

Abstract
The generation of mitochondrial superoxide (O2˙̄) by reverse electron transport (RET) at complex I causes oxidative damage in pathologies such as ischemia reperfusion injury, but also provides the precursor to H2O2 production in physiological mitochondrial redox signaling. Here, we quantified the factors that determine mitochondrial O2˙̄ production by RET in isolated heart mitochondria. Measuring mitochondrial H2O2 production at a range of proton-motive force (Δp) values and for several coenzyme Q (CoQ) and NADH pool redox states obtained with the uncoupler p-trifluoromethoxyphenylhydrazone, we show that O2˙̄ production by RET responds to changes in O2 concentration, the magnitude of Δp, and the redox states of the CoQ and NADH pools. Moreover, we determined how expressing the alternative oxidase from the tunicate Ciona intestinalis to oxidize the CoQ pool affected RET-mediated O2˙̄ production at complex I, underscoring the importance of the CoQ pool for mitochondrial O2˙̄ production by RET. An analysis of O2˙̄ production at complex I as a function of the thermodynamic forces driving RET at complex I revealed that many molecules that affect mitochondrial reactive oxygen species production do so by altering the overall thermodynamic driving forces of RET, rather than by directly acting on complex I. These findings clarify the factors controlling RET-mediated mitochondrial O2˙̄ production in both pathological and physiological conditions. We conclude that O2˙̄ production by RET is highly responsive to small changes in Δp and the CoQ redox state, indicating that complex I RET represents a major mode of mitochondrial redox signaling.
Keywords: mitochondria, mitochondrial membrane potential, redox signaling, reactive oxygen species (ROS), respiration, complex I, reverse electron transport, RET, coenzyme Q, superoxide
Introduction
Superoxide (O2˙̄)3 is the proximal reactive oxygen species (ROS) formed within mitochondria, with most O2˙̄ being very rapidly converted to H2O2 by manganese superoxide dismutase (MnSOD) within the matrix (1, 2). As well as contributing to oxidative damage, H2O2 acts as a redox signal, both within the mitochondria and in the cytosol (3–6). This mode of signal transduction arises via the reversible oxidation of protein thiols that pass on the modification to effector proteins as a redox relay (3–6). Whereas there are a number of potential mitochondrial sources of O2˙̄ (2, 7, 8), respiratory chain complex I is considered to be a major contributor (1). The production of O2˙̄ at complex I can be driven by reverse electron transport (RET) by a highly reduced coenzyme Q (CoQ) pool and a large proton-motive force (Δp), which together drive electrons backward through complex I and lead to a dramatic increase in O2˙̄ production (Fig. 1) (9). This process has been known since the 1960s (10) but was tacitly assumed to be an in vitro curiosity of no physiological relevance (1, 11, 12). However, there is now considerable evidence that RET at complex I is a physiological process that underlies mitochondrial redox signaling in a range of situations (11, 13) while also leading to pathological oxidative damage during ischemia–reperfusion injury (1, 14, 15).
Figure 1.

Mitochondrial O2˙̄ production from complex I by RET. The top panel shows conventional forward electron transport by mitochondria. The bottom panel shows mitochondrial O2˙̄ production by RET. This occurs when the Δp (a combination of the Δψ and the ΔpH) is high and the CoQ pool is reduced. Q, ubiquinone; QH2, ubiquinol.
A particularly intriguing aspect of RET is that it does not require damage to, or inhibition of, the respiratory chain (1, 9). As RET responds sensitively to physiological variables, O2˙̄ production by complex I can be modulated under physiological conditions (1, 9). Hence, there is considerable interest in understanding the mechanisms by which mitochondria regulate O2˙̄ production at complex I by RET as a physiological signaling process. Here, we quantified the factors that determine RET in isolated heart mitochondria. To do this, we measured H2O2 generation as a function of the membrane potential (Δψ) and of the reduction state of the CoQ and NADH pools. This was done by altering Δψ with the uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) and also oxidizing the CoQ pool by ectopic expression within heart mitochondria of the alternative oxidase (AOX) from Ciona intestinalis (16). Next, we assessed the dependence of RET O2˙̄ production on O2 concentration, [O2], and last, we determined how compounds known to alter mitochondrial ROS production affect RET. Together, these data provide a complete description of mitochondrial O2˙̄ production by RET and indicate how this process contributes to oxidative damage and redox signaling.
Results
Dependence of complex I O2˙̄ production by RET on Δp and CoQ redox state
To assess the factors that determine O2˙̄ production by RET at complex I, we used isolated heart mitochondria. H2O2 efflux from mitochondria is proportional to O2˙̄ production, assuming that O2˙̄ is converted to H2O2 by matrix MnSOD and that H2O2 degradation by peroxidases is similar across all conditions assessed (1). Whereas H2O2 movement across the plasma membrane is facilitated by aquaporins (17–19), these carriers are not present in mitochondria (20); hence, H2O2 efflux is probably unmediated, albeit facilitated by the large surface area of the inner membrane. Thus, measuring H2O2 efflux enables us to infer how O2˙̄ production by RET at complex I responds to mitochondrial state.
