

Abstract
Clear cell renal cell carcinoma (ccRCC) represents the most common kidney cancer worldwide. Increased cell proliferation associated with abnormal microRNA (miRNA) regulation are hallmarks of carcinogenesis. Ankyrin repeat and single KH domain 1 (ANKHD1) is a highly conserved protein found to interact with core cancer pathways in Drosophila; however, its involvement in RCC is completely unexplored. Quantitative PCR studies coupled with large-scale genomics data sets demonstrated that ANKHD1 is significantly up-regulated in kidneys of RCC patients when compared with healthy controls. Cell cycle analysis revealed that ANKHD1 is an essential factor for RCC cell division. To understand the molecular mechanism(s) utilized by ANKHD1 to drive proliferation, we performed bioinformatics analyses that revealed that ANKHD1 contains a putative miRNA-binding motif. We screened 48 miRNAs with tumor-enhancing or -suppressing activities and found that ANKHD1 binds to and regulates three tumor-suppressing miRNAs (i.e. miR-29a, miR-205, and miR-196a). RNA-immunoprecipitation assays demonstrated that ANKHD1 physically interacts with its target miRNAs via a single K-homology domain, located in the C terminus of the protein. Functionally, we discovered that ANKHD1 positively drives ccRCC cell mitosis via binding to and suppressing mainly miR-29a and to a lesser degree via miR-196a/205, leading to up-regulation in proliferative genes such as CCDN1. Collectively, these data identify ANKHD1 as a new regulator of ccRCC proliferation via specific miRNA interactions.
Keywords: kidney, cancer biology, microRNA (miRNA), microRNA mechanism, cell proliferation, ANKHD1, ccRCC, clear cell, miR-29a, miRNA-binding
Introduction
Renal cell carcinomas (RCCs)2 represent one of the commonest diagnosed cancers worldwide, with clear cell RCC (ccRCC) accounting for ∼80% of all RCCs. Molecularly, the tumor suppressor VHL (von Hippel–Lindau) is frequently inactivated by mutations (1), leading to increased proliferation, enhanced angiogenesis, and dedifferentiation of tumor cells. ccRCC pathogenesis is linked to abnormal regulation of certain microRNAs (miRNAs) (2, 3). The latter are a class of ∼22-nucleotide-long, single-stranded, noncoding, nucleic acid molecules involved in the regulation of proliferation (4, 5). In ccRCC, the tumor-suppressor miR-29a is strongly down-regulated and predicts metastasis (6). miR-29a, expressed by the kidney, is responsible for the regulation of extracellular matrix (7, 8) in addition to acting as a core regulator of cellular proliferation (9). The proteins that control the levels of miR-29a in ccRCC are currently unknown.
ANKHD1 (ankyrin repeat and KH domain-containing protein 1) is a K-homology (KH) domain containing protein first discovered in Drosophila as a genetic regulator of the epidermal growth factor/mitogen-activated protein kinase pathway (10). It was later shown to regulate additional signaling cascades including the cancer-associated HIPPO (11–14) and PINK/Parkin in Drosophila (15). Clinical epidemiological studies suggest that ANKHD1 is elevated and may be involved in acute leukemia (16) and prostate cancer (11). Despite these insights, the molecular mechanisms employed by ANKHD1 to exert these pleiotropic, cancer-associated, functions are as-yet unidentified.
Specifically, ANKHD1 contains a single KH domain, a motif predicted to bind single-stranded nucleic acids; however, its function has not been experimentally tested. Here, we report that ANKHD1 is elevated in patients with ccRCC and contributes to cancer cell proliferation via binding to and suppressing a number of tumor-suppressor miRNAs.
Results
ANKHD1 is overexpressed in RCC
Given that ANKHD1 regulates some cancer-associated biochemical pathways (10–15), we therefore first set out to examine the expression of ANKHD1 in normal and RCC human kidneys. Using a commercially available rabbit polyclonal antibody, we found that anti-ANKHD1 antibodies are strongly bound in tubular epithelial, parietal epithelial, and renal microvascular cells with no detectable signal in fibroblasts nor glomerular cells in the normal mouse kidney (representative pictures shown in Fig. S1A, n = 5 animals). Similar findings were observed in normal human kidney sections stained with an anti-ANKHD1 antibody (Fig. 1A, left panels). To examine whether ANKHD1 is present in kidney cancer cells, we analyzed its expression in 20 different RCC cancer patients (Fig. 1A, right panel, and Fig. S1B). Having established that ANKHD1 is expressed at the protein level in kidney cancer, we next wished to study whether kidneys of patients with renal cell carcinoma had altered ANKHD1 mRNA levels. We found a statistically significant elevation of ANKHD1 in RCC samples when compared with healthy control kidney tissues (Fig. 1C). The morphology of a normal (left) and a ccRCC kidney (right) was highlighted with hematoxylin and eosin (H&E) staining (Fig. 1B). Furthermore, ANKHD1 mRNA levels were not significantly different between early (I/II, nonmetastatic) and late stages (III/IV, metastatic) of cancer, suggesting that ANKHD1 is up-regulated early in the carcinogenesis process (Fig. 1D). Consistent with our findings, bioinformatics analyses of the National Institutes of Health Cancer Genome Atlas using cBioPortal (17, 18) showed that 33% of all ccRCC patients (of 538 patients) had significantly overexpressed ANKHD1 mRNA levels, with 16% of patients exhibiting overexpression due to locus amplification (17, 45), thus providing an explanation as to why ANKHD1 is elevated early in a large proportion of these patients. Taken together, these studies demonstrate that ANKHD1 is frequently up-regulated in RCC when compared with healthy noncancer renal tissues and may contribute to this disease.
Figure 1.

ANKHD1 is expressed by renal epithelial cells and is overexpressed in renal cell carcinoma patients. A, a human array of kidney tissues with twenty cases of RCC and three control noncancer tissues were stained with an anti-ANKHD1 antibody, and microscopy was performed using an upright Olympus microscope. B, hematoxylin and eosin (H&E) staining of the above matching tissues can be seen. C, qPCR was performed for ANKHD1 normalized to β-actin for noncancer kidney tissue when compared with renal cell carcinoma. D, subgroup analysis of the ANKHD1 expression in the RCC patient population was unable to identify any differences in the expression in the early (I/II) versus late stages of disease (III/IV). Error bars show means and S.E. There is a significant difference in expression in ANKHD1 message in RCCs when compared with healthy control subjects (B), whereas there is no difference in expression between the early and later stages of cancer (C), when analyzed by a Student's t test.
