

Abstract
The four R-spondins (RSPO1–4) strongly potentiate Wnt signaling and play critical roles in normal development, adult stem cell survival, and cancer development and aggressiveness. All four RSPOs have been suggested to potentiate Wnt signaling by binding to three related receptors, i.e. leucine-rich repeat–containing, G protein–coupled receptors 4, 5, and 6 (LGR4/5/6), and then inducing the clearance of two E3 ubiquitin ligases (RNF43 and ZNRF3) that otherwise would ubiquitinate Wnt receptors for degradation. Here, we show that RSPO1–4 have differential dependence on LGRs in potentiating Wnt/β-catenin signaling and that RSPO2 can enhance this pathway without any LGR. LGR4 knockout (LGR4KO) in HEK293 cells completely abrogated the Wnt/β-catenin signaling response to RSPO1 and RSPO4 and strongly impaired the response to RSPO3. RSPO2, however, retained robust activity albeit with decreased potency. Complete rescue of RSPO1–4 activity in LGR4KO cells required the seven-transmembrane domain of LGR4. Furthermore, an RSPO2 mutant with normal binding affinity to ZNRF3 but no or little binding to LGR4 or LGR5 still potentiated Wnt/β-catenin signaling in vitro, supported the growth of intestinal organoids ex vivo, and stimulated intestinal crypt growth in vivo. Mechanistically, RSPO2 could increase Wnt receptor levels in the absence of any LGR without affecting ZNRF3 endocytosis and stability. These findings suggest that RSPO1–4 use distinct mechanisms in regulating Wnt and other signaling pathways, which have important implications for understanding the pleiotropic functions of RSPOs and LGRs in both normal and cancer development.
Keywords: cell signaling, stem cells, 7-helix receptor, Wnt signaling, beta-catenin (β-catenin), LGR4, LGR5, organoids, R-spondin
Introduction
R-spondins (RSPOs)2 are a group of four related secreted proteins with critical roles in organ development and survival of adult stem cells as well as in cancer development. They were originally isolated as thrombospondin domain–containing genes with high expression in the roof plate of the neuronal tube and therefore referred to as R-spondins (1). The four RSPOs (RSPO1–4) are ∼50% identical to each other in amino acid sequence and constitute an overall similar structure with the N-terminal half containing two cysteine-rich, furin-like domains (Fu1 and Fu2) and the C-terminal half containing a thrombospondin (TSP)-like domain followed by a highly basic region (1, 2). Human genetic studies revealed that RSPO1 is essential for ovarian development, whereas RSPO4 is required for nail formation (3–5). In mice, knockout of either RSPO2 or RSPO3 is embryonically lethal, attributed to major defects in lung and limb (RSPO2) or placental development (RSPO3) (6–8). Aberrant expression of RSPO2 and RSPO3 through gain of expression of gene fusions was identified in subsets of colon and other solid tumors as a driving mechanism of oncogenesis (9). Overexpression of RSPO3 in lung adenocarcinomas due to transcriptional activation promotes tumor aggressiveness, and anti-RSPO3 antibodies are being tested in clinical trials for cancer treatment (10, 11).
The pleiotropic functions of RSPO1–4 in normal and cancer development were believed to be mediated by their robust potentiation of Wnt/β-catenin signaling. Initial studies showed that all RSPOs were able to enhance Wnt/β-catenin signaling through increasing phosphorylation of Wnt coreceptors LRP5/6 but could not activate the β-catenin pathway on their own (2, 12–14). Studies by us and others then demonstrated that RSPOs bound to a group of three related receptors leucine-rich repeat–containing, G protein–coupled receptors 4, 5, and 6 (LGR4, LGR5, and LGR6) with high affinity to potentiate Wnt signaling (15–18). Meanwhile, it was discovered that two related E3 ubiquitin ligases, RNF43 and ZNRF3, which are integral membrane proteins with an extracellular domain capable of binding to RSPOs and a large intracellular domain containing a RING-type ubiquitin ligase, could negatively regulate Wnt/β-catenin signaling via ubiquitination and degradation of the Wnt coreceptor Frizzled (FZD) (19, 20). These studies showed that RSPOs could clear the E3 ligases from the cell membrane through simultaneous binding to LGR4 and RNF43/ZNRF3 (19), leading to increased levels of FZD receptor and thus higher Wnt signaling. The binding affinities of RSPO1–4 for RNF43/ZNRF3 vary considerably, but they have similar potency and efficacy in potentiating Wnt/β-catenin signaling with the exception of RSPO4, which has lower efficacy (15, 21–23). Thus, the current model is that RSPO enhances Wnt/β signaling by facilitating LGR4/5 binding to RNF43/ZNRF3 and inducing their clearance. However, this is predicated on the hypothesis that RSPO requires LGR4/5 for binding and internalization of the E3 ligases (19, 24).
Conversely, we and others showed that LGR5 forms a supercomplex with the Wnt signalosome to potentiate Wnt/β-catenin signaling (16, 25). We demonstrated that both LGR4 and LGR5 interact with a signaling scaffold protein, IQGAP1, through their seven-transmembrane domain (7TM) (26, 27). Furthermore, LGR4 was shown to form a supercomplex with the Wnt signalosome through IQGAP1–Dishevelled interaction to increase LRP5/6 phosphorylation and therefore enhance Wnt/β-catenin signaling (26). Importantly, this LGR4/IQGAP1 pathway is contingent on RSPO-mediated inhibition of RNF43/ZNRF3 because the E3 ligases have a dominant effect on Wnt receptor level (26). Here, we report that, in the absence of LGR4/5, RSPO2 retains its ability to potentiate Wnt/β-catenin signaling, likely due to its intrinsic, high-affinity binding to ZNRF3. An LGR4 binding–defective RSPO2 mutant was able to support the growth of intestinal crypts ex vivo and in vivo. In contrast, RSPO1 and RSPO4 were completely dependent on LGR4 for potentiating Wnt/β-catenin signaling even though RSPO1 showed moderate binding to ZNRF3 in the absence of LGR4. Complete rescue of RSPO1–4–mediated Wnt/βcatenin activity in LGR4KO cells required the 7TM domain of LGR4 despite the extracellular domain (ECD) TM and full-length LGR4 having equal RSPO binding affinities. The findings suggest that LGR dependence of RSPOs is RSPO-specific and that both inhibition of RNF43/ZNRF3 and recruitment of IQGAP1 are essential for full activity of RSPO1–4.
Results
Knockout of LGR4 in the Wnt/β-catenin signaling reporter cell line HEK293-STF led to complete loss of response to RSPO1
HEK293-Super TOPFlash (STF) is a reporter cell line that stably harbors the firefly luciferase gene under the control of β-catenin/T-cell factor enhancer and has been widely used for the measurement of Wnt/β-catenin signaling (28). STF cells showed a robust, dose-dependent response to RSPO1, and this activity required the presence of Wnt ligands (Fig. S1, A and B). To characterize the relationship between RSPOs and LGR4 in potentiating Wnt/β-catenin signaling, we used the CRISPR/Cas9 method to knock out LGR4 in STF cells (29). Lenti-CRISPR2 containing an LGR4 single guide RNA sequence was introduced into STF cells, and two stable clones (designated C6 and C11) with loss of LGR4 protein were identified (Fig. 1A, upper panel). Analysis of the genomic DNA sequences of the two clones revealed that C11 had either a bp deletion or insertion located inside the guide sequence (Fig. S2) with no evidence of exon skipping (Fig. S3). Clone C6, however, contained various deletions of 7–80 nucleotides and a small percentage of wildtype (WT) sequences (Fig. S2), indicating that the colony was a mixture of cells when picked, and some of the cells were mutated in only one allele. Both clones no longer responded to RSPO1 stimulation at up to 10× the saturating concentration in the parental cells (Fig. 1B). All the following LGR4KO data presented here were obtained with 293-STF-LGR4KO C11 as it only contained two frameshift mutations in genomic DNA and lacked LGR4 expression. Western blot (WB) analysis of C11 showed that RSPO1 could no longer induce LRP6 phosphorylation (Fig. 1A, lower panel). Furthermore, transfection of recombinant LGR4 into either clone restored the RSPO1 response to its full potency and efficacy (Fig. S4A). In addition, loss of LGR4 had no apparent effect on the response to Wnt3a alone (Fig. S4B). These results suggest that STF cells depend on LGR4 to confer response to RSPO1, consistent with previous data that HEK293 cells do not express LGR5 or LGR6 at levels sufficient to confer response to RSPOs (15, 16, 18).
Figure 1.
