

Abstract
Major histocompatibility complex-I–β2m dimers (MHC-I) bind peptides derived from intracellular proteins, enabling the immune system to distinguish between normal cells and those expressing pathogen-derived or mutant proteins. The peptides bind to MHC-I in the endoplasmic reticulum (ER), and this binding is facilitated by the peptide loading complex (PLC), which contains calreticulin (CRT). CRT associates with MHC-I via a conserved glycan present on MHC-I and recruits it to the PLC for peptide binding. Somatic frameshift mutations in CRT (CRT-FS) drive the proliferation of a subset of myeloproliferative neoplasms, which are chronic blood tumors. All CRT-FS proteins have a C-terminal sequence lacking the normal ER-retention signal and possessing a net negative charge rather than the normal positive charge. We characterized the effect of CRT-FS on antigen presentation by MHC-I in human cells. Our results indicate that CRT-FS cannot mediate CRT's peptide loading function in the PLC. Cells lacking CRT exhibited reduced surface MHC-I levels, consistent with reduced binding of high-affinity peptides, and this was not reversed by CRT-FS expression. CRT-FS was secreted and not detectably associated with the PLC, leading to poor MHC-I recruitment, although CRT-FS could still associate with MHC-I in a glycan-dependent manner. The addition of an ER-retention sequence to CRT-FS restored its association with the PLC but did not rescue MHC-I recruitment or its surface expression, indicating that the CRT-FS mutants functionally compromise the PLC. MHC-I down-regulation permits tumor cells to evade immune surveillance, and these findings may therefore be relevant for designing effective immunotherapies for managing myeloproliferative neoplasms.
Keywords: major histocompatibility complex (MHC), antigen presentation, antigen processing, protein export, tumor immunology/immunotherapy, calreticulin, myeloproliferative neoplasms, peptide-loading complex, protein secretion
Introduction
Nucleated cells of most vertebrates express major histocompatibility complex (MHC)3 class I molecules on their surface. Each MHC-I molecule is a heterodimer composed of a transmembrane heavy chain and β2-microglobulin (β2m) that is associated with a short 8–11-amino acid peptide. These peptides are derived from cellular proteins and are presented on the surface of cells in a process called antigen presentation (1), which is integral to immune surveillance by cytotoxic CD8+ T lymphocytes. Cells presenting peptides derived from “self” proteins are generally ignored, whereas cells presenting peptides derived from foreign proteins (e.g. virally infected cells) can be recognized and destroyed. In the case of cancer cells, presentation of peptides derived from tumor-associated antigens or tumor-specific antigens by MHC-I may occur, facilitating their identification and killing by CD8+ T cells (1, 2).
In an attempt to evade immune surveillance, cancer cells employ various mechanisms to down-regulate the expression of MHC-I molecules or other proteins directly or indirectly involved in antigen processing and presentation (2, 3). Down-regulation is more common than total elimination of MHC-I expression because the latter renders the cell susceptible to the action of natural killer (NK) cells; reduction of surface MHC-I may allow evasion of NK-mediated killing, and reduced antigen presentation can prevent efficient detection by CD8+ T cells (3). In addition, β2m associated with MHC-I on the cell surface may inhibit phagocytosis of cells by macrophages even if they express other pro-phagocytic markers (4). The success of various immune-based therapies, such as DNA vaccines, checkpoint inhibitor antibody therapy, or dendritic cell therapy (5, 6), is dependent on effective antigen presentation by tumor cells. It is therefore important to understand the mechanisms by which antigen presentation is altered in tumor cells to aid their effective targeting and elimination.
Assembly and peptide loading of MHC-I molecules occurs in the ER, facilitated by the peptide loading complex (PLC) (1, 7). The core components of the PLC are ERp57, tapasin, and the dimeric transporter associated with antigen processing (TAP). ERp57 is a thiol-reductase that functions together with calreticulin (CRT), a lectin chaperone, in the quality control folding cycle used for many glycoproteins (8). Two tapasin molecules associate directly with TAP, and both are disulfide-linked to ERp57 (7, 9). Although CRT is a constituent of the PLC, its association depends on the presence of MHC-I molecules, which bind peptides that are derived primarily from cytosolic proteolysis and are imported into the ER by TAP. Tapasin facilitates peptide exchange, leading to the accumulation of MHC-I molecules associated with high-affinity peptides, and its mechanism of action has recently been illuminated by the structural analysis of its homolog TAPBPR in association with MHC-I (10, 11). TAPBPR is not a PLC component.
Cancer cells have been shown to down-regulate the expression of TAP, tapasin, CRT, ERp57, MHC heavy chain, or β2m by various mechanisms that ultimately lead to the loss or reduced expression of MHC-I on the cell surface (2, 3). This study is focused on the effects of disease-associated CRT mutations on antigen presentation by MHC-I. CRT is a multidomain protein with an N-terminal lectin domain, a central, proline-rich P-domain that interacts with ERp57, and an acidic C-terminal domain (CTD) that binds calcium ions and ends with an ER-retention sequence (KDEL) (Fig. 1A). The MHC-I heavy chain bears an N-linked glycan at a site that is conserved across species and, when monoglucosylated, is responsible for its association with CRT (12). This glycan is also a target of the ER-resident enzyme UDP-glucosyltransferase 1 (UGT1). UGT1 detects MHC-I loaded with a suboptimal, low-affinity peptide and reglucosylates the glycan, causing the MHC-I to reinteract with the PLC via CRT (13). This facilitates further iterations of peptide loading until an optimal peptide is loaded.
Figure 1.

Generation and characterization of CRT-null–, CRT-WT–, and CRT-FS–expressing cell lines. A, organization of the calreticulin gene and protein. Nine exons encode CRT, which is composed of an N-terminal lectin domain, a central, proline-rich P-domain, and an acidic CTD that contains an ER retention sequence, KDEL. Insertion and deletion mutants in exon 9 of the calreticulin gene lead to a +1-bp frameshift, resulting in a unique amino acid sequence of the C-terminal tail. B, CRT mRNA levels in CRT-null cells and CRT-null cells transduced with the indicated CRT constructs were assessed by qPCR via normalization to the housekeeping gene β-actin. CRT mRNA expression in HEK293T cells was used to calculate -fold change. C, Western blot analysis of expression of CRT-WT and CRT-FS in cell lysates using antibodies that recognize all forms of CRT (α-CRT) or only mutant CRT (α-CRT-FS). D, secretion of CRT was assessed by incubating cells expressing either CRT-WT or CRT-FS with 10 μg/ml BFA for 8 h, followed by Western blot analysis of CRT expression in cell lysates and culture supernatants. E, cells expressing CRT-FSDEL or CRT-FSINS were labeled with [35S]Met for 15 min followed by immunoprecipitation of CRT from the lysates or culture supernatant at the indicated time points of chase, and samples were visualized after separation on nonreducing SDS-polyacrylamide gels by autoradiography. F, interaction of CRT and MHC-I with the PLC in cells expressing CRT-WT or CRT-FS was carried out by immunoprecipitation of tapasin from cell lysates and Western blot analysis of co-immunoprecipitated proteins. #, a nonspecific band in the right panel at 49 kDa. G, MHC-I expressed on the surface of cells expressing no CRT or CRT-WT or CRT-FS was estimated by staining intact cells with the W6/32 antibody followed by FACS. Histograms from G are summarized in bar format. Qualitative data shown are a representation of at least three independent experiments. Quantitative data shown are mean ± S.D. (error bars) of three experiments. Statistical significance was evaluated using the unpaired Student's t test; *, p < 0.05; **, p < 0.01; ***, p < 0.005; ns, not significant.
Earlier reports have shown that CRT, in combination with other PLC components, is often down-regulated in colorectal (14) and bladder cancers (15), accompanied by a reduction in MHC-I expression. In 2013, a somatic, frameshift mutant of CRT (CRT-FS) was identified in myeloproliferative neoplasms (MPNs), a class of chronic blood diseases (16, 17). MPNs that are Philadelphia chromosome–negative and myeloproliferative leukemia virus oncogene–negative can be further categorized into three classes: polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). An activating mutation in Janus kinase 2 (JAK2V617F) is seen in 95% of PV patients, whereas only 50–60% of patients suffering from ET or PMF have this mutation. The two studies mentioned above discovered that the majority of ET or PMF patients with nonmutated JAK2 carry insertions or deletions in exon 9 of the CALR gene, all of which result in a 1-bp frameshift mutation that generates a novel C-terminal tail of CRT, of which 36 amino acids are common between all types of CALR mutations (16, 17). The amino acids in the mutant sequence are basic in nature compared with the acidic amino acids present in CRT-WT, and the ER-retention sequence, KDEL, present in WT is lost (Fig. 1A). The majority of patients are heterozygous for this mutation, but homozygous mutations have been reported in small cohorts (17, 18).
The C-terminal tail of CRT-FS mutants confers unique characteristics that promote tumorigenesis. The lack of a KDEL sequence causes mislocalization of CRT-FS, and the change in overall charge influences interactions with glycoprotein substrates. The mutant CTD promotes a glycan-mediated interaction of CRT-FS with the thrombopoietin receptor, causing ligand-independent activation of the JAK-STAT signaling pathway that drives neoplastic transformation (19–21). However, in at least one case, a loss-of-function phenotype is observed for CRT-FS with regard to its lectin chaperone activity. The glycoprotein substrate myeloperoxidase (MPO) requires CRT to fold properly, but, in the presence of CRT-FS, MPO is misfolded, leading to proteasomal degradation and thus a reduction in cellular MPO (18). CRT also mediates Ca2+ release from the ER as its C-terminal acidic tail serves as a low-affinity Ca2+ binding site. CRT-FS deletion mutants (CRT-FSDEL) have fewer acidic residues than CRT-FS insertion mutants (CRT-FSINS). Thus, cells expressing CRT-FSDEL exhibit abnormal cytosolic calcium signaling, which corresponds to a higher risk of myelofibrotic transformation (22). Two transgenic mouse models have been generated to study this mutation. In one, CRT-FSDEL was ubiquitously expressed in mice, which developed essential thrombocythemia (23). In the second, CRT-FSDEL conditional knockin mice were generated that recapitulated the thrombocytosis and myelofibrosis phenotype (24).
