

Abstract
The pyruvate dehydrogenase complex (PDC) is a key control point of energy metabolism and is subject to regulation by multiple mechanisms, including posttranslational phosphorylation by pyruvate dehydrogenase kinase (PDK). Pharmacological modulation of PDC activity could provide a new treatment for diabetic cardiomyopathy, as dysregulated substrate selection is concomitant with decreased heart function. Dichloroacetate (DCA), a classic PDK inhibitor, has been used to treat diabetic cardiomyopathy, but the lack of specificity and side effects of DCA indicate a more specific inhibitor of PDK is needed. This study was designed to determine the effects of a novel and highly selective PDK inhibitor, 2((2,4-dihydroxyphenyl)sulfonyl) isoindoline-4,6-diol (designated PS10), on pyruvate oxidation in diet-induced obese (DIO) mouse hearts compared with DCA-treated hearts. Four groups of mice were studied: lean control, DIO, DIO + DCA, and DIO + PS10. Both DCA and PS10 improved glucose tolerance in the intact animal. Pyruvate metabolism was studied in perfused hearts supplied with physiological mixtures of long chain fatty acids, lactate, and pyruvate. Analysis was performed using conventional 1H and 13C isotopomer methods in combination with hyperpolarized [1-13C]pyruvate in the same hearts. PS10 and DCA both stimulated flux through PDC as measured by the appearance of hyperpolarized [13C]bicarbonate. DCA but not PS10 increased hyperpolarized [1-13C]lactate production. Total carbohydrate oxidation was reduced in DIO mouse hearts but increased by DCA and PS10, the latter doing so without increasing lactate production. The present results suggest that PS10 is a more suitable PDK inhibitor for treatment of diabetic cardiomyopathy.
Keywords: pyruvate dehydrogenase complex (PDC), cardiomyopathy, nuclear magnetic resonance (NMR), diabetes, glucose metabolism, drug development, intermediary metabolism, fatty acid oxidation, mitochondria, hyperpolarization
Introduction
Type 2 diabetes (T2D)5 is a growing public health issue in many countries (1). It is generally accepted that carbohydrate oxidation is inhibited in the insulin-resistant heart, and is concomitant with the development of outright diabetic cardiomyopathy (2). The pyruvate dehydrogenase complex (PDC) connects glycolysis to the tricarboxylic acid (TCA) cycle for the production of NADH and FADH2, and subsequent oxidative phosphorylation, and plays a pivotal role in modulating substrate selection for the production of acetyl-CoA. Increased fatty acid availability and oxidation associated with T2D suppresses the activity of the PDC (2). The accumulation of “lipotoxic” metabolites may also lead to insulin resistance and cardiomyocyte apoptosis (3, 4). The PDC is negatively regulated by pyruvate dehydrogenase kinase isoforms 1 to 4 (PDK1–4) via phosphorylation of the E1α subunit (5). Because of its strategic location, the regulation of PDC activity is critical for whole body glucose homeostasis, but PDC activity is down-regulated in diabetic cardiomyopathy (6). Another pathogenic mechanism of diabetic cardiomyopathy derives from elevated peroxisome proliferator–activated receptor alpha (PPARα) expression in obesity and T2D, leading to the overexpression of PDK4 and decreased PDC activity (7, 8). Although it has been suggested that fuel overload may be causative in heart failure (HF) associated with diabetic cardiomyopathy (9), negative effects of insulin-sensitizing agents on HF are more likely to be related to plasma volume expansion than to dysregulated metabolism (10). For all these reasons, PDC activation remains a key therapeutic target for diabetic cardiomyopathy. PDC dysfunction is also a hallmark of other cardiovascular pathophysiologies including ischemia (11) and congestive heart failure (12).
Earlier studies have evaluated the utility of dichloroacetate (DCA), a widely studied pyruvate mimetic that inhibits PDKs by binding to the allosteric sites of PDK1, 2, and 4 (13). Treatment with DCA has been shown to improve diabetic cardiomyopathy (14), and ischemia and heart failure (15) in animal models. However, the potency of DCA is low and clinical translation is unlikely because DCA causes a peripheral neuropathy (16) along with carcinogenic effects (17). Treatment with another class of PDK inhibitors, AZD7545 (18), in normal rats caused cardiac steatosis and myocardial degeneration (19). The pathogenic effects of AZD7545 may be because of activation of PDK4 (13, 20). A new highly specific pan-PDK inhibitor, PS10, was recently developed (21), targeting the Bergerat ATP-binding fold in PDKs, which is uniquely conserved in the GHKL protein kinase family. PS10 restores glucose tolerance and reduces hepatic steatosis in diet-induced obese (DIO) mice (21).
In this study, we investigated the efficacy of PS10 in restoring pyruvate flux through the PDC in the DIO mouse heart. Quantitative metabolic flux measurements were used to directly assess differences in the mode of action of DCA and PS10. Conventional 13C and 1H magnetic resonance spectroscopy (MRS) isotopomer analysis measured the relative rates of fatty acid and carbohydrate oxidation (21–23). Because of low signal, conventional 13C MRS is not useful in vivo or for quickly assessing metabolic state of a tissue. For this reason we also investigated the utility of hyperpolarized (HP) 13C MRS to probe metabolism of [1-13C]pyruvate to [13C]bicarbonate through decarboxylation by the PDC with 2-s time resolution (22, 24). This technology has recently been used to image PDC activity in the human heart (25). The experiments here prove that PS10 up-regulates PDC flux without generating excess lactate production, as is the case with DCA. This suggests PS10 has significant potential as a therapeutic agent for PDC activation.
Results
Determination of PDK inhibitor dose for the MRS study
To compare the metabolic effects of each PDK inhibitor, we first assayed the optimal dose of each agent for restoration of glucose tolerance in DIO mice after 2 weeks of treatment. This dose was postulated to be optimal for metabolic comparison. After testing DCA at 100 (Fig. S1A), 200 (Fig. S1B), and 250 mg/kg/day, the 250-mg dose was chosen for maximal restoration of glucose homeostasis. Similar experiments led to a choice of 70 mg/kg/day for PS10. DIO mice fed with 60% high-fat diet for 18 weeks were administered either PS10 at 70 mg/kg/day or DCA at 250 mg/kg/day by intraperitoneal injection. These doses conferred the maximal restoration of glucose homeostasis. A DMSO injection was used as a control. A glucose tolerance test was carried out on control versus DIO animals. After 6.5 h of fasting, 1.5 g/kg of glucose was administered intraperitoneally. Plasma glucose levels were measured as indicated (Fig. 1). At these doses, both the PS10 and DCA groups show similar glucose tolerance response to glucose challenge (Fig. 1A). Compared with the control group, both PS10 and DCA treatments significantly improved glucose tolerance in DIO mice. We also compared effects of single-dose PDK inhibitors on the PDC activity in heart at the doses mentioned above. Treatment with both PDK inhibitors resulted in significant enhancements of PDC activity in DIO mouse hearts compared with DIO controls (Fig. 1B). The elevated PDC activity by both PDK inhibitors corresponded to a decrease of phosphorylation on the PDC E1α subunit in DIO mouse hearts (Fig. 1C).
Figure 1.