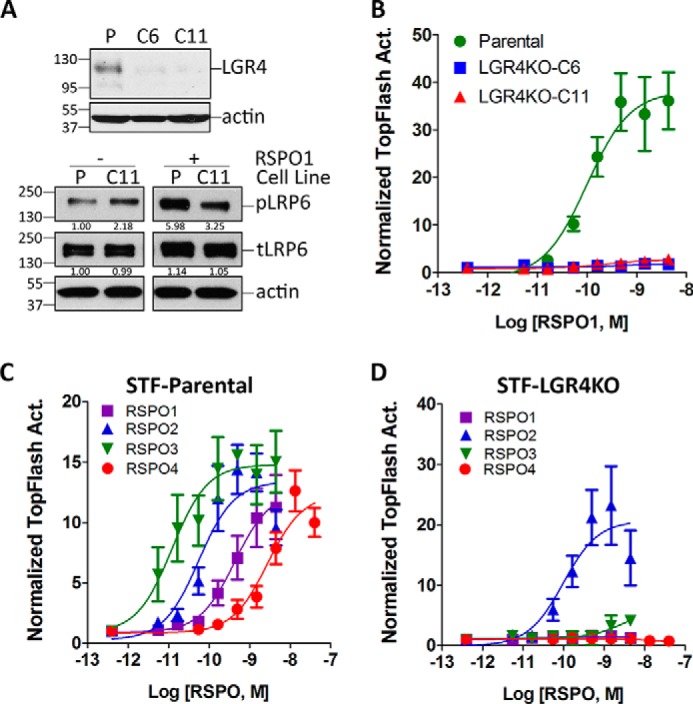
RSPO1–4 have differential dependence on LGR4 to potentiate Wnt/β-catenin signaling. A, upper panel, WB of LGR4 and actin in parental STF cells (P) and two clones (C6 and C11) with CRISPR-mediated KO of LGR4. Lower panel, WB of phospho-LRP6 (pLRP6), total LRP6 (tLRP6), and actin in parental and STF-LGR4KO C11 cells treated with RSPO1 and Wnt3a-CM for 5 h. Ratios of phospho-LRP6 and total LRP6 to actin, normalized to parental STF without RSPO1 treatment, are shown below the WB bands. B, TOPFlash results of RSPO1 response in parental STF cells and LGR4KO C6 and C11 cells. C, TOPFlash results of RSPO1–4 in parental STF cells. D, TOPFlash results of RSPO1–4 in STF-LGR4KO C11 cells. All TOPFlash activity (Act.) was normalized by the cell number first and then to the baseline (no ligand, Wnt3a-CM only). All error bars are S.E. (n = 3).
RSPO2 potentiates Wnt/β-catenin signaling in the absence of LGR4
Next, we compared the enhancement of Wnt/β-catenin signaling by recombinant, full-length RSPO1–4 in STF-parental and -LGR4KO C11 cells. As expected, all four RSPOs were able to potentiate Wnt/β-catenin signaling in STF-parental cells in the rank order of potency of RSPO3 > RSPO2 > RSPO1 > RSPO4 with similar maximum efficacy (Fig. 1C and Table 1), consistent with previously published results from multiple groups (15, 16, 21, 22). Surprisingly, RSPO2 was able to potentiate Wnt/β-catenin signaling in STF-LGR4KO cells with ∼⅓ of the potency of the parental cells (Fig. 1D and Table 1). RSPO3 began to show low activity at higher concentrations, whereas RSPO1 and RSPO4 were completely inactive (Fig. 1D). These results suggest that RSPO1 and RSPO4 are totally dependent on LGR4 to potentiate Wnt/β-catenin signaling, whereas RSPO2 has substantial activity without LGR4, seemingly consistent with reports showing poor affinity of RSPO1 and RSPO4 (low to high μm, respectively) and high affinity of RSPO2 (low nm) in binding to ZNRF3 (19, 22, 23, 30). Of note, HEK293 cells do not express RNF43 at a functionally relevant level (19, 20), and RSPO1–4 bind to both RNF43 and ZNRF3 with similar profiles (30). Intriguingly, although both RSPO2 and RSPO3 have similar affinities to ZNRF3 (23, 30), only RSPO2 could potentiate Wnt/β-catenin signaling with high potency and efficacy in LGR4KO cells. As all four RSPOs have similar high-affinity binding (low nm) to LGR4, these findings suggest that intrinsic differences among the four RSPOs in potentiating Wnt/β-catenin signaling cannot be simply attributed to their differential affinities to ZNRF3/RNF43.
Table 1.
Potency and efficacy of RSPOs in binding and functional assays
Mean and 95% confidence interval are given. ND, not determined; NC, not calculable.
| RSPO | Binding (KD) |
Function (EC50) |
|||
|---|---|---|---|---|---|
| LGR4-FL | LGR4-ECD | ZNRF3-ECD | STF | STF-LGR4KO | |
| nm | nm | ||||
| RSPO1 | ND | ND | ND | 0.43 (0.31–0.59) | NC |
| RSPO2 | ND | ND | ND | 0.05 (0.008–0.39) | 0.10 (0.01–0.68) |
| RSPO3 | ND | ND | ND | 0.02 (0.005–0.032) | NC |
| RSPO4 | ND | ND | ND | 2.75 (0.70–10.88) | NC |
| R1Fu-Fc | 19.50 (10.56–35.34) | 7.16 (5.54–9.26) | 11.48 (5.31 − 24.65) | 6.60 (4.21–10.42) | NC |
| R2Fu-Fc | 1.27 (0.88–1.82) | 0.35 (0.21–0.56) | 0.35 (0.14–0.86) | 0.30 (0.09–0.97) | 40.74 (13.49–125.80) |
| R3Fu-Fc | 2.06 (1.37–3.09) | 6.98 (3.42–14.23) | 2.34 (1.36–4.02) | 0.93 (0.23–3.85) | 18.62 (15.04–22.94) |
| R4Fu-Fc | 1.13 (0.81–1.57) | 1.05 (0.74–1.51) | NC | 0.78 (0.09–6.91) | NC |
| R2Fu-F109A | NC | ND | 1.95 (1.23–3.13) | 8.33 (2.44–28.5) | 21.04 (5.47–80.93) |
| R2Fu-RQ | 0.64 (0.31–1.31) | ND | 21.76 (10.95–43.24) | 8.63 (3.16–23.51) | NC |
Furin domains of RSPO1–4 exhibit differential dependence on LGR4
Previous determinations of RSPO binding to the E3 ligases were all carried out with recombinant, purified ECDs of RNF43 or ZNRF3 (22, 23, 30). As the furin domains of RSPO1–4 are both necessary and sufficient to confer potentiation of Wnt/β-catenin signaling (2, 31), we expressed and purified furin domain–Fc fusion proteins of each RSPO (R1–4Fu-Fc) and characterized their interactions with LGR4 and ZNRF3 expressed on cell membrane using live cells. Saturation binding analysis on HEK293T cells overexpressing LGR4 showed that furin domains of RSPO2/3/4 had similar binding affinity (KD ≈ 1 nm), whereas KD of R1Fu-Fc was 20 nm (Fig. 2A and Table 1). In binding to cells expressing ZNRF3-ECDTM (ZNRF3 truncated of the intracellular domain), the four furin domains displayed a wide range of affinity with the rank order of R2Fu > R3Fu ≫ R1Fu ⋙ R4Fu (Fig. 2B and Table 1). These results are largely consistent with previous binding data using purified ECDs of ZNRF3 and LGR4 (22, 23, 30). In STF-parental cells, the furin domains of RSPO1–4 all displayed similar dose-dependent activity as the full-length RSPOs but with higher EC50 values (Fig. 2C versus 1C). However, R4Fu-Fc showed an increase in potency (0.77 versus 2.75 nm) (Table 1). The decreased potency of the furin domains of RSPO1/2/3 is probably due to loss of binding to proteoglycans on the extracellular matrix by TSP domains, which have been shown to enhance RSPO availability at the cell surface (32). In STF-LGR4KO cells, furin domains of RSPO2 and RSPO3 were equally active, whereas those of RSPO1 and RSPO4 were completely inactive (Fig. 2D). The contrasting activity of full-length RSPO3 and its furin domain in STF-LGR4KO cells suggests that the TSP domain of RSPO3 may hinder its activity in the absence of LGR4.
Figure 2.