CRT has been shown to affect MHC-I function in mouse embryonic fibroblasts, where ablating CRT expression results in a reduction in endogenous antigen presentation and a decrease in surface MHC-I compared with cells expressing CRT (25–27). Here we demonstrate that ablation of CRT expression in human cells also causes inefficient peptide loading, leading to reduced levels of surface MHC-I and defective antigen presentation. The phenotype is rescued by the expression of CRT-WT, but not the CRT-FS mutants. In cells that co-express both CRT-WT and CRT-FSDEL, a small but significant reduction in surface MHC-I levels was observed.
Results
CRT-FS mutants are secreted
Expression of CRT in HEK293T cells was successfully ablated using the CRISPR/Cas9 approach, as confirmed by RT-PCR and Western blot analysis using a mouse anti-CRT antibody that recognizes both the WT and CRT-FS mutant forms (α-CRT) (Fig. 1, B and C). CRT-WT, CRT-FSDEL, or CRT-FSINS were re-expressed in the CRT-null cell line. RT-PCR analysis of these cells lines showed that CRT-WT and both CRT-FS mRNAs were present at similar levels (Fig. 1B). Western blot analysis indicated that whereas CRT-WT was present at levels similar to that of normal HEK293T cells, no CRT-FSDEL and CRT-FSINS was detected by α-CRT (Fig. 1C, top). However, the mutants could be detected using α-CRT-FS, a mutant-specific CRT rabbit antibody generated against the novel C-terminal peptide, implying that the mutants are present but poorly expressed within the cells (Fig. 1C, middle). We always observe that the level of CRT-FSDEL is lower in the cells than CRT-FSINS. Because CRT-FSDEL and CRT-FSINS lack the ER-retention KDEL sequence present on CRT-WT, a likely explanation is that they are secreted. The culture supernatants of cells expressing CRT-WT or the mutants were therefore examined by Western blotting. Both forms of CRT-FS were readily detected in the culture supernatant, and this was lost when the cells were incubated with brefeldin A (BFA) (Fig. 1D, left). In BFA-treated cells, CRT-FSDEL and CRT-FSINS were detected in the cell lysate (Fig. 1D, right), consistent with inhibition of their secretion by the classical ER-Golgi secretory pathway. We detected a small amount of CRT-WT in the culture supernatant that was not sensitive to BFA treatment (Fig. 1D, left). Lack of signal from the ER-resident chaperone GRP94 in the Western blot analysis of the culture supernatants indicated that there was no contamination by intracellular proteins due to cell death or lysis. We speculate that some CRT-WT is released from the cell via a nonclassical secretory pathway, as has been previously suggested (28, 29). Pulse-chase analysis was carried out to assess the kinetics of CRT-FSDEL and CRT-FSINS secretion. Both were detectable in the supernatant as early as 30 min after chase and half-maximal secretion occurred at 90 min. At 180 min, almost all of the intracellular pool of metabolically labeled CRT had disappeared (Fig. 1E).
CRT-FS mutants are not incorporated into the PLC
CRT-FSDEL and CRT-FSINS are largely secreted, lowering their effective concentration in the ER. To determine whether sufficient remained to detect association with the PLC, tapasin was immunoprecipitated from digitonin extracts of the cells followed by Western blotting to detect CRT associated with the PLC. Fig. 1F shows that neither CRT-FS mutant is detectably associated with tapasin. In addition, compared with cells expressing CRT-WT, reduced levels of MHC-I were associated with the PLC in cells expressing the CRT-FS mutants, suggesting that peptide loading in these cells may be affected, which in turn could impact surface MHC-I levels. In cells with neither CRT-WT nor the mutants, no MHC-I was detectably associated with the PLC.
Decreased surface MHC-I by cells expressing CRT-FS mutants
Flow cytometric analysis of surface MHC-I was carried out on HEK293T cells lacking CRT expression or expressing CRT-WT or the CRT-FS mutants. In the absence of CRT expression, a 50% decrease in surface MHC-I levels was seen compared with WT HEK293T cells (Fig. 1G). This was rescued by the expression of CRT-WT, but not CRT-FSDEL or CRT-FSINS. Because steady-state levels of the mutants within the cells are low, we wondered whether retaining them in the ER would rescue cell surface MHC-I expression. To test this, we generated cell lines expressing the mutants with an added C-terminal KDEL sequence (CRT-FSDEL+KDEL and CRT-FSINS+KDEL). As seen in Fig. 2A, the ER-retained CRT-FS mutants were detected in the cells. In a complementary experiment, CRT-WT lacking the KDEL sequence (CRT-WT−KDEL) was expressed in the CRT-null cells. CRT-WT−KDEL was poorly detected in cell lysates (Fig. 2B, right) but was readily detected in the supernatant (Fig. 2B, left). Upon BFA treatment, CRT-WT−KDEL was not secreted, and there was a concomitant increase in intracellular accumulation (Fig. 2B). Very few, if any, of the CRT-FS mutants with the added KDEL sequence were detected in the culture supernatants (Fig. 2B, left), and no significant increase in intracellular levels was observed in the presence of BFA (Fig. 2B, right). In the cell lysates, two bands of CRT-FS are sometimes detected by α-CRT, but only the upper band is detected by α-CRT-FS, suggesting that a low level of C-terminal clipping may result in two bands.
Figure 2.

The effect of retaining CRT-FS in the ER on peptide loading and surface MHC-I levels. A, Western blot analysis of the expression of CRT in cell lysates. B, Western blot analysis of the expression of CRT-WT and mutants in cell lysates and culture supernatant from control cells and cells treated with 10 μg/ml BFA for 8 h. C, MHC-I expressed on the surface of cells expressing no CRT or all CRT variants was estimated by staining intact cells with the W6/32 antibody followed by FACS. Histograms from C are summarized in bar format. D, the interaction of all CRT variants with the PLC was assessed by immunoprecipitation (IP) of tapasin, followed by Western blot analysis of associated proteins. E, purified GST and GST-fusion CRT proteins from E. coli used in the pulldown assay (left). Lysates from CRT-null cells were incubated with immobilized GST, GST-CRT-WT, GST-CRTY92A, and GST-CRT-FS. Associated proteins were analyzed by Western blotting. Qualitative data shown are a representation of at least three independent experiments. Quantitative data shown are mean ± S.D. (error bars) of at least three experiments. Statistical significance was evaluated using the unpaired Student's t test; *, p < 0.05; **, p < 0.01; ***, p < 0.005; ns, not significant.
Retaining the CRT-FS mutants in the ER failed to reverse the reduction in surface MHC-I (Fig. 2C). However, whereas the CRT-FS mutants were ineffective with or without an ER retention signal, CRT-WT−KDEL significantly, albeit partially, restored MHC-I surface expression (Fig. 2C). This suggested that there might be an inherent structural problem in the CRT-FS mutants that disrupts CRT function. To address this, we asked whether CRT-FSDEL+KDEL, CRT-FSINS+KDEL, or CRT-WT−KDEL could be incorporated into the PLC. Tapasin was immunoprecipitated from digitonin extracts of the cells, and co-immunoprecipitated components were analyzed by Western blotting (Fig. 2D). As before, CRT-WT was detected in association with the PLC, but the CRT-FS mutants lacking the KDEL sequence were not. Unexpectedly, the CRT-FS mutants plus the KDEL sequence were incorporated into the PLC, whereas CRT-WT−KDEL could not be detected. This suggests that increasing the local concentration of the CRT-FS mutants in the ER facilitated their interaction with the PLC, but the interaction was not functional because there was no increase in MHC-I associated with the PLC (Fig. 2D), and surface levels of MHC-I were not restored (Fig. 2C).
The glycan-dependent interaction between CRT and the MHC-I heavy chain is required for the recruitment of MHC-I to the PLC by CRT. Previous studies with other substrates have shown that these interactions may be altered for CRT-FS mutants (18–21). This suggests that the normal function of the N-terminal lectin domain of CRT-FS might be affected. To test this, we used a previously described pulldown assay that detects the interaction of CRT with MHC-I molecules bearing a monoglucosylated N-linked glycan (12). Purified immobilized GST-tagged CRT-WT, CRT-FSDEL, CRT-FSINS, and CRTY92A (a CRT mutant deficient in glycan binding (13)) (Fig. 2E, left) were incubated with detergent lysates of CRT-null cells, and MHC-I associated with the CRT variants was detected by Western blotting (Fig. 2E, right). We found that CRT-WT and the frameshift mutants interacted with MHC-I, whereas, as expected, CRTY92A did not. The frameshift mutants exhibited a reduced interaction with ERp57. In the case of CRT-WT−KDEL, although the loss of ER retention reduces the availability of CRT for PLC interaction, it retains its fundamental capacity to facilitate peptide loading in the ER. It appears that transient, labile interactions of CRT-WT−KDEL with tapasin-ERp57 dimers within the PLC lead to the partial restoration of MHC-I surface expression, but in the case of CRT-FS mutants, these interactions are ineffectual.
MHC-I delivery to the cell surface is reduced in cells expressing the CRT-FS mutants
When the integrity of the PLC is lost, for example in cells deficient in tapasin, assembly of stable MHC-I–peptide complexes is impaired, leading to reduced expression of MHC-I on the cell surface and reduced antigen presentation to CD8+ T cells (30–32). In murine cells, previous data suggest that MHC-I is exported at a faster rate in cells that lack CRT but that it is removed by endocytosis (26, 27). In our studies in human cells, the lack of MHC-I association with the PLC and lower surface MHC-I levels observed in HEK293T cells lacking CRT or expressing the CRT-FS mutants suggested that the critical role WT CRT plays in the assembly process is not replicated by the frameshift mutants. To further this analysis, we measured the rate of surface arrival of newly assembled MHC-I molecules. The cell lines were briefly exposed to a pH of 3.5, commonly referred to as “acid stripping,” which irreversibly denatures surface human MHC-I complexes such that they are no longer detectable by the mAb W6/32 (33). Reappearance of intact MHC-I from within the cell was monitored by flow cytometry. We also carried out this assay in the presence of an inhibitor of endocytosis, dynasore, to distinguish between MHC-I arriving at the surface versus MHC-I left behind after endocytosis at each time point. Dynasore is a cell-permeable inhibitor of dynamin, which is essential for clathrin-mediated endocytosis (34), a mechanism used for MHC-I removal from the cell surface (35).