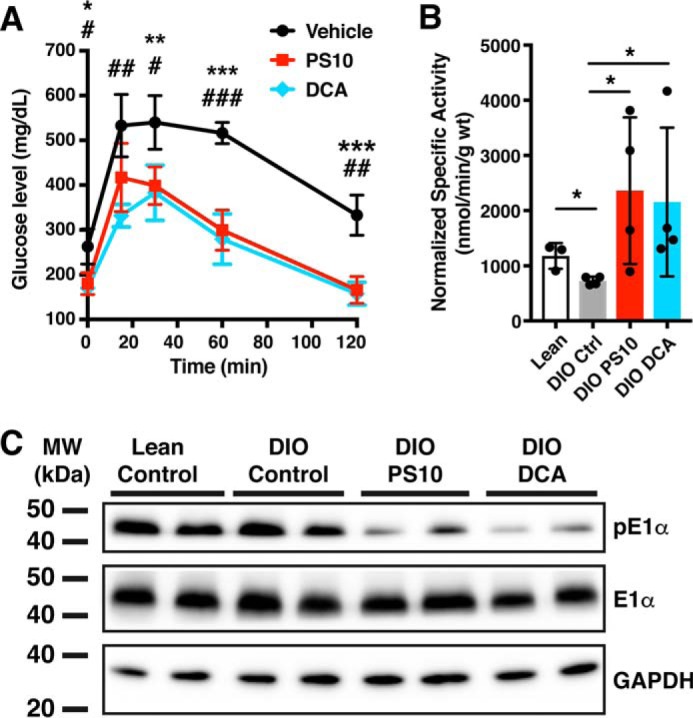
Pyruvate dehydrogenase kinase inhibitors improve glucose tolerance in diet-induced obese mice and activate pyruvate dehydrogenase complex activity by inhibition of pyruvate dehydrogenase kinases. A, the glucose tolerance test of PDK inhibitor–treated DIO mice after 2 weeks of treatment. n = 4 for PS10 (70 mg/kg) and control groups; n = 3 for DCA group (250 mg/kg). *, p values between PS10 and Control; #, p values between DCA and Control. B, the PDC activity of hearts treated with one dose of PDK inhibitors. n = 4 in each group. C, Western blotting of phospho-E1α subunit of PDC from hearts treated with one dose of PDK inhibitors. The data are presented as mean ± S.D. *, p < 0.05; **, p < 0.01, ***, p < 0.001.
Hyperpolarized [1-13C]pyruvate MRS on diet-induced obese mouse hearts
The activity of the PDC complex in functioning tissue was assayed directly using HP [1-13C]pyruvate. The experimental groups included a control set of DIO mice and additional sets of mice treated with either PS10 or DCA. A single dose of PS10 or DCA was administered intraperitoneally prior to heart extraction. Mouse hearts were perfused with Krebs-Henseleit buffer and 13C tracers (0.12 mm [3-13C]pyruvate, 1.2 mm [3-13C]lactate, and 0.4 mm [U-13C]free fatty acid) as described in “Experimental Procedures.” The representative 13C NMR spectra summed from 88 scans are presented in Fig. 2. Signals from 13CO2, [13C]bicarbonate, [1-13C]pyruvate, [1-13C]alanine, [1-13C]pyruvate hydrate, and [1-13C]lactate are easily detectable by NMR. We also were able to measure the conversion of [1-13C]pyruvate to four-carbon metabolites such as [1-13C]aspartate, [4-13C]aspartate, [1-13C]malate, and [4-13C]malate (Fig. 2). The [13C]bicarbonate signal was decreased in the DIO control group (Fig. 2B) comparing to lean group (Fig. 2A). Both PDK inhibitors, PS10 (Fig. 2C) and DCA (Fig. 2D), restored the [13C]bicarbonate signal in DIO hearts to levels higher than that in the lean control.
Figure 2.

Hyperpolarized [1-13C]pyruvate 13C magnetic resonance spectra from mouse hearts treated with PDK inhibitors. A, lean mouse. B, DIO mouse with DMSO control. C, DIO mouse with PS-10. D, DIO mouse with DCA. The numbers denote metabolite signals as follows: 1, [1-13C]aspartate; 2, [4-13C]aspartate; 3, [4-13C]malate; and 4, [1-13C]malate.
To visualize the kinetics of HP pyruvate, we plotted the area under the curve of each peak in the 13C spectrum against measurement time (Fig. 3). DIO control hearts trended toward lower [13C]bicarbonate production compared with lean control hearts (Fig. 3A). Both PS10 and DCA demonstrated significant activation of PDC flux generating [13C]bicarbonate (Fig. 3A, left) and 13CO2 (Fig. 3B, left). DCA induced an almost 2-fold increase for total [13C]bicarbonate signal compared with DIO control, whereas PS10 only brought the total signal back to a state similar to lean control (Fig. 3A, right). The result of 13CO2 matched that of [13C]bicarbonate as expected (Fig. 3B). However, DCA also triggered more [1-13C]lactate production than the other three groups (Fig. 3C, left). There is not a significant difference in [1-13C]lactate production among PS10-treated, lean control, and DIO control groups (Fig. 3C, right). We observed no difference in [1-13C]pyruvate signal (Fig. S2A) because the amount of pyruvate is fixed and in excess. The amount of [1-13C]alanine was similar in all groups (Fig. S2B), suggesting PDK inhibitors or high-fat diet did not change the interconversion of pyruvate to alanine. The products of pyruvate carboxylation (Fig. 4A), [1-13C]aspartate (Fig. S2C), [4-13C]aspartate (Fig. S2D), [1-13C]malate (Fig. S2E), and [4-13C]malate (Fig. S2F), were in general higher intensity in DIO control and DCA-treated compared with lean control and PS10 groups. These results strongly suggest that the flux through the malic enzyme (26), or pyruvate carboxylase (27), to generate malate or oxaloacetate, respectively, is lowered after PS10 treatment, but not in DIO control and DCA-treated groups. Positional enrichments of the four-carbon TCA intermediates cannot distinguish between flux through the malic enzyme versus pyruvate carboxylase.
Figure 3.
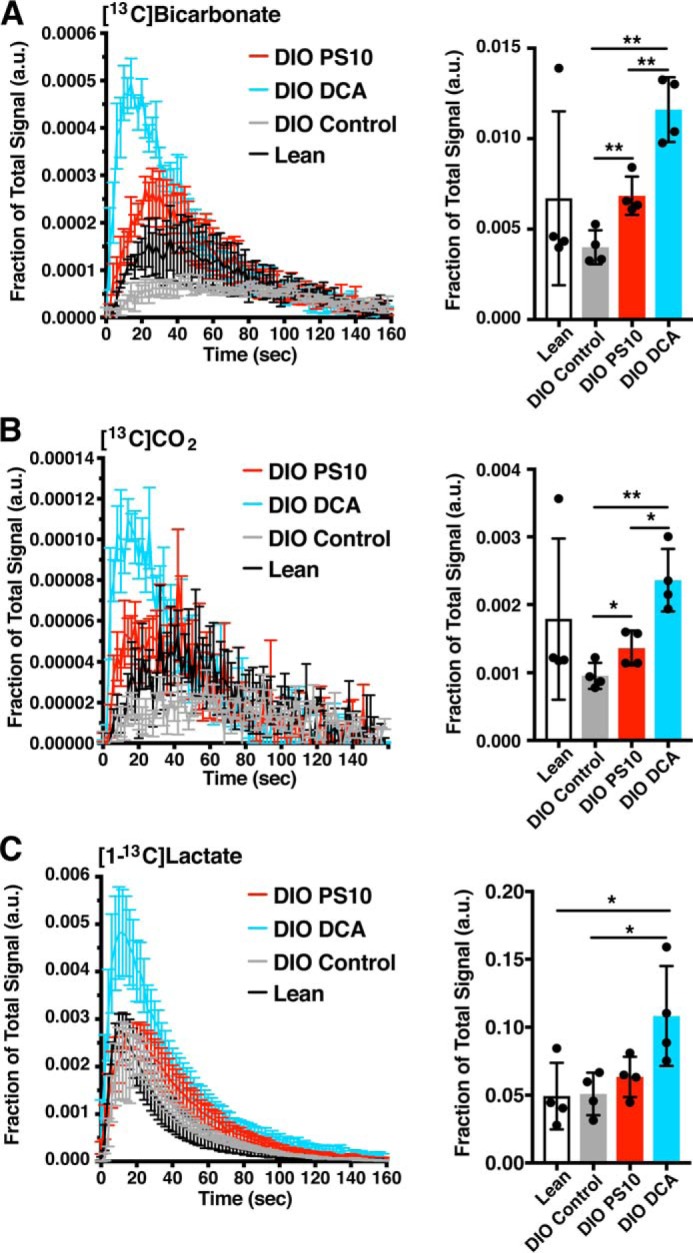
PDK inhibitors restore pyruvate flux through the PDC in DIO mouse hearts. The 13C signals versus the time of data acquisition for metabolic products of hyperpolarized [1-13C]pyruvate from mouse hearts with different treatment are indicated in the plots. The dose of PS10 was 70 mg/kg and DCA was 250 mg/kg. The integrated area under the curve (AUC) is presented on the right side. A, [13C]bicarbonate. B, 13CO2. C, [1-13C]lactate. Data are presented as mean ± S.E. in flux plots and mean ± S.D. in AUC quantification. n = 4 in each treatment group. *, p < 0.05; **, p < 0.01.