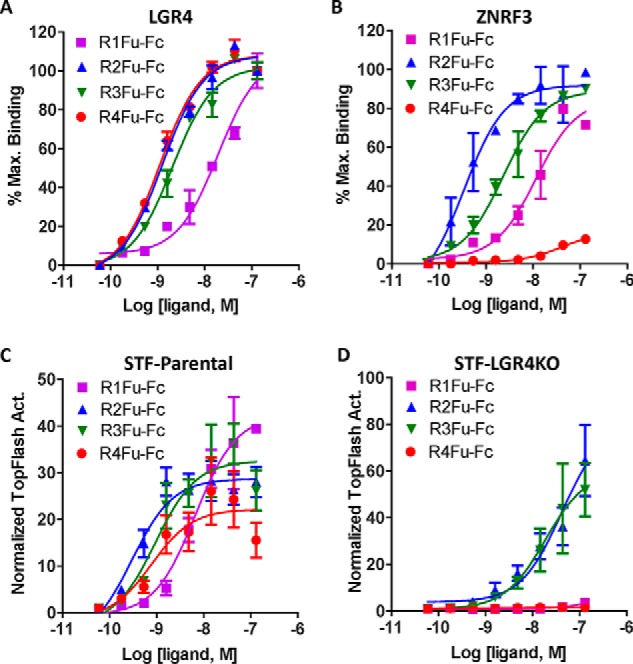
Furin domains of RSPO2 and RSPO3 potentiated Wnt/β-catenin signaling without LGR4. A, saturation binding analysis of the furin domains of RSPO1–4 fused to Fc on HEK293T cells stably expressing full-length human LGR4. B, saturation binding analysis of furin domains of RSPO1–4 fused on HEK293T cells transiently transfected with ZNRF3-ECDTM. C, TOPFlash results of furin-Fc fusion protein in parental STF cells. D, TOPFlash results of furin-Fc fusion proteins in STF-LGR4KO cells. All error bars represent S.E. Max., maximum; Act., activity.
LGR4 binding–defective RSPO2 mutant can potentiate Wnt/β-catenin signaling
Cocrystal structures showed that Fu-1 and Fu-2 domains of RSPOs bind to ZNRF3/RNF43 and LGR4, respectively, and the two domains are largely structurally independent (23, 33–36). To further characterize LGR4-independent activity of RSPO2, we generated and purified RSPO2 furin domain mutants in R2Fu-Fc (or R2Fu-WT) that are defective in binding to either LGR4 or ZNRF3. Phe-109 in RSPO2 Fu-2 domain is predicted to make direct contact with LGR4/5-ECD, and its mutation to Ala (R2Fu-F109A) led to total loss of binding to LGR4 and near total loss to LGR5 without affecting binding to ZNRF3 (Fig. 3, A and B, Fig. S5, and Table 1). Single mutations of Arg-65 and Glu-70 in RSPO2 Fu-1 domain were reported to have much lower binding affinity to purified ZNRF3-ECD (23). We generated a double mutant, R65A/Q70R, of R2Fu-Fc (R2Fu-RQ) and found that it still retained considerable binding to ZNRF3-overexpressing cells at higher concentrations, whereas its binding to LGR4-overexpressing cells remained unchanged when compared with the R2Fu-WT (Fig. 3, A and B, and Table 1). Wnt/β-catenin reporter assays of R2Fu-WT, R2Fu-F109A, and R2Fu-RQ in STF-parental cells showed that R2Fu-F109A and R2Fu-RQ were both active but displayed lower potency and efficacy (Fig. 3C). This suggests that the lack of LGR4 binding by R2Fu-F109A reduced the maximum TOPFlash activity by ∼½ when compared with WT. Likewise, mutation of E3 ligase–binding sites in R2Fu-RQ resulted in a similar decrease in TOPFlash activity. These results indicate that binding of RSPO2 to both LGR4 and E3 ligases is important to potentiate Wnt/β-catenin signaling. In contrast, R2Fu-WT and R2Fu-F109A showed similar TOPFlash activity in STF-LGR4KO cells (Fig. 3D) as both ligands only interacted with E3 ligases in the absence of LGR4. However, R2Fu-RQ TOPFlash activity was completely abolished even though it retained substantial binding affinity to ZNRF3 and unchanged affinity to LGR4 (Fig. 3, B and D). Taken together, these results revealed that, for RSPO2, binding to LGR4 is not essential for potentiation of Wnt/β-catenin signaling so long as high-affinity binding to ZNRF3 is retained.
Figure 3.
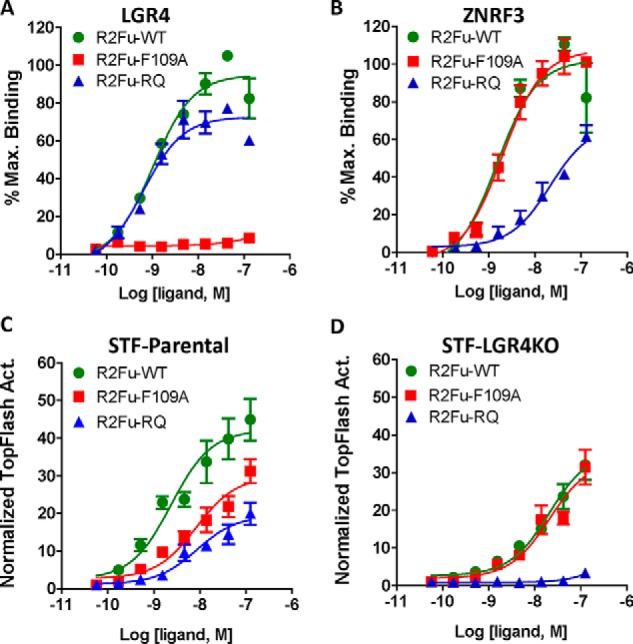
LGR4 binding–defective R2Fu mutant potentiated Wnt/β-catenin signaling. A and B, saturation binding of R2Fu-WT, -F109A, and -RQ to HEK293T cells stably expressing LGR4 (A) or transiently expressing ZNRF3-ECDTM (B). C and D, TOPFlash results of R2Fu-WT and the two mutants in parental STF cells (C) or STF-LGR4KO cells (D). All error bars represent S.E. Max., maximum; Act., activity.
Complete rescue of RSPO1–4 activity requires the 7TM domain of LGR4
Previously, we proposed that RSPO-LGR4 potentiated Wnt/β-catenin signaling through two parallel pathways: inhibition of ZNRF3/RNF43 and recruitment of IQGAP1 to enhance LRP6 phosphorylation with inhibition of the E3 ligases having a dominant effect (26). We reasoned that at least some of the differential activities of RSPO1–4 may be due to differences in engaging IQGAP1 and E3 ligase pathways. IQGAP1 recruitment requires the 7TM domain of LGR4, whereas ZNRF3/RNF43 binding is mediated by RSPOs bound to LGR4-ECD. Therefore, we examined whether RSPO1–4 would respond differentially in STF-LGR4KO cells transfected with either full-length LGR4 (LGR4-FL) or LGR4 mutant (LGR4-ECDTM) that expresses LGR4-ECD anchored to cell membrane via the single transmembrane domain of CD4 (26). Ligand binding analysis showed that LGR4-ECDTM expressed in HEK293T cells bound to the furin domains of RSPO1–4 with the same affinity as LGR4-FL–overexpressing cells (Fig. S6A and Table 1). When transfected into STF-LGR4KO cells, LGR4-FL was able to rescue RSPO1–4 activity to full potency and efficacy (Fig. 4, A–D, and Table 2). In contrast, LGR4-ECDTM only partially rescued RSPO1–3 with little effect on RSPO4 (Fig. 4, A–D, and Table 2). WB analysis of the transfected cells indicated that LGR4-FL and -ECDTM were expressed at comparable levels (Fig. S6B). Because LGR4-ECDTM binds to RSPO1–4 with the same high affinity as LGR4-FL, these results suggest that full activity of RSPO1–4 requires the 7TM domain and the engagement of both the IQGAP1 and E3 ligase pathways. Interestingly, LGR5-ECD anchored to cell membrane was also shown to be unable to rescue RSPO1 activity in Wnt/β-catenin signaling in HEK293 cells (34).
Figure 4.