Fig. 3A shows that only ∼10% of W6/32-reactive MHC-I remained on the cell surface after acid treatment. In the cells expressing CRT-WT, reconstitution to ∼50% of the level before stripping was apparent after 6 h. Reconstitution of surface MHC-I in cells lacking CRT reached only 30% of the “unstripped” level, and this was no better in the cells expressing the CRT-FS mutants with or without the KDEL retention signal. Note that this is only ∼15% of the level of surface MHC-I reached on cells reconstituted with CRT-WT. The reconstitution of surface MHC-I expression in the absence or presence of dynasore after acid stripping was the same, indicating that the levels of MHC-I measured are of newly synthesized chains that have arrived at the surface and not survivors of dynamin-dependent endocytosis. As a positive control, we showed that ligand-mediated endocytosis of epidermal growth factor receptor (EGFR) from the surface of HEK293T cells was blocked by dynasore, as expected (Fig. 3A). These findings are consistent with the hypothesis that the mutants cannot perform a critical peptide loading function. Notably, reconstitution of surface MHC-I was also impaired in the cells expressing CRT-WT−KDEL, consistent with the reduction in steady-state MHC-I surface expression in these cells and with the hypothesis that loss of ER retention results in a reduction in local concentration and impairment of PLC function (Fig. 2, C and D).
Figure 3.

Effect of CRT-FS on antigen presentation. A, the delivery of MHC-I to the surface of cells expressing various constructs of CRT was measured by acid-denaturing surface MHC-I at pH 3. 5, followed by neutralization and incubation at 37 °C in the absence (left) or presence (middle) of dynasore. The appearance of assembled, surface MHC-I over time was monitored by FACS using the W6/32 antibody. The activity of dynasore was verified by estimating surface EGFR on cells treated with EGF in the presence or absence of dynasore (right) by FACS. B, the thermostability of surface MHC-I in the various cell lines was assessed by incubating intact cells for 10 min at room temperature in PBS + 0.02% sodium azide, followed by incubation at the indicated temperatures for 10 min. The heat-resistant MHC-I remaining on the surface of cells was estimated by FACS using the W6/32 antibody. C, free heavy chain on the surface of cells at steady state was estimated by staining intact cells with the HC10 antibody followed by FACS analysis. D, free heavy chain on the surface of cells was generated by acid-denaturing surface MHC-I at pH 3.5 followed by neutralization. Levels of free heavy chain were estimated by FACS using the HC10 antibody. E, the stability of MHC-I complexes was also evaluated by a BFA-decay assay. The various cell lines were cultured overnight at 27 °C, treated with 10 μg/ml BFA, and incubated at 37 °C for the indicated periods of time. The decay of surface MHC-I over time was monitored by FACS using the W6/32 antibody. F, LMP1 mRNA levels in the seven cell lines were assessed by qPCR via normalization to the housekeeping gene β-actin. LMP1 mRNA expression in LMP1-expressing WT HEK293T cells was used to calculate -fold change. G, presentation of an Epstein–Barr virus–derived, high-affinity peptide (YLLEMLWRL) was estimated in cells expressing various CRT constructs and the LMP1 protein by staining intact cells with the MHC-I–YLLEMLWRL–specific antibody, followed by FACS. H, surface expression of the MHC-I allele that presents the peptide YLLEMLWRL, HLA-A2, was estimated by staining intact cells with the HLA-A2–specific antibody BB7.2 followed by FACS. Total surface MHC-I was determined by staining intact cells with the W6/32 antibody followed by FACS. Data shown are mean ± S.D. (error bars) of at least three experiments. Statistical significance was evaluated using the unpaired Student's t test: *, p < 0.05; **, p < 0.01; ***, p < 0.005; ns, not significant. Statistical significance in A, B, and E is from comparing data from cells expressing CRT-WT and the other six cell lines.
Surface MHC-I in cells expressing CRT-FS mutants is associated with suboptimal peptides
MHC-I is detectable on the surface of cells expressing no CRT or the CRT-FS mutants with or without the KDEL sequence, but at less than 50% of the level on cells expressing CRT-WT. Cells expressing CRT-WT−KDEL do only slightly better (Fig. 2C). Impaired peptide loading can result in reduced MHC-I surface expression if the associated peptides are of reduced affinity; MHC-I complexes associated with suboptimal peptides are generally less stable than those associated with high-affinity peptide. We addressed this issue by determining the relative thermal stabilities of the MHC-I molecules at the surface of the various cell lines (36). Because this assay measures a purely physical parameter, we incubated the cells with sodium azide, a potent mitochondrial inhibitor that impedes various ATP-dependent cellular processes, including endocytosis (37). Cells were exposed to a range of temperatures for 10 min, followed by flow cytometric analysis of surviving surface W6/32-reactive MHC-I. Fig. 3B shows that the MHC-I–peptide complexes present on the surface of cells expressing CRT-WT are more resistant to thermal denaturation than those on cells expressing CRT-FS, CRT-FS+KDEL, or CRT-WT−KDEL. These data indicate that MHC-I molecules in cells expressing the various CRT mutants, including CRT-WT−KDEL, are associated with suboptimal peptides.
From the above experiments, we conclude that the rate of surface arrival of MHC-I in cells that do not express CRT-WT is reduced and that at steady state, they are associated with suboptimal peptides. To ask whether low-affinity peptide–MHC-I complexes dissociate on the surface of the cells, we looked for liberated free MHC-I heavy chains using the mAb HC10 (38). As seen in Fig. 3C, the level of free heavy chain on the surface of cells was very low and was the same across all the cell lines. It seems likely that HC10-reactive heavy chains liberated by dissociation of β2m and peptide are cleared too rapidly for detection at steady state. However, free heavy chains were detectable immediately after acid stripping, and the pattern of expression across the cell lines was similar to that of W6/32-reactive MHC-I before stripping (Fig. 3D).
Cells in which peptide loading is affected assemble a high percentage of unstable MHC-I–peptide complexes that do not reach the cell surface (39, 40). These complexes can be stabilized by incubation at 27 °C, leading to surface accumulation of MHC-I. When the temperature is raised to 37 °C, the unstable MHC-I complexes decay at a faster rate than stable MHC-I complexes. Fig. 3E shows the rate of decay of W6/32-reactive MHC-I expressed in the various cell lines after overnight incubation at 27 °C and subsequent incubation at 37 °C, performed in the presence of BFA to prevent the arrival of newly synthesized molecules at the cell surface. In CRT-null cells, cells expressing the CRT-FS mutants with or without the ER retention signal, or cells expressing CRT-WT−KDEL, MHC-I molecules are cleared from the cell surface much more rapidly than in cells expressing CRT-WT, suggesting that a larger fraction of their MHC-I molecules are associated with lower-affinity peptides. All of these data are consistent with an impaired ability of the CRT mutants to mediate MHC-I peptide loading.
Presentation of a defined antigenic peptide is decreased in cells expressing the CRT-FS mutants
The data so far argue that the CRT-FS mutants lack the quality control function of WT CRT required for normal assembly of high-affinity MHC-I–peptide complexes. We set out to determine whether the mutants affected assembly of MHC-I complexes containing a defined antigenic epitope. Rather than using a CD8+ T cell for this analysis, we used a mAb specific for an MHC-I–peptide complex, which provides much better quantitation. Latent membrane protein 1 (LMP1) is an Epstein–Barr virus protein containing an epitope that is efficiently presented by HLA-A2 (41). A TCR-like mAb generated against HLA-A2 associated with this peptide (YLLEMLWRL) was used to measure surface expression of HLA-A2–YLLEMLWRL complexes (41). All seven cell lines were transduced with expression constructs expressing a vector control or LMP1, and presentation of the YLLEMLWRL peptide was analyzed by flow cytometry. Fig. 3F shows that LMP1 mRNA was expressed at similar levels in the cell lines. In LMP1-expressing cells, HLA-A2–YLLEMLWRL complexes were detectable at higher levels when CRT-WT was expressed, whereas in cells lacking CRT or expressing CRT-FS, CRT-FS+KDEL, or CRT-WT−KDEL, presentation of YLLEMLWRL was considerably lower (Fig. 3G). The presentation of the LMP1-derived peptide mirrors the expression of HLA-A2 in the cell lines, which in turn mimics the pattern of total MHC-I expression (Fig. 3H).
MHC-I assembly in cells co-expressing CRT-WT and CRT-FS mutants
The experiments described above examined the effect of CRT-FS on MHC class I expression and antigen presentation in cell lines lacking endogenous CRT expression, which is seen in a small group of patients (17, 18). However, a majority of the patients carry a WT CRT gene in addition to a CRT-FS mutant gene. To examine the phenotypic consequences of this, WT HEK293T cells expressing CRT-FS were generated and characterized, along with HEK293T cells overexpressing CRT-WT. As expected, CRT-FS expression was low in cell lysates, except in the presence of BFA (Fig. 4A, left). The mutants were secreted and easily detected in the culture supernatant (Fig. 4A, right) in the absence of BFA. Only in HEK293T cells expressing CRT-FSDEL along with endogenous CRT was a significant, although small, reduction in surface MHC-I molecules observed (Fig. 4B). These MHC-I molecules were associated with suboptimal peptides, as determined by the BFA-decay assay (Fig. 4C).
Figure 4.

