

Abstract
Prostaglandin (PG) E2 is an important lipid mediator that is involved in several pathophysiological processes contributing to fever, inflammation, and pain. Previous studies have shown that early and continuous application of nonsteroidal anti-inflammatory drugs significantly reduces pain behavior in the spared nerve injury (SNI) model for trauma-induced neuropathic pain. However, the role of PGE2 and its receptors in the development and maintenance of neuropathic pain is incompletely understood but may help inform strategies for pain management. Here, we sought to define the nociceptive roles of the individual PGE2 receptors (EP1–4) in the SNI model using EP knockout mice. We found that PGE2 levels at the site of injury were increased and that the expression of the terminal synthase for PGE2, cytosolic PGE synthase was up-regulated in resident positive macrophages located within the damaged nerve. Only genetic deletion of the EP3 receptor affected nociceptive behavior and reduced the development of late-stage mechanical allodynia as well as recruitment of immune cells to the injured nerve. Importantly, EP3 activation induced the release of CC-chemokine ligand 2 (CCL2), and antagonists against the CCL2 receptor reduced mechanical allodynia in WT but not in EP3 knockout mice. We conclude that selective inhibition of EP3 might present a potential approach for reducing chronic neuropathic pain.
Keywords: prostaglandin, G protein-coupled receptor (GPCR), neuroinflammation, pain, chemokine, CCL2, mast cells, neuropathic pain, peripheral neuron, Prostaglandin E2
Introduction
Neuropathic pain often occurs after nerve injury caused by metabolic (e.g. diabetes), mechanical, chemical, or virus-induced damage of neurons. It has a high tendency for chronification, and the therapeutic options are often unsatisfactory. Mediators, comprising cytokines, chemokines, and lipids (i.e. prostanoids), are released at the site of injury by immune cells and are fundamental for the development and maintenance of neuropathic pain (1, 2). Although nonsteroidal anti-inflammatory drugs (NSAIDs),2 which inhibit cyclooxygenases (COX) and as a consequence decrease the synthesis of prostanoids, are sometimes used for treatment, their therapeutic significance is relatively small. However, animal studies showed that early and continuous application of NSAIDs significantly reduces mechanical allodynia in the spared nerve injury (SNI) (3), the chronic constriction injury (4), and the partial sciatic nerve ligation (5) models for trauma-induced neuropathic pain. Accordingly, many prostanoid-generating enzymes, as well as prostanoid receptors, are up-regulated after trauma-induced nerve injury, supporting a functional role of prostanoids in the pathomechanism of neuropathic pain (3, 6, 7). For example, COX-2 is up-regulated in macrophages (8) and Schwann cells (9) in injured nerves following various types of injury.
Prostaglandin E2 (PGE2) is known as an important lipid mediator that is involved in several pathophysiological processes contributing to fever, inflammation, and pain (10). It can activate four G protein–coupled receptors named EP1, EP2, EP3, and EP4, which are Gq (EP1), Gs (EP2 and EP4), or Gi (EP3) coupled. Because of the different signaling pathways regulated by these receptors, as well as their variable expression patterns in the different tissues, PGE2 can exhibit opposing effects ranging from proinflammatory to anti-inflammatory (10, 11). The variability in signaling events and downstream effectors raises the question of whether it could be more useful to target single EP receptors instead of inhibiting the PGE2 synthesis itself and, therefore, stimulation of all EP receptors. In regard to neuropathic pain, mRNA for all four EP receptors was found in the nervous system of mice with a strong expression in peripheral sensory neurons (5, 10). Partial sciatic nerve ligation, a model for trauma-induced neuropathic pain, induced the up-regulation of all four EP receptors in the injured nerve (12, 13). However, in addition to pharmacological evidence for a pronociceptive role of EP4 (14) and EP1 (15), so far little is known about the specific importance of the four EP receptors in the development and maintenance of neuropathic pain.
Here, we aimed to define the nociceptive role of the individual PGE2 receptors in the SNI model for trauma-induced neuropathic pain using knockout mice for the four receptors. We found increased PGE2 levels at the site of injury and enhanced expression of the terminal synthase for PGE2, cytosolic PGE synthase (cPGES), in IBA1-positive macrophages, which were located within the damaged nerve. Interestingly, of the four PGE2 receptors, only deletion of the EP3 receptor decreased late stage trauma-induced mechanical allodynia. EP3 activation can induce the release of C–C motif-chemokine ligand 2 (CCL2), and pharmacological inhibition of its receptor C–C motif chemokine receptor 2 (CCR2) reduced mechanical allodynia in WT but not in EP3 knockout mice.
Results
EP3 deletion diminishes mechanical allodynia in the SNI model for neuropathic pain
First we investigated whether PGE2 levels in sciatic nerve, DRGs (L3–L5) and spinal cord (L3–L5) are altered 14 days after inducing the nerve injury according to the SNI model when a stable mechanical allodynia was established (16). Although LC–MS/MS analysis showed no change of the PGE2 levels in DRGs and the spinal cord, PGE2 levels were increased at the site of injury in the sciatic nerve (Fig. 1A). Next, we compared the paw withdrawal latencies after mechanical stimulation of WT mice with mice deficient for either EP1, EP2, EP3, or EP4. All mouse strains showed an initial reduction of mechanical thresholds 4 days after nerve injury. This reduction was ongoing in the WT mice for the whole time period of the experiment, reaching a minimum of the mechanical thresholds 28 days after SNI. Mice deficient for EP1 (Fig. 1B), EP2 (Fig. 1C), or EP4 (Fig. 1E) showed a similar reduction of the mechanical thresholds compared with WT mice. However, EP3-deficient mice developed a milder form of mechanical allodynia in which the mechanical thresholds did not further decrease after day 4 and were, starting at day 10, significantly higher than in WT mice (Fig. 1D).
Figure 1.

Deletion of the EP3 receptor reduces mechanical allodynia in the SNI model for neuropathic pain. A, LC–MS/MS analysis of PGE2 levels in the sciatic nerve, DRGs (L3–L5) and spinal cord (L3–L5) in naïve mice and 14 days after SNI. The data are shown as means ± S.E. (n = 5). **, p ≤ 0.01; two-tailed t test. B–E, C57BL/6N mice and EP receptor knockout mice underwent SNI surgery. Mechanical thresholds were measured before and at the indicated times after SNI. The data are shown as means ± S.E. B–D, WT, n = 15. B, EP1−/−, n = 7. C, EP2−/−, n = 8. D, EP3−/−, n = 13. E, EP4−/−, n = 6. *, p < 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; two-way ANOVA with Bonferroni post hoc test. BL, baseline; d, days; PW, paw withdrawal.
Although it is known that COX-2 is up-regulated in macrophages (8) and Schwann cells (9) after nerve injury, the localization of the PGE2 synthases downstream of COX-2 and, therefore, the localization of PGE2 synthesis have not been described in injured nerves. Because day 10 after the SNI operation marks the beginning of the reduced pain behavior in EP3-deficient mice, we confirmed that PGE2 levels are significantly elevated at this day at the injured nerve (Fig. 2A). 10 days after the SNI operation the expression of the PGE2 synthase cPGES was higher in the injured sciatic nerve, whereas the other terminal synthase for PGE2, mPGES-1, was not detectable at the site of injury (Fig. 2B). Densitometric analysis showed a significantly elevated expression of cPGES in the injured sciatic nerve as compared with uninjured sciatic nerves (Fig. 2C). After having identified the enzymatic source of PGE2, we investigated which cell types express cPGES. Using the MELC technique for multiple sequential immunohistochemical staining, we identified four markers, which colocalized with cPGES: IBA1, CD11b, CD45, and F4-80 (Fig. 2, D and E). Importantly, IBA1/F4-80 double-positive cells are microglia of the central nervous system or resident macrophages of the peripheral nervous system (17–19). Thus, the combination of these cell markers together with the absence of Ly6C suggests that cPGES is expressed in resident macrophages.