Figure 4.

The proton NMR spectrum of alanine, lactate, and 13C labeling patterns of glutamate from the mouse hearts with different PDK inhibitor treatments. A, sources of 13C label on acetyl-CoA and glutamate in the TCA cycle. The ratio of labeled pyruvate/lactate over unlabeled endogenous sources is defined as B/A. The ratio of labeled pyruvate/lactate over labeled fatty acid is defined as B/C. B, the proton spectrum of methyl group on alanine and lactate. The main doublet (center two lines, 12C bonded 1H resonances) and 13C directly bonded satellite doublets (on two ends) are indicated. The 4JCH manifests as doublets close to the 12C-bonded resonances around 1.33 ppm and 1.47 ppm. C and D, the 13C signals of label enrichment on glutamate C4 (C) and glutamate C3 (D) are shown. The coupling pattern is shown as S, singlet; D, doublet; T, triplet; Q, quartet. The subscript numbers indicate carbons of the coupling. HP, hyperpolarized [1-13C]pyruvate. Endo, unlabeled pyruvate derived from endogenous sources. Note that no assumption of steady state is needed for this analysis.
Proton and proton-decoupled 13C NMR spectra of perfused mouse hearts
To confirm the hyperpolarization results, substrate selection was measured in the mouse hearts using 13C isotopomer analysis (28). An appropriate choice of labeled substrates allows carbohydrate versus fatty acid utilization to be easily analyzed. We chose [3-13C]pyruvate, [3-13C]lactate, and [U-13C]free fatty acids as tracers to study substrate competition (Fig. 4A). During ∼40 min of perfusion, oxygen consumption rates were similar in all groups with slight, but not significant decrease in the PS10 group (Fig. S3). The flow rate and heart rate were unchanged across groups. All hearts were freeze-clamped immediately after the 3-min hyperpolarization data acquisition.
From the acid-soluble extract of hearts after perfusion and HP [1-13C]pyruvate injection, we acquired the proton spectra of alanine and lactate, and proton-decoupled 13C NMR spectrum of glutamate (Fig. 4). The 13C labeling in glutamate and glutamine reveals substrate utilization and competition (29). The proton signals of the alanine methyl group are composed of the doublet generated by 12C-bonded methyl protons and two satellite doublets because of carbon-proton coupling (1JCH = 124 Hz) of the 13C methyl group (indicated in Fig. 4B). The peaks adjacent to the main doublet come from the long-range carbon-proton coupling (4JCH ∼ 4 Hz), indicating the interaction of 13C labeling on C1 arising from injection of the [1-13C]pyruvate. In the glutamate C4 peaks (Fig. 4C), the doublet of C4-C5 coupling and the quartet of C3-C4/C4-C5 (C4Q) carbon-carbon coupling reflect a strong contribution of fatty acid oxidation to acetyl-CoA production (30). Note that this analysis is made without the assumption of a steady state, but rather indicates the fractions of labeled acetyl-CoA that contributed to the production of glutamate at the instant metabolism was quenched by freeze-clamping. An increasing fraction of D34 and C4S indicates more carbohydrate oxidation. It is clear that both the C4 quartet and D45 are stronger in the DIO control group, with decreased pyruvate anaplerosis in the DCA-treated hearts as evidenced by the increased fraction of C4Q (Fig. 4C). The 13C labeling in glutamate C3 demonstrates similar results of higher PC flux with stronger doublet and triplet of C2-C3/C3-C4 coupling in DIO control group compared to other groups (Fig. 4D). Direct analysis of glutamine C3 and glutamine C4 (Fig. S3) supported similar conclusions.
We incorporate the fractional enrichment of the alanine methyl group and direct analysis of glutamate 13C labeling pattern to calculate the substrate utilization (oxidation). In this calculation, we defined “carbohydrate” as the sum of endogenous glycogen and exogenous labeled pyruvate (both 1- and 3-13C) and lactate (3-13C). We observed a trend toward decreased carbohydrate contribution as a fraction of the total acetyl-CoA production in DIO control hearts (Fig. 5A) compared with lean control group, whereas the FFA contribution was trending higher in DIO control (Fig. 5B) compared with lean controls. In DIO mouse hearts, PS10 was able to reverse the changes in substrate preference with significantly increased carbohydrate contribution (Fig. 5A) and significantly decreased FFA contribution compared with DIO control (Fig. 5B). Interestingly, the carbohydrate contribution to the total acetyl-CoA pool was smaller in the DCA group than the PS10 group (Fig. 5A), whereas the fatty acid contribution was not suppressed in the DCA group compared with the PS10 group (Fig. 5B). These results indicate that under our non–steady state experimental conditions, PS10 efficiently modulated PDC activity, whereas DCA did not.
Figure 5.
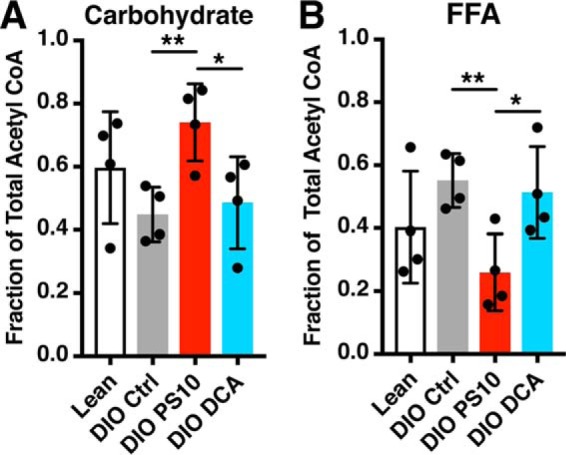
The carbohydrate contribution to total acetyl-CoA pool is increased, and the free fatty acid contribution is decreased in hearts treated with PS10. The fractional contribution of each substrate to acetyl-CoA production was calculated with a direct analysis of the glutamate isotopomers, without assumption of a metabolic steady state. A, the carbohydrate contribution to total acetyl-CoA production in mouse hearts with different treatment. B, the free fatty acid contribution to total acetyl-CoA pool in hearts with different treatment. Data are presented as mean ± S.D. n = 4 for all groups. *, p < 0.05; **, p < 0.01.
Using the relative efficiency of the substrates for the production of NADH per mole of O2 consumed (31) and the measured O2 consumption, the TCA flux and quantitative contributions of carbohydrates and fatty acids were calculated using the fractional substrate selection obtained from the non–steady state analysis (Fig. 6). The TCA flux in PS10 group trended to be lower than the other groups, but the difference did not reach a significant level with n = 4 (Fig. 6A). There was no difference in absolute turnover between the lean and DIO control groups based on our spectral data. Both PS10 and DCA group had similar carbohydrate contributions to TCA flux as the lean control group (Fig. 6B). However, PS10 significantly down-regulated the FFA contribution to total TCA flux compared not only to the DIO control but also to the DCA group (Fig. 6C).
Figure 6.