Effect of co-expression of CRT-FS and CRT-WT on MHC-I function. A, Western blot analysis of the expression of CRT-WT and mutants in lysates and culture supernatant from HEK293T cells treated or not with 10 μg/ml BFA for 8 h. B, the effect of co-expressing CRT-FS on surface MHC-I levels was measured by staining intact cells with W6/32 antibody followed by FACS. Histograms from B are summarized in bar format. C, the stability of MHC-I complexes was also evaluated by a BFA-decay assay. The various cell lines were cultured overnight at 27 °C, treated with 10 μg/ml BFA, and incubated at 37 °C for the indicated periods of time. The reduction in surface MHC-I over time was monitored by staining intact cells with the W6/32 antibody followed by FACS. Qualitative data shown are a representation of three independent experiments. Quantitative data shown are mean ± S.D. (error bars) of three experiments. Statistical significance was evaluated using the unpaired Student's t test: *, <p 0.05; **, p < 0.01; ***, p < 0.005; ns, not significant. Statistical significance in C is from comparing data from cells expressing CRT-FSDEL and cells expressing VC.
Discussion
The classical pathway of peptide loading and antigen presentation by MHC-I is highly conserved. The component proteins of the PLC are key players in the process, and together they ensure that optimal peptides, generally of high affinity, are loaded onto MHC-I in the ER for subsequent presentation at the cell surface. In agreement with previous results obtained with mouse cells, we find that CRT is important for normal peptide binding by MHC-I molecules in human cells (25–27). This is manifested in an ∼50% reduction in surface MHC-I expression when CRT is deleted. In both mouse and human cells, surface expression was restored when full-length CRT was expressed (25). In mouse cells, export of MHC-I from the ER is slower in cells that lack CRT expression, although this may be allele-dependent; the export of H-2 Kb, but not H-2 Db, molecules is reduced in the absence of CRT (27). In this case, MHC-I complexes containing suboptimal peptides may escape the MHC-I quality-control mechanisms in the ER, traffic rapidly to the surface, and be removed by endocytosis (27). Interestingly, expression of an ER-retained form of CRT lacking the CTD in CRT-null mouse cells did not rescue surface MHC-I levels, indicating the importance of the CTD in facilitating MHC-I peptide binding (25).
The C-terminal domain of human CRT is ∼113 amino acids, but the CRT-FS mutants do not lack the entire CTD. Even the CRT-FSDEL used in our study lacks only 17 amino acids of the 113-amino acid-long CTD. However, the sequence is significantly altered (Fig. 1A). To determine whether the CRT-FS mutants are functional, we asked whether expressing them in the CRT-negative cells would restore surface MHC-I levels. A cohort of MPN patients that express CRT-FS but lack CRT in their proliferating myeloid cells has been described (17, 18), making this analysis clinically relevant. We found that the CRT-FS mutants are nonfunctional, as the decrease in surface MHC-I in CRT-null cells was not reversed by expression of either CRT-FSDEL or CRT-FSINS (Fig. 1G). The secretion of CRT-FS and consequent low level of ER retention was not solely responsible for the defect because the addition of a C-terminal KDEL sequence to CRT-FS could not rescue the surface levels of MHC-I, even partially (Fig. 2C). Thus, cells expressing CRT-FS seemed to phenocopy CRT-null cells with regard to the effect on MHC-I, emphasizing the importance of the C-terminal region in CRT function.
In cells that lack β2m, and therefore lack MHC-I–β2m dimers, CRT is not found associated with the residual PLC core, which consists of tapasin, ERp57, and TAP (31). Under normal circumstances, CRT is thought to enter the PLC with MHC-I and dissociate from the complex when MHC-I has acquired a peptide (31). In CRT-null mouse cells, significantly lower association of MHC-I with the PLC is seen, indicating that its recruitment to the PLC depends on CRT (26, 27). Because CRT-FS mutants are able to bind MHC-I molecules in solution (Fig. 2E), they should, in principle, be able to interact with MHC-I in cells. Hence, the decrease in the generation of stable MHC-I molecules at steady state we observed (Fig. 1G) could have been attributed to the lack of a detectable CRT-FS interaction with the PLC (Fig. 1F). However, expression of the CRT-FS mutants does result in a low level of MHC-I interaction with the PLC, whereas none is detectable in the complete absence of CRT. This suggested that the mutants probably associate with the PLC transiently, recruiting associated MHC-I molecules as previously suggested for WT CRT (13), but that the reduction in local concentration in the ER caused by their secretion minimizes the efficiency of the process. However, the addition of C-terminal KDEL sequences to the CRT-FS mutants increased their ER retention and restored their interaction with the PLC but did little to enhance MHC-I association with the PLC and failed to increase MHC-I surface expression. In contrast, CRT-WT lacking the KDEL sequence was secreted and, like the CRT-FS mutants, was not detectably associated with the PLC, but it partially rescued surface MHC-I levels (Fig. 2C). This differs from studies in mouse cells, where CRT lacking the KDEL sequence was retained within the cell and incorporated into the PLC to the same extent at CRT-WT (27) but did not rescue surface MHC-I levels, even partially. In human cells, it appears that CRT-WT−KDEL retains the fundamental CRT function within the PLC, whereas the CRT-FS mutants do not. In this case, the large reduction in the local concentration of CRT-WT when the ER-retention signal is removed may make it the limiting component in MHC-I–peptide complex assembly, leading to the reduced MHC-I surface expression observed. However, CRT-FS can neither facilitate MHC-I peptide loading nor rescue normal surface MHC-I expression.
The electrostatic charge of the altered C terminus of CRT-FS has been shown to affect protein interactions mediated by other domains of the mutant (19–21), and this may also impede PLC function. The incorporation of CRT depends on three interactions: (a) with the glycan of MHC-I, (b) with ERp57 (which is covalently linked to tapasin), and (c) with tapasin (7, 42). The N-terminal lectin domain of CRT-FS is responsible for its chaperone activity and appears to be functional, although the preference of glycosylated substrates it interacts with may be altered. CRT-FS, but not CRT-WT, binds to glycosylated thrombopoietin receptor to drive proliferation (19) but does not appear to fulfill its chaperone function in the folding of MPO (18). Fig. 2E shows that both CRT-FS and CRT-WT can interact with the heavy chain of MHC-I. Whereas MHC-I is glycosylated, the lectin specificity of CRT is for monoglucosylated glycans that represent only a small fraction of total glycosylated MHC-I. We therefore see only a small percentage of the input interacting with CRT. CRT-FS is also able to interact with ERp57, an interaction dependent on the P-domain, albeit to a lesser extent than CRT-WT. At least in this context, it appears that the mutant CTD does not influence the interactions of the lectin domain but partially affects the interactions of the P-domain. The crystal structure of CRT has been solved (43), but it does not include the C-terminal tail. However, the cryo-EM structure of the PLC indicates that the CTD of CRT (in pink in Fig. 5A, left) is located in proximity to the C-terminal domain of tapasin, shown in cyan (7). Using the reported structure of the PLC (Protein Data Bank code 6ENY), we looked more closely at the interactions between CRT and tapasin (Fig. 5A, right). The CTD of CRT was modeled as an α-helix and appears to be accommodated in a groove in tapasin, with possible interactions between the loops that make up the edges of the groove. The deposited cryo-EM structure is of low resolution, so mapping the interactions between the side chains was not possible. However, we could determine the surface charge of the two molecules (Fig. 5B). The helix of the CTD of CRT is negatively charged (indicated in red), whereas the tapasin loops and groove carry a more positive charge (blue). In CRT-FS, these acidic residues have mostly been replaced by basic residues, and this may disrupt interactions between CRT-FS and tapasin, interfering with PLC function. We speculate that the modified CTD of CRT-FS impacts its interaction with tapasin, and this, along with the reduced interaction with ERp57, impairs a functionally important interaction between CRT-FS and the PLC.
Figure 5.
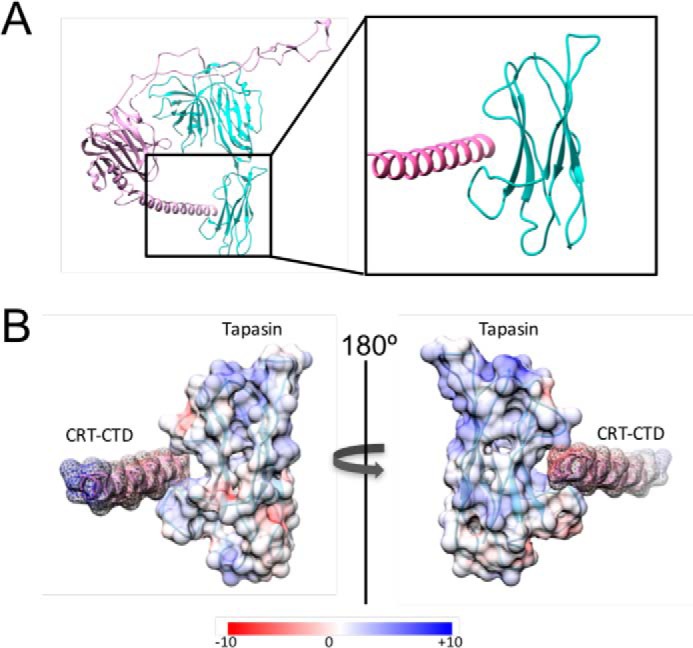
Structural analysis of the interaction between CRT and tapasin. A, the structure of the PLC was obtained from the Protein Data Bank (entry 6ENY), and a cartoon representation of the structure of CRT in pink and tapasin in cyan was rendered in UCSF Chimera. The region of interaction between CRT and tapasin has been enlarged on the right. B, surface charge of the rodlike α-helix of CRT and the cradle-like structure of tapasin was calculated using coulombic surface coloring. Red, negative charge; blue, positive charge.