Figure 2.

cPGES but not mPGES-1 is expressed in macrophages at the injury site. A, LC–MS/MS analysis of PGE2 in the ipsi- and contralateral sciatic nerves 10 days after the SNI operation. The data are shown as means ± S.E. (n = 6). **, p ≤ 0.01 one-tailed t test. B, representative Western blotting for cPGES and mPGES-1 in ipsilateral and contralateral sciatic nerves 10 days after the SNI operation; bone marrow–derived macrophages stimulated for 24 h with 0.1 μg of LPS served as positive control. C, densitometric analysis of four independent Western blotting experiments as shown in B. The data are shown as means ± S.E. **, p ≤ 0.01, two-tailed t test. D and E, MELC analysis of sciatic nerve 10 days after the SNI operation. The white squares indicate the regions shown for cPGES, IBA1, CD11b, CD45, and F4-80. White arrows indicate cells expressing these five markers. The radar chart (E) displays the percentage of cPGES-expressing cells (100%) that are positive for the respective stated marker (n = 40).
Immune cell recruitment to the site of injury is decreased in EP3-deficient mice
Because recruitment of myeloid immune cells is a prerequisite in the development of trauma-induced neuropathic pain (2), we tested whether or not the recruitment of immune cells to the site of injury is impaired in EP3-deficent mice. Indeed, 10 days after the nerve injury, the number of all CD45-positive cells and more specifically the number of neutrophils, monocytes, macrophages, B cells, NK cells, and T cells were significantly increased in WT mice (Fig. 3, A and B). Importantly, in EP3-deficient mice the numbers of several immune cell populations were significantly lower as compared with WT mice (Fig. 3B). No significant difference between WT and EP3-deficient mice was observed at the contralateral side.
Figure 3.

Immune cell recruitment to the site of injury is decreased in EP3−/− mice. A and B, FACS analysis with cells isolated from the site of injury from WT and EP3−/− mice 10 days after the SNI operation. The gating strategy is shown in A. The data are shown as means ± S.E. (WT, n = 7; EP3−/− ipsilateral, n = 6; EP3−/− contralateral, n = 5; two-way ANOVA/Bonferoni post hoc test). *,#, p ≤ 0.05; **,##, p ≤ 0.01; ***, p < 0.001. #, compared with contralateral.
Next, we studied whether or not deletion of EP3 in myeloid cells affects the development of neuropathic pain. Therefore, irradiated WT mice received bone marrow from WT or EP3-deficient mice and were tested for successful transplantation by genotyping blood samples (Fig. 4A). 4 weeks after transplantation, the mice underwent the SNI operation and were tested for the mechanical paw withdrawal latencies. We found that independently of EP3 expression in the myeloid cells, the mice developed the same mechanical pain intensity, demonstrating that EP3 deletion in myeloid cells is not sufficient to reduce the mechanical allodynia (Fig. 4B). Furthermore, FACS analysis showed no significant difference in the amount of immune cells at the site of injury between the chimera types (Fig. 4C), suggesting that EP3 expression on the recruited immune cells does not play a role in trauma-induced neuropathic pain. Taken together, the data show that EP3 expression on recruited immune cells is not important for nociception in the SNI model, whereas EP3 expression on nonmyeloid cells (e.g. neuronal cells) or myeloid cells, such as mast cells, which are not affected by the irradiation, are involved in SNI-induced nociception.
Figure 4.
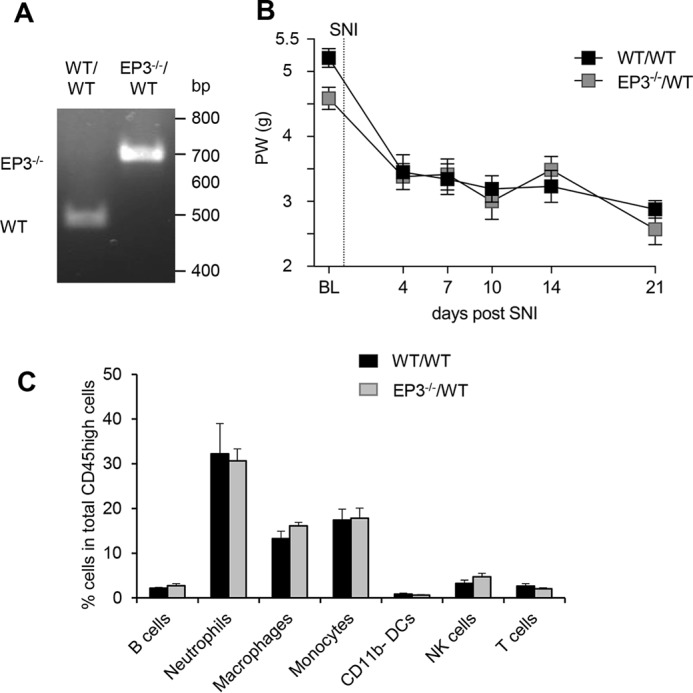
EP3-deficiency on myeloid cells does not affect mechanical allodynia or immune cell recruitment in the SNI model. A, genotyping of white blood cells from irradiated WT mice having received bone marrow from wild-type (WT/WT) or EP3−/− (EP3−/−/WT) mice. The lower PCR band (450 bp) shows WT and the upper and 650-bp EP3 knockouts. B, C57Bl/6N received bone marrow from either WT or EP3-deficient mice before they underwent SNI surgery. Mechanical allodynia was determined before and at the indicated times after surgery. The data are shown as means ± S.E. (WT, n = 7; EP3−/−, n = 10). C, immune cell numbers determined by FACS analysis at the site of injury at the sciatic nerve in bone marrow–transplanted mice. The data are shown as means ± S.E. (WT, n = 5; EP3−/−, n = 7).
EP3 activation induces the release of CCL2 from neurons
One of the mediators regulated by EP3 is the chemokine CCL2 (20), which is also known to contribute to neuropathic pain development after injury of the sciatic nerve caused by its ability to recruit immune cells (21). Thus, we performed real-time RT-PCR 10 days after the SNI operation using mRNA from naïve and injured nerves and found that CCL2 mRNA levels were indeed increased in injured nerves (Fig. 5A) with a 4.4-fold increase in the injured versus the uninjured sciatic nerve. Relative mRNA levels for the CCL2 receptor CCR2 were also significantly up-regulated with a 1.5-fold increase (Fig. 5A). To identify cells that release CCL2 at the injury site, we performed immunohistochemical staining with sciatic nerve tissue of WT mice after SNI. CCL2 immunostaining colocalized with the mast cell marker CD117 (Fig. 5B), as well as with isolectin B4, a marker for sensory neurons (Fig. 5C).
Figure 5.