Three factors are critical in determining O2˙̄ production by RET: the Δp and the redox states of the mitochondrial CoQ and NADH pools (Fig. 1) (1). The O2 concentration is also likely to affect O2˙̄ production (1); however, these experiments were in air-saturated incubation medium ([O2] ∼200 μm); the dependence of O2˙̄ production on [O2] was determined later. The dependence of mitochondrial H2O2 production on these three variables was measured in parallel (Fig. 2). Δp was measured as Δψ from the distribution of the lipophilic methyltriphenylphosphonium (TPMP) cation (21) (Fig. 2B). Whereas this does not measure the pH gradient (ΔpH) across the inner membrane, ΔpH is a smaller (∼10–20%) component of Δp than Δψ and here is assumed to be invariant. The redox state of the CoQ pool was assessed by measuring its percentage reduction by reverse-phase HPLC (Fig. 2C). The redox state of the NADH pool was inferred from NAD(P)H fluorescence. Although this cannot distinguish between NADH and NADPH, the mitochondrial NADPH pool is thought to contribute less to changes in this variable (22); hence, this method gives a reasonable assessment of the NADH pool redox state (Fig. 2D).
Figure 2.
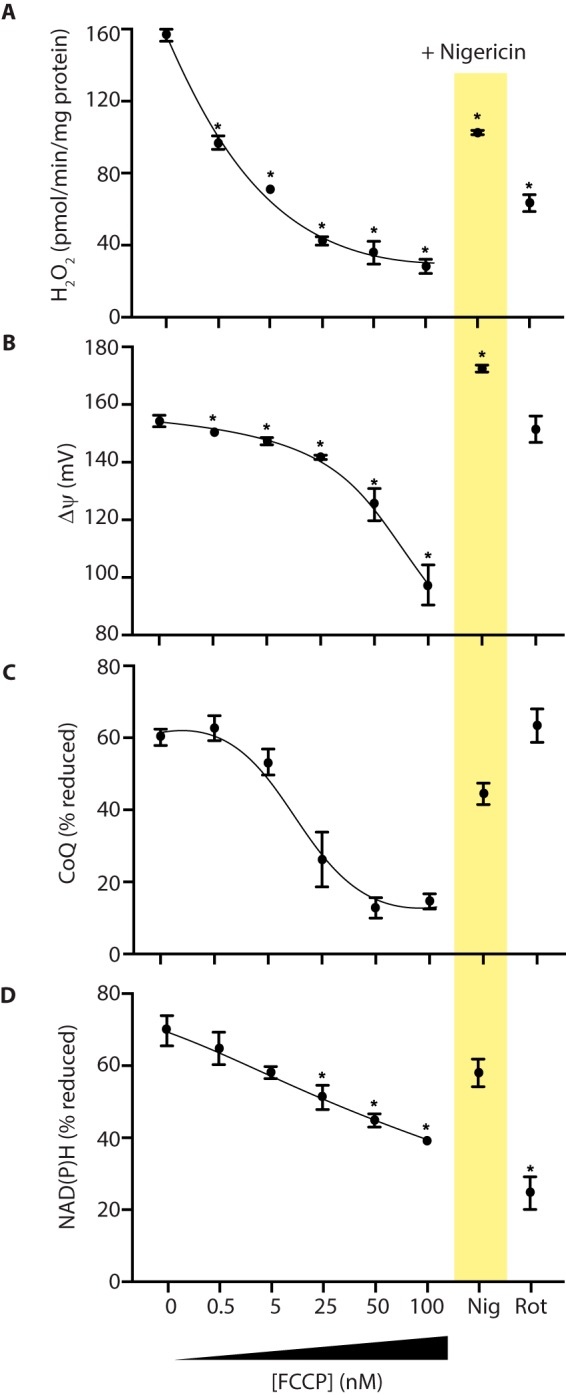
Parallel measurement of mitochondrial H2O2 efflux, Δψ, and the redox status of the NAD(P)H and CoQ pools. Rat heart mitochondria were incubated in the presence of potassium succinate. Where indicated, FCCP, nigericin (1 μm), or rotenone (5 μm) was added, and the indicated variables were measured. A, H2O2 efflux. B, Δψ. C, CoQ redox status. D, NAD(P)H redox status. Data are the mean ± S.E. (error bars) from 3–4 independent mitochondrial preparations. *, p < 0.05 compared with succinate-only conditions by ANOVA.
The addition of succinate to heart mitochondria led to extensive H2O2 production (Fig. 2A), a large Δψ (Fig. 2B), and highly reduced CoQ (Fig. 2C) and NAD(P)H (Fig. 2D) pools. The addition of the complex I inhibitor rotenone decreased H2O2 production without affecting Δψ (Fig. 2B) or the CoQ pool (Fig. 2C), but there was oxidation of the NAD(P)H pool (Fig. 2D). These findings are consistent with H2O2 production originating from complex I by RET (1). To explore the dependence of RET on Δψ and the redox state of the CoQ and NAD(P)H pools, we measured these variables with increasing amounts of the uncoupler FCCP. The gradual decrease in H2O2 production that resulted (Fig. 2A) was associated with a decrease in Δψ (Fig. 2B) and oxidation of the CoQ (Fig. 2C) and NAD(P)H (Fig. 2D) pools.
The matrix pH has been suggested to alter RET directly at complex I, independently of its role as the ΔpH component of Δp (23, 24). Hence, we next used the K+/H+ exchanger nigericin to decrease the matrix pH from ∼7.7 to that of the incubation medium (pH 7.4), thereby abolishing the ΔpH component of Δp. Importantly, the magnitude of Δp will not change, due to a compensatory increase of ∼20 mV in Δψ (Fig. 2B). Nigericin resulted in an oxidation of the CoQ (Fig. 2C) and NAD(P)H pools (Fig. 2D) and a decrease in H2O2 efflux (Fig. 2A). Together, these data show a strong dependence of RET at complex I on the magnitude of Δp and on the CoQ and NAD(P)H redox states.
