [Google Scholar]
- 64. Henzel W. J., Watanabe C., and Stults J. T. (2003) Protein identification: The origins of peptide mass fingerprinting. J. Am. Soc. Mass Spectrom. 14, 931–942 10.1016/S1044-0305(03)00214-9 [DOI] [PubMed] [Google Scholar]
- 65. McGrath M. E., Sprengeler P. A., Hirschbein B., Somoza J. R., Lehoux I., Janc J. W., Gjerstad E., Graupe M., Estiarte A., Venkataramani C., Liu Y., Yee R., Ho J. D., Green M. J., Lee C. S., et al. (2006) Structure-guided design of peptide-based tryptase inhibitors. Biochemistry 45, 5964–5973 10.1021/bi060173m [DOI] [PubMed] [Google Scholar]
- 66. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 10.1107/S0907444909047374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., and Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 10.1107/S0907444911007773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.