ANKHD1 controls cancer cell proliferation
To better understand the function of ANKHD1 and examine whether it promotes or inhibits cancer-associated phenotypes, we initially tested its role in two independent renal cancer lines derived from ccRCC patients, RCC4 and 786-O. Two independent siRNAs targeting ANKHD1 mRNA were used, and their efficiency was tested by qPCR and Western blotting (Fig. S2, A and B). Cells exhibiting a reduction of ANKHD1 by 70% or greater were used in all subsequent experiments. Given that ccRCCs are characterized by their proliferative nature, we decided to study whether ANKHD1 controls the ability of RCC cells to divide using propidium iodine–stained cells by flow cytometry. In both renal cell lines tested and in each independent experiment, ANKHD1 silencing resulted in a significant reduction in the number of cells in the G2–M phase, fewer cells in the S phase, and a statistically significant increase in the G1 phase (Fig. 2A). Ultimately, for cancer cells to divide and propagate, they need to undergo mitosis; therefore to investigate whether ANKHD1 knockdown reduces mitosis, we studied phosphoserine 10 histone H3 (19) (subsequently referred to as pH3), which is a specific marker of mitosis. Loss of ANKHD1 function, using two independent siRNAs in multiple experiments, resulted in a statistically significant reduction in the number of pH3-positive cells in both 786-O cells (Fig. 2, B and D) and RCC4 cells (Fig. 2, C and E). Taken together, these results indicate that ANKHD1 is a positive regulator of proliferation in RCC-derived cancer cells in vitro, thus suggesting it contributes to disease.
Figure 2.

ANKHD1 is required for the proliferation of 786-O and RCC4 renal cancer cells. A, either 786-O (left panel) or RCC4 (right panel) cells transfected with ANKHD1 siRNA or nontargeting RNAi control were fixed and stained with propidium iodine. The numbers of cells in the different stages of the cell cycle were counted by performing flow cytometry cell cycle–based analyses. Three independent transfection experiments are shown with S.E. There is a statistically significant increase in the G1 phase and a decrease in the S/M phase of the cell cycle when the data were analyzed by a one-way ANOVA with Bonferroni corrections. B and C, either 786-O cells (B) or RCC4 (C) were treated with the indicated siRNAs and stained with antibody that recognizes phosphoserine 10 histone H3 (pH3). pH3-positive cells were gated, and the percentage of positive cells was identified. Isotype fluorotope–matched control was used as a negative control. D and E, a statistically significant reduction in mitotic cells is observed in both 786-O cells (D) and RCC4 cells (E). All experiments were performed three times, and p values of less than 0.05 were considered significant, using a one-way ANOVA. **, p ≤ 0.01; ***, p ≤ 0.001.
ANKHD1 reduces the expression of a subset of miRNAs
We next set out to determine the molecular mechanisms underlying the pro-proliferative activity of ANKHD1. ANKHD1 contains a single KH domain from amino acids 1694 to 1764. Given that other KH domain-containing proteins, such as KSRP (20), RCF3 (21), and Bicc1 (22), were previously shown to bind miRNAs, we set out to test whether ANKHD1, similarly, controls proliferation through interactions with miRNAs. To identify ANKHD1 relevant miRNAs, we measured the levels of 48 cancer-associated miRNAs using a hybridization array in cancer cells with and without ANKHD1 silencing. We found ANKHD1 siRNA resulted in enhanced expression of five miRNAs by greater than 2-fold (Fig. 3A, red box), when compared with cells treated with a nontargeting siRNA, whereas four miRNAs (namely, −342, 1, 223, and −182) showed more than 2-fold decreased when ANKHD1 was silenced (Fig. 3A, black box).
Figure 3.
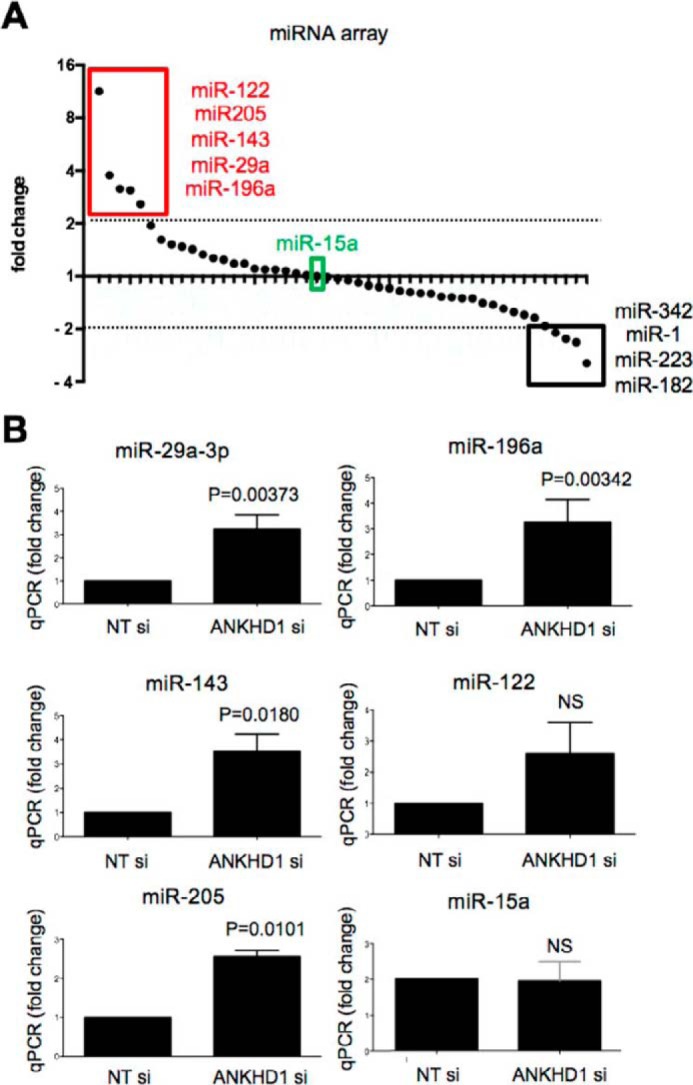
ANKHD1 regulates expression of a subset of tumor-suppressing miRNAs. A, RNA from cells treated with either nontargeting siRNA or ANKHD1 siRNA was hybridized in a 48-miRNA hybridization plate. The levels of these miRNAs are shown following normalization to U6. miRNAs showing more than 2-fold increase are highlighted in the red box, and those exhibiting more than 2-fold decrease are shown in the black box. miR-15, which did not show a change, is highlighted in green. B, the expression of specific miRNA identified in A were verified by carrying out independent transfections followed by qPCR in RCC4 cells transfected with either nontargeting siRNA or ANKHD1 siRNA. All experiments were performed three times, and p values of less than 0.05 were considered significant using a Student's t test.