Effect of the AOX on O2˙̄ production by RET
To analyze the effects of CoQ pool redox state on O2˙̄ production by RET, independently of effects on Δp, we utilized mice expressing the AOX from C. intestinalis (16). AOX transfers electrons from CoQH2 directly to O2, bypassing complex IV, and thus acts as a safety valve to prevent the excessive reduction of the CoQ pool (25, 26). The AOX protein was present in heart mitochondria from AOX+/− knock-in mice (Fig. 3A) and was catalytically active, as respiration in mitochondria from WT mice was inhibited by cyanide, whereas mitochondria from AOX mice continued to respire, but this residual respiration was sensitive to the AOX inhibitor N-propyl gallate (Fig. 3B). Mitochondrial H2O2 efflux during succinate oxidation was decreased by AOX expression as shown previously (16) (Fig. 3C). This was not due to a decrease in Δψ (Fig. 3D); however, the CoQ pool was more oxidized in mitochondria containing AOX (27) (Fig. 3E). These findings show that the expression of AOX affects O2˙̄ production by RET at complex I by oxidizing the CoQ pool and further support the importance of CoQ pool redox state in determining mitochondrial O2˙̄ production by RET.
Figure 3.
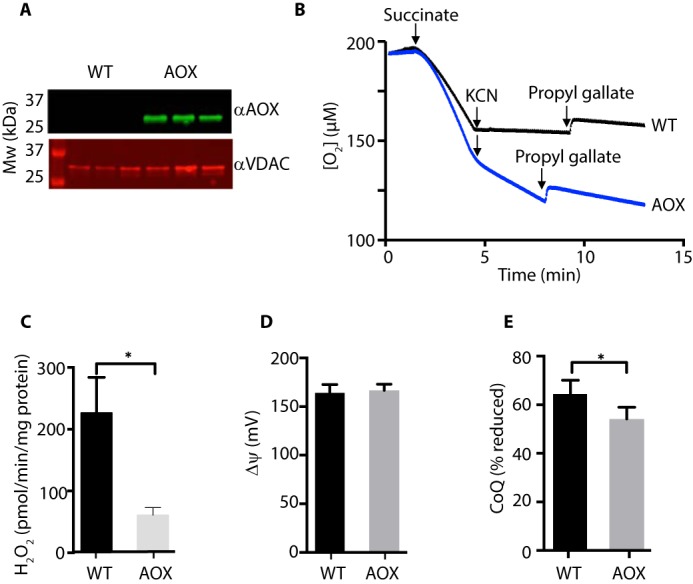
Effect of AOX expression on mitochondrial H2O2 production. A, AOX expression in mouse heart mitochondria. Heart mitochondria from AOX+/− mice or WT littermate controls were analyzed by Western blotting for AOX using the mitochondrial outer membrane protein voltage-dependent anion channel as a loading control. Mitochondria from three separate AOX+/− and WT mice were assessed. B, O2 consumption by mitochondria from AOX+/− and WT mice. Mitochondria (200 μg of protein/ml) were incubated at 37 °C in an oxygen electrode and respiration was initiated by the addition of succinate (10 mm) followed by KCN (1 mm) and propyl gallate (50 μm). Traces are typical of experiments repeated with at least three independent mitochondrial preparations for each condition. C, H2O2 production by heart mitochondria from AOX+/− and WT mice. Mitochondria (200 μg of protein/ml) were incubated with succinate (10 mm), and H2O2 production was assessed. n = 3 (WT) or 6 (AOX). D, Δψ of heart mitochondria from AOX and WT mice. Mitochondria (500 μg of protein/ml) were incubated at 37 °C for 5 min with succinate (10 mm), and Δψ was assessed. n = 4. E, CoQ redox state of heart mitochondria from AOX and WT mice. Mitochondria (1 mg of protein/ml) were incubated at 37 °C for 2 min with succinate (10 mm), and the CoQ redox state was assessed. n = 4. *, p < 0.05; Error bars, S.E.
Effect of [O2] on O2˙̄ production by complex I RET
As the generation of O2˙̄ by mitochondria requires that O2 react with a protein-bound electron carrier, the rate of O2˙̄ production will probably depend on the [O2] (1). To assess this dependence, we measured mitochondrial H2O2 efflux at different [O2] (Fig. 4). As the apparent Km of cytochrome oxidase for O2 is very low (< 1 μm) (28), Δp and the redox state of the CoQ and NADH pools will not vary with [O2]. The assessment of H2O2 efflux at different [O2] relies on the conversion of Amplex Red to its fluorescent product resorufin catalyzed by horseradish peroxidase (HRP); hence, we first demonstrated that the fluorescence of resorufin (Fig. 4A, inset) and the conversion of Amplex Red to resorufin (Fig. 4A) were both independent of [O2]. We then measured mitochondrial H2O2 generation by RET and showed that it was proportional to [O2] and was abolished by excess FCCP (Fig. 4B). In contrast, production of H2O2 by NADH-linked substrates in the presence of rotenone (Fig. 4C) or from complex III upon inhibition of succinate respiration by antimycin (Fig. 4C) also showed increased H2O2 production with [O2], but in this case ROS production reached a plateau as [O2] increased. Thus, production of O2˙̄ by RET at complex I is proportional to [O2].
Figure 4.