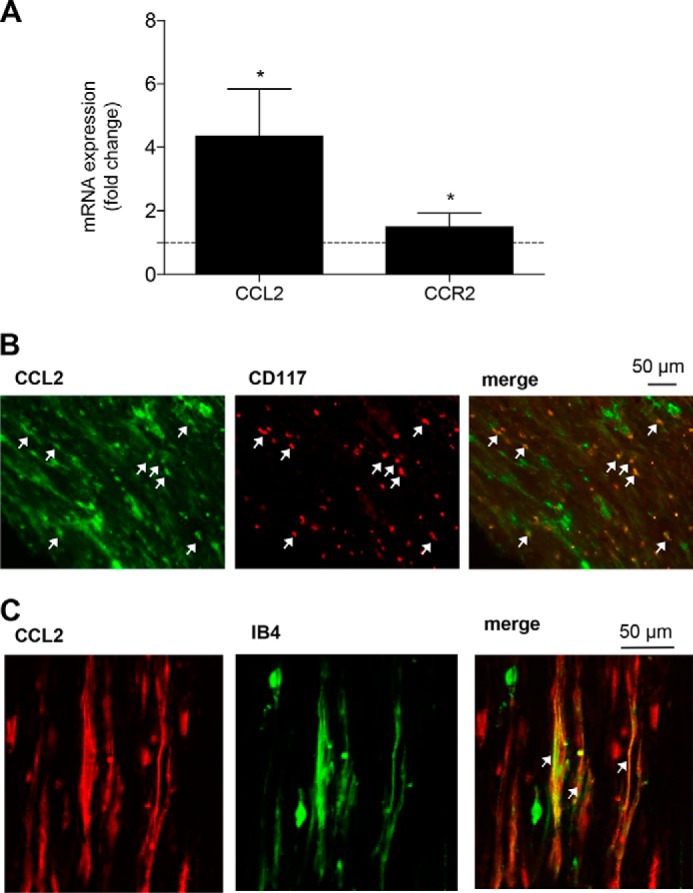
CCL2 expression in mast cells and neurons is up-regulated at the site of injury. A, real-time RT-PCR for CCL2 and CCR2 mRNA at the site of injury 10 days after the SNI operation. The data are shown as means ± S.E. of the fold stimulation compared with uninjured sciatic nerves (injured nerve, n = 4; uninjured nerve, n = 5). *, p ≤ 0.05, two-tailed t test. B and C, colocalization of CCL2 with the mast cell marker CD117 (B) and the neuronal marker IB4 (C) expression in the injured nerve. The white arrows indicate double-positive cells.
Because CCL2 mRNA expression is up-regulated at the site of injury and CCL2 can be detected in mast cells and sensory neurons, we hypothesized that EP3 activation might induce CCL2 release from these cells. Indeed, treatment of neuronal DRG cultures from WT mice with the EP3 receptor agonist sulprostone resulted in a significantly increased release of CCL2 as compared with DRGs from EP3-deficient mice (Fig. 6A). To study whether or not EP3 also mediates the release of CCL2 from mast cells, bone marrow–derived mast cells were treated with PMA as positive control and increasing concentrations of sulprostone. Importantly, stimulation with sulprostone induced CCL2 release only in WT but not in EP3-deficient mast cells (Fig. 6, B and C). Then we studied whether or not mast cells are involved in the mechanical allodynia response in the SNI model and therefore tested mast cell–deficient mice (Mcpt5–DTA–Cre mice (22)). Cre-positive mast cell–deficient mice had a significantly decreased number of monocyte-derived macrophages at the injury site (Fig. 6D), whereas other immune cell populations, such as resident macrophages, were not affected (data not shown). More importantly, mast cell–deficient mice developed a normal mechanical allodynia (Fig. 6E). Thus, although mast cells seem to mediate the recruitment of macrophages to the site of injury, these macrophages are apparently not relevant for the SNI-induced mechanical allodynia.
Figure 6.
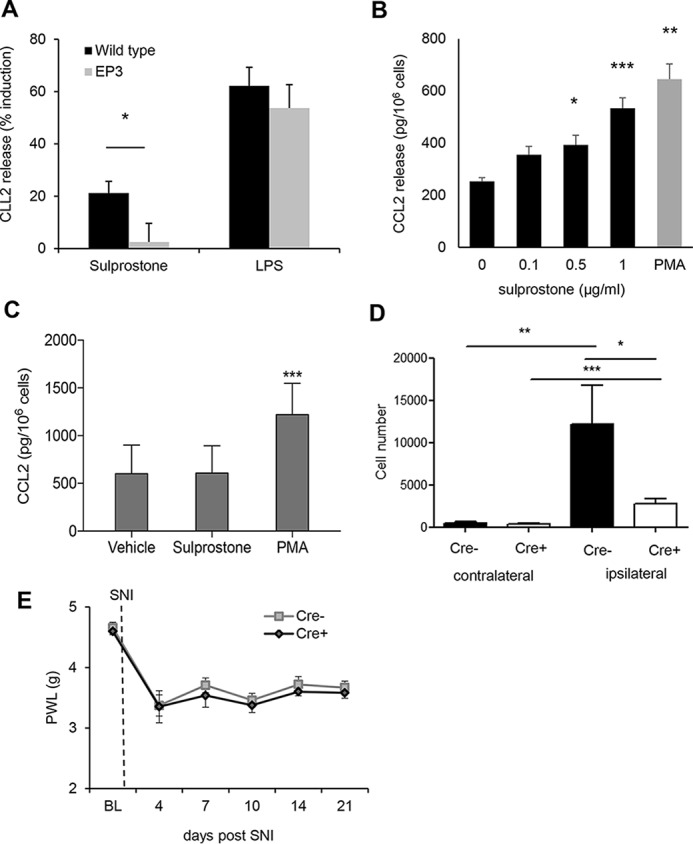
EP3 activation induces the release of CCL2 from DRG neurons and mast cells. A, CCL2 release from DRG cultures of WT and EP3−/− mice was determined by ELISA after 6 h of incubation with vehicle, sulprostone (1 μg/ml), or LPS (25 ng/ml). The data are shown as means ± S.E. (WT, n = 8; EP3−/−, n = 5). *, p ≤ 0.05; Student's t test. B, CCL2 release from mast cells of WT mice was determined by ELISA after 6 h of incubation with vehicle, sulprostone or PMA (0.1 μm). The data are shown as means ± S.E. (n = 5). **, p ≤ 0.01; ***, p ≤ 0.001; ***, p ≤ 0.001; one-way ANOVA with Bonferroni multiple comparison. C, CCL2 release from mast cells of EP3−/− mice was determined by ELISA after 6 h incubation with vehicle, sulprostone (1 μg/ml), or PMA (0.1 μm). The data are shown as means ± S.E. (n = 5). ***, p ≤ 0.001; one-way ANOVA with Bonferroni multiple comparison. D, FACS analysis with cells isolated from the site of injury from WT and Mcpt5–DTA–Cre−/+ mice 14 days after the SNI operation. The data are shown as means ± S.E. (n = 5). *, p ≤ 0.05; **, p ≤ 0.01; ***, p < 0.001; two-way ANOVA/Bonferoni post hoc test. E, Cre-positive and -negative Mcpt5 DTA mice underwent SNI surgery. Mechanical thresholds were measured before surgery and at the indicated times after SNI. The data are shown as means with ± S.E. (n = 6). PWL, paw withdrawal latency.
Antinociceptive effects of CCR2 antagonists are absent in EP3-deficient mice
Because our data indicate that EP3 stimulation leads to CCL2 release in neurons and mast cells, we investigated whether or not EP3-controlled CCL2 release contributes to the allodynia in the SNI model. Therefore, mechanical allodynia of WT and EP3 knockout mice was determined before and after the SNI operation. After the mice developed a sustained allodynia (day 18/19), the CCR2 antagonist INCB3344 was injected intravenously once daily on two consecutive days (0.18 μmol/injection) as described previously (23). Vehicle-treated mice showed no change in the intensity of the SNI-induced nociceptive behavior (Fig. 7). Importantly, application of the CCR2 antagonist significantly increased mechanical allodynia in WT mice, whereas it had no effect on the threshold of EP3-deficient mice (Fig. 7). Thus, the findings demonstrate that the injury-induced EP3-mediated mechanical allodynia is depending on the CCL2-CCR2 axis and that signaling in this model for neuropathic pain the CCL2-CCR2 is downstream of EP3.