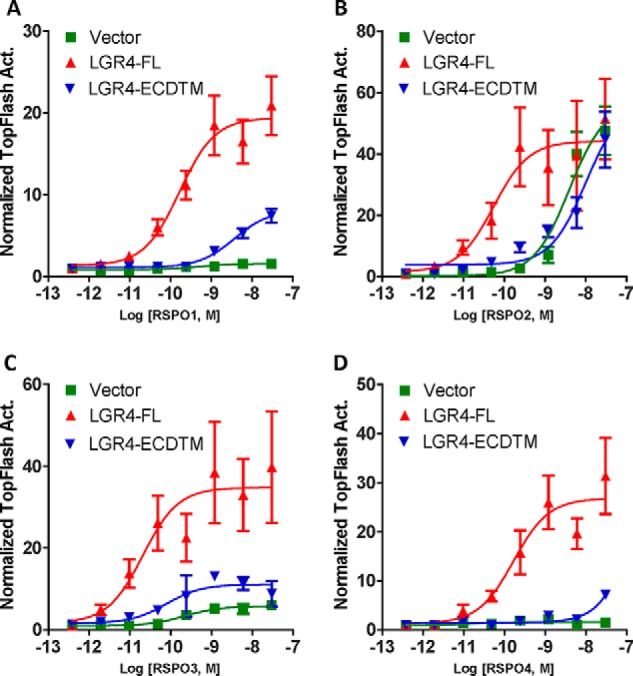
The 7TM domain of LGR4 is required to fully rescue RSPO1–4. A–D, TOPFlash assay results of STF-LGR4KO C11 cells transiently transfected with vector control, LGR4-FL, or LGR4-ECDTM anchored to the cell membrane and then stimulated with RSPO1 (A), RSPO2 (B), RSPO3 (C), and RSPO4 (D). All error bars represent S.E. Act., activity.
Table 2.
Potency and efficacy of RSPOs in functional assays in transiently transfected LGR4KO cells
Mean and 95% confidence interval are given. NC, not calculable.
| RSPO | Vector control | LGR4-FL | LGR4-ECD |
|---|---|---|---|
| RSPO1 | NC | 0.17 (0.07–0.42) | 3.68 (2.38–5.67) |
| RSPO2 | 3.93 (1.39–11.11) | 0.05 (0.01–0.24) | 10.07 (2.35–43.17) |
| RSPO3 | NC | 0.02 (0.003–0.132) | 0.08 (0.01–0.55) |
| RSPO4 | NC | 0.17 (0.03–0.84) | NC |
LGR4 binding–defective RSPO2 mutant supports organoid growth ex vivo and intestinal growth in vivo
Based on our in vitro studies, LGR4-independent potentiation of Wnt/β-catenin signaling by RSPO2 is expected to support intestinal growth ex vivo and in vivo. In this context, we first tested effects of R2Fu-WT and R2Fu-F109A on intestinal organoid growth ex vivo. Organoid cultures were established from the neonates of Lgr5 heterozygous mice (Lgr5-EGFP-IRES-creERT2) and tested with various concentrations of R2Fu-WT and R2Fu-F109A. In the absence of any RSPO ligand in the culture, there was no visible organoid growth by day 5 (Fig. 5, left panel) as expected. In contrast, both R2Fu-WT– and R2Fu-F109A–supplemented cultures exhibited organoid growths starting at day 3 and continuing to day 5 (Fig. 5, right panels). R2Fu-WT was able to support organoid growth at the lowest concentration tested, 0.1 μg/ml, whereas R2Fu-F109A required a higher dose to observe significant growth of organoids (Fig. 5), consistent with our in vitro TOPFlash activity where the EC50 of R2FuWT was at least 10-fold lower than that of R2Fu-F109A. Of note, R2Fu-F109A also showed little binding to LGR5 as expected (Fig. S5), so this organoid-supporting activity was not due to residual LGR5-mediated function. Then, we tested whether the F109A mutant was able to stimulate intestinal crypt growth in vivo. R2Fu-WT and R2Fu-F109A were administered into normal C57 mice during an 8-day period for a total of three doses, and the intestinal growth was examined via histology and Ki67 and olfactomedin 4 (Olfm4) staining. Hematoxylin and eosin staining of the intestines showed that both R2Fu-WT and R2Fu-F109A increased crypt length compared with the PBS-administered intestine (Fig. 6A). Ki67 staining confirmed that animals treated with R2Fu-WT and R2Fu-F109A showed increased numbers of proliferating cells in their intestinal crypts (Fig. 6B). To further confirm the activation of the Wnt signaling pathway by R2Fu-WT and R2Fu-F109A, we examined the expression of Olfm4, a validated Wnt/β-catenin signaling–response gene and marker of murine intestinal stem cells (37). As shown in Fig. 6C, Olfm4 staining revealed that the number of stem cells was increased in both R2Fu-WT– and R2Fu-F109A–injected cells compared with the PBS-injected control, confirming activation of Wnt/β-catenin signaling and increased proliferation of stem cells. Overall, these results indicate that the RSPO2 furin domain is able to potentiate Wnt/β-catenin signaling and stimulate intestinal stem cell growth without binding to LGR4, albeit with lower potency.
Figure 5.

LGR4 binding–defective RSPO2 mutant supported organoid growth ex vivo. A and B, micrographs of intestinal organoids cultured with R2Fu-WT or R2Fu-F109A mutant at the indicated concentrations on day 3 (A) or day 5 (B). Scale bars, 250 μm.
Figure 6.
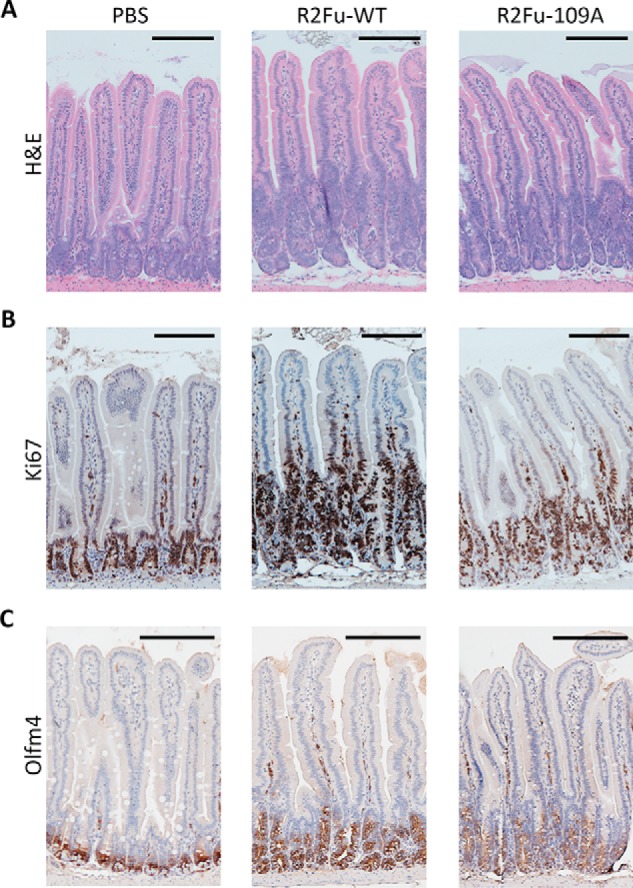
LGR4 binding–defective RSPO2 mutant stimulated intestinal crypt growth in vivo. Shown are micrographs of hematoxylin and eosin (H&E) staining (A) or Ki67 staining (B), or Olfm4 staining (C) of representative intestine sections. Scale bars (top right corners), 200 μm.
RSPO2 increased levels of Wnt coreceptor FZD without LGR4
We then dissected the mechanism of how RSPO2 may potentiate Wnt/β-catenin signaling without binding to its primary receptor, LGR4. First, to confirm that the canonical Wnt/β-catenin signaling pathway was activated by both R2Fu-WT and R2Fu-F109A, we examined phosphorylation of LRP6 and levels of nonmembrane-bound β-catenin in STF-parental and -LGR4KO cells in response to R2Fu-WT or R2Fu-F109A treatment in the presence of Wnt. Phosphorylated LRP6 and β-catenin levels were elevated to a similar extent in STF-parental and -LGR4KO cells by both R2Fu-WT and R2Fu-F109A (Fig. 7A), confirming that LGR4 is not necessary to activate the canonical Wnt/β-catenin signaling pathway by RSPO2.
Figure 7.