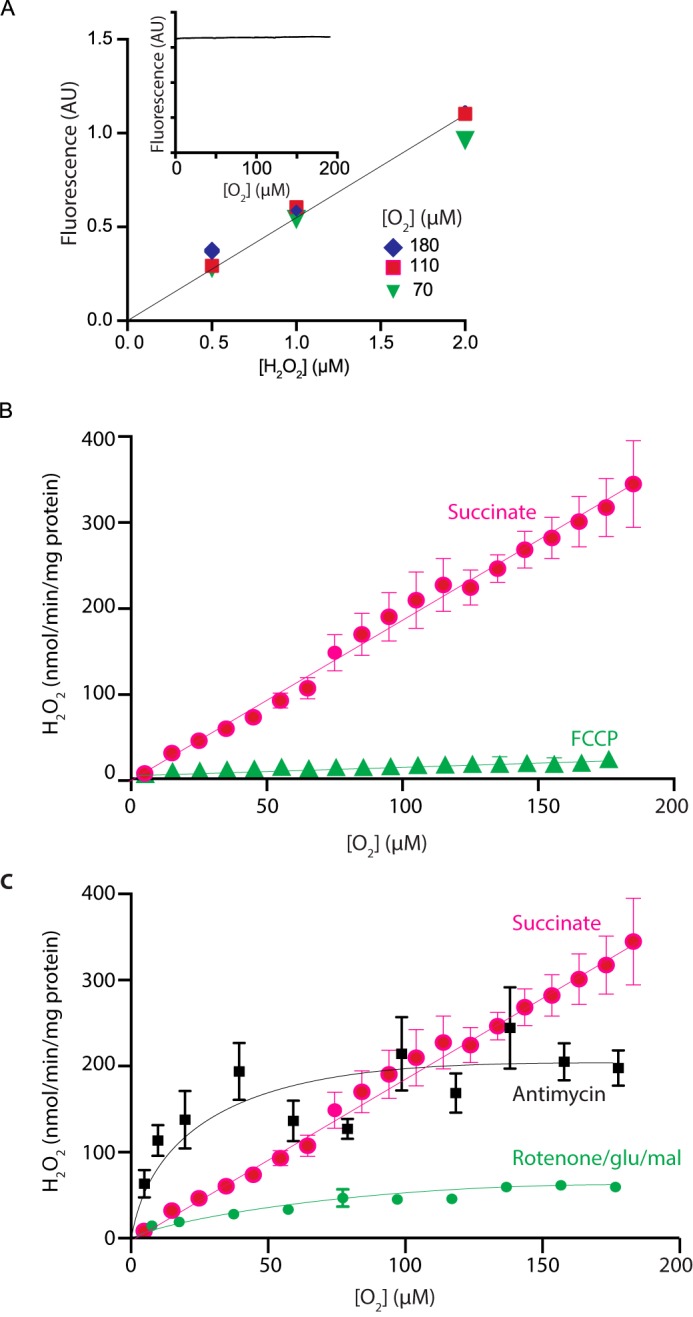
Dependence on mitochondrial H2O2 generation on [O2]. A, conversion of Amplex Red to resorufin by HRP at different [O2]. The [O2] of KCl buffer containing Amplex Red, HRP, and SOD was decreased by bubbling with N2. Then known amounts H2O2 were injected, and fluorescence was measured. The inset shows the fluorescence of resorufin (25 μm), which was added to an incubation of mitochondria respiring on succinate in the presence of rotenone (5 μm), and resorufin fluorescence was measured as the [O2] decreased due to mitochondrial respiration. B, dependence on [O2] of mitochondrial H2O2 generation by RET. Rat heart mitochondria were incubated in an oxygen electrode, and H2O2 generation was measured at various [O2] set by bubbling with N2, and ROS production was measured over a dynamic range of O2 tensions. Where indicated, 500 nm FCCP was present. Data are the mean ± S.E. (error bars). n = 11. C, comparison of [O2] dependence of different modes of mitochondrial H2O2 generation. Mitochondria were assessed as in B, except that the respiratory substrate was glutamate/malate (5 mm each) in the presence of rotenone (4 μg/ml). n = 6, or mitochondria were respiring on succinate with antimycin (1 μm) present. Data are the mean ± S.E. (n = 5) and compared with the trace from C.
Effects of therapeutic compounds on complex I RET
A number of potentially therapeutic compounds are thought to act, at least in part, by decreasing mitochondrial ROS production. Therefore, we set out to assess some of these compounds to determine whether they altered RET at complex I. The compounds tested were as follows: MitoQ, a mitochondria-targeted antioxidant based on ubiquinone (29); decylTPP, which contains the mitochondria-targeting triphenylphosphonium (TPP) cation and which is frequently used as a control compound to correct for nonspecific effects of MitoQ (21); SS31, a peptide composed of d-Arg-Dmt-Lys-Phe-NH2 (30) (where Dmt represents 2,6-dimethyltyrosine), whose therapeutic effects are thought to be due to its interactions with mitochondria; CN-POBS, an inhibitor of mitochondrial O2˙̄ production by RET at complex I (12); and the antidiabetic biguanides metformin and phenformin, which are known to interact with complex I (31, 32).
We first assessed whether these compounds affected H2O2 efflux by RET at complex I. All of the compounds, except SS31, decreased mitochondrial O2˙̄ production by RET to some extent (Fig. 5A). As RET at complex I is very sensitive to the magnitude of Δψ and redox state of the CoQ pool, these compounds may affect RET indirectly by altering these bioenergetic variables rather than by directly interacting with complex I to block RET-dependent O2˙̄ production. Consistent with this interpretation, all of the compounds decreased Δψ (Fig. 5B), and some affected the redox states of the CoQ and NAD(P)H pools (Fig. 5, C and D). Together, these findings suggest that the effects of these compounds on RET at complex I may be indirect due to effects on Δψ and perhaps on the redox states of the NAD(P)H and CoQ pools.