Figure 7.
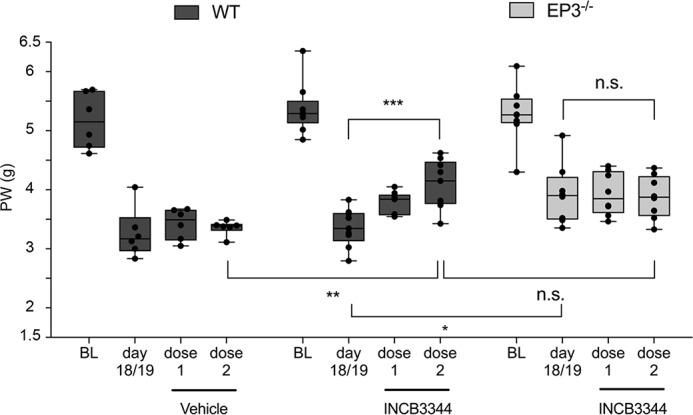
The CCR2-antagonist INCB3344 increases paw withdrawal (PW) latencies in WT, but not in EP3−/− mice. Mechanical allodynia was first determined before the SNI operation. BL, baseline. When the mice had developed sustained neuropathic pain on day 18/19 after injury, vehicle or the CCR2-antagonist INCB3344 was applied intravenously on two consecutive days (dose 1 and dose 2) with a dose of 0.1 mg (0.18 μmol) per injection. Mechanical allodynia was determined 2 h after application. The data are shown as means ± S.E. (vehicle, n = 6; WT + INCB3344, n = 9; EP3−/− + INCB3344, n = 8). *, p ≤ 0.05; ***, p ≤ 0.001; two-way ANOVA with Bonferroni post hoc test.
Discussion
Previous studies demonstrated the involvement of PGE2 in the development and maintenance of neuropathic pain and showed that early and continuous treatment with NSAIDS prevents at least partly the occurrence of trauma-induced neuropathic pain (15, 24, 25). Here, we show that of the four PGE2 receptors, only the deletion of EP3 reduces the development of mechanical allodynia in mice after peripheral nerve injury and that this effect is mediated via the CCL2-CCR2 axis.
In contrast to our observation of a pronociceptive effect of EP3, it was previously reported that EP3 activation can decrease nociception. In this report intrathecal application of an EP3 agonist induced antinociception in a model for acute inflammation through EP3 receptors expressed on spinal cord neurons (26). However, in the SNI mode used in our study, PGE2 levels increased only in the periphery but not in the spinal cord. This finding suggests strongly that central EP3-mediated effects are not involved nociception in the SNI model. Thus, inhibition EP3 in the periphery might present an alternative approach to reduce long-lasting neuropathic pain.
In regard to the importance of peripheral events for the development of neuropathic pain, it has been shown in various animal models for peripheral nerve injury that immune cell recruitment to the injury site is required for the development of neuropathic pain (1, 2, 12). Notably, EP3−/− mice showed a decreased recruitment of immune cells to the sciatic nerve after the SNI operation.
This decreased recruitment did not depend on the activation of EP3 receptors on myeloid cells, because the selective deletion of EP3 on these cells did not alter SNI-induced mechanical allodynia. Fittingly, only 6% of macrophages in injured nerves of rats with partial nerve ligation express EP3 (12), whereas EP3 mRNA is found in ∼50% of murine DRG neurons (26). Accordingly, real-time RT-PCR analysis of the injured nerve showed an apparent decrease of EP3 mRNA in the injured nerve (data not shown), which is probably due to the massive recruitment of immune cells, which do not express EP3. In accordance with previous reports, we found that EP3 activation induces the release of CCL2 from neurons and mast cells. This neuronal CCL2 expression was already reported in models for trauma-induced neuropathic pain (21, 22). We also found CCL2 expression in mast cells, which have been described to release CCL2 after activation of EP3 (20). However, in mast cell–deficient mice was only the number of monocyte-derived macrophages decreased, and there was no apparent effect on the SNI-induced mechanical allodynia. It should be noted that although we did not detect by immunohistochemistry CCL2 in other cells than neurons and mast cells, we cannot exclude the possibility that other immune cells also express CCL2 at the site of injury, because a rapid release of CCL2 may lower intracellular CCL2 expression levels under the detection limit.
The significance of the CCL2/CCR2 signaling pathway for the development of neuropathic pain was demonstrated in the chronic constriction injury model using CCR2-deficient mice, which did not develop mechanical allodynia (27), and the use of the CCR2 antagonist INCB3344, which partly reversed mechanical allodynia (23). Fittingly we found that the antinociceptive effect INCB3344 is absent in EP3-deficient mice in the SNI model. This observation shows that CCR2 is a downstream effector of EP3. This is in accordance with our other results showing that EP3 activation induces the release of the CCR2 activator CCL2 from mast cells and peripheral neurons. An additive effect should be seen if EP3 influences the pain behavior in ways that are independent of CCL2 or if the CCL2 release in this model would also occur independent from EP3. This does not seem to happen according to our data.
Currently two modes of actions are discussed for the pronociceptive effect of CCL2. On one hand, chemotaxis of immune cells can be mediated by direct binding of CCL2 to its cognate receptor CCR2 located on the cell surface of myeloid immune cells, directing these cells to the site of inflammation. Additionally, CCL2 has been shown to facilitate immune cell infiltration to nervous tissue by increasing the blood–brain barrier permeability (28). As mentioned above, the recruitment of the immune cells to the site of injury is a requirement for the development of trauma-induced neuropathic pain, and the number of infiltrating cells for several immune cell types was significantly decreased at the site of injury in EP3-deficient mice. On the other hand, CCR2 is expressed in peripheral myeloid cells, DRG neurons, and microglia (26) and potentially in second order neurons in lamina II of the spinal cord (29, 30). Here, several pronociceptive electrophysiological effects of CCL2, such as enhancement of glutamate receptor function or reduction of GABAergic signaling (29, 31), have been described. Thus, CCL2 released from DRG neurons may stimulate second order (spinal) neurons to enhance nociception. In addition, CCL2 released from neurons might activate spinal microglia (32), which are important contributors to neuropathic pain.
Experimental procedures
Animals
6–8-week-old male C57BL/6N (Janvier, Le Genest, France) and knockout mice with C57BL/6N background (33–35) were compared with strain-, age-, and sex-matched controls. Mcpt5–DTA–Cre mice were originally described by the group of Prof. Roers at Technische Universität Dresden (22). In all experiments the ethics guidelines of the German Public Health Service were obeyed, and the procedures were approved by the local ethics committee.
Animal experimentation
The investigator was unaware of the treatment or the genotypes during all behavioral experiments. The mice were anesthetized, and the sciatic nerve (nervus ischiadicus) with its three branches was exposed by blunt dissection. The common peroneal and the tibial branches were tightly ligated with 6-0 silk thread and cut distally from the ligature (16). Mechanical thresholds of the plantar side of a hind paw were determined using a plantar aesthesiometer (Dynamic Plantar Aesthesiometer, Ugo Basile). Here, a steel rod (0.5-mm diameter) was pushed against the paw with ascending force (0–5 g over a 10-s period; time resolution, 0.1 s) until a strong and immediate withdrawal occurred. The paw withdrawal was taken to be the mean of at least three consecutive trials with at least 20 s in between. Cutoff time was set at 20 s. Baselines were taken prior to the surgery.