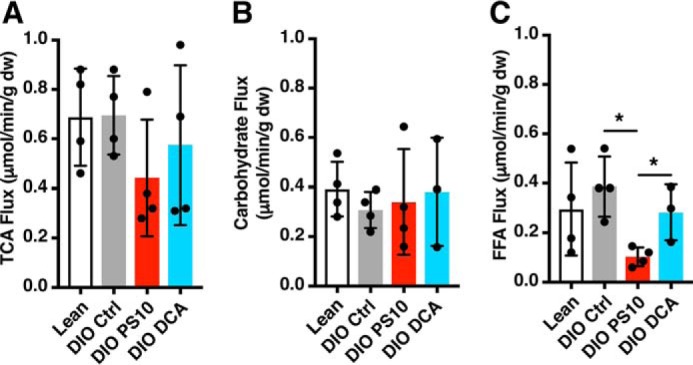
PS10 and DCA did not alter TCA flux in DIO mouse hearts as measured with a non–steady state isotopomer analysis. A, TCA flux of hearts with different treatment. B, the absolute carbohydrate contribution to TCA cycle flux in the mouse hearts with different treatment. C, the absolute free fatty acid contribution to TCA cycle flux in the mouse hearts with different treatment. Data are presented as mean ± S.D. n = 4 all groups. *, p < 0.05.
In addition to isotopomer analysis of the glutamate, we chose to perform a steady state analysis of the carbon-13 labeling in glutamine. Glutamine turns over much more slowly than glutamate in the heart, and there was an expectation that it might give a readout of oxidative metabolism prior to the injection of the HP pyruvate. Figs. S4 and S5 showed data similar to those obtained in the glutamate, but in this analysis the fractional contribution of carbohydrates to total TCA flux was significantly raised in the case of PS10 (Fig. S5D).
Discussion
The pathogenesis of diabetic cardiomyopathy is complex (32). There are three major factors related to energy production that lead to early-stage development of diabetic cardiomyopathy. First, hyperinsulinemia triggers insulin resistance in T2D, and concurrent suppression of GLUT4 and overexpression of PDK4 (33). The reciprocal regulation of these two proteins leads to increased fatty acid oxidation and decreased glucose oxidation (34). Second, excess circulating free fatty acid is a common symptom of T2D, which leads to enhanced fatty acid oxidation. The increased fatty acid oxidation impedes normal physiology, resulting in accumulation of toxic metabolites (such as ceramides or acylcarnitines), which further exacerbate insulin resistance in hearts (35, 36). Third, because of overexpression of PDK4, PDC activity is inhibited. This leads to impaired pyruvate oxidation and accumulation of lactate, eventually triggering apoptosis (37, 38). The profound dysregulation in myocardial energy metabolism ultimately manifests redox stress and the loss of myocytes. It is noteworthy that control DIO mouse hearts displayed concurrent up-regulation of FFA and pyruvate oxidation, presumably in the face of increased PDK4 activation, when the availability of the substrates was held constant. This suggests that modulation of substrate availability in diabetic cardiomyopathy, i.e. supplementing the diet with pyruvate, might have cardioprotective effects. This method of treatment would be distinct from glitazone administration, which is known to have complicated feedback effects on heart function (9). Increased pyruvate availability could down-regulate FFA oxidation and subsequent proinflammatory reactive oxygen species (ROS) signaling (39).
PS10 inhibits all PDK isoforms (21) and restores PDC activity in the DIO mouse hearts (Fig. 1, B and C). Additionally, we have demonstrated that PS10 enhances pyruvate flux through the PDC in DIO mouse hearts using hyperpolarized [1-13C]pyruvate (Figs. 2 and 3A). Elevated pyruvate flux through the PDC increases energy production and mitigates accumulation of lactate in DIO hearts. From the isotopomer analysis, we calculated substrate selection in the PS10-treated DIO mouse hearts. PS10 lowers the fatty acid contribution and promotes pyruvate/lactate utilization for acetyl-CoA production over the control DIO mouse group. Fatty acid oxidation normally accounts for the majority (70–90%) of ATP production in hearts, but complete glucose oxidation is slightly more efficient in terms of ATP production per mole of O2 utilization (40). Under our perfusion conditions, PS10 did not augment oxygen consumption (Fig. S4). However, based on increased glucose/pyruvate contribution to acetyl-CoA production in PS10-treated DIO mouse hearts (Fig. 5A), slightly more efficient energy production might take place in the presence of this PDK inhibitor. Because the magnitude of this effect is small in the presence of physiological mixtures of substrates, establishing a clear difference in O2 consumption in the Langendorff heart is difficult (31). We hypothesize that a working heart would perform better than the Langendorff model upon the administration of PS10. In addition, because an increase in glucose/pyruvate oxidation would suppress fatty acid oxidation (41), we anticipate that there might be less accumulation of lipotoxic metabolites from fatty acid oxidation in PS10-treated hearts. Much like the hypothesis that pyruvate supplementation would have beneficial effects on heart metabolism in this etiology, PS10 should have a similar effect of balancing FFA oxidation versus carbohydrate oxidation. This should preserve heart function without exacerbating complications associated with fuel overload (9).
Considerations for data interpretation
It should be noted that the fractional sources of acetyl-CoA (Fig. 5), as measured by non–steady state analysis, differ significantly from well-known values of myocardial substrate selection. The nature of the hyperpolarization experiment necessitates the introduction of a bolus of pyruvate, bypassing regulation by GLUT4, and flooding the mitochondria with oxidizable substrate (42). The data of Fig. 5 are essentially a snapshot of substrate selection following equilibration to steady state with the [3-13C]pyruvate/lactate and the [U-13C]fatty acids, followed by a 4-min exposure of the myocardium to [1-13C]pyruvate. The high values for total carbohydrate contribution to acetyl-CoA production are therefore associated with the HP pyruvate bolus, not with underlying metabolic function. Nonetheless, every analysis performed here points to a single conclusion: PS10 increases fractional carbohydrate oxidation in the normal myocardium and in our model of diabetic cardiomyopathy.
DCA is the most commonly used PDK inhibitor since the 1970s (43). Because of its small molecular weight, there are multiple off-target effects associated with DCA. Therefore, it is not surprising that we observed a different mode of fuel utilization between DCA-treated and PS10-treated DIO mouse hearts. In our recent publication (44), the plasma lactate/pyruvate ratio changed from 38.4 in control DIO mice to 42.5 in the PS10-treated DIO mice. We did not measure the plasma pyruvate level in DCA-treated DIO mice. A study about the metabolic effects of dichloroacetate in the diabetic dog (45) demonstrated that the plasma lactate/pyruvate ratio changed from 27.1 to 34.1 in diabetic dogs within 2 h following a single oral dose (150 mg/kg) of DCA. These observations suggest that both DCA and PS10 treatments lead to a more reduced plasma state. However, only DCA produced a measurable change in lactate production as measured with HP pyruvate (Fig. 3). We believe the increasing lactate production from pyruvate induced by DCA is one of its secondary effects. The unmodulated FFA oxidation (fatty acid contribution to total acetyl-CoA pool) observed by isotopomer analysis (Figs. 4, C and D, 5B, and 6C) suggests that in addition to the intended PDK inhibition, DCA exerts secondary, negative effects on energy homeostasis. The multisite action of DCA makes it less useful than PS10 for the modulation of PDC activity. It is interesting to note that DCA up-regulated HP bicarbonate production (14) more than PS10, but did not enforce a similar change in the direct analysis of the glutamate isotopomers (Fig. 4, C and D). Similar discordant results for hyperpolarization experiments versus isotopomer analysis have been observed in the perfused mouse heart before (46). The total signal intensity observed in hyperpolarization experiments depends directly upon the T1 of the observed species. The T1 in turn is affected by many parameters, one of which is the tumbling rate of the molecule. Binding to an enzyme essentially reduces molecular tumbling to zero, and can be a source of significant relaxation. We suspect that changes in the T1 of the observed products associated with the length of time they are bound to the PDC underlies the appearance of the significantly larger [13C]bicarbonate signal for DCA hearts, but confirmation of this hypothesis would be exceedingly difficult. The 13C NMR data of Fig. 4C are exceedingly strong evidence that pyruvate oxidation is significantly more up-regulated in the PS10 heart versus the DCA heart.