Figure 5.
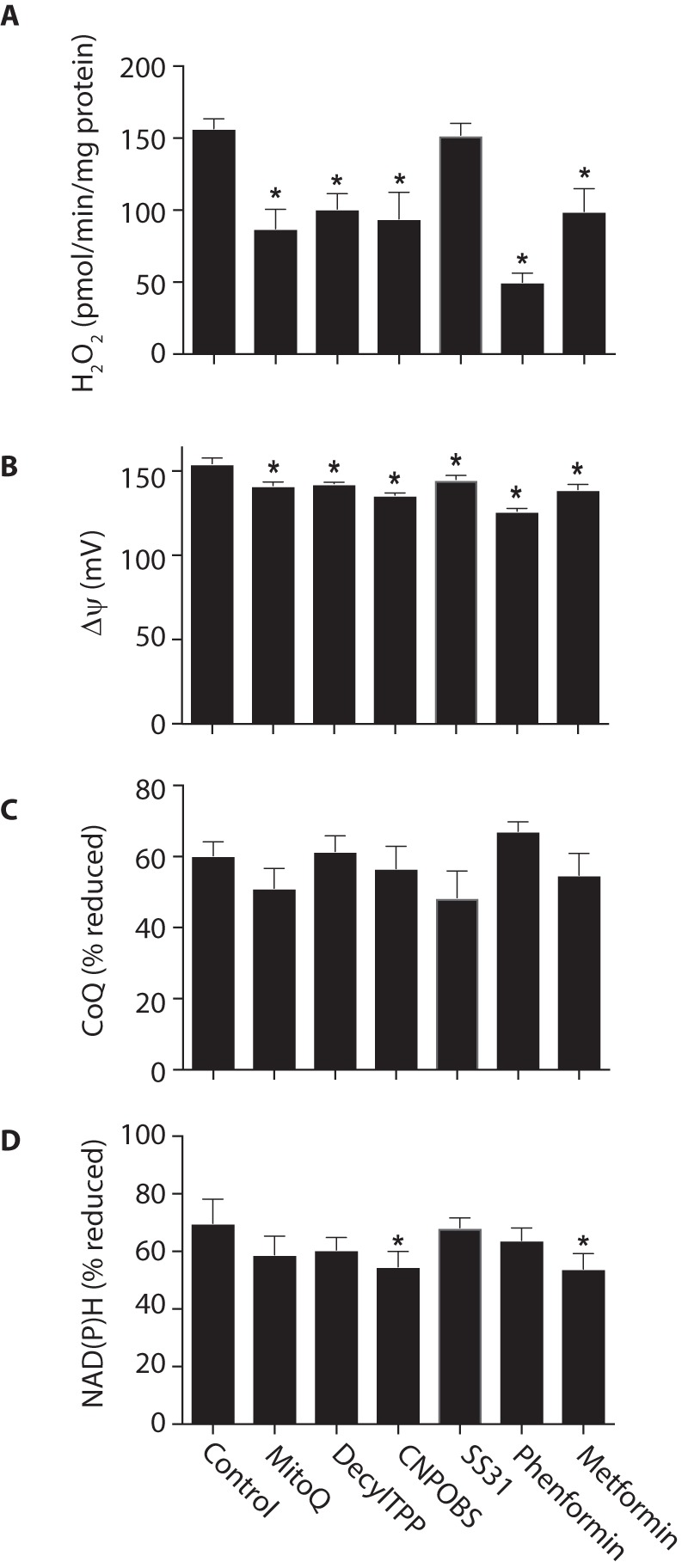
Effects of compounds that interact with mitochondria on H2O2 generation by RET. Rat heart mitochondria were incubated for 10 min in the presence of 10 mm succinate and 1 μm MitoQ, decylTPP, CN-POBS, SS31, phenformin, and metformin and assessed as in Fig. 2. Data are the mean ± S.E. (error bars), n = 4. A, H2O2 efflux. B, Δψ. C, CoQ redox status. D, NAD(P)H redox status. *, p < 0.05.
Dependence of RET on Δψ and the redox state of the CoQ pool
Measuring H2O2 production by mitochondria in parallel with Δψ and the redox state of the CoQ pool indicated that O2˙̄ production by RET at complex I was very sensitive to both variables (Figs. 2 and 3). To illustrate the dependence of RET on Δψ and redox state of the CoQ pool, we replotted the data from Fig. 2 to show H2O2 production as a function of both Δψ and CoQ redox state (Fig. 6). This 3D plot makes evident the very steep dependence of H2O2 production on Δψ and on CoQ redox state, confirming the exquisite sensitivity of O2˙̄ production by RET at complex I to these two physiological variables.
Figure 6.