The CCR2 antagonist INCB3344 (MedChem Express, Sollentuna, Sweden) was dissolved in DMSO and diluted with PBS to a final concentration of 1 mg/ml. To dissolve precipitated substance it was kept in a water bath at 37 °C for 30 min and sonicated. 100 μl (0.1 mg) were intravenously injected into the tail vein of C57BL/6N and EP3-deficient mice. The antagonist was injected once a day, and the mechanical thresholds were measured 2 h after the application. 100 μl of 0.25% DMSO in PBS was used as vehicle control.
LC–MS/MS
LC–MS/MS analysis of PGE2 from spinal tissue, DRGs, and sciatic nerves was performed as described previously (36). Briefly, at different time points after SNI surgery the sciatic nerves (∼1 cm proximal starting at the ligation site), the DRGs, and the dorsal part of the spinal cord were prepared. Then the tissue was weighed and liquid–liquid–extracted using 200 μl of PBS, 600 μl of ethyl acetate, 100 μl of 150 mm EDTA, and 20 μl of internal standard solution (25 ng/ml of [2H4]PGE2 in methanol). The samples were homogenized using Mixer Mill MM400 (Retsch, Haan, Germany) and centrifuged at 20,000 × g for 5 min. The extraction was repeated using 600 μl of ethyl acetate, and organic layers were collected, evaporated at 45 °C under a gentle stream of nitrogen, and stored at −80 °C until measurement. The residues were reconstituted with 50 μl of acetonitrile/water/formic acid (20:80:0.0025, v/v/v), and 20 μl were injected into the LC–MS/MS system. Chromatographic separation was done using a Synergi Hydro-RP 2.0 × 150 mm (Phenomenex, Aschaffenburg, Germany), 4-μm column coupled to a precolumn of the same material, and 0.0025% formic acid (A) and acetonitrile with 0.0025% formic acid (B) as mobile phases. The elution gradient is shown in Table 1.
Table 1.
Elution gradient
| Time | Flow rate | A | B |
|---|---|---|---|
| μl/min | % | % | |
| 0 min | 300 | 90 | 10 |
| 1 min | 300 | 90 | 10 |
| 2 min | 300 | 60 | 40 |
| 3 min | 300 | 60 | 40 |
| 4 min | 300 | 50 | 50 |
| 6 min | 300 | 50 | 50 |
| 8 min | 300 | 10 | 90 |
| 9 min | 300 | 10 | 90 |
| 10 min | 300 | 90 | 10 |
| 16 min | 300 | 90 | 10 |
Sample analysis was performed using LC–electrospray ionization–MS/MS consisting of a QTrap 5500 hybrid triple quadrupole-ion trap mass spectrometer (AB Sciex, Darmstadt, Germany) equipped with a Turbo V source operating in negative electrospray ionization mode, an Agilent 1200 binary HPLC pump and degasser (Agilent, Waldbron, Germany), and an HTC Pal autosampler (Chromtech, Idstein). A cooling stack was used to store the samples at 6 °C in the autosampler. High purity nitrogen for the mass spectrometer was produced by a NGM 22-LC/MS nitrogen generator (cmc Instruments, Eschborn, Germany). The mass spectrometer was operated in the negative ion mode with an electrospray voltage of −4500 V at 450 °C. Multiple reaction monitoring was used for quantification. The mass transitions used were m/z 351.1 → m/z 315.0 for PGE2 and m/z 355.1 → m/z 275.1 for [2H4]PGE2 all with a dwell time of 50 ms. All quadrupoles were working at unit resolution. Quantitation was performed with Analyst Software V1.5 (Applied Biosystems, Darmstadt, Germany) using the internal standard method (isotope-dilution MS). Ratios of analyte peak area and internal standard peak area (y axis) were plotted against concentration (x axis), and calibration curves for each prostaglandin were calculated by least-square regression with 1/concentration2 weighting.
Western blotting
Ipsilateral and contralateral sciatic nerves were prepared 10 days after SNI operation and immediately quick-frozen in liquid nitrogen. The nerves were homogenized by ultrasonication (3 × 10 s) in a suitable volume of PhosphoSafe buffer (Merck Millipore) and afterward centrifuged at 18,000 × g for 4 min at 4 °C to isolate the cytosolic fraction. The protein concentration was determined, and 20–30 μg protein were loaded on a 12% SDS-polyacrylamide gel for analyses. cPGES (catalog no. 10209) and mPGES (catalog no. 160145) were detected with polyclonal antibodies from Cayman Chemical (Ann Arbor, MI) used at dilutions of 1:200 and 1:6000, respectively. Bone marrow–derived macrophages stimulated with 0.1 μg/ml LPS for 24 h served as positive control. ERK-2 served as loading control (sc154; Santa Cruz, Heidelberg, Germany, dilution 1:1000). The bands were visualized using HRP-coupled secondary antibodies (Sigma–Aldrich) and an ECL reagent (Thermo Fisher Scientific) and densitometrically analyzed using ImageJ software.
Multiepitope–ligand cartography (MELC)
The MELC technology is an immunohistological imaging method that allows the visualization of 20–40 proteins on the same sample and has been described previously (37, 38). Briefly, tissues were embedded in tissue freezing medium (Leica Microsystems, Nussloch, Germany), and cryosections of 10-μm thickness were applied on silane-coated coverslips, fixed in 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.1% Triton X-100 in PBS for 15 min, and blocked with 3% BSA in PBS for 1 h. The sample was placed on the stage of a Leica DM IRE2, and a picture was taken. Then by a robotic process, the sample was incubated for 15 min with bleachable fluorescence-labeled antibodies (Table 2) and rinsed with PBS. Afterward, the phase contrast and fluorescence signals were imaged by a cooled charge-coupled device camera (Apogee KX4; Apogee Instruments, Roseville, CA, 2048 × 2048 pixels; final pixel size was 286 × 286 nm). To delete fluorescence signals, a bleaching step was performed. A postbleaching image was recorded and the next antibody was applied. The postbleaching image was subtracted from the following fluorescence image during the data analysis. Using the corresponding phase contrast images, fluorescence images produced by each antibody were aligned pixel-wise. Images were corrected for illumination faults using flat-field correction. After the MELC run, the tissue slices were stained with Diff-Quick (Dade Behring).
Table 2.
Antibodies used for MELC and FACS analysis
7-AAD, 7-aminoactinomycin D; MHC, major histocompatibility complex.
| Target/marker | Source | Distributor | Clone |
|---|---|---|---|
| 7-AAD | Calbiochem | ||
| cPGES | Mouse | Cayman Chemical | JJ6 |
| CD4 | Rat | SouthernBiotech | L3T4 |
| CD8a | Rat | BD Pharmingen | 53-6.7 |
| CD11b | Rat | AbD Serotec | M1/70.15 |
| CD11c | Hamster | Miltenyi Biotec | N418 |
| CD25 | Rat | Miltenyi Biotec | 7D4 |
| CD45 | Rat | Miltenyi Biotec | 30F11.1 |
| CD56 (N-Cam) | Rat | R&D Systems | 809220 |
| CD117 | Rat | Miltenyi Biotec | 3C11 |
| F4-80 | Rat | Biolegend | BM8 |
| Fibroblasts | Rat | Santa Cruz | ER-TR7 |
| IB4 | Lectin | Sigma–Aldrich | |
| IBA1 | Mouse | Gene Tex Inc | 1022-5 |
| Ly6C | Rat | Miltenyi Biotec | 1G7.G10 |
| Ly6G | Rat | eBioscience | RB6-8C5 |
| NK1.1 | Mouse | BD Pharmingen | PK136 |
| MHC II | Human | Miltenyi Biotec | REA813 |
To analyze coexpression, the relative immunofluorescence intensities for all antibodies were determined in a single pixel using the TIC Experiment Viewer software (Meltec, Magdeburg, Germany) as described previously (39). Briefly, the minimum intensity (value 0) was set individually for each antibody to eliminate background staining. Maximum intensity (value 1) was set automatically for each antibody using the brightest pixel in its image. A protein was defined as present in an individual pixel when its signal reached at least 10% of the maximal signal strength seen for this protein.