Although the infusion of HP pyruvate essentially prevented application of steady state modeling to analysis of the glutamate isotopomers, the glutamine pool in the heart turns over considerably slower than the glutamate pool. TCA flux using a steady state analysis of the glutamine should be more indicative substrate selection prior to the HP pyruvate injection (47, 48). Although the non–steady state analysis of the glutamate must be regarded as the truest representation of oxidative metabolism at the moment metabolism was quenched, it was hoped that a steady state analysis of the glutamine would augment the glutamate-based analysis.
We integrated the signals of multiplets from the C3 and C4 of glutamine (Fig. S4), oxygen consumption rate, and heart weights to determine quantitative TCA turnover (Figs. S4 and S5). Using the analysis of glutamine, the fractional oxidation of carbohydrate in lean controls was ∼50%, closer to previously measured values but still very high (49). Despite differences in the quantitative results, the trends seen in Figs. 5 and 6 using the non–steady state analysis of glutamate are confirmed by the steady state analysis of the glutamine (Fig. S5). Indeed, PS10 causes a significant up-regulation of carbohydrate oxidation compared with both lean and DIO controls, a trend that did not reach significance in the non–steady state analysis. The overall lack of significant changes for DCA treatment are most puzzling. Further experiments without transitory exposure to pyruvate would produce more unequivocal results.
In summary, the present findings demonstrate that 13C MRI coupled with hyperpolarized tracers is likely a practical noninvasive method for specific detection of carbohydrate oxidation via the TCA cycle in the human heart. We believe the results here are robust enough to translate to human studies (25). We have demonstrated that PS10 exhibits beneficial properties of correcting defects in pathogenic bioenergetics in diabetic cardiomyopathy. PS10 increases fractional carbohydrate oxidation (Figs. 3 and 6). Glucose is the most efficient producer of reducing equivalents for oxidative phosphorylation per mole of O2 consumed (31). This should have benefits for cardiac energetics in diabetic cardiomyopathy. In addition, our hyperpolarization data suggest that it down-regulates pyruvate carboxylation (Fig. S2). This leads to reduced anaplerosis, which should also provide more efficient energy production per mole of O2 consumed by the myocardium (Fig. 7). With its high selectivity for PDK isoforms, PS10 is an excellent drug lead for treating diabetic cardiomyopathy.
Figure 7.
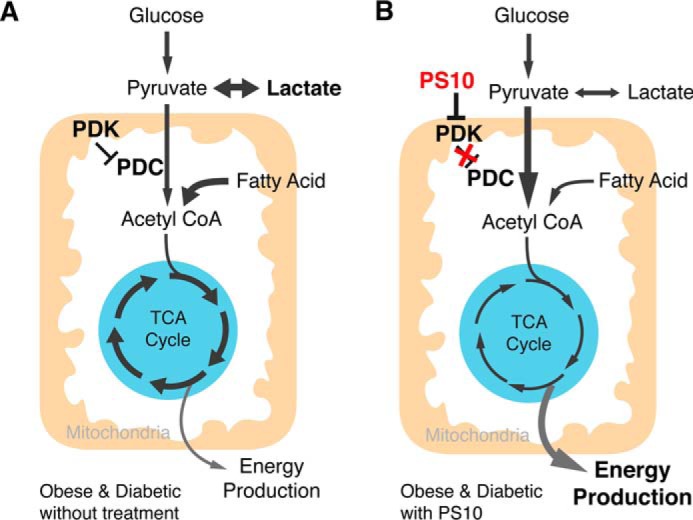
A working model of PS10 activating pyruvate dehydrogenase complex in obese mouse hearts. A, in obese/type 2 diabetes stage, PDKs are elevated to inhibit PDC activity, followed by reducing energy production from glucose, accumulation of lactate and increase of anaplerosis. B, with the treatment of PDK inhibitors, like PS10, PDC is active to utilize more glucose as fuel to increase energy production, and reduces both lactate accumulation and anaplerosis.
Experimental procedures
Animals
C57BL/6J male mice at 6 to 8 weeks old were obtained from a local breeding colony at UT Southwestern Medical Center (Dallas, TX). DIO mice were obtained by feeding the animals with a 60% high-fat diet, which contained 32% saturated and 68% unsaturated fat (catalogue no.: D12492, Research Diet Inc. New Brunswick, NJ) for 18 weeks. Age-matched C57BL/6J mice in the lean control group were fed with normal chow diet (no. 2916, Envigo, Madison, WI) for the same period of time before treatment. Mice were housed in an air-conditioned animal facility with 12-hour light-to-dark cycle. The protocol of accommodation and treatment was approved by the Institutional Animal Care and Use Committee at UT Southwestern Medical Center.
Chemicals
[1-13C]Pyruvic acid, sodium [3-13C]pyruvate, sodium [3-13C]lactate, and [U-13C]free fatty acids (all 99% enriched) were obtained from Cambridge Isotope Laboratories. The trityl radical OX063 was obtained from Oxford Molecular Biotools (Oxford, UK). All other chemicals were obtained from Sigma-Aldrich.
Treatment of PDK inhibitors
DIO mice were randomized into three groups: vehicle-, DCA-, and PS10-treated. PS10 was dissolved in 100% DMSO and then diluted to make a 10% DMSO aqueous solution containing 17.5% (w/v) (2-hydroxypropyl)-β-cyclodextrin for delivery. Animals were dosed at midday by intraperitoneal (IP) injections at 70 mg/kg of PS10 using 1-ml syringe and 30-gauge needle. The dose of DCA was 250 mg/kg in the same buffer solution. The treatment time for PS10 was 8 h and 1 h for DCA. At the treatment time indicated above after the injection, animals were euthanized using carbon dioxide asphyxiation followed by cervical dislocation and dissection. Hearts were removed and snap frozen in liquid nitrogen for assays. Average ischemia time before organ harvest was about 1 to 2 min. The time interval after injection was determined based on time-course experiments shown in Fig. S6 and literature (50).
Assay for PDC activity
Liquid nitrogen–stored tissue samples were removed and thawed on ice. Individual heart (200–300 mg) tissue samples were manually homogenized in an ice-chilled glass homogenizer containing 1 ml of the homogenization buffer. The homogenization buffer contained 30 mm potassium Pi, pH 7.5, 3 mm EDTA, 5 mm DTT, 1 mm benzamidine, 3% fetal bovine serum, 5% Triton X-100, and 1 mm leupeptin. Samples were transferred to ice-cold 10-ml polycarbonate tubes and spun in an ultracentrifuge at 25,000 × g for 10 min to pellet cell and tissue debris. Supernatants were removed and stored on ice until diluted (1:3 for muscle, 1:5 for liver, and 1:20 for kidneys and heart tissues) with a dilution buffer containing 50 mm HEPES, pH 7.5, 1.0 mm DTT, 0.1% Triton X-100, 5 mm DCA, 50 mm sodium fluoride, 3% fetal bovine serum, and 1 mm leupeptin. The diluted samples (50 μl) were placed in each well of a 24-well plate containing 310 μl of the reaction mixture. A micro-bridge (Hampton Research) was preset into each well holding one piece of 2 m NaOH-soaked filter wick. The reaction mixture contained 30 mm potassium Pi, pH 7.5, 0.4 mm CoA, 3 mm NAD+, 5% fetal bovine serum, 2 mm thiamine diphosphate, 2 mm MgCl2, and 65 μg of recombinant human E3. [1-14C]Pyruvate (PerkinElmer) was added to each well to initiate the reaction, with the wells sealed with a clear Mylar adhesive film. The assay plates were incubated at 37 °C for 10 min. Fifty μl of a 20% TCA solution was added to each well to stop the reaction. Assay plates were incubated further at 37 °C for 45 min. 14CO2 trapped on 2 m NaOH-soaked filter wicks were counted in a liquid scintillation counter.