We then carried out qPCR analysis of miR-29a-3p, miR-196a, miR-143, miR-122, and miR-205 and found that silencing of ANKHD1 resulted in a statistically significant up-regulation of all miRNAs identified in the array expect for miR-122 (Fig. 3B). miR-122, although it showed an 11-fold increase in the array (Fig. 3A), did not significantly change in our independent qPCR validation experiments (Fig. 3B), suggesting that it is likely to represent a false positive in the initial assay. As a control, we also tested miR-15a (Fig. 3B), which did not show a significant change, as expected from the array results (Fig. 3A, green box). Collectively, our findings indicate that ANKHD1 controls expression of a subset of miRNAs, including miR-29a-3p, miR-205, and miR-196a.
ANKHD1 physically associates with miR-29a-3p, -196a, and -205 via its KH domain
ANKHD1 contains two clusters of ankyrin repeats, which have previously been shown to mediate protein-protein interactions, and a single KH domain, a motif predicted to bind to single-stranded nucleic acids (23). Therefore, theoretically, if ANKHD1 interacts with miRNAs, this interaction may be mediated via its KH domain, or alternatively, ANKHD1 may associate with a miRNA-binding protein complex via its ankyrin domains. In the first instance, we tested whether endogenous full-length ANKHD1 is capable of binding miRNAs by undertaking endogenous RNA immunoprecipitations (RIPs). The efficiency of the ANKHD1 pulldown was checked by Western blotting indicating that ANKHD1 is pulled down effectively when an anti-ANKHD1 antibody is used but not with IgG control; the identity of ANKHD1 was also validated by a single siRNA against ANKHD1 (Fig. S2C). ANKHD1 RIPs demonstrated a statistically significant enrichment of miR-29a-3p, miR-196a, and miR-205 in ANKHD1 pulldowns when compared with IgG control antibodies (Fig. 4A), indicating that ANKHD1 physically associates with these miRNAs. However, not surprisingly, ANKHD1 antibodies were not able to pulldown miR-122, one of the miRNAs with suspected false-positive results (Fig. S2D), confirming the specificity of the RNA-immunoprecipitation approach. To test whether the full-length ANKHD1 molecule binds to miRNAs directly, via its KH domain, or indirectly, via its ankyrin repeats, we generated and overexpressed FLAG-tagged constructs. A full-length FLAG-tagged WT ANKHD1 construct (FL-WT) was used to generated a truncation construct that we termed KH-WT; this construct lacked the N-terminal region of the protein but maintained the C terminus with an intact KH domain (KH-WT) (Fig. 4B). We transfected these constructs into cells and performed RIP assays showing that KH-WT was able to physically associate with miR-29a-3p (Fig. 4D) and miR-196a (Fig. 4E) to a level comparable with that observed with FL-WT ANKHD1 (compare second and fourth bars), thus indicating that the N-terminal portion of the protein (which contains the ankyrin repeats) is not required for this interaction. Having shown that the ankyrin domains are redundant for the miRNA interaction, we next examined whether there was a functional unit within the KH domain that directly mediated the nucleic acid binding. Previously a GXXG loop within KH domains of other proteins was identified to be critical for mediating miRNA interactions (24). We searched the KH sequence of ANKHD1 and found a canonical GXXG loop that exhibited 100% amino acid conservation between humans, mice, and Drosophila (Fig. S2E), suggesting an important biological role. Using site-directed mutagenesis, we genetically engineered a 2-amino acid mutation within this loop, which generated a GDDG structure (FL-Mut), previously shown to exhibit reduced nucleic acid binding ability without compromising protein stability (24, 25); we confirmed the mutation by sequencing (Fig. 4C). We next overexpressed both FL-WT and FL-Mut versions of ANKHD1 and performed RIP assays. We found that the FL-Mut construct possessed reduced RNA binding ability (Fig. 4, D and E), thus suggesting that ANKHD1 binds its targets miRNAs via the GXXG loop of its KH domain.
Figure 4.

ANKHD1 binds to miR-29a-3p, -196a, and -205 via its single KH domain. A, endogenous RIPs for miR-29a-3p, -196a, and -205 were performed using anti-ANKHD1 or IgG control antibodies. The results are presented as fold enrichment over IgG. All experiments were performed three times, and p values of less than 0.05 were considered significant using a Student's t test. B, a full-length FLAG-tagged ANKHD1 construct (FL-WT) was used to generate a truncation mutation, a FLAG-tagged version of ANKHD1 that lacks the ankyrin repeats (KH-WT, 16 kDa). The canonical GXXG loop was mutated to GDDG to produce a FLAG-tagged full-length mutant construct (FL-Mut). C, sequencing was carried out to confirm the mutation in the FL-Mut construct. D and E, RIP pulldowns were performed in RCC4 cells following transfection of either FL-WT or FL-mut or KH-WT constructs. qPCR analysis of either miR-29a-3p (D) or miR196a (E) revealed that these physically interacted with the full-length protein, as well as the KH-only proteins, but were unable to interact with the mutant construct (FL-mut). F, RCC4 were transfected either with an empty vector (EV) or ANKHD1 full-length (FL-WT) or the full-length mutant (FL-Mut), and the cells were stained with pH3 and subjected to flow cytometry to measure mitosis. The experiment was performed three times, and a p value of less than 0.05 was considered as significant using a one-way ANOVA. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Functionally, to test whether an intact ANKHD1–GXXG loop is required to mediate the proliferative functions of ANKHD1, we forced expressed FL-WT or mutant FL-Mut ANKHD1 in RCC4 cells. Consistent with previous knockdown experiments, forced expression of FL-WT was sufficient to increase the proportion of cells undergoing mitosis, whereas transfection with the FL-Mut reduced proliferation compared with empty vector (Fig. 4F, EV), thus suggesting that ANKHD1 mediates its pro-proliferative actions via its GXXG loop. Taken together, these data demonstrate that ANKHD1 physically associates with its target miRNAs via its single KH domain and that mutations in this region are able to abolish miRNAs binding and in turn reduce the ability of ANKHD1 to enhance proliferation of ccRCC cells.