R2Fu-Fc potentiated Wnt/β-catenin signaling without inducing degradation of ZNRF3. A, WB results of phospho-LRP6 (pLRP6), total LRP6 (tLRP6), actin, nonmembrane-bound β-catenin, and tubulin in parental STF and LGR4KO cells treated with vehicle (lane 1), R2Fu-WT (lane 2), or R2Fu-F109A (lane 3). Ratios of phospho-LRP6 and total LRP6 to actin and ratios of β-catenin to tubulin, all normalized to parental STF without treatment, are listed below the WB bands. B, WB results of FZD5, ZNRF3, and actin in STF-parental and -LGR4KO cells treated with vehicle (lane 1), R2Fu-WT (lane 2), or R2Fu-F109A (lane 3). Ratios of FZD5 and ZNRF3 to actin, normalized to parental STF without treatment, are listed below the WB bands. C, confocal microscopy images of STF-LGR4KO C11 cells transiently transfected with ZNRF3 and incubated with vehicle (panel a), R2Fu-WT (panels b–d), or R2Fu-F109A (panels e–g) for 1 h at 37 °C. Immunostaining was performed with Cy3-labeled Myc (ZNRF3) and Alexa Fluor 488–labeled anti-human antibody (R2Fu-Fc). Scale bars, 10 μm.
Previously, RSPOs were shown to increase levels of Wnt coreceptor FZDs by bridging LGR4 and RNF43/ZNRF3 to form a heterotrimer and induce the clearance of the E3 ligases from the membrane (19), although the details remained poorly understood. We tested whether RSPO2 activates the Wnt/β-catenin pathway by stabilizing FZD and found that both WT and the F109A mutant of R2Fu-Fc increased levels of FZD5 to a similar extent when ZNRF3-WT and FZD5 were cotransfected into either STF-parental or -LGR4KO cells (Fig. 7B). Interestingly, R2Fu-Fc treatment did not cause changes in levels of ZNRF3 (Fig. 7B), suggesting that increased FZD levels were not due to RSPO-induced degradation of ZNRF3. We then examined whether R2Fu-WT and mutant affect endocytosis of ZNRF3 and found that ZNRF3 was constitutively internalized in STF-LGR4KO cells, and treatment with either WT or F109A mutant of R2Fu had no gross effect on endocytosis (Fig. 7C). These results indicate that RSPO2 alone is able to potentiate Wnt/β-catenin signaling through stabilization of FZD without affecting degradation or endocytosis of ZNRF3.
Discussion
The four RSPOs share a similar overall structure with a highly conserved furin domain at the N terminus that is both necessary and sufficient to potentiate Wnt/β-catenin signaling and a less conserved C-terminal TSP domain that binds syndecans and other proteoglycans and may regulate noncanonical Wnt signaling. RSPO1–4 have divergent, nonoverlapping expression with each RSPO having unique, tissue-specific roles in organ development and carcinogenesis. RSPOs bind to LGR4–6 with high affinity and to RNF43/ZNRF3 with a wide range of affinities (23, 30). However, it has been assumed that RSPOs potentiate Wnt/β-catenin signaling predominantly by a similar mechanism, i.e. using LGR4–6 to bind to and clear RNF43/ZNRF3 by LGR-mediated endocytosis to increase Wnt receptor level. Here, we present compelling data that indicate that RSPO1–4 have distinct requirements for LGRs in potentiating Wnt/β-catenin signaling and that endocytosis-mediated clearance from cell membrane by LGR4 is not essential for inhibition of RNF43/ZNRF3.
For RSPO1–3, the furin domains alone displayed much lower potency when compared with full-length proteins despite similar affinity in binding to LGR4, consistent with data from other reports (14, 32). The TSP domains of RSPO1–3 were shown to bind to proteoglycans, such as heparin sulfate proteoglycans, and syndecans with high affinity (2, 32, 38). Without the TSP domains, abundance of RSPOs at the membrane is probably lower than that of the full-length protein, and thus deletion of TSP domains leads to lower potency without affecting efficacy. Interestingly, the furin domain alone of RSPO1 was nearly as active as full-length RSPO1 in cells overexpressing LGR4 (21), further implying that receptor density plays a large role in potency determination. In addition, the furin domains of both RSPO2 and RSPO3 bind to ZNRF3/RNF43 with relatively high affinity (low to sub-nm) and have similar potency and efficacy in parental STF cells; however, only full-length RSPO2 could potentiate Wnt/β-catenin signaling with high potency and efficacy in STF-LGR4KO cells. The much-reduced potency and efficacy of the full-length RSPO3 in LGR4KO cells suggest that the TSP domain and C-terminal basic regions of RSPO3 inhibit RSPO3 function structurally without LGR4 expression. Unfortunately, none of the RSPOs have full-length crystal structures available to verify this speculation. These findings clearly indicate that the furin and TSP domains of different RSPOs affect each other in unique ways that require further investigation.
The prevailing model of RSPO function is that potency and efficacy in the potentiation of Wnt/β-catenin signaling are totally determined by its binding affinity to RNF43/ZNRF3 (22, 23). Here, we found that the correlation between ZNRF3-binding affinity and potency/efficacy in signaling is unique for each RSPO, and the potency and efficacy do not always track with their binding affinity to the E3 ligases. RSPO1 furin domain (R1Fu) and RSPO2 furin domain mutant (F2Fu-RQ) bind to ZNRF3 with approximately the same affinity (Table 1); however, R1Fu showed much better efficacy in signaling potentiation (Fig. 2C versus 3C). Also, R4Fu binds to ZNRF3 with poor affinity and low maximum binding compared with other RSPOs; however, it was nearly as effective as all others in signaling (Fig. 2, B and C). If binding to ZNRF3 solely determines RSPO's strength in signaling, these results imply that binding to LGR4 would somehow induce a conformational change in RSPO4 to increase its affinity for ZNRF3. However, several structural studies showed that RSPO binding to LGR4/5 has no effect on its binding to ZNRF3 (23, 34, 36). Another argument for ZNRF3 binding affinity being the determining factor is that RSPO mutations located in the ZNRF3-binding motif all led to severe reduction or total loss of activity (23, 24, 34, 36). However, the ZNRF3-binding region of RSPOs is also involved in 2:2 heterodimer formation of RSPO-LGR5 and presumably of RSPO-LGR4, which may be critical for LGR4 activation (34, 36, 39). Clearly, further studies will be required to delineate the relationship between RSPO binding to ZNRF3 versus binding to LGR in trans and its effect on potentiating Wnt signaling.
Another unequivocal aspect of our study is that the 7TM domain of LGR4 is essential for the full potency and efficacy of RSPO1–4, presumably through engagement of the IQGAP1 pathway. A similar result was obtained with LGR5 using a glycosylphosphatidylinositol-anchored ECD of LGR5 (34). This cannot be simply due to the lack of endocytosis by the LGR5-ECD alone because an LGR5 mutant with impaired endocytosis was actually more effective in mediating RSPO activity (25, 40). Furthermore, we found that full-length ZNRF3 is capable of undergoing endocytosis spontaneously, and treatment with R2Fu had no obvious effect on the process (Fig. 7C) or on the protein level of ZNRF3 (Fig. 7B). However, ZNRF3-induced degradation of FZD5 was prevented by RSPO2 even in the absence of LGR4 (Fig. 7B). These results imply that RSPOs with high-affinity binding to ZNRF3 are able to inhibit the interaction of ZNRF3 with its substrate (FZD) or its E3 ligase activity without involving LGRs and the endocytic pathway.
While this manuscript was in preparation, Lebensohn and Rohatgi (41) reported that RSPO2 and RPSO3 were able to potentiate Wnt/β-catenin signaling without LGR4, largely consistent with our findings presented here. The report focused on characterization of RSPO3 and its mutant using a haploid cell line derived from HEK293 cells and presented evidence that RSPO3 binding to proteoglycan played a major role in rescuing its activity. One key difference between this report and ours is that we found that full-length RPSO3 had low activity, whereas the furin domain of RSPO3 had higher activity in diploid LGR4KO cells. This difference may be due to the use of different cell lines. In the STF cells we used, binding of RSPO3 to proteoglycan must have prevented its interaction with ZNRF3 in the absence of LGR4.
In conclusion, we showed that the four R-spondins have unique requirements for LGR4, and their furin and TSP domains affect each other in RSPO-specific ways to potentiate Wnt/β-catenin signaling. Of particular surprise is that RSPO2 has substantial activity without LGR4 and that RSPO3 may also be able to function similarly depending on the extracellular matrix context. Additionally, LGR4-mediated endocytosis is not required for blockade of ZNRF3/RNF43 by RSPO to increase Wnt receptor levels. These findings have important implications in understanding the pleiotropic functions and mechanisms of RSPOs and LGR in normal development and disease pathophysiology as well as in the development of therapeutics. This study raises important questions about the independent functions and mechanisms of RSPOs and LGR4 in regulating Wnt signaling and potentially other cell signaling pathways.