Western blotting
SDS-PAGE gels were run using 15–20 μg of protein lysate per lane. Western blots were transferred to PVDF membranes for 2 h at 200 mV. PVDF membranes were blocked with 5% nonfat dried milk and then probed using polyclonal antibodies to pyruvate dehydrogenase/decarboxylase E1α and to phosphorylated E1α (pE1α). The E1α antibody was obtained from MitoSciences/Abcam (Cambridge, MA). Antibodies against the phosphorylated serine (pSer-293) residue of the E1α subunit were purchased from EMD Millipore/Calbiochem Biochemical (Billerica, MA). One ml of Luminata Forte Western HRP (EMD Millipore) substrate reagent was pipetted across the membrane for signal detection in a FluorChem E system (Cell Biosciences, Santa Clara, CA).
Glucose tolerance test
Mice fasted for 6.5 h before the tolerance test. Eight h after PS10 intraperitoneal administration or 1 h after DCA intraperitoneal administration, 1.5 g/kg of glucose was delivered to mice. Tail vein serum samples were collected immediately before and 15, 30, 60, and 120 min after the glucose challenge. Glucose levels in serum samples were determined by a blood glucose meter (Contour Blood Glucose Meter, Bayer, Whippany, NJ).
Heart perfusions
The protocol was similar to the one published earlier (22). In brief, hearts from mice with different treatment were rapidly excised under general anesthesia (ketamine/xylazine mixture) and perfused at 37 °C and 80 cm H2O pressure for 40 min. The perfusion solution was a modified Krebs-Henseleit buffer containing 25 mm NaHCO3, 118 mm NaCl, 4.7 mm KCl, 1.2 mm MgSO4, 1.2 mm KH2PO4, 0.5 mm Na2EDTA, 2.5 mm CaCl2, and 0.75% BSA. Oxidizable substrates available in the perfusion solution were 0.12 mm [3-13C]pyruvate, 1.2 mm [3-13C]lactate, and 0.4 mm [U-13C]free fatty acid. To mimic in vivo plasma concentration of the PDK inhibitors used, both inhibitors were dissolved in the perfusion buffer at 1 mg/ml for PS10 and 10 mg/ml for DCA to maintain compound concentrations in hearts during perfusion. These concentrations match the circulating concentrations achieved for the glucose tolerance test. The hearts were placed in a 10-mm NMR tube and inserted into the bore of a 600 MHz NMR spectrometer (Bruker AVIII HD) equipped with a 10-mm CPDUL CryoProbe (Bruker). The magnet was shimmed using 23Na signal from the heart surrounded by 23Na-free sucrose flush. The 23Na resonance was shimmed to a half height line width of ∼10 Hz.
Heart rates, coronary flow, myocardial oxygen consumptions, and developed pressures were measured at 30 min. The hearts were originally perfused for 40 min with the fatty acid, lactate, and pyruvate mixture to reach a physiological steady state before being injected with hyperpolarized 13C-pyruvate described below. After injection with HP[1-13C]pyruvate, 13C NMR observations were continued for about 3 min. Hearts were then removed from the magnet and freeze-clamped for later analysis.
Polarization of [1-13C]pyruvate and 13C NMR spectroscopy of the isolated heart
[1-13C]Pyruvic acid (2 μl) was polarized neat at ∼1.4 K using trityl OX063 (15 mm) as a radical in the presence of a gadolinium chelate (2 mm Gd3+) in a HyperSense Polariser (Oxford Instruments). The electron irradiation frequency was 94.112 GHz. After ∼90 min, the polarized sample was rapidly dissolved in hot PBS (5 ml). All of the hyperpolarized solution was mixed with 15 ml of substrate-free KH solution and delivered directly into the heart by catheter over a period of around 60 s. 13C NMR acquisition was initiated a few seconds before injection of the hyperpolarized solution. A series of 13C NMR spectra were collected every 2 s until all of the hyperpolarized 13C signal had decayed. Generally, 90 spectra were collected over 180 s. The data were zero filled before Fourier transformation and the relative peak areas were measured by integration using ACD/SpecManager (ACD/Labs, Toronto, Ontario, Canada). Peak areas of all metabolites were normalized to the total peak areas of all 13C resonances and plotted as a function of time. The perfused hearts were rapidly arrested and freeze-clamped for perchloric acid extracts and subsequent isotopomer analyses by high-resolution 13C NMR.
Analysis and metabolic flux measurement by high-resolution NMR
The freeze-clamped hearts were pulverized in liquid nitrogen and extracted with perchloric acid (4–6% v/v), neutralized, freeze-dried, and resuspended in 0.2 ml D2O containing 1 mm Na2EDTA and 0.5 mm 2,2-dimethyl-2-silapentanesulfonic acid as an internal standard for proton-decoupled 13C NMR spectroscopy (47, 48). Flux through PDC and citric acid cycle flux was determined using 13C NMR isotopomer analysis and normalization by O2 consumption. The relative rates of oxidation of each energy source, pyruvate, free fatty acid, or endogenous stores, were measured from the proton-decoupled 13C NMR spectrum of glutamate or glutamine, and proton spectrum of alanine. The hearts were freeze-clamped about 4 or 5 min after introduction of hyperpolarized [1-13C]pyruvate. At the time of freeze-clamping there were two sources of unlabeled acetyl-CoA ([1-13C]pyruvate and glycogen), two sources of [2-13C]acetyl-CoA ([3-13C]lactate and [3-13C]pyruvate in the perfusate), and one source of [1,2-13C]acetyl-CoA ([U-13C]long chain fatty acids). The fraction of acetyl-CoA derived from each source was defined as A, B, and C, respectively. The ratio B/A was determined from the 1H NMR spectrum of alanine based on the satellites because of single-bond JCH. This measurement assumes high activity of lactate dehydrogenase and alanine aminotransferase and interconversion of lactate, alanine, and pyruvate within seconds to minutes. The B/C ratio was determined as [2-13C]acetyl-CoA/[1, 2-13C]acetyl-CoA, which was calculated by the direct (non–steady state) analysis of glutamate C4 labeling pattern from the 13C NMR spectrum (31, 51). Glutamine is in slower exchange with TCA cycle intermediates, and hence steady state isotopomer analysis was used to analyze glutamine spectra (47, 48). After correcting for generation of NADH outside the citric acid cycle, the relative rate of flux through each pathway was converted to absolute flux with the measured oxygen consumption (31).
Statistics
Data are reported as mean ± S.D. or mean ± S.E. as indicated in figure legends. Statistical significance between any two groups was tested by unpaired two-tailed Student's t test in GraphPad Prism 7.0 software. *, p < 0.05; **, p < 0.01, ***, p < 0.001.
Author contributions
C.-Y. W., S. S., S. C. B., D. C., and M. E. M. conceptualization; C.-Y. W., S. S., W. G., R. M. W., G. S., C. K., and M. E. M. data curation; C.-Y. W. software; C.-Y. W., S. S., W. G., R. M. W., G. S., C. M., C. K., and M. E. M. formal analysis; C.-Y. W., S. S., R. M. W., G. S., C. M., D. C., and M. E. M. investigation; C.-Y. W. visualization; C.-Y. W. and S. S. writing-original draft; C.-Y. W., S. C. B., and M. E. M. project administration; C.-Y. W., R. M. W., C. M., A. D. S., D. C., and M. E. M. writing-review and editing; W. G., R. M. W., G. S., C. K., A. D. S., D. C., and M. E. M. methodology; R. M. W., M. L., X. Q., C. K., A. D. S., D. C., and M. E. M. resources; R. M. W., C. M., C. K., D. C., and M. E. M. validation; S. C. B., C. M., A. D. S., D. C., and M. E. M. supervision; C. M., A. D. S., D. C., and M. E. M. funding acquisition; M. L. and X. Q. small molecule synthesis; D. C. co-corresponding author.
Supplementary Material
Acknowledgments
We acknowledge Dr. Xiaodong Wen and Thomas Hever in the Advanced Imaging Research Center (AIRC) of UT Southwestern (UTSW) for their technical support in the perfusion experiments.
This work was supported by National Institutes of Health Grants DK62306, R37-HL034557, and P41-EB015908 and by Welch Foundation Grants I-1286 and I-1804. This work was also supported by National Institutes of Health Grants R01s DK105346, HD087306, DK112865, P41122698, and U24DK097209 and by NSF DMR 1644779 (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6.