ANKHD1 regulates proliferation of cancer cells via miR-29a-3p
Given the significant alterations in miRNA levels mediated by ANKHD1, we hypothesized that ANKHD1 may exert its pro-proliferative effects by regulating one or more of the miRNAs identified above. Firstly, to test whether miR-29a is significantly reduced in RCC, as previously suggested, we performed qPCR analysis, which confirmed that miR-29a is indeed down-regulated in RCC (Fig. S2F). As a starting point, we searched the Mirtarbase database for experimentally validated mRNA targets of miR-29a-3p, miR-196a, and miR-205 (Table S1). We then queried the annotation of these targets using the David functional annotation tool and found that “regulation of cell proliferation” was a statistically significantly enriched gene ontology term for miR-29a-3p (nine target genes) and miR-205 (four target genes) but not for miR-196a (Table S1). Because miR-29a-3p is associated with the greatest number of proliferation-regulating genes, we started by testing whether it may mediate the proliferative effects of ANKHD1. We firstly knocked down ANKHD1 in RCC4 cells transfected with either a miScript microRNA inhibitor targeting miR-29a-3p or an empty vector control (Fig. 5A). As expected, silencing of ANKHD1 reduced the proportion of pH3-positive RCC4 when compared with nontarget RNAi-treated cells (Fig. 5B, NT si). Importantly, transfection of a miR-29a-3p inhibitor did not affect the proliferation of RCC4 cells on its own (Fig. 5B); however, co-transfection of the miR-29a-3p inhibitor together with knocking out ANKHD1 was capable of completely relieving the block in proliferation mediated by silencing of ANKHD1, thus restoring cell division to basal levels (Fig. 5, A and B). This suggested that miRNA-29a-3p suppression is required for ANKHD1 to promote mitosis in ccRCC cells. To investigate whether ANKHD1 regulates proliferation via miR-29a-3p in an additional RCC-derived cell-line, 786-O cells were transfected either with ANKHD1 siRNA on its own or co-transfected with an miR-29a-3p inhibitor. Consistent with previous observations in RCC4 cells, co-transfection of miR-29a-3p inhibitor in 786-O cells was also sufficient to enhance proliferation when ANKHD1 was concomitantly silenced, suggesting that the suppression of miR-29a-3p is a key mechanism employed by ANKHD1 to drive proliferation in RCC-derived cancer cells.
Figure 5.

ANKHD1 regulates proliferation via miR-29a-3p/miR-205 and to a lesser degree miR-196a. A, RCC4 cells were transfected with either nontarget (NT) or ANKHD1 siRNA (ANKHD1 si) and co-transfected with an miR-29a-3p inhibitor. The number of RCC4 cells undergoing mitosis was studied by flow cytometry measuring number of pH3-positive cells. B, experiments were repeated three times, and a p value of less than 0.05 was considered as significant using a one-way ANOVA. C, ANKHD1 was also silenced in 786-O cells using two independent siRNAs (top panels), and the number of mitoses was measured. Inhibition of miR-29a (left panels), miR-196a (middle panels), or miR-205 (right panels) was studied. D, to study how ANKHD1 control proliferation of 786-O cells, ANKHD1 was silenced using two independent siRNA, and the expression of CCDN1, VEGFA, and KLF4 was measured by qPCR. In addition to test whether the modulation of the target mRNAs is via miR-29a, the cells were co-transfected with a miR-29a-3p inhibitor. The results are normalized against HPRT expression and are shown as fold change when compared with nontarget siRNA-transfected cells. The experiments were repeated three times, and a p value of less than 0.05 was considered as significant using a one-way ANOVA. NS, not significant. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
In addition to miR-29a-3p, ANKHD1 also regulates miR-205 and 196a. To examine whether the proliferative effects of ANKHD1 are mediated solely via miR-29a-3p or whether other miRNAs also play a role, we silenced ANKHD1 and transfected either with miR-196a inhibitor or an miR-205 inhibitor. We found that miR-205/miR-196a inhibition caused a marginal increase in proliferation (Fig. 5C). These results collectively suggest that the main mechanism employed by ANKHD1 to drive RCC proliferation is suppression of miR-29a-3p and to a lesser degree miR-205/196a.
Finally, to test how silencing of ANKHD1 may be driving reduced proliferation of RCC cells, we silenced ANKHD1 using two-independent siRNAs and tested by qPCR the expression of CCDN1, VEGFA, KLF4, and CDC42, four well-characterized targets of miR-29a-3p. We found that ANKHD1 loss of function led to a statistically significant reduction of each of these genes (apart from CDC42, Fig. S1E), a phenomenon that could be restored by inhibiting miR-29a-3p (Fig. 5D). These data together suggest that ANKHD1 is a positive regulator of proliferation in renal cancer cells via suppressing primarily miR-29a-3p and to a lesser degree via miR-205/miR-196a, thus altering a number of known proliferation altering genes such as CCDN1, leading to increased RCC proliferation.
Discussion
Here we presented the first study assigning control of cancer cell proliferation as a molecular function of ANKHD1 in ccRCC. We showed that ANKHD1 controls renal cancer cell proliferation by engaging with and altering the levels of a subset of tumor-suppressing miRNAs. We demonstrated that ANKHD1 physically forms a complex with three target miRNAs (namely miR-29a-3p, miR-205, and miR-196a) via its KH domain, which leads to a significant reduction in the levels of these miRNAs. Finally, we provide the first demonstration that ANKHD1 mRNA and protein are significantly enhanced in human ccRCC when compared with control and may play an important role in human ccRCC.