Experimental procedures
Plasmids and cloning
Plasmids encoding HA-LGR4-FL (human LGR4 full length) and HA-LGR4-ECDTM in pIRES-puro3 were described previously (15). Full-length rat FZD5 with a FLAG tag at the N terminus was cloned into pIRES-puro3 using standard procedures. Myc-ZNRF3-ECDTM and Myc-ZNRF3-WT were gifts from Dr. Feng Cong (Novartis Institute for Biomedical Research) (19). Furin domains of human RSPO1 (AA 21–144), RSPO2 (AA 37–143), RSPO3 (AA 21–144), and RSPO4 (AA 32–137) fused to human IgG1-Fc at the C terminus via a (GGGGS)3 linker were cloned into pCEP4 vector with a signal peptide using the In-Fusion HD cloning kit (Clontech). Mutations in RSPO2 furin domain were also generated using the In-Fusion HD cloning kit. All plasmids were verified by DNA sequencing.
Recombinant proteins and antibodies
Recombinant full-length human RSPO1–4 were purchased from R&D Systems. For Western blot analysis, anti-HA (Invitrogen catalog number 71-5500), anti-FLAG (Sigma catalog number F7425), anti-Myc (Cell Signaling Technology catalog number 2276), anti-β-actin (Cell Signaling Technology catalog number 4970), anti-phosphorylated and total LRP6 (Cell Signaling Technology catalog numbers 2568 and 3395), anti-tubulin (Cell Signaling Technology catalog number 2146), and anti-β-catenin (Cell Signaling Technology catalog number 9562) were used. For immunocytochemistry, anti-HA-Alexa Fluor 488 (Cell Signaling Technology catalog number 2350), anti-Myc-Cy3 (Sigma catalog number C6594), and anti-human Alexa Fluor 488 or Alexa Fluor 555 (Invitrogen catalog numbers A11013 and A21433) were used. For intestine staining, a hematoxylin and eosin staining kit (Vector Laboratories catalog number G-3502), anti-Ki67 (Cell Signaling Technology catalog number 12202), and anti-Olfm4 (Cell Signaling Technology catalog number 39141) were used.
Cell culture, transient transfection, and generation of stable STF LGR4KO cells
HEK293T and HEK293-STF cells were purchased from ATCC and cultured in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin at 37 °C with 95% humidity and 5% CO2. HEK293-LGR4– and HEK293-LGR4-ECD–overexpressing cells were generated and cultured as described previously (15). For transient transfection, ∼80% confluent cells were transfected with DNA: FuGENE HD (Promega) at a ratio of 1:3 for all transient transfections presented. For CRISPR-based knockout of LGR4 in STF cells, the guide sequence GGATGACAACAGCTTGACGG (corresponding to nucleotides 480–499 of human LGR4 ORF) was cloned into the vector Lenti-CRISPR2 as described (29). Lentiviral particles of Lenti-CRISPR2 containing the LGR4 guide sequence were used to infect STF cells and were selected with puromycin at 1 μg/ml. Single colonies were isolated and analyzed for LGR4 protein level by Western blotting using the anti-LGR4 antibody 7E6, which we described previously (42). Genomic DNA sequences of the two LGR4KO clones, C6 and C11, were determined as follows: the target single guide RNA region (exon 5 of LGR4) was amplified from their respective genomic DNA by PCR using a forward primer (TTGTGAGACTTGAACTGACTCA; located in intron 4) and a reverse primer (AGGGAGATTCAACAATTATCATCA; located in intron 5). The PCR products were cloned into pCR2.1 vector using TA cloning kit (Invitrogen), and a total of 16 plasmid clones (six for C6 and ten for C11) were sequenced.
Expression and purification of Fc-tagged RSPO furin domain proteins
HEK293F cells were purchased from Invitrogen and cultured in FreestyleTM 293 Expression medium at 37 °C with 95% humidity and 7% CO2. To produce human Fc-tagged RSPO furin domain proteins, HEK293F cells were transfected with pCEP4 vectors harboring each DNA sequence for RSPO furin domains with Fc tags. Briefly, 1 μg/ml DNA/2.5 × 106 cells was transfected with 0.5 μg/μl polyethylenimine, and cells were supplemented with 2.2 mm valproic acid 24 h post-transfection. Supernatant was retrieved 8 days later, and target proteins were applied to a column with CaptivA protein A affinity resin (Repligen) for purification. The column was washed with 20 mm phosphate buffer (0.2 m NaH2PO4 and 0.2 m Na2HPO4), and the target proteins were eluted in 100 mm glycine buffer, pH 2.6, and 1 m Tris buffer, pH 9.0. Finally, the buffer was exchanged to PBS using a 30-kDa Amicon Ultra-15 centrifugal filter (Millipore). Protein concentrations were determined using A280, and purities were verified by Coomassie staining.
Western blotting
If treated, STF-parental, -LGR4KO, or transiently transfected LGR4KO cells were incubated with 0.1 μg/ml RSPO1 for 5 h or 1 μg/ml R2Fu-Fc overnight in the presence of Wnt. Cells were lysed with radioimmune precipitation assay buffer (50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 1 mm DTT, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) supplemented with protease and phosphatase inhibitors and reduced at 37 °C for 1 h. Horseradish peroxidase–conjugated secondary rabbit or mouse antibodies (Cell Signaling Technology) were used following the standard ECL protocol. For FZD5 blots, ubiquitin-, FZD5-, and ZNRF3-WT–cotransfected cells were treated with 1 μg/ml R2Fu-WT or R2Fu-F109A in the presence of Wnt, incubated overnight, and lysed with immunoprecipitation lysis buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, and 5% glycerol) supplemented with protease and phosphatase inhibitors. WB analysis results were quantified by ratios of each band to its loading control (actin or tubulin) and normalized to a control condition.
Wnt/β-catenin signaling TOPFlash reporter enzyme assay
HEK293T-STF parental, STF-LGR4KO C6 and C11, and transiently transfected STF-LGR4KO C11 cells were subjected to a Wnt3a TOPFlash activity assay in various conditions. For the Wnt dependence test, STF-parental and -LGR4KO cells were diluted in various concentrations of Wnt3a-conditioned medium (CM). RSPO ligands were diluted by 3× or 5× serial dilution in DMEM or 1:5 Wnt3a CM:DMEM, added to a half-well white 96-well plate with 5000 cells/well, and incubated overnight at 37 °C with 95% humidity and 5% CO2. In the case of treatment with the porcupine inhibitor, LGK974 was added 2 days before cells were treated with RSPO1. Then 1× Alamar Blue was added to each well, and emission reading at 585 nm was used to estimate the live cell number. 1:1 luciferase reagent:DMEM was then added to each well at room temperature for 15 min, and the luciferase level was measured using an Envision Multimode plate reader (PerkinElmer Life Sciences). The TOPFlash activity was formulated by dividing the luciferase reading by the Alamar Blue count and normalized to the baseline, which contained Wnt3a-CM only. Dose-response curves (log(agonist) versus response (three parameters)) were fitted using GraphPad Prism, and EC50 was retrieved. All experiments were repeated at least three times with duplicates or triplicates in each experiment.
Whole-cell binding analysis
HEK293-LGR4, HEK293-LGR4-ECDTM, and HEK293-LGR5 stable cell lines or HEK293T cells transiently transfected with Myc-ZNRF3-ECDTM were seeded on poly-d-lysine–coated black, clear-bottom 96-well plates and incubated overnight. The plates were chilled on ice before binding assays started. Fc-tagged RSPO furin domain proteins were diluted by 3× serial dilution, added to the cells, and incubated on ice for 1 h. After fixation of cells with 4.2% paraformaldehyde, cells were incubated with anti-human Alexa Fluor 555 antibody. Emission at 550 nm was measured using a plate reader (Tecan). Dose-response curves (log(agonist) versus response (three parameters)) were fitted using GraphPad Prism to retrieve half-maximum binding (KD). All experiments were repeated at least three times with duplicates or triplicates in each experiment.
Immunofluorescence analysis
For ZNRF3 localization, STF-LGR4KO C11 cells, transfected with Myc-ZNRF3-WT, were treated with anti-Myc-Cy3 along with 1 μg/ml R2Fu-WT or R2Fu-F109A in 1:5 Wnt3a-CM:DMEM with Wnt3a-CM only as a control. The treated cells were incubated at 37 °C with 95% humidity and 5% CO2 for 1 h and fixed with 4.2% paraformaldehyde followed by permeabilization with 0.1% saponin. Then the secondary antibody, anti-human Alexa Fluor 488 was used to label Fc-tagged RSPO2. Cells were imaged under a confocal microscope and analyzed by Leica LAS AF Lite software.