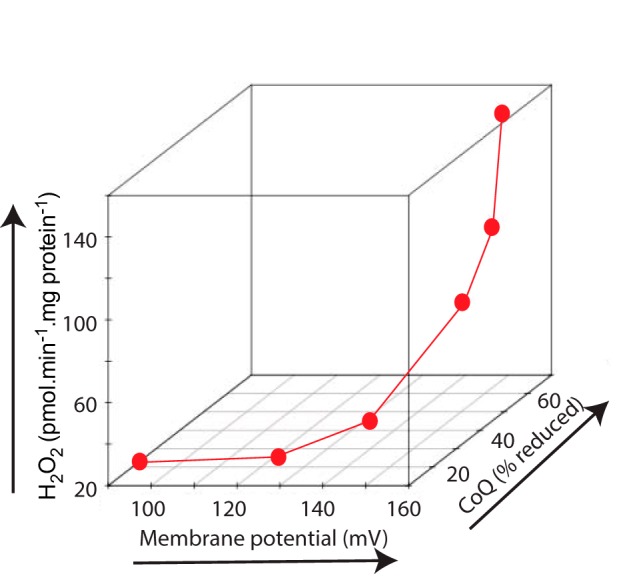
Dependence of mitochondrial H2O2 production by RET upon Δψ and the redox state of the CoQ pool. The data from Fig. 2 (A–C) are plotted together to show the relationship between mitochondrial H2O2 production, Δψ, and CoQ redox state.
The thermodynamic driving force for RET at complex I
Whereas Fig. 6 shows clearly that decreasing Δψ and oxidizing the CoQ pool lowers RET, it does not provide a quantitative basis to allow us to infer whether the effects of nigericin shown in Fig. 2A and those of the various compounds shown in Fig. 5A are due to direct interactions with complex I itself or are indirect effects due to altering the driving forces of RET. Therefore, we next determined how O2˙̄ production during RET depends on the overall thermodynamic driving force across complex I.
The direction of electron flow at complex I is determined by the balance of thermodynamic driving forces across the complex (Fig. 1). During forward electron transfer at complex I, two electrons are passed from NADH to CoQ.
The driving force for the transfer of two electrons from NADH to CoQ is ΔEh.
| (1) |
This driving force, 2ΔEh, is used to pump four protons across the mitochondrial inner membrane against the Δp. Hence, for forward electron movement to occur, the thermodynamic requirement is as follows.
| (2) |
However, when the Δp is high and/or the ΔEh is decreased, RET can occur provided the following is true.
| (3) |
We can thus calculate the thermodynamic driving force (ΔG) for RET, where F is the Faraday constant (33).
| (4) |
Rearranging to express the thermodynamic driving force for RET as a positive number in V gives the following.
| (5) |
From this, we can use the data from the FCCP titration in Fig. 2 to calculate −ΔG/F, the driving force for RET (see “Experimental procedures”). This analysis yields a plot of H2O2production by RET as a function of the thermodynamic driving force across complex I (Fig. 7A). This shows that when −ΔG/F < 0, there is a residual background level of ROS production, but as soon as the driving force for RET passes a threshold and −ΔG/F > 0, there is a dramatic and steep increase in O2˙̄ production by RET. This analysis confirms that the O2˙̄ production by RET requires a sufficient thermodynamic force to reverse electron transport at complex I and further shows the steep dependence of O2˙̄ production on this driving force.
Figure 7.
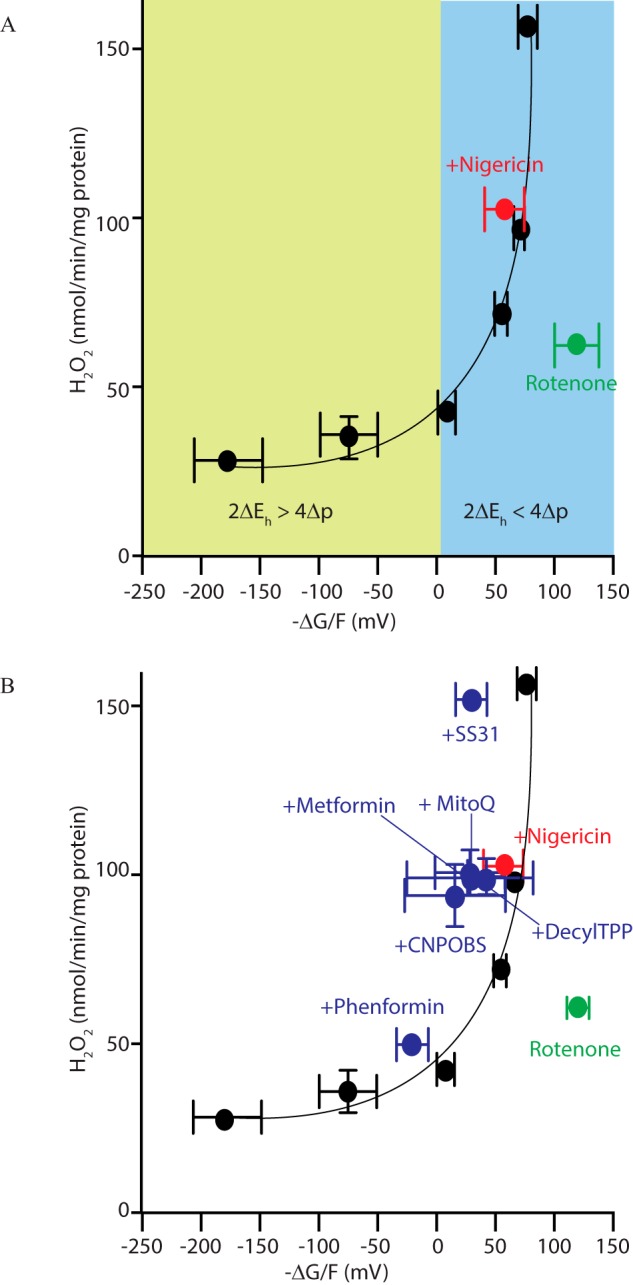
Thermodynamic driving forces across complex I during RET. A, dependence of mitochondrial H2O2 efflux on the thermodynamic driving force for RET across complex I. Data from Fig. 2 were used to calculate −ΔG/F, as this was varied by titration of the uncoupler FCCP (black circles) or in the presence of FCCP (red circle) or rotenone (green circle). Data are the mean ± S.E. (error bars) (n = 3–4). B, the effects of compounds that interact with mitochondria on H2O2 efflux and the thermodynamic driving force for RET. Data from Fig. 5 were used to calculate −ΔG/F, and this was plotted against mitochondrial H2O2 efflux (blue circles), along with the data from A.