- T2D
- type 2 diabetes
- DIO
- diet-induced obese
- HF
- heart failure
- HP
- hyperpolarized
- MRS
- magnetic resonance spectroscopy
- PDC
- pyruvate dehydrogenase complex
- PDK
- pyruvate dehydrogenase kinase
- TCA
- tricarboxylic acid
- DCA
- dichloroacetate
- PS10
- 2((2,4-dihydroxyphenyl)sulfonyl) isoindoline-4,6-diol.
References
- 1. Hu F. B. (2011) Globalization of diabetes: The role of diet, lifestyle, and genes. Diabetes Care 34, 1249–1257 10.2337/dc11-0442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fukushima A., and Lopaschuk G. D. (2016) Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta 1861, 1525–1534 10.1016/j.bbalip.2016.03.020 [DOI] [PubMed] [Google Scholar]
- 3. Regan T. J., Lyons M. M., Ahmed S. S., Levinson G. E., Oldewurtel H. A., Ahmad M. R., and Haider B. (1977) Evidence for cardiomyopathy in familial diabetes mellitus. J. Clin. Invest. 60, 884–899 10.1172/JCI108843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aneja A., Tang W. H., Bansilal S., Garcia M. J., and Farkouh M. E. (2008) Diabetic cardiomyopathy: Insights into pathogenesis, diagnostic challenges, and therapeutic options. Am. J. Med. 121, 748–757 10.1016/j.amjmed.2008.03.046 [DOI] [PubMed] [Google Scholar]
- 5. Harris R. A., Bowker-Kinley M. M., Huang B., and Wu P. (2002) Regulation of the activity of the pyruvate dehydrogenase complex. Adv. Enzyme Regul. 42, 249–259 10.1016/S0065-2571(01)00061-9 [DOI] [PubMed] [Google Scholar]
- 6. Kuo T. H., Giacomelli F., Wiener J., and Lapanowski-Netzel K. (1985) Pyruvate dehydrogenase activity in cardiac mitochondria from genetically diabetic mice. Diabetes 34, 1075–1081 [DOI] [PubMed] [Google Scholar]
- 7. Wu P., Sato J., Zhao Y., Jaskiewicz J., Popov K. M., and Harris R. A. (1998) Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. Biochem. J. 329, 197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang B., Wu P., Bowker-Kinley M. M., and Harris R. A. (2002) Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-α ligands, glucocorticoids, and insulin. Diabetes 51, 276–283 10.2337/diabetes.51.2.276 [DOI] [PubMed] [Google Scholar]
- 9. Taegtmeyer H., Beauloye C., Harmancey R., and Hue L. (2013) Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am. J. Physiol. Heart Circ. Physiol. 305, H1693–H1697 10.1152/ajpheart.00854.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Standl E., Schnell O., and McGuire D. K. (2016) Heart failure considerations of antihyperglycemic medications for type 2 diabetes. Circ. Res. 118, 1830–1843 10.1161/CIRCRESAHA.116.306924 [DOI] [PubMed] [Google Scholar]
- 11. Lopaschuk G. D., and Stanley W. C. (1997) Glucose metabolism in the ischemic heart. Circulation 95, 313–315 10.1161/01.CIR.95.2.313 [DOI] [PubMed] [Google Scholar]
- 12. Lei B., Lionetti V., Young M. E., Chandler M. P., d'Agostino C., Kang E., Altarejos M., Matsuo K., Hintze T. H., Stanley W. C., and Recchia F. A. (2004) Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J. Mol. Cell Cardiol. 36, 567–576 10.1016/j.yjmcc.2004.02.004 [DOI] [PubMed] [Google Scholar]
- 13. Kato M., Li J., Chuang J. L., and Chuang D. T. (2007) Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 15, 992–1004 10.1016/j.str.2007.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Le Page L. M., Rider O. J., Lewis A. J., Ball V., Clarke K., Johansson E., Carr C. A., Heather L. C., and Tyler D. J. (2015) Increasing pyruvate dehydrogenase flux as a treatment for diabetic cardiomyopathy: A combined 13C hyperpolarized magnetic resonance and echocardiography study. Diabetes 64, 2735–2743 10.2337/db14-1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bersin R. M., and Stacpoole P. W. (1997) Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am. Heart J. 134, 841–855 10.1016/S0002-8703(97)80007-5 [DOI] [PubMed] [Google Scholar]
- 16. Stacpoole P. W., Harwood H. J. Jr., Cameron D. F., Curry S. H., Samuelson D. A., Cornwell P. E., and Sauberlich H. E. (1990) Chronic toxicity of dichloroacetate: Possible relation to thiamine deficiency in rats. Fundam. Appl. Toxicol. 14, 327–337 10.1016/0272-0590(90)90212-3 [DOI] [PubMed] [Google Scholar]
- 17. Bull R. J. (2000) Mode of action of liver tumor induction by trichloroethylene and its metabolites, trichloroacetate and dichloroacetate. Environ. Health Perspect. 108, Suppl. 2, 241–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mayers R. M., Butlin R. J., Kilgour E., Leighton B., Martin D., Myatt J., Orme J. P., and Holloway B. R. (2003) AZD7545, a novel inhibitor of pyruvate dehydrogenase kinase 2 (PDHK2), activates pyruvate dehydrogenase in vivo and improves blood glucose control in obese (fa/fa) Zucker rats. Biochem. Soc. Trans. 31, 1165–1167 10.1042/bst0311165 [DOI] [PubMed] [Google Scholar]
- 19. Jones H. B., Reens J., Johnson E., Brocklehurst S., and Slater I. (2014) Myocardial steatosis and necrosis in atria and ventricles of rats given pyruvate dehydrogenase kinase inhibitors. Toxicol. Pathol. 42, 1250–1266 10.1177/0192623314530195 [DOI] [PubMed] [Google Scholar]
- 20. Wynn R. M., Kato M., Chuang J. L., Tso S. C., Li J., and Chuang D. T. (2008) Pyruvate dehydrogenase kinase-4 structures reveal a metastable open conformation fostering robust core-free basal activity. J. Biol. Chem. 283, 25305–25315 10.1074/jbc.M802249200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tso S. C., Qi X., Gui W. J., Wu C. Y., Chuang J. L., Wernstedt-Asterholm I., Morlock L. K., Owens K. R., Scherer P. E., Williams N. S., Tambar U. K., Wynn R. M., and Chuang D. T. (2014) Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J. Biol. Chem. 289, 4432–4443 10.1074/jbc.M113.533885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Merritt M. E., Harrison C., Storey C., Jeffrey F. M., Sherry A. D., and Malloy C. R. (2007) Hyperpolarized 13C allows a direct measure of flux through a single enzyme-catalyzed step by NMR. Proc. Natl. Acad. Sci. U.S.A. 104, 19773–19777 10.1073/pnas.0706235104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lapidot A., and Gopher A. (1994) Cerebral metabolic compartmentation. Estimation of glucose flux via pyruvate carboxylase/pyruvate dehydrogenase by 13C NMR isotopomer analysis of v-[U-13C]glucose metabolites. J. Biol. Chem. 269, 27198–27208 [PubMed] [Google Scholar]
- 24. Schroeder M. A., Atherton H. J., Ball D. R., Cole M. A., Heather L. C., Griffin J. L., Clarke K., Radda G. K., and Tyler D. J. (2009) Real-time assessment of Krebs cycle metabolism using hyperpolarized 13C magnetic resonance spectroscopy. FASEB J. 23, 2529–2538 10.1096/fj.09-129171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cunningham C. H., Lau J. Y. C., Chen A. P., Geraghty B. J., Perks W. J., Roifman I., Wright G. A., and Connelly K. A. (2016) Hyperpolarized 13C metabolic MRI of the human heart: Initial experience. Circ. Res. 119, 1177–1182 10.1161/CIRCRESAHA.116.309769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sundqvist K. E., Hiltunen J. K., and Hassinen I. E. (1989) Pyruvate carboxylation in the rat heart. Role of biotin-dependent enzymes. Biochem. J. 257, 913–916 10.1042/bj2570913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Russell R. R. 3rd, and Taegtmeyer H. (1991) Pyruvate carboxylation prevents the decline in contractile function of rat hearts oxidizing acetoacetate. Am. J. Physiol. Heart Circ. Physiol. 261, H1756–H1762 10.1152/ajpheart.1991.261.6.H1756 [DOI] [PubMed] [Google Scholar]