It is not only the quantity of MHC-I at the surface of cells expressing the CRT-FS mutants that is affected, but the “quality” as well, as seen in Figs. 3 and 4C. The presentation of a known, high-affinity peptide is significantly lower in these cells compared with cells expressing CRT-WT. This is of particular relevance, as immunotherapeutic interventions that harness the CD8+ T-cell arm of the adaptive immune system depend on antigen presentation by MHC-I. It is conceivable that the unique C-terminal tail of CRT-FS could serve as a neo-antigen. However, a recent report (44) found CD4+ T cells but not CD8+ T cells that targeted disease cells. This may be due to poor presentation of MHC-I–restricted epitopes on these cells, leading to ineffectual recognition and killing. Indeed, when CRT-FSDEL is expressed together with CRT-WT in HEK293T cells, there was a 20% reduction of surface MHC-I, although this is not seen with CRT-FSINS. The stability of assembled MHC-I complexes was also reduced, reflecting the association of MHC-I with suboptimal peptides (Fig. 4C). This suggests that there may be an impairment of MHC-I peptide loading even in the majority of CRT-FS–positive tumors that retain expression of the WT protein. As both CRT-WT and CRT-FS can interact with MHC-I, there may be competition between the mutant and WT proteins for MHC-I in the ER. When CRT-FSDEL interacts with MHC-I, it prevents a functional interaction of the latter with the PLC, preventing peptide editing. Of course, the lack of a CD8+ T cell response may also depend on the MHC-I alleles expressed.
In the model cells expressing both CRT-WT and CRT-FS, CRT-FSDEL appears to be a better competitor than CRT-FSINS. The major difference between the two proteins is that the amino acid composition of the C-terminal tail of CRT-FSDEL is more dramatically affected than CRT-FSINS (22). This has been shown to confer unique properties to these proteins, as expression of CRT-FSDEL in patients correlates with a worse prognosis of overall survival than expression of CRT-FSINS (22, 45). This may be due to a difference in their ability to buffer Ca2+ flux. How this difference affects protein conformation and antigen loading is worth investigating. Notably, an increase in surface MHC-I levels and an increase in the stability of MHC-I–peptide complexes was observed in HEK293T cells overexpressing CRT-WT, indicating that the efficiency of MHC-I peptide loading and presentation depends on levels of CRT-WT expression. Whether this is unique to HEK293T cells or is universal remains to be determined.
When CRT-FS is expressed alone or with CRT-WT, CRT-FS is secreted (Figs. 1D and 2B). Although past studies have found CRT-FS in the extracellular media (46, 47), we have shown conclusively that CRT-FS is secreted due to the loss of the ER-retention sequence and that the secretion occurs via the classical ER-Golgi secretory pathway, not due to cell damage or lysis. Extracellular CRT-WT is considered a pro-phagocytic marker that induces immunogenic cell death (48), a phenomenon linked to better prognosis of overall disease-free survival in cancer patients. However, cells expressing CRT-FS appear to evade clearance by macrophages and establish disease. Either the altered CTD may influence the pro-phagocytic property of CRT or the process of extracellular localization for CRT-WT, which is still unclear, may confer this property. It is also plausible that the reduced but not absent surface MHC-I and associated β2m can still serve as anti-phagocytic signals, repressing phagocytosis by macrophages (4). Clinical symptoms of MPN in patients carrying the CRT mutations are less severe than those with the JAK kinase mutation (45). Chronic inflammation is one aspect of this disease, and qualitative differences are seen in cytokine expression in ET, PV, and PMF, independent of the mutation causing the disease (49). Mislocalized CRT-FS is believed to have only an autocrine function (50) and therefore does not affect the cytokine profile. However, a recent study shows that treating primary monocytes with conditioned medium from cells expressing CRT-FS can stimulate cytokine expression, indicating a potential paracrine effect (46). Thus, secreted CRT-FS may have the potential to modulate the host's immune system, allowing expressing cells to evade detection and promote their survival.
Our overall interpretation of the data is presented in Fig. 6. CRT-FS is secreted from cells and poorly incorporated into the PLC. This leads to a decrease in surface MHC-I levels and reduced association with optimal peptides. CRT is a known marker for inflammatory cell death and is pro-phagocytic, but despite the increased secretion of the mutant forms combined with the lower expression of MHC-I, an anti-phagocytic signal, these cells are still able to evade immune surveillance and cause disease. Understanding the mechanistic basis of how this is achieved will provide great insight into the pathophysiology of MPNs, paving the way for effective therapeutic intervention for this group of chronic blood neoplasms.
Figure 6.
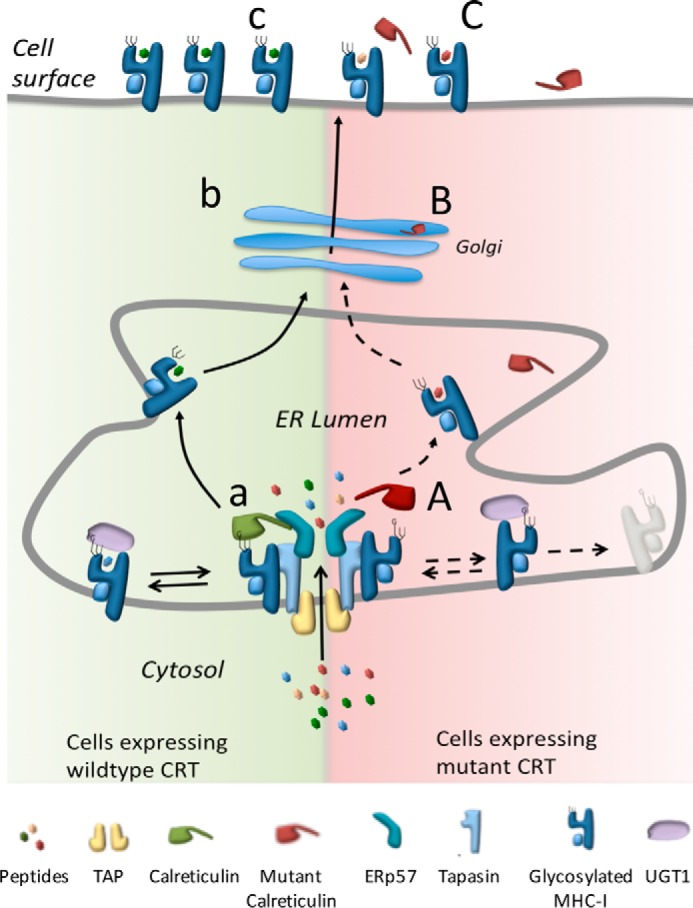
Effect of tumor-associated CRT mutations on antigen presentation by MHC-I. Peptides derived from the cytosol are transported into the ER lumen by the peptide TAP. Left, the PLC and TAP facilitate peptide loading onto MHC-I. In cells expressing nonmutant CRT, CRT associates with MHC-I via the glucose present on its glycan and also interacts with the core PLC. UGT1 can act on the MHC-I glycan to preserve the glucose and facilitate reassociation with the PLC (a). MHC-I loaded with an appropriate peptide is trafficked via the Golgi (b) to the surface of cells (c). Right, in cells expressing CRT-FS, interaction with the PLC is reduced (A) as CRT-FS is secreted (B), but the resulting PLC is nonfunctional. Interaction of MHC-I with other proteins, such as UGT1, may be disrupted, causing inefficient loading and/or increased degradation of empty MHC-I complexes, resulting in lower surface MHC-I levels and an altered repertoire of peptides presented on the surface of cells expressing CRT-FS (C).
Experimental procedures
Generation of DNA constructs
pspCas9(BB)-2A-GFP vector (Addgene, catalog no. 48138) was used to knock out calreticulin expression. The gRNAs were designed and generated based on the protocol outlined in Zhang and co-workers (51). Briefly, guide RNAs (gRNAs) targeting exon 1 and 2 of the CALR were designed using the gene tool (http://crispr.mit.edu/)4 (51, 52). Off-target effects were analyzed, and two 17-mers were selected (Table 1). The gRNAs were cloned into pspCas9(BB)-2A-GFP vector and verified by sequencing. Plasmids encoding the calreticulin mutants were a kind gift from Dr. Kralovics at the CeMM Research Center for Molecular Medicine (17). The most frequently occurring deletion mutant (52-bp deletion) and insertion mutant (5-bp insertion) was used in our study. From these plasmids, the cDNA encoding WT and mutant calreticulin was excised using BglII and EcoRI and ligated into pMXS vector with a puromycin selection cassette digested with BamHI and EcoRI. Mutant CRT with KDEL and CRT-WT lacking KDEL were generated by Gibson assembly (53) using the primers in Table 1. All constructs were verified by sequencing. Retroviral transduction of CRT-null cells was carried out to generate stable cell lines expressing the following constructs: pMXS alone or vector control (VC), CRT-WT, CRT-FS, CRT-FS+KDEL, and CRT-WT−KDEL. Transduced cells were selected by treatment with 2 μg/ml puromycin. Plasmid encoding LMP1, pCMMP-LMP1-IRES-eGFP, was purchased from Addgene (catalog no. 36955). Plasmid lacking LMP1 expression was generated by removing the DNA fragment encoding LMP1 by BamHI enzyme digestion and religation. For bacterial expression of CRT-WT, pGEX2TCRT available in the laboratory was used (12). For the expression of GST-tagged CRT-FS, the cDNA encoding the CTD of CRT-WT was replaced by the cDNA encoding the CTD of CRT-FS.
Table 1.