A benefit of describing H2O2 production by RET at complex I as a function of its overall thermodynamic driving force is that it enables us to quantify whether compounds that affect O2˙̄ production by RET do so by altering the drivers of this process or by acting directly on complex I. A compound that only affects O2˙̄ production by RET indirectly through altering Δp and ΔEh would lie on the curve shown in Fig. 7A. In contrast, a compound that directly affected complex I independently of the thermodynamic drivers of RET, would lie below this curve. We first applied this analysis to nigericin, which decreases H2O2 production by RET (Fig. 2A) but which also led to a more oxidized CoQ pool (Fig. 2C) without affecting Δp. Carrying out this analysis, including accounting for changes in matrix pH on Eh of the NADH and CoQ pools (see “Calculations”) indicated that the decrease in H2O2 production by nigericin was due to its effects on the CoQ pool redox state (Fig. 7A). In contrast, the decrease of H2O2 production by the complex I inhibitor rotenone (Fig. 2A) was not due to changes in the thermodynamic driving forces for RET, as these data lay below the trend line in Fig. 7A.
If the compounds that affect mitochondrial ROS production assessed in Fig. 5 decrease mitochondrial ROS production independently of the drivers of RET, then they should lie below the trend line shown in Fig. 7A, as was the case for rotenone. When the data from Fig. 5 were analyzed to show the effect of these compounds on the thermodynamic driving forces for RET, the results lay above the trend line, showing the dependence of H2O2 production on the thermodynamic driving forces across complex I (Fig. 7B). Hence, these data suggest that the effects of the compounds analyzed in Fig. 5 on ROS production by RET are more likely to be accounted for by their effects on Δp and/or ΔEh rather than due to specific inhibitory effects on complex I.
Discussion
We investigated the dependence of O2˙̄ production by RET at complex I within isolated mitochondria on Eh of the NAD(P)H and CoQ pools, Δp, matrix pH, and [O2]. This approach confirmed that O2˙̄ production by RET at complex I is favored by a high Δp and a reduced CoQ pool, with exquisite sensitivity to small changes in these two drivers (Fig. 6). This analysis was extended to show that O2˙̄ production by RET at complex I was also highly responsive to small changes in the overall thermodynamic driving force for RET across the complex (Fig. 7A).
Two sites have been proposed for O2˙̄ production by complex I during RET: the FMN of the complex I NADH-binding site (9, 34, 35) or the CoQ-binding site (36, 37). We favor the FMN site as the source of O2˙̄ production by complex I during RET. This is because the penetration of O2 to the CoQ site is difficult to envisage from the structure of complex I (38, 39). In addition, the generation of the negatively charged O2˙̄ from the CoQ site would require its thermodynamically unfavorable formation within the hydrophobic core of the membrane bilayer (38, 39). Furthermore, the FMN site is well established as a source of O2˙̄ production by rotenone-inhibited complex I (38, 39). During this process, NADH reduces the FMN to FMNH−, which is then readily accessed by O2 to form O2˙̄ (38, 39). In addition, any O2˙̄ formed at this site is released directly into the aqueous phase (9, 35, 40). The relationship between [O2] and RET was linear over the physiological [O2] range, consistent with O2˙̄ production being driven by the second-order reaction between O2 and FMNH− on complex I when it is free from bound NAD+ or NADH (9, 35, 40).
During rotenone inhibition, the FMN/FMNH− ratio is set by a rapid pre-equilibration with the matrix NAD+/NADH pool and thereby determines the rate of O2˙̄ production (9, 35). We favor FMNH− as the donor of an electron to O2 for O2˙̄ production by complex I during RET. However, if this is the case then the greater O2˙̄ production during RET compared with rotenone-inhibited complex I has to be explained, because under both conditions, O2˙̄ production is determined by the FMN/FMNH− ratio (9, 23, 24, 37). The most likely reason why RET-driven O2˙̄ production is greater than that upon rotenone inhibition is because the large thermodynamic driving force for electron movement backward through complex I during RET holds the FMN/FMNH− ratio at a more negative Eh than is possible by equilibrium with the NAD+/NADH pool (9). The more negative midpoint potential of the FMN/FMNH− couple (Em,7.5 = −380 mV) (41) compared with the NAD+/NADH couple (Em,7.5 = −335 mV) is consistent with this hypothesis. Other factors that could contribute to the elevated O2˙̄ production during RET compared with rotenone inhibition include differential access of O2 to the FMNH− due to alterations in the NADH and NAD+ binding or differences in the activity of the peroxidases that degrade H2O2 within the mitochondrial matrix. One further consideration is that during O2˙̄ production, FMNH− donates one electron to O2 to form a semiquinone radical, FMN•, which is then thought to rapidly redistribute its unpaired electron throughout the iron–sulfur centers on complex I (40). However, the FMN• radical also reacts very rapidly with O2 to form O2˙̄, so if electron redistribution were slowed during RET, the enhanced lifetime of FMN• would also enhance O2˙̄ production (40).