Previous studies have convincingly demonstrated that miR-29a, miR-196a, and miR-205 possess tumor-suppressor activities and are frequently, and significantly, down-regulated in RCCs (6, 26–31), a finding that correlates with the up-regulation of ANKHD1, demonstrated here. Furthermore, given that inhibition of miRNA-29a is sufficient to restore the proliferation of RCC cells lacking ANKHD1, we suggest that miR-29a–targeted genes are likely to be responsible for the ANKHD1-mediated proliferation of ccRCCs. miR-29a has previously been shown to inhibit many proliferation-regulating genes including BCL2, CDC42, CCDN1, CCNT2, CDK2, CDK6, NASP, VEGFA, and KLF4 (32–37). Indeed, we were able to show that ANKHD1 regulates CCDN1, VEGFA, and KLF4 via miR-29a-3p, further implicating suppression of miR-29a-3p as the main mechanism employed by ANKHD1 to control proliferation of renal cancer cells.
The other two miRNAs identified as binding partners of ANKHD1 are miR-196a and miR-205. We demonstrated that ANKHD1 silencing leads to an increase in miR-196a and 205, and it physically interacts with these miRNAs. Our bioinformatics functional annotation of miR-196a, using validated targets, did not reveal a major proliferation-regulating role for miR-196a. Interestingly, miR-196a was previously shown to have a role in autophagy (38). This is particularly interesting because ANKHD1 has been previously shown to control autophagy in Drosophila (15). Thus, we speculate that ANKHD1 may have important, but as-yet-unidentified, additional molecular functions in mammalian cells, including ccRCC, one of which could be the control of autophagy. We therefore hypothesize that these additional molecular functions of ANKHD1 may be mediated via miRNA molecules, some of which are identified in this study (e.g. miR-196a/205).
Despite a clear role in the regulation of miRNA levels, the mechanism employed by ANKHD1 to suppress the levels of its target miRNAs is currently unclear. Although a simple “trapping” model may appear attractive, we reasoned that miRNAs tightly bound to ANKHD1, in an inactive complex, would still be detectable in TRIzol-derived total RNA extracted from whole cells, and we would therefore detect them in our qPCR studies. Rather the accumulation of miRNAs following ANKHD1 knockdown suggests that ANKHD1 may promote destruction of the identified miRNAs or alternatively control their levels by altering their rate of transcription; however, the precise mechanism underlying this process is as-yet unknown.
Interestingly, ANKHD1 has been shown to physically interact with factor-inhibiting hypoxia-inducible factor (HIF) (39), a molecule that alters HIF transcriptional activity; HIF is involved in the development of ccRCC (40, 41). Moreover, HIF controls cell cycle in hypoxia partly via controlling p21 among other targets (42). Interestingly ANKHD1 has been shown to interact with p21 (43), a finding that we confirmed in our study (Fig. S1, C and D). Therefore, ANKHD1 may alter proliferation and other related cancer phenotypes via multiple mechanisms, including the regulation of miRNAs and also potentially via HIF signaling.
In conclusion, this study combines functional, biochemical, and bioinformatics approaches to collectively demonstrate that ANKHD1 controls proliferation of renal cell carcinoma cells via binding to and modulating a specific subset of ccRCC-relevant tumor-suppressor miRNAs. Further studies using miRNA modulating agents (mimics or inhibitors) and ANKHD1 knockout animal models are required to determine the genetic role of ANKHD1 in this cancer.
Experimental procedures
Cell culture and transfections
Human renal cancer cells 786-O were purchased from ATCC, and human renal clear cell carcinoma cells (RCC4) with empty vector were purchased from Sigma and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 2 mm GlutaMAX without antibiotics at 37 °C in a humidified incubator with 5% CO2. The cells were trypsinized and reversed transfected with 20 pmol of RNAi with RNAi MAX transfection reagent following the manufacturer's instructions (Invitrogen). Two independent siRNAs were used for ANKHD1, with the following catalogue numbers: D-014405-01 and D-014405-03. In every siRNA experiment, the control cells were treated with nontarget RNAi complexes (D-001210-01). Anti-miR29a-3p inhibitor was purchased from Qiagen (3p, MIN0000086), and cells were transfected using 20 pmol of inhibitor with RNAi MAX. For plasmid transfection, a forward transfection, 12 h after seeding was performed with Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions.
RNA immunoprecipitation
RIPs were performed using Magna RIP kit (upstate, 70–700), following a modified version of the manufacturer's instructions. Briefly, ∼2.5 × 107 cells were used per immunoprecipitation; after washing the cell monolayer, the cells were collected by centrifugation and lysed in RIP lysis buffer containing RNase inhibitor and incubated on ice for 5 min. The cells were then placed at −80 °C to allow cell lysis to take place. Immunoprecipitation using either rabbit anti-ANKHD1 antibody (Sigma, HPA008718) or anti-FLAG M2 antibody (Sigma, A2220) or relevant rabbit or mouse IgG was used in a pulldown experiment for 4 h at 4 °C with rotation. Then, after thorough washing of the beads, total RNA was extracted using TRIzol and reversed transcribed immediately. Any remaining RNA was stored at −80 °C.
Western blotting, immunoprecipitation, and antibodies
Following specific treatment, a confluent monolayer of cells was washed twice in ice-cold Tris-PBS (1× TBS), TBS was aspirated, and the cells were lysed using ice-cold Lysis Buffer (50 mm Tris (pH 7.4) 250 mm NaCl, 0.3% Triton X-100, 1 mm ETDA) supplemented with protease inhibitor mixture (Roche). The cells were then subjected to freeze-thaw and mild sonication. Whole cell lysates were then incubated with appropriate antibodies or isotype-specific IgG control for 4 h at 4 °C followed by overnight incubation with Dynabeads protein G (Life Technologies). The beads were then washed four times in cold lysis buffer and boiled in 2× Laemmli sample buffer for 5 min. Samples eluted from the beads or whole cells lysates were resolved by SDS-PAGE and transferred using the Mini-PROTEAN system (Bio-Rad). Primary antibodies were rabbit polyclonal anti-ANKHD1 (HPA008718, Sigma), and mouse monoclonal anti-β-actin (ab8226, Abcam).