FACS analysis
Polychromatic flow cytometry was performed as described (40). Briefly, single-cell suspensions were generated from sciatic nerves (∼1-cm length starting at the ligation site, proximal), which were cut into pieces of ∼1 mm3, by digestion with 3 mg/ml collagenase IA (Sigma), 1 units/ml DNase I (Promega) for 30 min at 37 °C, followed by filtration through a 70-μm nylon mesh (BD Biosciences). The cells were transferred to FACS tubes, and nonspecific antibody binding to FC-γ receptors was blocked with mouse BD Fc block (BD Biosciences) for 20 min on ice. This was followed by incubation with an antibody mixture for 30 min on ice comprising antibodies listed in Table 2 or for multiparameter FACS analysis CD45-Vioblue, HLA-DR-APC (Miltenyi Biotec, Bergisch-Gladbach, Germany), F4/80-PE-Cy7 (eBioscience, Frankfurt, Germany), CD3-PE-CF594, CD11b-BV605, CD11c-AlexaFluor700, CD19-APC-H7, Ly6C-PerCP-Cy5.5, and Ly6G-APC–Cy7, NK1.1-PE (BD Biosciences). The samples were acquired with a LSRII/Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software 7.6.5 (Treestar, Ashland, OR). All antibodies were titrated to determine optimal concentrations. Antibody-capturing CompBeads (BD Biosciences) were used for single-color compensation to create multi-color compensation matrices. For gating, fluorescence minus one controls were used. The instrument calibration was controlled daily using cytometer setup and tracking beads (BD Biosciences).
Bone marrow transplantation and generation of chimeric mice
Bone marrow was isolated from WT or EP3−/− mice. Recipients WT mice were irradiated with sublethal doses of 9.5 Gy (Biobeam 2000 137CS γ-irradiation source; Eckert & Ziegler Strahlen und Medizintechnik AG, Berlin, Germany), injected with at least 4 × 106 bone marrow cells from donor mice via the tail vein, and kept for 4 weeks to allow humoral reconstitution as described previously (3, 41). The genotype of white blood cells was determined after the experiment by extracting genomic DNA using the Extract-N-Amp blood PCR kit (Sigma–Aldrich). Genotyping was done as described previously by PCR (42) using the following EP3 primers: GCTGGCTCTGGTGGTGACCTT and AAGCCAGGCGAACTGCAATTAGAA.
Real-time PCR
Sciatic nerves were dissected, quick-frozen in liquid nitrogen, and stored at −80 °C. Total RNA of the DRGs was isolated using a mirVana miRNA isolation kit (Ambion) according to the manufacturer's instructions and quantified with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). The cDNA was synthesized from 54 ng of RNA in the nerve and 138 ng of RNA in DRGs using a first-strand cDNA synthesis kit (Thermo Scientific) according to the manufacturer's instructions. Real-time RT-PCR was performed using an ABI Prism 7500 Sequence Detection System (Applied Biosystems) with Maxima SYBR Green qPCR Master Mix ROX and primers for GAPDH (CAATGTGTCCGTCGTGGATCT, GTCCTCAGTGTAGCCCAAGAT), CCL2 (GCCCCACTCACCTGCTGCTAC, GGTTCTGATCTCATTTGGTTCCG), and CCR2 (ATCCACGGCATACTATCAACATCTC, GACAAGGCTCACCATCATCGTAG). Water controls were included to ensure specificity. Relative expression of target gene levels were determined using the ΔΔCt method, with Ct indicating the cycle number at which the signal of the PCR product crosses a defined threshold set within the exponential phase of the PCR. The amount of sample RNA was normalized to GAPDH.
DRG neuron culture
C57BL/6N and EP3-deficient mice were sacrificed, and the spinal cord was exposed and removed. Connective tissue and nerve processes were removed to collect the DRGs. DRGs were transferred into ice-cold Hanks' balanced salt solution containing Ca2+ and Mg2+. After 3 min of centrifugation at 1000 rpm, the cells were incubated with 3 ml of collagenase (500 units/ml) and dispase (2.5 units/ml) for 75 min at 37 °C and 5% CO2. Following two washing steps with neurobasal medium containing 10% fetal calf serum and 1% penicillin/streptomycin, the cells were digested with trypsin for 10 min. Afterward the cells were washed twice, resuspended in the washing medium, and distributed onto a 48-well plate coated with poly-l-lysine. After 2 h of incubation, the medium was changed to neurobasal medium containing (+ glutamin, + gentamicin + penicillin/streptomycin, + B27) without FCS, and the cells were then incubated overnight. The following conditions were used for stimulation: LPS (25 ng/ml), sulprostone (1 μg/ml), and vehicle (0.1% DMSO). For CCL2 quantitation, the Legend Max mouse MCP-1/CCL2 ELISA kit (BioLegend, San Diego, CA) was used following the manufacturer's instructions.
Bone marrow–derived mast cells
Femur and tibia of hind legs from adult mice were extracted from muscle tissue. Bone ends were cut, and bone marrow was extracted by centrifugation at 10,000 × g for 10 s. The cells of a single animal were resuspended in 40 ml of RPMI 1640 medium (Thermo Fisher, Darmstadt, Germany) containing 10% fetal calf serum, 4 nm l-glutamine, 1% penicillin/streptomycin, 1 mm sodium pyruvate, 1% minimum Eagle's medium nonessential amino acid solution, 50 μm 2-mercaptoethanol, and 10 μg/liter IL-3 (PeproTech, Hamburg, Germany). Twice a week, 40 ml of medium was added, and during the first week the culture flasks were exchanged to remove macrophages adherent to the flasks. After 5 weeks mast cells were centrifuged (1000 × g for 5 min) and resuspended in 3 ml of fresh medium. 500 μl of cell suspension was pipetted into a 12-well plate and stimulated with the following conditions: 0.1 μm phorbol 12-myristate 13-acetate (PMA), 2.148 μm sulprostone (1 μg/ml), or vehicle (0.1% DMSO). After 2 h of stimulation, supernatant was collected and stored at −20 °C for later quantitation of CCL2. FACS analysis showed that >90% of the cells were CD117+.
Statistics
Experiments with only two treatment groups were analyzed for statistical significance using Student's t test. Experiments with more than two groups were analyzed using two-way analysis of variance (ANOVA) with the Bonferroni post hoc test. Significance was accepted at p < 0.05.
Author contributions
E.-M. T., K. K., and K. S. conceptualization; E.-M. T., A. W., N. T., and K. S. formal analysis; E.-M. T., G. G., and K. S. supervision; E.-M. T., K. K., A. W., N. T., C.-D. S., Y. S., N. F., S. P., and K. S. investigation; E.-M. T., K. K., N. T., R. M. N., Y. S., N. F., B. B., S. P., and K. S. methodology; E.-M. T., K. K., A. W., and S. P. writing-original draft; E.-M. T., G. G., and K. S. project administration; E.-M. T., K. K., A. W., N. T., C.-D. S., R. M. N., Y. S., N. F., B. B., G. G., S. P., and K. S. writing-review and editing; R. M. N. and G. G. resources.
Acknowledgments
We are grateful to Praveen Mathoor and Annett Wilken Schmitz for excellent technical help.