Primers used in the study
Shown are sequences of primers used to generate CRT-null lines (KO CRT), the CRT-FS mutants with the KDEL sequence, and the CRT-WT lacking the KDEL sequence (cloning) and used in qPCR analysis.
| Primer no. | Primer name | Sequence (5′ to 3′) | Purpose |
|---|---|---|---|
| 1 | CRTgRNA-1f | GCCTGCCGTCTACTTCA | KO CRT |
| 2 | CRTgRNA-2r | TTTGGATTCGATCCAGC | KO CRT |
| 3 | CRT-Fwd | CTAGCTAGTTAATTAAGGATCCATGCTGCTATCCGTGCCGC | Cloning |
| 4 | CRT-FS + KDELr | CTCGAGGCCTGCAGGAATTCCTACAGCTCGTCCTTGGCCTCAGTCCAGCCC | Cloning |
| 5 | CRT-WT-KDELr | CTCGAGGCCTGCAGGAATTCCTAGGCCTGGCCGGGGAC | Cloning |
| 6 | CRT-RTf | GAATACAACATCTGGTTTGGTCCCGAC | qPCR |
| 7 | CRT-RTr | CAATCAGTGTGTACAGGTGTGTAAAC | qPCR |
| 8 | LMP1-RTf | ACAAAACTGGTGGACTC | qPCR |
| 9 | LMP1-RTr | GTCTGCCCTCGTTGGA | qPCR |
| 10 | β-Actin-RTf | GGCATCGTGATGGACTCCG | qPCR |
| 11 | β-Actin-RTr | GCTGGAAGGTGGACAGCGA | qPCR |
Antibodies
A rabbit polyclonal antibody (α-CRT-FS) specific to the mutant C-terminal tail (RMRRMRRTRRKMRRKMSPARPRTSCREAC) was generated by GenScript. Commercially available antibodies that detect both CRT-WT and CRT-FS used in the study include FMC75 (ADI-SPA-601, Enzo Life Sciences) for Western blotting and PA3-900 (Thermo Fisher) for immunoprecipitation. Rat anti-human GRP94 (ADI-SPA-850) was purchased from Enzo Life Sciences). The anti-human tapasin antibodies PaSta1 (54) and R.Gp48N (55) and the anti-human MHC I antibodies BB7.2 (flow cytometry) (56) and HC10 (Western blotting and flow cytometry) (38) have been reported previously. Mouse anti-human HLA ABC (W6/32) conjugated to Alexa Fluor 647 (MCA81A647, Bio-Rad) was used in flow cytometric analysis of MHC-I. Mouse anti-human EGFR antibody conjugated to Alexa Fluor 488 (catalog no. 352907, Biolegend) was used in the flow cytometric analysis of EGFR. Recombinant human EGF was purchased from Biolegend (catalog no. 585506). The TCR-like mAb that detects HLA-A2–YLLEMLWRL complexes by flow cytometry was a kind gift from Prof. Paul A. MacAry (41). All secondary antibodies used for flow cytometry (goat anti-mouse IgG coupled to Alexa Fluor 488, Alexa Fluor 647, or FITC and goat anti-rabbit IgG coupled to Alexa Fluor 647) were purchased from Invitrogen, and those for immunoblotting (goat anti-mouse and anti-rat IgG coupled to horseradish peroxidase and goat anti-mouse, anti-rat, and anti-rabbit IgG coupled to alkaline phosphatase) were purchased from Jackson ImmunoResearch.
Reagents
Reagents used were as follows: Lipofectamine 2000 (Invitrogen), Brefeldin A (Biolegend), Triton X-100 (AmericanBio), digitonin (Calbiochem), EDTA-free protease inhibitor mixture (Roche Applied Science), l-[35S]methionine, and l-[35S]cysteine (PerkinElmer Life Sciences), protein molecular weight markers, [Methyl-14C] Methylated (PerkinElmer Life Sciences), TRIzol (Invitrogen), Bradford dye for protein assay (Bio-Rad), B-PERTM bacterial protein extraction reagent (Thermo Scientific), GSH-Sepharose 4B (GE Healthcare), SimplyBlueTM SafeStain (Invitrogen), and RestoreTM PLUS Western blotting stripping buffer (Thermo Scientific).
Cells
HEK293T cells were grown in Dulbecco's modified Eagle's medium (Sigma) supplemented with 10% fetal calf serum (HyClone) and 1% penicillin/streptomycin (Invitrogen). Where indicated, cells were incubated in FreeStyle 293 expression medium (Thermo Fisher Scientific) or CO2-independent medium (Gibco), supplemented with 10% fetal calf serum (HyClone) and 1% penicillin/streptomycin (Invitrogen).
Generation of CRT-null cells
Plasmids were introduced into HEK293T cells using cationic lipid for transient transfection. Two sequential rounds of transfection were carried out, and ablation of CRT expression was verified by Western blot analysis at each stage. From the bulk population after round two, clonal selection by limiting dilution was carried out, and one clone was selected for the generation of all subsequent cell lines.
Generation of stable cells by retroviral transduction
In all cases, retrovirus was generated in 293T cells transfected with 4 μg/ml of the packaging vector pCL-Ampho (Imgenex) and the construct of interest and 20 μg/ml Lipofectamine 2000 (Invitrogen) in Opti-MEM (Invitrogen), followed by three rounds of retroviral transduction of the recipient cell line with 8 μg/ml Polybrene (Millipore). Stably expressing cells were obtained by selection in complete medium supplemented with 2 μg/ml puromycin (Sigma).
RNA extraction, RT-PCR, and qPCR
Cells were harvested in TRIzol, and RNA was extracted using the Direct-zol RNA MiniPrep kit (Zymo Research). cDNA synthesis was carried out using the iScriptTM cDNA synthesis kit (Bio-Rad). Power SYBR® Green PCR Master Mix (Invitrogen) was used for qPCR analysis, and the reactions were set up by the epMotion M5073 robot (Eppendorf) and carried out in the CFX384 TouchTM real-time PCR detection system (Bio-Rad). Primers for CRT, LMP1 (57), and β-actin (58) are listed in Table 1.
Western blotting of cell extracts and culture supernatant
Cell lysates were prepared by incubating cells in 1% Triton X-100 in 10 mm Tris, pH 7.4, 150 mm sodium chloride (TBS) + protease inhibitors at 4 °C for 1 h. Clarified lysates were diluted 10-fold to determine protein concentration using the Bio-Rad Bradford dye, as per the manufacturer's instructions. For analysis of culture supernatant, equal number of cells from each cell line were grown in FreeStyle 293 expression medium (Thermo Fisher Scientific). Proteins (30 μg of lysates and 30 μl of media) were separated by SDS-PAGE followed by Western blot analysis. Blots were stripped and reprobed following treatment with RestoreTM PLUS Western blotting stripping buffer.
PLC immunoprecipitation
To immunoprecipitate tapasin, cells were incubated in 1% digitonin in TBS + protease inhibitors + 2 mm CaCl2 at 4 °C for 1 h. Clarified lysates were then either prepared for SDS-PAGE by the addition of Laemmli sample buffer or immunoprecipitated with PaSta1 for 1 h at 4 °C. After three washes in 0.1% digitonin lysis buffer and elution in nonreducing sample buffer, Laemmli sample buffer was added to the eluates, and the immunoprecipitates were separated by SDS-PAGE followed by Western blotting. Input and lysates were run on two gels, as it was not feasible to strip a single blot several times to probe with the five different antibodies used in the analysis.
Metabolic labeling and immunoprecipitation
Cells were incubated in cysteine- and methionine-free Dulbecco's modified Eagle's medium supplemented with 5% dialyzed fetal bovine serum (Invitrogen), 1% Glutamax, 1% penicillin/streptomycin for 1 h, followed by labeling with [35S]methionine/cysteine (PerkinElmer Life Sciences) in the same medium for 15 min. Cells were then spun down and resuspended in complete medium, and cells and supernatant were collected at 0, 30, 60, 120, and 180 min of chase. Cell lysates were prepared in 1% Triton X-100 in TBS (10 mm Tris, pH 7.4, 150 mm sodium chloride) + protease inhibitors and precleared with normal rabbit IgG, followed by immunoprecipitation with rabbit anti-human calreticulin (PA3900). For the supernatant, 1% Triton X-100 and protease inhibitors were added, followed by preclearing with normal rabbit IgG, followed by immunoprecipitation with rabbit anti-human calreticulin (PA3900). The immunoprecipitates were washed three times with TBS + 0.5% Triton X-100, and proteins were visualized by autoradiography after separation on nonreducing SDS-polyacrylamide gels.
Expression and purification of GST-tagged proteins from bacteria
GST or GST-tagged proteins were expressed in Escherichia coli BL21 GOLD (DE3) following induction with isopropyl 1-thio-β-d-galactopyranoside (250 μm) for 3 h at 37 °C, followed by incubation at 16 °C overnight. Cells were pelleted and lysed in B-PER buffer + protease inhibitors by incubation at room temperature for 20 min. Soluble, GST-tagged proteins were isolated from the clarified lysate by interaction with GSH beads for 1 h at 4 °C. Associated proteins were eluted in TBS + 10 mm reduced GSH (pH 8.0), followed by size-exclusion chromatography. The GST-tagged proteins migrated as a dimer. Purity of protein was checked by SDS-PAGE, followed by Coomassie Blue staining.
GST-pulldown assay
Lysates from CRT-null HEK293T cells were prepared by solubilizing cells in interaction buffer (TBS + 5 mm CaCl2 + 1% Triton X-100 + protease inhibitors). Interaction with 400 μg of protein was carried out at 4 °C for 4 h with 30 μg of GST-CRT on GSH beads or an equivalent amount of GST associated with GSH beads. Following interaction, beads were washed three times with the interaction buffer without protease inhibitors and once with buffer without detergent. Proteins associated with the beads were subjected to SDS-PAGE followed by Western blotting.
Flow cytometry
Cells were detached with PBS-EDTA, and cells were sequentially labeled with primary and secondary antibody (unless the primary antibody was fluorescently labeled) in FACS staining buffer (PBS + 0.5% BSA + 0.02% sodium azide) for 1 h at 4 °C, followed by three washes with FACS buffer, and analyzed using FACSCalibur (BD Biosciences) and FlowJo software (Tree Star). Staining with propidium iodide was carried out to distinguish dead from live cells.
Acid denaturation of surface MHC-I
Surface MHC-I was denatured by resuspending cells for 2 min in medium at pH 3.5 (using sodium citrate), followed by neutralization with sodium phosphate buffer at pH 10.5. Complete medium was added to the cells, followed by incubation at 37 °C for 0, 0.5, 1, 2, 4, and 6 h in the absence or presence of 100 μm dynasore. MHC-I on the surface of cells was estimated by flow cytometry. Several samples, from both the untreated and dynasore-treated cell suspensions, were randomly selected to estimate surface levels of EGFR by FACS. Cells were also treated (or not) with 40 ng/ml recombinant EGF ligand for 30 min to promote receptor endocytosis (59). Staining with propidium iodide was carried out to distinguish dead from live cells.
Thermostability assay
Cells were resuspended in FACS staining buffer for 10 min at room temperature followed by exposure to 37, 45, 55, and 65 °C for 10 min. The amount of MHC-I on the surface of cells was assessed by flow cytometry. Staining with propidium iodide was carried out to distinguish dead and live cells.