Flow cytometry
The cells were reverse transfected with 20 nm of either ANKHD1 siRNAs or nontarget siRNA and grown in 6-well plates for 72 h. On the day of the assay, the cells were lifted off the 6-well plate using Accutase solution (A6964, Sigma), resuspended in DMEM with 10% FBS, and centrifuged at 200 × g for 5 min to pellet cells. Supernatant was discarded, and cell pellets were resuspended in 50 μl of 1× TBST, followed by the addition of 1 ml of ice-cold 100% methanol. The cells were then immediately placed in the freezer and left for 30 min to allow adequate fixation. Following fixation, the cells were washed twice in 1× TBST, and then either isotype control antibody (9078, Cell Signaling Technology), no antibody, or phosphorylated histone H3-Pacific Blue–conjugated antibody (1:125) (8552, Cell Signaling Technology) was added to cells and left to incubate in the dark at room temperature for 60 min. After this incubation, the cells were transferred into flow cytometry tubes and were analyzed using LSRII running BD FACS Diva software. 10,000 events were recorded in the forward and side scatter, and from these a gate was placed on single cells to eliminate doublets and clumps from analysis. From these, a clear positive population could be seen, and a percentage of positive cells was obtained by gating this population. The percentage of positive cells of the 10,000 events was plotted on Prism 6.0 from three different biological replicates, and a Student's t test was carried out. p values of 0.05 or lower were considered significant.
Quantitative PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) following the manufacturer's instructions. Total RNA was then reverse transcribed using miScript RT kit (Qiagen, 218193). qPCR was carried out using and iQ SYBR green supermix reagents (Bio-Rad) following the manufacturer's instructions on a CFX-96 or CFX-384 Touch real-time PCR detection system (Bio-Rad). A change in expression levels was calculated relative to either β-actin housekeeping gene (for mRNA) or U6 (for miRNA) using the δδCT method. The primers used are ANKHD1 (forward, 5′-AGCGGTACGGGCGGAG-3′; and reverse, 5′-AAATAAATGATTCAACCTCGGACAC-3′) and β-actin (forward, 5′-ATCATTGCTCCTCCTGAGCG-3′; and reverse, 5′-GACAGCGAGGCCAGGATG-3′).
miRNA hybridization plate and human RCC cancer tissue arrays
To identify targets of ANKHD1, a miRNA hybridization plate array was used (Signosis, MA-1001). 20 μg of RNA (either from cells with nontarget RNA or siRNA against ANKHD1) was added to each well and incubated overnight; the array was then analyzed using the manufacturer's instructions. To study the mRNA levels of ANKHD1 or miR29a in human RCC patients and healthy controls, a tissue scan kidney cancer tissue qPCR array was used (Origene, HKRT102), and qPCR was performed following manufacturer's instructions (using the primers stated above). To study protein levels of ANKHD1 in RCC, an array of 20 independent cases of RCC and 60 cores (each case repeated three times) was utilized (BC07013, US Biomax). The tissues were deparaffinized, and immunohistochemistry was performed as previously described (44). These studies abide by the Declaration of Helsinki principles, and tissues were collected under Health Insurance Portability and Accountability Act–approved protocols.
Statistics
All experiments were repeated at least three times unless otherwise stated. GraphPad Prism 6 was used to input data, and either nonparametric, two-tailed, Mann–Whitney tests were performed, or for two-sample comparison a Student's t test was used. p values less than 0.05 were recorded as significant.
Author contributions
M. F. and M. P. Z. conceptualization; M. F. and M. P. Z. resources; M. F. data curation; M. F. and M. P. Z. formal analysis; M. P. Z. supervision; M. F. and M. P. Z. funding acquisition; M. F. validation; M. F. investigation; M. F. visualization; M. F. methodology; M. F. writing-original draft; M. F. and M. P. Z. project administration; M. F. and M. P. Z. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Paul C. Evans for carefully reading the manuscript and providing critical feedback and Lindsay Farrell for technical support.
This work was supported by a Kidney Research UK Fellowship and a Thomas-Berry and Simpson Fellowship (to M. F.) and a Cancer Research UK Senior Fellowship (to M. P. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1 and S2.
- RCC
- renal cell carcinoma
- ccRCC
- clear cell RCC
- miRNA
- microRNA
- KH
- K-homology
- qPCR
- quantitative PCR
- RIP
- RNA immunoprecipitation
- HIF
- hypoxia-inducible factor
- ANOVA
- analysis of variance.