This work was supported by Deutsche Forschungsgemeinschaft Grants SCHO817/3-2, GRK2336 AVE, and SFB1039 (TPA08, B04, B06, Z01) and by funds from the Else Kröner Fresenius Foundation as part of the Else Kröner Graduate School (to E.-M. T.) and as part of the Translational Research Innovation–Pharma Graduate School (to K. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- NSAID
- nonsteroidal anti-inflammatory drug
- CCL2
- chemokine (CC-motif) ligand 2
- CCR2
- chemokine (CC motif) receptor 2
- cPGES
- cytosolic PGE synthase
- DRG
- dorsal root ganglia
- LPS
- lipopolysaccharide
- SNI
- spared nerve injury
- PG
- prostaglandin
- COX
- cyclooxygenase
- PMA
- phorbol 12-myristate 13-acetate
- ANOVA
- analysis of variance.
References
- 1. Grace P. M., Rolan P. E., and Hutchinson M. R. (2011) Peripheral immune contributions to the maintenance of central glial activation underlying neuropathic pain. Brain Behav Immun. 25, 1322–1332 10.1016/j.bbi.2011.04.003 [DOI] [PubMed] [Google Scholar]
- 2. Scholz J., and Woolf C. J. (2007) The neuropathic pain triad: neurons, immune cells and glia. Nat. Neurosci. 10, 1361–1368 10.1038/nn1992 [DOI] [PubMed] [Google Scholar]
- 3. Schuh C. D., Brenneis C., Zhang D. D., Angioni C., Schreiber Y., Ferreiros-Bouzas N., Pierre S., Henke M., Linke B., Nüsing R., Scholich K., and Geisslinger G. (2014) Prostacyclin regulates spinal nociceptive processing through cyclic adenosine monophosphate-induced translocation of glutamate receptors. Anesthesiology 120, 447–458 10.1097/ALN.0b013e3182a76f74 [DOI] [PubMed] [Google Scholar]
- 4. Schäfers M., Marziniak M., Sorkin L. S., Yaksh T. L., and Sommer C. (2004) Cyclooxygenase inhibition in nerve-injury- and TNF-induced hyperalgesia in the rat. Exp. Neurol. 185, 160–168 10.1016/j.expneurol.2003.09.015 [DOI] [PubMed] [Google Scholar]
- 5. Ma W., Chabot J. G., Vercauteren F., and Quirion R. (2010) Injured nerve-derived COX2/PGE2 contributes to the maintenance of neuropathic pain in aged rats. Neurobiol Aging 31, 1227–1237 10.1016/j.neurobiolaging.2008.08.002 [DOI] [PubMed] [Google Scholar]
- 6. Muja N., and DeVries G. H. (2004) Prostaglandin E2 and 6-keto-prostaglandin F(1α) production is elevated following traumatic injury to sciatic nerve. Glia 46, 116–129 10.1002/glia.10349 [DOI] [PubMed] [Google Scholar]
- 7. Ma W., and Quirion R. (2005) Up-regulation of interleukin-6 induced by prostaglandin E from invading macrophages following nerve injury: an in vivo and in vitro study. J. Neurochem. 93, 664–673 10.1111/j.1471-4159.2005.03050.x [DOI] [PubMed] [Google Scholar]
- 8. Ma W., and Eisenach J. C. (2002) Morphological and pharmacological evidence for the role of peripheral prostaglandins in the pathogenesis of neuropathic pain. Eur. J. Neurosci. 15, 1037–1047 10.1046/j.1460-9568.2002.01940.x [DOI] [PubMed] [Google Scholar]
- 9. Takahashi M., Kawaguchi M., Shimada K., Konishi N., Furuya H., and Nakashima T. (2004) Cyclooxygenase-2 expression in Schwann cells and macrophages in the sciatic nerve after single spinal nerve injury in rats. Neurosci. Lett. 363, 203–206 10.1016/j.neulet.2004.03.040 [DOI] [PubMed] [Google Scholar]
- 10. Grösch S., Niederberger E., and Geisslinger G. (2017) Investigational drugs targeting the prostaglandin E2 signaling pathway for the treatment of inflammatory pain. Expert Opin. Investig. Drugs 26, 51–61 10.1080/13543784.2017.1260544 [DOI] [PubMed] [Google Scholar]
- 11. Ricciotti E., and FitzGerald G. A. (2011) Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 986–1000 10.1161/ATVBAHA.110.207449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ma W., and Eisenach J. C. (2003) Four PGE2 EP receptors are up-regulated in injured nerve following partial sciatic nerve ligation. Exp. Neurol. 183, 581–592 10.1016/S0014-4886(03)00182-1 [DOI] [PubMed] [Google Scholar]
- 13. Woodhams P. L., MacDonald R. E., Collins S. D., Chessell I. P., and Day N. C. (2007) Localisation and modulation of prostanoid receptors EP1 and EP4 in the rat chronic constriction injury model of neuropathic pain. Eur. J. Pain 11, 605–613 10.1016/j.ejpain.2006.09.005 [DOI] [PubMed] [Google Scholar]
- 14. Cruz Duarte P., St-Jacques B., and Ma W. (2012) Prostaglandin E2 contributes to the synthesis of brain-derived neurotrophic factor in primary sensory neuron in ganglion explant cultures and in a neuropathic pain model. Exp. Neurol. 234, 466–481 10.1016/j.expneurol.2012.01.021 [DOI] [PubMed] [Google Scholar]
- 15. Syriatowicz J. P., Hu D., Walker J. S., and Tracey D. J. (1999) Hyperalgesia due to nerve injury: role of prostaglandins. Neuroscience 94, 587–594 10.1016/S0306-4522(99)00365-6 [DOI] [PubMed] [Google Scholar]
- 16. Decosterd I., and Woolf C. J. (2000) Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 10.1016/S0304-3959(00)00276-1 [DOI] [PubMed] [Google Scholar]
- 17. Shechter R., London A., Varol C., Raposo C., Cusimano M., Yovel G., Rolls A., Mack M., Pluchino S., Martino G., Jung S., and Schwartz M. (2009) Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 6, e1000113 10.1371/journal.pmed.1000113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Streit W. J., Schulte B. A., Balentine D. J., and Spicer S. S. (1985) Histochemical localization of galactose-containing glycoconjugates in sensory neurons and their processes in the central and peripheral nervous system of the rat. J. Histochem. Cytochem. 33, 1042–1052 10.1177/33.10.4045182 [DOI] [PubMed] [Google Scholar]
- 19. Streit W. J., and Kreutzberg G. W. (1987) Lectin binding by resting and reactive microglia. J. Neurocytol. 16, 249–260 10.1007/BF01795308 [DOI] [PubMed] [Google Scholar]
- 20. Nakayama T., Mutsuga N., Yao L., and Tosato G. (2006) Prostaglandin E2 promotes degranulation-independent release of MCP-1 from mast cells. J. Leukoc. Biol. 79, 95–104 10.1189/jlb.0405226 [DOI] [PubMed] [Google Scholar]
- 21. White F. A., Sun J., Waters S. M., Ma C., Ren D., Ripsch M., Steflik J., Cortright D. N., Lamotte R. H., and Miller R. J. (2005) Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc. Natl. Acad. Sci. U.S.A. 102, 14092–14097 10.1073/pnas.0503496102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dudeck A., Dudeck J., Scholten J., Petzold A., Surianarayanan S., Köhler A., Peschke K., Vöhringer D., Waskow C., Krieg T., Müller W., Waisman A., Hartmann K., Gunzer M., and Roers A. (2011) Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 34, 973–984 10.1016/j.immuni.2011.03.028 [DOI] [PubMed] [Google Scholar]