BFA-decay assay
Cells were plated in CO2-independent medium and incubated at 27 °C overnight to allow accumulation of surface MHC-I complexes, after which they were incubated at 37 °C for 0, 0.5, 1, 2, 4, or 6 h in the presence of 10 μg/ml BFA. Surface MHC-I levels were determined by flow cytometry. Staining with propidium iodide was carried out to distinguish dead from live cells.
Structural analysis
The deposited cryo-EM structure of the PLC, Protein Data Bank ID 6ENY, was analyzed using UCSF Chimera (60).
Author contributions
N. A. and P. C. conceptualization; N. A. data curation; N. A. investigation; N. A. methodology; N. A. writing-original draft; N. A. and P. C. writing-review and editing; P. C. funding acquisition; P. C. project administration.
Acknowledgments
We thank Susan Mitchell for help with protein purification, Yue Li for help in preparing Fig. 5, Robert Kralovics for DNA constructs, and Paul A. MacAry for antibodies.
This work was supported by National Institutes of Health Grant RO1-AI097206 (to P. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MHC
- major histocompatibility complex
- β2m
- β2-microglobulin
- NK
- natural killer
- PLC
- peptide loading complex
- TAP
- transporter associated with antigen processing
- CRT
- calreticulin
- CTD
- C-terminal domain
- UGT1
- UDP-glucosyltransferase 1
- CRT-FS
- CRT with frameshift mutations
- MPN
- myeloproliferative neoplasm
- PV
- polycythemia vera
- ET
- essential thrombocythemia
- PMF
- primary myelofibrosis
- JAK
- Janus kinase
- STAT
- signal transducers and activators of transcription
- MPO
- myeloperoxidase
- BFA
- brefeldin A
- ER
- endoplasmic reticulum
- EGFR
- epidermal growth factor receptor
- gRNA
- guide RNA
- VC
- vector control
- qPCR
- quantitative PCR
- GST
- glutathione S-transferase
- TCR
- T-cell receptor.
References
- 1. Blum J. S., Wearsch P. A., and Cresswell P. (2013) Pathways of antigen processing. Annu. Rev. Immunol. 31, 443–473 10.1146/annurev-immunol-032712-095910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leone P., Shin E. C., Perosa F., Vacca A., Dammacco F., and Racanelli V. (2013) MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 105, 1172–1187 10.1093/jnci/djt184 [DOI] [PubMed] [Google Scholar]
- 3. Beatty G. L., and Gladney W. L. (2015) Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 21, 687–692 10.1158/1078-0432.CCR-14-1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barkal A. A., Weiskopf K., Kao K. S., Gordon S. R., Rosental B., Yiu Y. Y., George B. M., Markovic M., Ring N. G., Tsai J. M., McKenna K. M., Ho P. Y., Cheng R. Z., Chen J. Y., Barkal L. J., Ring A. M., Weissman I. L., and Maute R. L. (2018) Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 19, 76–84 10.1038/s41590-017-0004-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hofmann S., Babiak A., and Greiner J. (2011) Immunotherapy for myeloproliferative neoplasms (MPN). Curr. Cancer Drug Targets 11, 72–84 10.2174/156800911793743682 [DOI] [PubMed] [Google Scholar]
- 6. Nagler E., Xavier M. F., and Frey N. (2017) Updates in immunotherapy for acute myeloid leukemia. Translational Cancer Res. 6, 86–92 10.21037/tcr.2017.02.14 [DOI] [Google Scholar]
- 7. Blees A., Januliene D., Hofmann T., Koller N., Schmidt C., Trowitzsch S., Moeller A., and Tampé R. (2017) Structure of the human MHC-I peptide-loading complex. Nature 551, 525–528 [DOI] [PubMed] [Google Scholar]
- 8. Oliver J. D., van der Wal F. J., Bulleid N. J., and High S. (1997) Interaction of the thiol-dependent reductase ERp57 with nascent glycoproteins. Science 275, 86–88 10.1126/science.275.5296.86 [DOI] [PubMed] [Google Scholar]
- 9. Panter M. S., Jain A., Leonhardt R. M., Ha T., and Cresswell P. (2012) Dynamics of major histocompatibility complex class I association with the human peptide-loading complex. J. Biol. Chem. 287, 31172–31184 10.1074/jbc.M112.387704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang J., Natarajan K., Boyd L. F., Morozov G. I., Mage M. G., and Margulies D. H. (2017) Crystal structure of a TAPBPR-MHC I complex reveals the mechanism of peptide editing in antigen presentation. Science 358, 1064–1068 10.1126/science.aao5154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thomas C., and Tampé R. (2017) Structure of the TAPBPR-MHC I complex defines the mechanism of peptide loading and editing. Science 358, 1060–1064 10.1126/science.aao6001 [DOI] [PubMed] [Google Scholar]
- 12. Wearsch P. A., Jakob C. A., Vallin A., Dwek R. A., Rudd P. M., and Cresswell P. (2004) Major histocompatibility complex class I molecules expressed with monoglucosylated N-linked glycans bind calreticulin independently of their assembly status. J. Biol. Chem. 279, 25112–25121 10.1074/jbc.M401721200 [DOI] [PubMed] [Google Scholar]
- 13. Wearsch P. A., Peaper D. R., and Cresswell P. (2011) Essential glycan-dependent interactions optimize MHC class I peptide loading. Proc. Natl. Acad. Sci. U.S.A. 108, 4950–4955 10.1073/pnas.1102524108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dierssen J. W., de Miranda N. F., Ferrone S., van Puijenbroek M., Cornelisse C. J., Fleuren G. J., van Wezel T., and Morreau H. (2007) HNPCC versus sporadic microsatellite-unstable colon cancers follow different routes toward loss of HLA class I expression. BMC Cancer 7, 33 10.1186/1471-2407-7-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cathro H. P., Smolkin M. E., Theodorescu D., Jo V. Y., Ferrone S., and Frierson H. F. Jr. (2010) Relationship between HLA class I antigen processing machinery component expression and the clinicopathologic characteristics of bladder carcinomas. Cancer Immunol. Immunother. 59, 465–472 10.1007/s00262-009-0765-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nangalia J., Massie C. E., Baxter E. J., Nice F. L., Gundem G., Wedge D. C., Avezov E., Li J., Kollmann K., Kent D. G., Aziz A., Godfrey A. L., Hinton J., Martincorena I., Van Loo P., et al. (2013) Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 369, 2391–2405 10.1056/NEJMoa1312542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klampfl T., Gisslinger H., Harutyunyan A. S., Nivarthi H., Rumi E., Milosevic J. D., Them N. C., Berg T., Gisslinger B., Pietra D., Chen D., Vladimer G. I., Bagienski K., Milanesi C., Casetti I. C., et al. (2013) Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 369, 2379–2390 10.1056/NEJMoa1311347 [DOI] [PubMed] [Google Scholar]
- 18. Theocharides A. P., Lundberg P., Lakkaraju A. K., Lysenko V., Myburgh R., Aguzzi A., Skoda R. C., and Manz M. G. (2016) Homozygous calreticulin mutations in patients with myelofibrosis lead to acquired myeloperoxidase deficiency. Blood 127, 3253–3259 10.1182/blood-2016-02-696310 [DOI] [PubMed] [Google Scholar]
- 19. Araki M., Yang Y., Masubuchi N., Hironaka Y., Takei H., Morishita S., Mizukami Y., Kan S., Shirane S., Edahiro Y., Sunami Y., Ohsaka A., and Komatsu N. (2016) Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 127, 1307–1316 10.1182/blood-2015-09-671172 [DOI] [PubMed] [Google Scholar]
- 20. Chachoua I., Pecquet C., El-Khoury M., Nivarthi H., Albu R. I., Marty C., Gryshkova V., Defour J. P., Vertenoeil G., Ngo A., Koay A., Raslova H., Courtoy P. J., Choong M. L., Plo I., et al. (2016) Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 127, 1325–1335 10.1182/blood-2015-11-681932 [DOI] [PubMed] [Google Scholar]
- 21. Elf S., Abdelfattah N. S., Chen E., Perales-Patón J., Rosen E. A., Ko A., Peisker F., Florescu N., Giannini S., Wolach O., Morgan E. A., Tothova Z., Losman J. A., Schneider R. K., Al-Shahrour F., and Mullally A. (2016) Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 6, 368–381 10.1158/2159-8290.CD-15-1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pietra D., Rumi E., Ferretti V. V., Di Buduo C. A., Milanesi C., Cavalloni C., Sant'Antonio E., Abbonante V., Moccia F., Casetti I. C., Bellini M., Renna M. C., Roncoroni E., Fugazza E., Astori C., et al. (2016) Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 30, 431–438 10.1038/leu.2015.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shide K., Kameda T., Yamaji T., Sekine M., Inada N., Kamiunten A., Akizuki K., Nakamura K., Hidaka T., Kubuki Y., Shimoda H., Kitanaka A., Honda A., Sawaguchi A., Abe H., et al. (2017) Calreticulin mutant mice develop essential thrombocythemia that is ameliorated by the JAK inhibitor ruxolitinib. Leukemia 31, 1136–1144 10.1038/leu.2016.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J., Prins D., Park H. J., Grinfeld J., Gonzalez-Arias C., Loughran S., Dovey O. M., Klampfl T., Bennett C., Hamilton T. L., Pask D. C., Sneade R., Williams M., Aungier J., Ghevaert C., et al. (2018) Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stem cell self-renewal advantage. Blood 131, 649–661 10.1182/blood-2017-09-806356 [DOI] [PubMed] [Google Scholar]
- 25. Liu C., Fu H., Flutter B., Powis S. J., and Gao B. (2010) Suppression of MHC class I surface expression by calreticulin's P-domain in a calreticulin deficient cell line. Biochim. Biophys. Acta 1803, 544–552 10.1016/j.bbamcr.2010.03.001 [DOI] [PubMed] [Google Scholar]