- 28. Malloy C. R., Thompson J. R., Jeffrey F. M. H., and Sherry A. D. (1990) Contribution of exogenous substrates to acetyl coenzyme-A: Measurement by C-13 NMR under non-steady-state conditions. Biochemistry 29, 6756–6761 10.1021/bi00481a002 [DOI] [PubMed] [Google Scholar]
- 29. Malloy C. R., Sherry A. D., and Jeffrey F. M. (1987) Carbon flux through citric acid cycle pathways in perfused heart by 13C NMR spectroscopy. FEBS Lett. 212, 58–62 10.1016/0014-5793(87)81556-9 [DOI] [PubMed] [Google Scholar]
- 30. Zhao G., Jeoung N. H., Burgess S. C., Rosaaen-Stowe K. A., Inagaki T., Latif S., Shelton J. M., McAnally J., Bassel-Duby R., Harris R. A., Richardson J. A., and Kliewer S. A. (2008) Overexpression of pyruvate dehydrogenase kinase 4 in heart perturbs metabolism and exacerbates calcineurin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 294, H936–H943 10.1152/ajpheart.00870.2007 [DOI] [PubMed] [Google Scholar]
- 31. Malloy C. R., Jones J. G., Jeffrey F. M., Jessen M. E., and Sherry A. D. (1996) Contribution of various substrates to total citric acid cycle flux and anaplerosis as determined by 13C isotopomer analysis and O2 consumption in the heart. MAGMA 4, 35–46 10.1007/BF01759778 [DOI] [PubMed] [Google Scholar]
- 32. Fang Z. Y., Prins J. B., and Marwick T. H. (2004) Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr. Rev. 25, 543–567 10.1210/er.2003-0012 [DOI] [PubMed] [Google Scholar]
- 33. Garvey W. T., Hardin D., Juhaszova M., and Dominguez J. H. (1993) Effects of diabetes on myocardial glucose transport system in rats: Implications for diabetic cardiomyopathy. Am. J. Physiol. 264, H837–H844 10.1152/ajpheart.1993.264.3.H837 [DOI] [PubMed] [Google Scholar]
- 34. Sugden M. C., Lall H. S., Harris R. A., and Holness M. J. (2000) Selective modification of the pyruvate dehydrogenase kinase isoform profile in skeletal muscle in hyperthyroidism: Implications for the regulatory impact of glucose on fatty acid oxidation. J. Endocrinol. 167, 339–345 10.1677/joe.0.1670339 [DOI] [PubMed] [Google Scholar]
- 35. Yazaki Y., Isobe M., Takahashi W., Kitabayashi H., Nishiyama O., Sekiguchi M., and Takemura T. (1999) Assessment of myocardial fatty acid metabolic abnormalities in patients with idiopathic dilated cardiomyopathy using 123I BMIPP SPECT: Correlation with clinicopathological findings and clinical course. Heart 81, 153–159 10.1136/hrt.81.2.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakayama H., Morozumi T., Nanto S., Shimonagata T., Ohara T., Takano Y., Kotani J., Watanabe T., Fujita M., Nishio M., Kusuoka H., Hori M., and Nagata S. (2001) Abnormal myocardial free fatty acid utilization deteriorates with morphological changes in the hypertensive heart. Jpn Circ. J. 65, 783–787 10.1253/jcj.65.783 [DOI] [PubMed] [Google Scholar]
- 37. Hall J. L., Stanley W. C., Lopaschuk G. D., Wisneski J. A., Pizzurro R. D., Hamilton C. D., and McCormack J. G. (1996) Impaired pyruvate oxidation but normal glucose uptake in diabetic pig heart during dobutamine-induced work. Am. J. Physiol. 271, H2320–H2329 10.1152/ajpheart.1996.271.6.H2320 [DOI] [PubMed] [Google Scholar]
- 38. Chatham J. C., Gao Z. P., Bonen A., and Forder J. R. (1999) Preferential inhibition of lactate oxidation relative to glucose oxidation in the rat heart following diabetes. Cardiovasc. Res. 43, 96–106 10.1016/S0008-6363(99)00056-5 [DOI] [PubMed] [Google Scholar]
- 39. Du X., Edelstein D., Obici S., Higham N., Zou M. H., and Brownlee M. (2006) Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J. Clin. Invest. 116, 1071–1080 10.1172/JCI23354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mjos O. D. (1971) Effect of free fatty acids on myocardial function and oxygen consumption in intact dogs. J. Clin. Invest. 50, 1386–1389 10.1172/JCI106621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Randle P. J. (1998) Regulatory interactions between lipids and carbohydrates: The glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 14, 263–283 10.1002/(SICI)1099-0895(199812)14:4%3C263::AID-DMR233%3E3.0.CO%3B2-C [DOI] [PubMed] [Google Scholar]
- 42. Moreno K. X., Sabelhaus S. M., Merritt M. E., Sherry A. D., and Malloy C. R. (2010) Competition of pyruvate with physiological substrates for oxidation by the heart: Implications for studies with hyperpolarized [1-13C]pyruvate. Am. J. Physiol. Heart Circ. Physiol. 298, H1556–H1564 10.1152/ajpheart.00656.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Whitehouse S., and Randle P. J. (1973) Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (short communication). Biochem. J. 134, 651–653 10.1042/bj1340651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu C. Y., Tso S. C., Chuang J. L., Gui W. J., Lou M., Sharma G., Khemtong C., Qi X., Wynn R. M., and Chuang D. T. (2018) Targeting hepatic pyruvate dehydrogenase kinases restores insulin signaling and mitigates ChREBP-mediated lipogenesis in diet-induced obese mice. Mol. Metab. 10.1016/j.molmet.2018.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ribes G., Valette G., and Loubatieres-Mariani M. M. (1979) Metabolic effects of sodium dichloroacetate in normal and diabetic dogs. Diabetes 28, 852–857 10.2337/diab.28.9.852 [DOI] [PubMed] [Google Scholar]
- 46. Purmal C., Kucejova B., Sherry A. D., Burgess S. C., Malloy C. R., and Merritt M. E. (2014) Propionate stimulates pyruvate oxidation in the presence of acetate. Am. J. Physiol. Heart Circ. Physiol. 307, H1134–H1141 10.1152/ajpheart.00407.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Malloy C. R., Sherry A. D., and Jeffrey F. M. (1988) Evaluation of carbon flux and substrate selection through alternate pathways involving the citric acid cycle of the heart by 13C NMR spectroscopy. J. Biol. Chem. 263, 6964–6971 [PubMed] [Google Scholar]
- 48. Malloy C. R., Sherry A. D., and Jeffrey F. M. (1990) Analysis of tricarboxylic acid cycle of the heart using 13C isotope isomers. Am. J. Physiol. 259, H987–H995 10.1152/ajpheart.1990.259.3.H987 [DOI] [PubMed] [Google Scholar]
- 49. Stanley W. C., Recchia F. A., and Lopaschuk G. D. (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 85, 1093–1129 10.1152/physrev.00006.2004 [DOI] [PubMed] [Google Scholar]
- 50. Wells P. G., Moore G. W., Rabin D., Wilkinson G. R., Oates J. A., and Stacpoole P. W. (1980) Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans. Diabetologia 19, 109–113 10.1007/BF00421855 [DOI] [PubMed] [Google Scholar]
- 51. Sherry A. D., Malloy C. R., Zhao P., and Thompson J. R. (1992) Alterations in substrate utilization in the reperfused myocardium: A direct analysis by 13C NMR. Biochemistry 31, 4833–4837 10.1021/bi00135a014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.