- 23. Van Steenwinckel J., Auvynet C., Sapienza A., Reaux-Le Goazigo A., Combadière C., and Melik Parsadaniantz S. (2015) Stromal cell-derived CCL2 drives neuropathic pain states through myeloid cell infiltration in injured nerve. Brain Behav. Immun. 45, 198–210 10.1016/j.bbi.2014.10.016 [DOI] [PubMed] [Google Scholar]
- 24. Schuh C. D., Pierre S., Weigert A., Weichand B., Altenrath K., Schreiber Y., Ferreiros N., Zhang D. D., Suo J., Treutlein E. M., Henke M., Kunkel H., Grez M., Nüsing R., Brüne B., et al. (2014) Prostacyclin mediates neuropathic pain through interleukin 1beta-expressing resident macrophages. Pain 155, 545–555 10.1016/j.pain.2013.12.006 [DOI] [PubMed] [Google Scholar]
- 25. Ma W., and Quirion R. (2008) Does COX2-dependent PGE2 play a role in neuropathic pain? Neurosci. Lett. 437, 165–169 10.1016/j.neulet.2008.02.072 [DOI] [PubMed] [Google Scholar]
- 26. Natura G., Bär K. J., Eitner A., Boettger M. K., Richter F., Hensellek S., Ebersberger A., Leuchtweis J., Maruyama T., Hofmann G. O., Halbhuber K. J., and Schaible H. G. (2013) Neuronal prostaglandin E2 receptor subtype EP3 mediates antinociception during inflammation. Proc. Natl. Acad. Sci. U.S.A. 110, 13648–13653 10.1073/pnas.1300820110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abbadie C., Lindia J. A., Cumiskey A. M., Peterson L. B., Mudgett J. S., Bayne E. K., DeMartino J. A., MacIntyre D. E., and Forrest M. J. (2003) Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc. Natl. Acad. Sci. U.S.A. 100, 7947–7952 10.1073/pnas.1331358100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stamatovic S. M., Shakui P., Keep R. F., Moore B. B., Kunkel S. L., Van Rooijen N., and Andjelkovic A. V. (2005) Monocyte chemoattractant protein-1 regulation of blood–brain barrier permeability. J. Cereb. Blood Flow Metab. 25, 593–606 10.1038/sj.jcbfm.9600055 [DOI] [PubMed] [Google Scholar]
- 29. Gao Y. J., Zhang L., Samad O. A., Suter M. R., Yasuhiko K., Xu Z. Z., Park J. Y., Lind A. L., Ma Q., and Ji R. R. (2009) JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J. Neurosci. 29, 4096–4108 10.1523/JNEUROSCI.3623-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jung H., Bhangoo S., Banisadr G., Freitag C., Ren D., White F. A., and Miller R. J. (2009) Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J. Neurosci. 29, 8051–8062 10.1523/JNEUROSCI.0485-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gosselin R. D., Varela C., Banisadr G., Mechighel P., Rostene W., Kitabgi P., and Melik-Parsadaniantz S. (2005) Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J. Neurochem. 95, 1023–1034 10.1111/j.1471-4159.2005.03431.x [DOI] [PubMed] [Google Scholar]
- 32. Thacker M. A., Clark A. K., Bishop T., Grist J., Yip P. K., Moon L. D., Thompson S. W., Marchand F., and McMahon S. B. (2009) CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur. J. Pain 13, 263–272 10.1016/j.ejpain.2008.04.017 [DOI] [PubMed] [Google Scholar]
- 33. Takeuchi K., Ukawa H., Furukawa O., Kawauchi S., Araki H., Sugimoto Y., Ishikawa A., Ushikubi F., and Narumiya S. (1999) Prostaglandin E receptor subtypes involved in stimulation of gastroduodenal bicarbonate secretion in rats and mice. J. Physiol. Pharmacol. 50, 155–167 [PubMed] [Google Scholar]
- 34. Hizaki H., Segi E., Sugimoto Y., Hirose M., Saji T., Ushikubi F., Matsuoka T., Noda Y., Tanaka T., Yoshida N., Narumiya S., and Ichikawa A. (1999) Abortive expansion of the cumulus and impaired fertility in mice lacking the prostaglandin E receptor subtype EP2. Proc. Natl. Acad. Sci. U.S.A. 96, 10501–10506 10.1073/pnas.96.18.10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Segi E., Sugimoto Y., Yamasaki A., Aze Y., Oida H., Nishimura T., Murata T., Matsuoka T., Ushikubi F., Hirose M., Tanaka T., Yoshida N., Narumiya S., and Ichikawa A. (1998) Patent ductus arteriosus and neonatal death in prostaglandin receptor EP4-deficient mice. Biochem. Biophys. Res. Commun. 246, 7–12 10.1006/bbrc.1998.8461 [DOI] [PubMed] [Google Scholar]
- 36. Linke B., Schreiber Y., Zhang D. D., Pierre S., Coste O., Henke M., Suo J., Fuchs J., Angioni C., Ferreiros-Bouzas N., Geisslinger G., and Scholich K. (2012) Analysis of sphingolipid and prostaglandin synthesis during zymosan-induced inflammation. Prostaglandins Other Lipid Mediat. 99, 15–23 10.1016/j.prostaglandins.2012.06.002 [DOI] [PubMed] [Google Scholar]
- 37. Linke B., Pierre S., Coste O., Angioni C., Becker W., Maier T. J., Steinhilber D., Wittpoth C., Geisslinger G., and Scholich K. (2009) Toponomics analysis of drug-induced changes in arachidonic acid-dependent signaling pathways during spinal nociceptive processing. J. Proteome Res. 8, 4851–4859 10.1021/pr900106v [DOI] [PubMed] [Google Scholar]
- 38. Pierre S., Maeurer C., Coste O., Becker W., Schmidtko A., Holland S., Wittpoth C., Geisslinger G., and Scholich K. (2008) Toponomics analysis of functional interactions of the ubiquitin ligase PAM (protein associated with Myc) during spinal nociceptive processing. Mol. Cell. Proteomics 7, 2475–2485 10.1074/mcp.M800201-MCP200 [DOI] [PubMed] [Google Scholar]
- 39. Pierre S., Linke B., Suo J., Tarighi N., Del Turco D., Thomas D., Ferreiros N., Stegner D., Frölich S., Sisignano M., Meyer Dos Santos S., deBruin N., Nüsing R. M., Deller T., Nieswandt B., et al. (2017) GPVI and thromboxane receptor on platelets promote proinflammatory macrophage phenotypes during cutaneous inflammation. J. Invest. Dermatol. 137, 686–695 10.1016/j.jid.2016.09.036 [DOI] [PubMed] [Google Scholar]
- 40. Ley S., Weigert A., Weichand B., Henke N., Mille-Baker B., Janssen R. A., and Brüne B. (2013) The role of TRKA signaling in IL-10 production by apoptotic tumor cell-activated macrophages. Oncogene 32, 631–640 [DOI] [PubMed] [Google Scholar]
- 41. Degousee N., Simpson J., Fazel S., Scholich K., Angoulvant D., Angioni C., Schmidt H., Korotkova M., Stefanski E., Wang X. H., Lindsay T. F., Ofek E., Pierre S., Butany J., Jakobsson P. J., et al. (2012) Lack of microsomal prostaglandin E2 synthase-1 in bone marrow–derived myeloid cells impairs left ventricular function and increases mortality after acute myocardial infarction. Circulation 125, 2904–2913 10.1161/CIRCULATIONAHA.112.099754 [DOI] [PubMed] [Google Scholar]
- 42. Murata T., Ushikubi F., Matsuoka T., Hirata M., Yamasaki A., Sugimoto Y., Ichikawa A., Aze Y., Tanaka T., Yoshida N., Ueno A., Oh-ishi S., and Narumiya S. (1997) Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature 388, 678–682 10.1038/41780 [DOI] [PubMed] [Google Scholar]