Ex vivo intestinal crypt organoid culture and intestine growth in vivo
All animal experiments were performed in accordance with a protocol approved by the Animal Protocol Review Committee of the University of Texas Health Science Center at Houston. B6.129P2-Lgr5tm1(cre/ERT2)Cle/J mice were purchased from The Jackson Laboratory. Mouse intestinal crypt organoid cultures were established from B6.129P2-Lgr5tm1(cre/ERT2)Cle/J mice as described previously (27, 43). In brief, small intestines were harvested from neonates and washed to remove contaminants and villi. Intestinal fragments were incubated in EDTA for 30 min, strained, and pelleted. Crypts were resuspended in Matrigel with DMEM/F-12 containing 10 mm HEPES, GlutaMAX, 1× B27, 1× N2, 1 mm N-acetylcysteine, 50 ng/ml mouse epidermal growth factor, 100 ng/ml mouse Noggin, and penicillin/streptomycin. The medium was supplemented with 0.1, 0.5, or 2 μg/ml R2Fu-WT and R2Fu-F109A for 5 days. For intestine growth in vivo, female C57BL/6 mice were purchased from The Jackson Laboratory and administered with R2Fu-WT or R2Fu-F109A at 3 mg/kg by intraperitoneal injection on days 1, 3, and 5. PBS was used as a vehicle control. On day 8, the mice were sacrificed, and intestines were collected and fixed for histology. Paraffin sections of intestines were either stained with hematoxylin and eosin for phenotype characterization or immunostained with Ki67 and Olfm4 antibodies for detection of proliferating cells and intestinal stem cells using standard protocols for immunohistochemistry analysis.
Author contributions
S. P., J. C., K. C., and Q. J. L. conceptualization; S. P., J. C., W. A. Y., L. W., K. C., and Q. J. L. data curation; S. P., J. C., K. C., and Q. J. L. formal analysis; S. P., K. C., and Q. J. L. validation; S. P., J. C., W. A. Y., L. W., K. C., and Q. J. L. investigation; S. P., K. C., and Q. J. L. visualization; S. P., W. A. Y., L. W., K. C., and Q. J. L. methodology; S. P., K. C., and Q. J. L. writing-original draft; S. P., K. C., and Q. J. L. writing-review and editing; Q. J. L. resources; Q. J. L. software; Q. J. L. supervision; Q. J. L. funding acquisition; Q. J. L. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Feng Zhang (Massachusetts Institute of Technology) for the Lenti-CRISPR2 plasmid and Dr. Feng Cong (Novartis) for kindly providing the ZNRF3-ECDTM and ZNRF3-WT plasmids.
This work was supported in part by Cancer Prevention and Research Institute of Texas Grants RP160235 and RP170245 and the Janice D. Gordon endowment for bowel cancer research (to Q. J. L). Q. J. L. is a cofounder of Wntrix, and J. C. is a full-time employee of Wntrix.
This article contains Figs. S1–S6.
- RSPO
- R-spondin
- LGR
- leucine-rich repeat–containing, G protein–coupled receptor
- Fu
- furin-like domain
- TSP
- thrombospondin
- FZD
- Frizzled
- TM
- transmembrane domain
- ECD
- extracellular domain
- STF
- Super TOPFlash
- CRISPR
- clustered regularly interspaced short palindromic repeats
- WB
- Western blot
- RQ
- R65A/Q70R
- FL
- full-length
- Olfm4
- olfactomedin 4
- AA
- amino acids
- CM
- conditioned medium
- KO
- knockout.
References
- 1. Kamata T., Katsube K., Michikawa M., Yamada M., Takada S., and Mizusawa H. (2004) R-spondin, a novel gene with thrombospondin type 1 domain, was expressed in the dorsal neural tube and affected in Wnts mutants. Biochim. Biophys. Acta 1676, 51–62 10.1016/j.bbaexp.2003.10.009 [DOI] [PubMed] [Google Scholar]
- 2. Nam J. S., Turcotte T. J., Smith P. F., Choi S., and Yoon J. K. (2006) Mouse cristin/R-spondin family proteins are novel ligands for the Frizzled 8 and LRP6 receptors and activate β-catenin-dependent gene expression. J. Biol. Chem. 281, 13247–13257 10.1074/jbc.M508324200 [DOI] [PubMed] [Google Scholar]
- 3. Parma P., Radi O., Vidal V., Chaboissier M. C., Dellambra E., Valentini S., Guerra L., Schedl A., and Camerino G. (2006) R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat. Genet. 38, 1304–1309 10.1038/ng1907 [DOI] [PubMed] [Google Scholar]
- 4. Bergmann C., Senderek J., Anhuf D., Thiel C. T., Ekici A. B., Poblete-Gutierrez P., van Steensel M., Seelow D., Nürnberg G., Schild H. H., Nürnberg P., Reis A., Frank J., and Zerres K. (2006) Mutations in the gene encoding the Wnt-signaling component R-spondin 4 (RSPO4) cause autosomal recessive anonychia. Am. J. Hum. Genet. 79, 1105–1109 10.1086/509789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blaydon D. C., Ishii Y., O'Toole E. A., Unsworth H. C., Teh M. T., Rüschendorf F., Sinclair C., Hopsu-Havu V. K., Tidman N., Moss C., Watson R., de Berker D., Wajid M., Christiano A. M., and Kelsell D. P. (2006) The gene encoding R-spondin 4 (RSPO4), a secreted protein implicated in Wnt signaling, is mutated in inherited anonychia. Nat. Genet. 38, 1245–1247 10.1038/ng1883 [DOI] [PubMed] [Google Scholar]
- 6. Aoki M., Kiyonari H., Nakamura H., and Okamoto H. (2008) R-spondin2 expression in the apical ectodermal ridge is essential for outgrowth and patterning in mouse limb development. Dev. Growth Differ. 50, 85–95 10.1111/j.1440-169X.2007.00978.x [DOI] [PubMed] [Google Scholar]
- 7. Bell S. M., Schreiner C. M., Wert S. E., Mucenski M. L., Scott W. J., and Whitsett J. A. (2008) R-spondin 2 is required for normal laryngeal-tracheal, lung and limb morphogenesis. Development 135, 1049–1058 10.1242/dev.013359 [DOI] [PubMed] [Google Scholar]
- 8. Aoki M., Mieda M., Ikeda T., Hamada Y., Nakamura H., and Okamoto H. (2007) R-spondin3 is required for mouse placental development. Dev. Biol. 301, 218–226 10.1016/j.ydbio.2006.08.018 [DOI] [PubMed] [Google Scholar]
- 9. Seshagiri S., Stawiski E. W., Durinck S., Modrusan Z., Storm E. E., Conboy C. B., Chaudhuri S., Guan Y., Janakiraman V., Jaiswal B. S., Guillory J., Ha C., Dijkgraaf G. J., Stinson J., Gnad F., et al. (2012) Recurrent R-spondin fusions in colon cancer. Nature 488, 660–664 10.1038/nature11282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gong X., Yi J., Carmon K. S., Crumbley C. A., Xiong W., Thomas A., Fan X., Guo S., An Z., Chang J. T., and Liu Q. J. (2015) Aberrant RSPO3-LGR4 signaling in Keap1-deficient lung adenocarcinomas promotes tumor aggressiveness. Oncogene 34, 4692–4701 10.1038/onc.2014.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chartier C., Raval J., Axelrod F., Bond C., Cain J., Dee-Hoskins C., Ma S., Fischer M. M., Shah J., Wei J., Ji M., Lam A., Stroud M., Yen W. C., Yeung P., et al. (2016) Therapeutic targeting of tumor-derived R-spondin attenuates β-catenin signaling and tumorigenesis in multiple cancer types. Cancer Res. 76, 713–723 10.1158/0008-5472.CAN-15-0561 [DOI] [PubMed] [Google Scholar]
- 12. Kazanskaya O., Glinka A., del Barco Barrantes I., Stannek P., Niehrs C., and Wu W. (2004) R-spondin2 is a secreted activator of Wnt/β-catenin signaling and is required for Xenopus myogenesis. Dev. Cell 7, 525–534 10.1016/j.devcel.2004.07.019 [DOI] [PubMed] [Google Scholar]
- 13. Wei Q., Yokota C., Semenov M. V., Doble B., Woodgett J., and He X. (2007) R-spondin1 is a high affinity ligand for LRP6 and induces LRP6 phosphorylation and β-catenin signaling. J. Biol. Chem. 282, 15903–15911 10.1074/jbc.M701927200 [DOI] [PubMed] [Google Scholar]