References
- 1. Cockman M. E., Masson N., Mole D. R., Jaakkola P., Chang G. W., Clifford S. C., Maher E. R., Pugh C. W., Ratcliffe P. J., and Maxwell P. H. (2000) Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 275, 25733–25741 10.1074/jbc.M002740200 [DOI] [PubMed] [Google Scholar]
- 2. Chen J., Zhang D., Zhang W., Tang Y., Yan W., Guo L., and Shen B. (2013) Clear cell renal cell carcinoma associated microRNA expression signatures identified by an integrated bioinformatics analysis. J. Transl. Med. 11, 169 10.1186/1479-5876-11-169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marcucci G., Mrózek K., Radmacher M. D., Garzon R., and Bloomfield C. D. (2011) The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood 117, 1121–1129 10.1182/blood-2010-09-191312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Avraham R., and Yarden Y. (2012) Regulation of signalling by microRNAs. Biochem. Soc. Trans. 40, 26–30 10.1042/BST20110623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mamoori A., Gopalan V., Smith R. A., and Lam A. K. (2016) Modulatory roles of microRNAs in the regulation of different signalling pathways in large bowel cancer stem cells. Biol. Cell 108, 51–64 10.1111/boc.201500062 [DOI] [PubMed] [Google Scholar]
- 6. Heinzelmann J., Henning B., Sanjmyatav J., Posorski N., Steiner T., Wunderlich H., Gajda M. R., and Junker K. (2011) Specific miRNA signatures are associated with metastasis and poor prognosis in clear cell renal cell carcinoma. World J. Urol. 29, 367–373 10.1007/s00345-010-0633-4 [DOI] [PubMed] [Google Scholar]
- 7. Liu Y., Taylor N. E., Lu L., Usa K., Cowley A. W. Jr., Ferreri N. R., Yeo N. C., and Liang M. (2010) Renal medullary microRNAs in Dahl salt-sensitive rats: miR-29b regulates several collagens and related genes. Hypertension 55, 974–982 10.1161/HYPERTENSIONAHA.109.144428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Rooij E., Sutherland L. B., Thatcher J. E., DiMaio J. M., Naseem R. H., Marshall W. S., Hill J. A., and Olson E. N. (2008) Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. U.S.A. 105, 13027–13032 10.1073/pnas.0805038105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao Z., Wang L., Song W., Cui H., Chen G., Qiao F., Hu J., Zhou R., and Fan H. (2015) Reduced miR-29a-3p expression is linked to the cell proliferation and cell migration in gastric cancer. World J. Surg. Oncol. 13, 101 10.1186/s12957-015-0513-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith R. K., Carroll P. M., Allard J. D., and Simon M. A. (2002) MASK, a large ankyrin repeat and KH domain-containing protein involved in Drosophila receptor tyrosine kinase signaling. Development 129, 71–82 [DOI] [PubMed] [Google Scholar]
- 11. Machado-Neto J. A., Lazarini M., Favaro P., Franchi G. C. Jr., Nowill A. E., Saad S. T., and Traina F. (2014) ANKHD1, a novel component of the Hippo signaling pathway, promotes YAP1 activation and cell cycle progression in prostate cancer cells. Exp. Cell Res. 324, 137–145 10.1016/j.yexcr.2014.04.004 [DOI] [PubMed] [Google Scholar]
- 12. Sansores-Garcia L., Atkins M., Moya I. M., Shahmoradgoli M., Tao C., Mills G. B., and Halder G. (2013) Mask is required for the activity of the Hippo pathway effector Yki/YAP. Curr. Biol. 23, 229–235 10.1016/j.cub.2012.12.033,10.1016/j.sbi.2013.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sidor C. M., Brain R., and Thompson B. J. (2013) Mask proteins are cofactors of Yorkie/YAP in the Hippo pathway. Curr. Biol. 23, 223–228 10.1016/j.cub.2012.11.061 [DOI] [PubMed] [Google Scholar]
- 14. Tumaneng K., Schlegelmilch K., Russell R. C., Yimlamai D., Basnet H., Mahadevan N., Fitamant J., Bardeesy N., Camargo F. D., and Guan K. L. (2012) YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol. 14, 1322–1329 10.1038/ncb2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu M., Li X., Tian X., and Wu C. (2015) Mask loss-of-function rescues mitochondrial impairment and muscle degeneration of Drosophila pink1 and parkin mutants. Hum. Mol. Genet. 24, 3272–3285 10.1093/hmg/ddv081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Traina F., Favaro P. M., Medina Sde S., Duarte Ada S., Winnischofer S. M., Costa F. F., and Saad S. T. (2006) ANKHD1, ankyrin repeat and KH domain containing 1, is overexpressed in acute leukemias and is associated with SHP2 in K562 cells. Biochim. Biophys. Acta 1762, 828–834 10.1016/j.bbadis.2006.07.010 [DOI] [PubMed] [Google Scholar]
- 17. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cancer Genome Atlas Research Network (2013) Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49 10.1038/nature12222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin S., Yuan Z. F., Han Y., Marchione D. M., and Garcia B. A. (2016) Preferential phosphorylation on old histones during early mitosis in human cells. J. Biol. Chem. 291, 15342–15357 10.1074/jbc.M116.726067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trabucchi M., Briata P., Garcia-Mayoral M., Haase A. D., Filipowicz W., Ramos A., Gherzi R., and Rosenfeld M. G. (2009) The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459, 1010–1014 10.1038/nature08025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karlsson P., Christie M. D., Seymour D. K., Wang H., Wang X., Hagmann J., Kulcheski F., and Manavella P. A. (2015) KH domain protein RCF3 is a tissue-biased regulator of the plant miRNA biogenesis cofactor HYL1. Proc. Natl. Acad. Sci. U.S.A. 112, 14096–14101 10.1073/pnas.1512865112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Piazzon N., Maisonneuve C., Guilleret I., Rotman S., and Constam D. B. (2012) Bicc1 links the regulation of cAMP signaling in polycystic kidneys to microRNA-induced gene silencing. J Mol. Cell. Biol. 4, 398–408 10.1093/jmcb/mjs027 [DOI] [PubMed] [Google Scholar]
- 23. Valverde R., Edwards L., and Regan L. (2008) Structure and function of KH domains. FEBS J. 275, 2712–2726 10.1111/j.1742-4658.2008.06411.x [DOI] [PubMed] [Google Scholar]