- 26. Gao B., Adhikari R., Howarth M., Nakamura K., Gold M. C., Hill A. B., Knee R., Michalak M., and Elliott T. (2002) Assembly and antigen-presenting function of MHC class I molecules in cells lacking the ER chaperone calreticulin. Immunity 16, 99–109 10.1016/S1074-7613(01)00260-6 [DOI] [PubMed] [Google Scholar]
- 27. Howe C., Garstka M., Al-Balushi M., Ghanem E., Antoniou A. N., Fritzsche S., Jankevicius G., Kontouli N., Schneeweiss C., Williams A., Elliott T., and Springer S. (2009) Calreticulin-dependent recycling in the early secretory pathway mediates optimal peptide loading of MHC class I molecules. EMBO J. 28, 3730–3744 10.1038/emboj.2009.296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Byrne J. C., Ní Gabhann J., Stacey K. B., Coffey B. M., McCarthy E., Thomas W., and Jefferies C. A. (2013) Bruton's tyrosine kinase is required for apoptotic cell uptake via regulating the phosphorylation and localization of calreticulin. J. Immunol. 190, 5207–5215 10.4049/jimmunol.1300057 [DOI] [PubMed] [Google Scholar]
- 29. Feng M., Chen J. Y., Weissman-Tsukamoto R., Volkmer J. P., Ho P. Y., McKenna K. M., Cheshier S., Zhang M., Guo N., Gip P., Mitra S. S., and Weissman I. L. (2015) Macrophages eat cancer cells using their own calreticulin as a guide: roles of TLR and Btk. Proc. Natl. Acad. Sci. U.S.A. 112, 2145–2150 10.1073/pnas.1424907112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garbi N., Tan P., Diehl A. D., Chambers B. J., Ljunggren H. G., Momburg F., and Hämmerling G. J. (2000) Impaired immune responses and altered peptide repertoire in tapasin-deficient mice. Nat. Immunol. 1, 234–238 10.1038/79775 [DOI] [PubMed] [Google Scholar]
- 31. Sadasivan B., Lehner P. J., Ortmann B., Spies T., and Cresswell P. (1996) Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity 5, 103–114 10.1016/S1074-7613(00)80487-2 [DOI] [PubMed] [Google Scholar]
- 32. Ortmann B., Copeman J., Lehner P. J., Sadasivan B., Herberg J. A., Grandea A. G., Riddell S. R., Tampé R., Spies T., Trowsdale J., and Cresswell P. (1997) A critical role for tapasin in the assembly and function of multimeric MHC class I-TAP complexes. Science 277, 1306–1309 10.1126/science.277.5330.1306 [DOI] [PubMed] [Google Scholar]
- 33. Barnstable C. J., Jones E. A., and Crumpton M. J. (1978) Isolation, structure and genetics of HLA-A, -B, -C and -DRw (Ia) antigens. Br. Med. Bull. 34, 241–246 10.1093/oxfordjournals.bmb.a071504 [DOI] [PubMed] [Google Scholar]
- 34. Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., and Kirchhausen T. (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 10, 839–850 10.1016/j.devcel.2006.04.002 [DOI] [PubMed] [Google Scholar]
- 35. Stahlschmidt W., Robertson M. J., Robinson P. J., McCluskey A., and Haucke V. (2014) Clathrin terminal domain-ligand interactions regulate sorting of mannose 6-phosphate receptors mediated by AP-1 and GGA adaptors. J. Biol. Chem. 289, 4906–4918 10.1074/jbc.M113.535211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leonhardt R. M., Fiegl D., Rufer E., Karger A., Bettin B., and Knittler M. R. (2010) Post-endoplasmic reticulum rescue of unstable MHC class I requires proprotein convertase PC7. J. Immunol. 184, 2985–2998 10.4049/jimmunol.0900308 [DOI] [PubMed] [Google Scholar]
- 37. Cooper A., and Shaul Y. (2006) Clathrin-mediated endocytosis and lysosomal cleavage of hepatitis B virus capsid-like core particles. J. Biol. Chem. 281, 16563–16569 10.1074/jbc.M601418200 [DOI] [PubMed] [Google Scholar]
- 38. Stam N. J., Spits H., and Ploegh H. L. (1986) Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J. Immunol. 137, 2299–2306 [PubMed] [Google Scholar]
- 39. Everett M. W., and Edidin M. (2007) Tapasin increases efficiency of MHC I assembly in the endoplasmic reticulum but does not affect MHC I stability at the cell surface. J. Immunol. 179, 7646–7652 10.4049/jimmunol.179.11.7646 [DOI] [PubMed] [Google Scholar]
- 40. Kelly A., Powis S. H., Kerr L. A., Mockridge I., Elliott T., Bastin J., Uchanska-Ziegler B., Ziegler A., Trowsdale J., and Townsend A. (1992) Assembly and function of the two ABC transporter proteins encoded in the human major histocompatibility complex. Nature 355, 641–644 10.1038/355641a0 [DOI] [PubMed] [Google Scholar]
- 41. Sim A. C., Too C. T., Oo M. Z., Lai J., Eio M. Y., Song Z., Srinivasan N., Tan D. A., Pang S. W., Gan S. U., Lee K. O., Loh T. K., Chen J., Chan S. H., and MacAry P. A. (2013) Defining the expression hierarchy of latent T-cell epitopes in Epstein-Barr virus infection with TCR-like antibodies. Sci. Rep. 3, 3232 10.1038/srep03232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dong G., Wearsch P. A., Peaper D. R., Cresswell P., and Reinisch K. M. (2009) Insights into MHC class I peptide loading from the structure of the tapasin-ERp57 thiol oxidoreductase heterodimer. Immunity 30, 21–32 10.1016/j.immuni.2008.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pocanschi C. L., Kozlov G., Brockmeier U., Brockmeier A., Williams D. B., and Gehring K. (2011) Structural and functional relationships between the lectin and arm domains of calreticulin. J. Biol. Chem. 286, 27266–27277 10.1074/jbc.M111.258467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Holmström M. O., Martinenaite E., Ahmad S. M., Met Ö., Friese C., Kjaer L., Riley C. H., Thor Straten P., Svane I. M., Hasselbalch H. C., and Andersen M. H. (2018) The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia 32, 429–437 [DOI] [PubMed] [Google Scholar]
- 45. Patel U., Luthra R., Medeiros L. J., and Patel K. P. (2017) Diagnostic, prognostic, and predictive utility of recurrent somatic mutations in myeloid neoplasms. Clin. Lymphoma Myeloma Leuk. 17S, S62–S74 [DOI] [PubMed] [Google Scholar]
- 46. Garbati M. R., Welgan C. A., Landefeld S. H., Newell L. F., Agarwal A., Dunlap J. B., Chourasia T. K., Lee H., Elferich J., Traer E., Rattray R., Cascio M. J., Press R. D., Bagby G. C., Tyner J. W., et al. (2016) Mutant calreticulin-expressing cells induce monocyte hyperreactivity through a paracrine mechanism. Am. J. Hematol. 91, 211–219 10.1002/ajh.24245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han L., Schubert C., Köhler J., Schemionek M., Isfort S., Brümmendorf T. H., Koschmieder S., and Chatain N. (2016) Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J. Hematol. Oncol. 9, 45 10.1186/s13045-016-0275-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Obeid M., Tesniere A., Ghiringhelli F., Fimia G. M., Apetoh L., Perfettini J. L., Castedo M., Mignot G., Panaretakis T., Casares N., Métivier D., Larochette N., van Endert P., Ciccosanti F., Piacentini M., et al. (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 10.1038/nm1523 [DOI] [PubMed] [Google Scholar]
- 49. Hermouet S., Bigot-Corbel E., and Gardie B. (2015) Pathogenesis of myeloproliferative neoplasms: role and mechanisms of chronic inflammation. Mediators Inflamm. 2015, 145293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Araki M., and Komatsu N. (2017) Novel molecular mechanism of cellular transformation by a mutant molecular chaperone in myeloproliferative neoplasms. Cancer Sci. 108, 1907–1912 10.1111/cas.13327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guo D., Li X., Zhu P., Feng Y., Yang J., Zheng Z., Yang W., Zhang E., Zhou S., and Wang H. (2015) Online high-throughput mutagenesis designer using scoring matrix of sequence-specific endonucleases. J. Integr. Bioinform. 12, 35–48 [DOI] [PubMed] [Google Scholar]
- 53. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A. 3rd, Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 54. Dick T. P., Bangia N., Peaper D. R., and Cresswell P. (2002) Disulfide bond isomerization and the assembly of MHC class I-peptide complexes. Immunity 16, 87–98 10.1016/S1074-7613(02)00263-7 [DOI] [PubMed] [Google Scholar]
- 55. Lehner P. J., Surman M. J., and Cresswell P. (1998) Soluble tapasin restores MHC class I expression and function in the tapasin-negative cell line .220. Immunity 8, 221–231 10.1016/S1074-7613(00)80474-4 [DOI] [PubMed] [Google Scholar]
- 56. Parham P., and Brodsky F. M. (1981) Partial purification and some properties of BB7.2: a cytotoxic monoclonal antibody with specificity for HLA-A2 and a variant of HLA-A28. Hum. Immunol. 3, 277–299 10.1016/0198-8859(81)90065-3 [DOI] [PubMed] [Google Scholar]
- 57. Liu H. P., Chen C. C., Wu C. C., Huang Y. C., Liu S. C., Liang Y., Chang K. P., and Chang Y. S. (2012) Epstein-Barr virus-encoded LMP1 interacts with FGD4 to activate Cdc42 and thereby promote migration of nasopharyngeal carcinoma cells. PLoS Pathog. 8, e1002690 10.1371/journal.ppat.1002690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leonhardt R. M., Lee S. J., Kavathas P. B., and Cresswell P. (2007) Severe tryptophan starvation blocks onset of conventional persistence and reduces reactivation of Chlamydia trachomatis. Infect. Immun. 75, 5105–5117 10.1128/IAI.00668-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Al-Akhrass H., Naves T., Vincent F., Magnaudeix A., Durand K., Bertin F., Melloni B., Jauberteau M. O., and Lalloué F. (2017) Sortilin limits EGFR signaling by promoting its internalization in lung cancer. Nat. Commun. 8, 1182 10.1038/s41467-017-01172-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]