- 14. Kim K. A., Wagle M., Tran K., Zhan X., Dixon M. A., Liu S., Gros D., Korver W., Yonkovich S., Tomasevic N., Binnerts M., and Abo A. (2008) R-spondin family members regulate the Wnt pathway by a common mechanism. Mol. Biol. Cell 19, 2588–2596 10.1091/mbc.e08-02-0187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carmon K. S., Gong X., Lin Q., Thomas A., and Liu Q. (2011) R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 11452–11457 10.1073/pnas.1106083108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Lau W., Barker N., Low T. Y., Koo B. K., Li V. S., Teunissen H., Kujala P., Haegebarth A., Peters P. J., van de Wetering M., Stange D. E., van Es J. E., Guardavaccaro D., Schasfoort R. B., Mohri Y., et al. (2011) Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 476, 293–297 10.1038/nature10337 [DOI] [PubMed] [Google Scholar]
- 17. Glinka A., Dolde C., Kirsch N., Huang Y. L., Kazanskaya O., Ingelfinger D., Boutros M., Cruciat C. M., and Niehrs C. (2011) LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep. 12, 1055–1061 10.1038/embor.2011.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruffner H., Sprunger J., Charlat O., Leighton-Davies J., Grosshans B., Salathe A., Zietzling S., Beck V., Therier M., Isken A., Xie Y., Zhang Y., Hao H., Shi X., Liu D., et al. (2012) R-spondin potentiates Wnt/β-catenin signaling through orphan receptors LGR4 and LGR5. PLoS One 7, e40976 10.1371/journal.pone.0040976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hao H. X., Xie Y., Zhang Y., Charlat O., Oster E., Avello M., Lei H., Mickanin C., Liu D., Ruffner H., Mao X., Ma Q., Zamponi R., Bouwmeester T., Finan P. M., et al. (2012) ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 485, 195–200 10.1038/nature11019 [DOI] [PubMed] [Google Scholar]
- 20. Koo B. K., Spit M., Jordens I., Low T. Y., Stange D. E., van de Wetering M., van Es J. H., Mohammed S., Heck A. J., Maurice M. M., and Clevers H. (2012) Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488, 665–669 10.1038/nature11308 [DOI] [PubMed] [Google Scholar]
- 21. Moad H. E., and Pioszak A. A. (2013) Reconstitution of R-spondin:LGR4:ZNRF3 adult stem cell growth factor signaling complexes with recombinant proteins produced in E. coli. Biochemistry 52, 7295–7304 10.1021/bi401090h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Warner M. L., Bell T., and Pioszak A. A. (2015) Engineering high-potency R-spondin adult stem cell growth factors. Mol. Pharmacol. 87, 410–420 10.1124/mol.114.095133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zebisch M., Xu Y., Krastev C., MacDonald B. T., Chen M., Gilbert R. J., He X., and Jones E. Y. (2013) Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat. Commun. 4, 2787 10.1038/ncomms3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie Y., Zamponi R., Charlat O., Ramones M., Swalley S., Jiang X., Rivera D., Tschantz W., Lu B., Quinn L., Dimitri C., Parker J., Jeffery D., Wilcox S. K., Watrobka M., et al. (2013) Interaction with both ZNRF3 and LGR4 is required for the signalling activity of R-spondin. EMBO Rep. 14, 1120–1126 10.1038/embor.2013.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carmon K. S., Lin Q., Gong X., Thomas A., and Liu Q. (2012) LGR5 interacts and cointernalizes with Wnt receptors to modulate Wnt/β-catenin signaling. Mol. Cell. Biol. 32, 2054–2064 10.1128/MCB.00272-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carmon K. S., Gong X., Yi J., Thomas A., and Liu Q. (2014) RSPO-LGR4 functions via IQGAP1 to potentiate Wnt signaling. Proc. Natl. Acad. Sci. U.S.A. 111, E1221–E1229 10.1073/pnas.1323106111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carmon K. S., Gong X., Yi J., Wu L., Thomas A., Moore C. M., Masuho I., Timson D. J., Martemyanov K. A., and Liu Q. J. (2017) LGR5 receptor promotes cell-cell adhesion in stem cells and colon cancer cells via the IQGAP1-Rac1 pathway. J. Biol. Chem. 292, 14989–15001 10.1074/jbc.M117.786798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu Q., Wang Y., Dabdoub A., Smallwood P. M., Williams J., Woods C., Kelley M. W., Jiang L., Tasman W., Zhang K., and Nathans J. (2004) Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 116, 883–895 10.1016/S0092-8674(04)00216-8 [DOI] [PubMed] [Google Scholar]
- 29. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan K. S., Janda C. Y., Chang J., Zheng G. X. Y., Larkin K. A., Luca V. C., Chia L. A., Mah A. T., Han A., Terry J. M., Ootani A., Roelf K., Lee M., Yuan J., Li X., et al. (2017) Non-equivalence of Wnt and R-spondin ligands during Lgr5+ intestinal stem-cell self-renewal. Nature 545, 238–242 10.1038/nature22313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kazanskaya O., Ohkawara B., Heroult M., Wu W., Maltry N., Augustin H. G., and Niehrs C. (2008) The Wnt signaling regulator R-spondin 3 promotes angioblast and vascular development. Development 135, 3655–3664 10.1242/dev.027284 [DOI] [PubMed] [Google Scholar]
- 32. Ren Z., van Andel H., de Lau W., Hartholt R. B., Maurice M. M., Clevers H., Kersten M. J., Spaargaren M., and Pals S. T. (2018) Syndecan-1 promotes Wnt/β-catenin signaling in multiple myeloma by presenting Wnts and R-spondins. Blood 131, 982–994 10.1182/blood-2017-07-797050 [DOI] [PubMed] [Google Scholar]
- 33. Chen P. H., Chen X., Lin Z., Fang D., and He X. (2013) The structural basis of R-spondin recognition by LGR5 and RNF43. Genes Dev. 27, 1345–1350 10.1101/gad.219915.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Peng W. C., de Lau W., Forneris F., Granneman J. C., Huch M., Clevers H., and Gros P. (2013) Structure of stem cell growth factor R-spondin 1 in complex with the ectodomain of its receptor LGR5. Cell Rep. 3, 1885–1892 10.1016/j.celrep.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 35. Xu J. G., Huang C., Yang Z., Jin M., Fu P., Zhang N., Luo J., Li D., Liu M., Zhou Y., and Zhu Y. (2015) Crystal structure of LGR4-Rspo1 complex: insights into the divergent mechanisms of ligand recognition by leucine-rich repeat G-protein-coupled receptors (LGRs). J. Biol. Chem. 290, 2455–2465 10.1074/jbc.M114.599134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zebisch M., and Jones E. Y. (2015) Crystal structure of R-spondin 2 in complex with the ectodomains of its receptors LGR5 and ZNRF3. J. Struct. Biol. 191, 149–155 10.1016/j.jsb.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van der Flier L. G., Haegebarth A., Stange D. E., van de Wetering M., and Clevers H. (2009) OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology 137, 15–17 10.1053/j.gastro.2009.05.035 [DOI] [PubMed] [Google Scholar]
- 38. Ohkawara B., Glinka A., and Niehrs C. (2011) Rspo3 binds syndecan 4 and induces Wnt/PCP signaling via clathrin-mediated endocytosis to promote morphogenesis. Dev. Cell 20, 303–314 10.1016/j.devcel.2011.01.006 [DOI] [PubMed] [Google Scholar]
- 39. Xu K., Xu Y., Rajashankar K. R., Robev D., and Nikolov D. B. (2013) Crystal structures of Lgr4 and its complex with R-spondin1. Structure 21, 1683–1689 10.1016/j.str.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Snyder J. C., Rochelle L. K., Lyerly H. K., Caron M. G., and Barak L. S. (2013) Constitutive internalization of the leucine-rich G protein-coupled receptor-5 (LGR5) to the trans-Golgi network. J. Biol. Chem. 288, 10286–10297 10.1074/jbc.M112.447540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lebensohn A. M., and Rohatgi R. (2018) R-spondins can potentiate WNT signaling without LGRs. eLife 7, e33126 10.7554/eLife.33126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yi J., Xiong W., Gong X., Bellister S., Ellis L. M., and Liu Q. (2013) Analysis of LGR4 receptor distribution in human and mouse tissues. PLoS One 8, e78144 10.1371/journal.pone.0078144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sato T., Vries R. G., Snippert H. J., van de Wetering M., Barker N., Stange D. E., van Es J. H., Abo A., Kujala P., Peters P. J., and Clevers H. (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.