- 24. Hollingworth D., Candel A. M., Nicastro G., Martin S. R., Briata P., Gherzi R., and Ramos A. (2012) KH domains with impaired nucleic acid binding as a tool for functional analysis. Nucleic Acids Res. 40, 6873–6886 10.1093/nar/gks368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. García-Mayoral M. F., Hollingworth D., Masino L., Díaz-Moreno I., Kelly G., Gherzi R., Chou C. F., Chen C. Y., and Ramos A. (2007) The structure of the C-terminal KH domains of KSRP reveals a noncanonical motif important for mRNA degradation. Structure 15, 485–498 10.1016/j.str.2007.03.006 [DOI] [PubMed] [Google Scholar]
- 26. He H., Wang L., Zhou W., Zhang Z., Wang L., Xu S., Wang D., Dong J., Tang C., Tang H., Yi X., and Ge J. (2015) MicroRNA expression profiling in clear cell renal cell carcinoma: identification and functional validation of key miRNAs. PLoS One 10, e0125672 10.1371/journal.pone.0125672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nishikawa R., Chiyomaru T., Enokida H., Inoguchi S., Ishihara T., Matsushita R., Goto Y., Fukumoto I., Nakagawa M., and Seki N. (2015) Tumour-suppressive microRNA-29s directly regulate LOXL2 expression and inhibit cancer cell migration and invasion in renal cell carcinoma. FEBS Lett. 589, 2136–2145 10.1016/j.febslet.2015.06.005 [DOI] [PubMed] [Google Scholar]
- 28. Chen Z., Tang Z. Y., He Y., Liu L. F., Li D. J., and Chen X. (2014) miRNA-205 is a candidate tumor suppressor that targets ZEB2 in renal cell carcinoma. Oncol. Res. Treat. 37, 658–664 10.1159/000368792 [DOI] [PubMed] [Google Scholar]
- 29. Hirata H., Hinoda Y., Shahryari V., Deng G., Nakajima K., Tabatabai Z. L., Ishii N., and Dahiya R. (2015) Long noncoding RNA MALAT1 promotes aggressive renal cell carcinoma through Ezh2 and interacts with miR-205. Cancer Res. 75, 1322–1331 10.1158/1538-7445.AM2015-1322,10.1158/0008-5472.CAN-14-2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Y., Jin L., Chen D., Liu J., Su Z., Yang S., Gui Y., Mao X., Nie G., and Lai Y. (2016) Tumor suppressive miR-196a is associated with cellular migration, proliferation and apoptosis in renal cell carcinoma. Mol. Med. Rep. 14, 560–566 10.3892/mmr.2016.5251 [DOI] [PubMed] [Google Scholar]
- 31. White N. M., Khella H. W., Grigull J., Adzovic S., Youssef Y. M., Honey R. J., Stewart R., Pace K. T., Bjarnason G. A., Jewett M. A., Evans A. J., Gabril M., and Yousef G. M. (2011) miRNA profiling in metastatic renal cell carcinoma reveals a tumour-suppressor effect for miR-215. Br. J. Cancer 105, 1741–1749 10.1038/bjc.2011.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen X., Hu Z., Wang W., Ba Y., Ma L., Zhang C., Wang C., Ren Z., Zhao Y., Wu S., Zhuang R., Zhang Y., Hu H., Liu C., Xu L., et al. (2012) Identification of ten serum microRNAs from a genome-wide serum microRNA expression profile as novel noninvasive biomarkers for nonsmall cell lung cancer diagnosis. Int. J. Cancer 130, 1620–1628 10.1002/ijc.26177 [DOI] [PubMed] [Google Scholar]
- 33. Zhao J. J., Lin J., Lwin T., Yang H., Guo J., Kong W., Dessureault S., Moscinski L. C., Rezania D., Dalton W. S., Sotomayor E., Tao J., and Cheng J. Q. (2010) microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood 115, 2630–2639 10.1182/blood-2009-09-243147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tang W., Zhu Y., Gao J., Fu J., Liu C., Liu Y., Song C., Zhu S., Leng Y., Wang G., Chen W., Du P., Huang S., Zhou X., Kang J., et al. (2014) MicroRNA-29a promotes colorectal cancer metastasis by regulating matrix metalloproteinase 2 and E-cadherin via KLF4. Br. J. Cancer 110, 450–458 10.1038/bjc.2013.724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma W., Xie S., Ni M., Huang X., Hu S., Liu Q., Liu A., Zhang J., and Zhang Y. (2012) MicroRNA-29a inhibited epididymal epithelial cell proliferation by targeting nuclear autoantigenic sperm protein (NASP). J. Biol. Chem. 287, 10189–10199 10.1074/jbc.M111.303636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen L., Xiao H., Wang Z. H., Huang Y., Liu Z. P., Ren H., and Song H. (2014) miR-29a suppresses growth and invasion of gastric cancer cells in vitro by targeting VEGF-A. BMB Rep. 47, 39–44 10.5483/BMBRep.2014.47.1.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu L., Xu Y., Jing Z., Wang X., Zha X., Zeng C., Chen S., Yang L., Luo G., Li B., and Li Y. (2014) Altered expression pattern of miR-29a, miR-29b and the target genes in myeloid leukemia. Exp. Hematol. Oncol. 3, 17 10.1186/2162-3619-3-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brest P., Lapaquette P., Souidi M., Lebrigand K., Cesaro A., Vouret-Craviari V., Mari B., Barbry P., Mosnier J. F., Hébuterne X., Harel-Bellan A., Mograbi B., Darfeuille-Michaud A., and Hofman P. (2011) A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat. Genet. 43, 242–245 10.1038/ng.762 [DOI] [PubMed] [Google Scholar]
- 39. Cockman M. E., Webb J. D., Kramer H. B., Kessler B. M., and Ratcliffe P. J. (2009) Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell. Proteomics 8, 535–546 10.1074/mcp.M800340-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maxwell P. H., Pugh C. W., and Ratcliffe P. J. (2001) Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 11, 293–299 10.1016/S0959-437X(00)00193-3 [DOI] [PubMed] [Google Scholar]
- 41. Maxwell P. H., Wiesener M. S., Chang G. W., Clifford S. C., Vaux E. C., Cockman M. E., Wykoff C. C., Pugh C. W., Maher E. R., and Ratcliffe P. J. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 10.1038/20459 [DOI] [PubMed] [Google Scholar]
- 42. Goda N., Ryan H. E., Khadivi B., McNulty W., Rickert R. C., and Johnson R. S. (2003) Hypoxia-inducible factor 1α is essential for cell cycle arrest during hypoxia. Mol. Cell. Biol. 23, 359–369 10.1128/MCB.23.1.359-369.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dhyani A., Machado-Neto J. A., Favaro P., and Saad S. T. (2015) ANKHD1 represses p21 (WAF1/CIP1) promoter and promotes multiple myeloma cell growth. Eur. J. Cancer 51, 252–259 10.1016/j.ejca.2014.11.012 [DOI] [PubMed] [Google Scholar]
- 44. Fragiadaki M., Lannoy M., Themanns M., Maurer B., Leonhard W. N., Peters D. J., Moriggl R., and Ong A. C. (2017) STAT5 drives abnormal proliferation in autosomal dominant polycystic kidney disease. Kidney Int. 91, 575–586 10.1016/j.kint.2016.10.039 [DOI] [PubMed] [Google Scholar]
- 45. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.