A further point to note is that the term RET is often interpreted as requiring NAD+ reduction at the FMN site of complex I. This is not the case, as O2˙̄ production by RET at complex I occurs when the NAD+/NADH pool is highly reduced and there is no net electron flow from complex I into this pool (e.g. Fig. 2D). Thus, we favor a model in which O2 reacts with a FMNH− to generate O2˙̄, whereas the FMN still exchanges electrons with the matrix NAD+/NADH pool, although there is no net electron transfer (Fig. 8).
Figure 8.
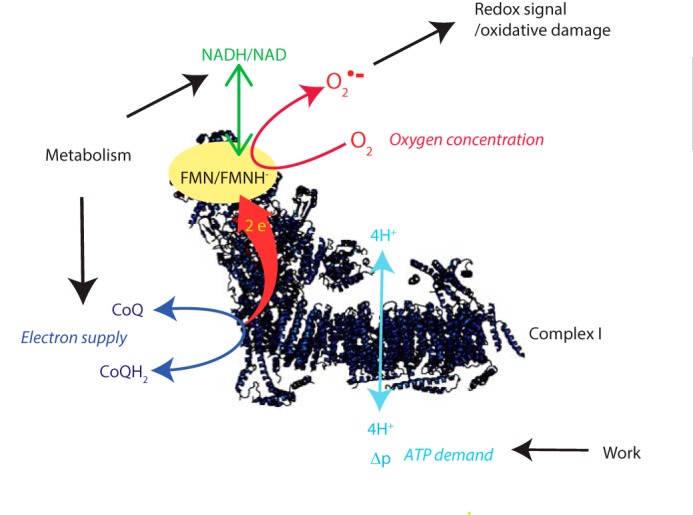
Model of the factors that determine the production of O2˙̄ by RET at complex I.
The generation of O2˙̄ by RET at complex I could be described as a function of the thermodynamic driving forces, Δp and ΔEh, across the complex. As well as illustrating the factors that drive RET, this analysis allowed us to integrate the effects of the forces driving RET. Applying this to the decrease in RET when the matrix pH decreased suggested that the lower rate of RET could be accounted for by the change in the ΔEh between the CoQ and NADH pools and may not be due to a direct effect of pH on complex I itself (24). Furthermore, this approach suggests that compounds such as MitoQ and metformin that affect mitochondrial ROS metabolism may do so indirectly, rather than by specific interactions with complex I. However, it is important to note that the calculation of −ΔG/F requires a number of assumptions and combines several technically challenging experimental measurements; hence, systematic errors will affect accuracy and precision. In addition, we have assumed that the CoQ pool interacts to the same extent with all complexes, and any effects of supercomplex formation on this were not considered (42). Nevertheless, our work does indicate novel approaches for determining how compounds impact mitochondrial ROS production and has implications for the interpretation of experiments using these compounds. For example, MitoQ is widely used in vitro and in vivo, where it acts as a chain-breaking antioxidant decreasing oxidative damage (29, 43). As the excessive accumulation of hydrophobic TPP compounds disrupts mitochondrial function (44, 45), controls with compounds with matched physicochemical properties are essential to correct for nonspecific effects. Thus, some “antioxidant” effects of lipophilic cations may be due to mild disruption of Δp that lowers mitochondrial O2˙̄ production by RET. Similarly, some of the beneficial effects of less hydrophobic cations, such as metformin in vivo, may be associated with limiting O2˙̄ production by RET, in addition to stimulating AMP-activated protein kinase by inhibiting complex I (46, 47).
Mitochondrial O2˙̄ production by RET was initially investigated in vitro, and at that time its relevance to in vivo physiology was not considered (10). Recently, mitochondrial ROS production by RET has been demonstrated in multiple situations in vivo. For example, during ischemia, high levels of succinate accumulate, and it is oxidized upon reperfusion to drive a burst of ROS via RET (14, 48). More generally, RET at complex I occurs as a redox-signaling pathway in inflammation (13), contributes to lifespan in flies (49), and is part of the oxygen-sensing mechanism of the carotid body (49). The linear dependence of ROS production by RET on O2 (Fig. 4), which has been shown previously by others (50–53), further adds to its appeal as a potential component of the carotid body O2 sensor (49). One possibility is that O2˙̄ production by RET at complex I accounts for most of the redox signaling from the mitochondrion to the rest of the cell (3–6). The appeal of RET as a mitochondrial redox signal is illustrated in Figs. 6 and 7, which show the tremendous sensitivity of RET to Δp and the redox status of the CoQ pool. The magnitude of Δp is directly linked to ATP demand, whereas the redox state of the CoQ pool reflects electron supply to the respiratory chain. Thus, RET provides a sensitive mechanism for the real-time feedback of the most critical aspects of mitochondrial status to the rest of the organelle and to the cell (Fig. 8).
In summary, we have provided a thermodynamic underpinning to O2˙̄ production by RET at complex I. This analysis highlights the potential for this mechanism to play a key role in mitochondrial redox signaling and demonstrates how to investigate the effects of RET on various physiological and pathological processes.
Experimental procedures
Mitochondria isolation
All procedures were performed in accordance with the UK Guide for the Care and Use of Laboratory Animals (PPL: 70/7538). Rat hearts were collected from 10–12-week-old female Wistar rats (Charles River). C57Bl/6 mice carrying a single copy of the C. intestinalis AOX gene in the Rosa26 locus were generated as described (16). AOX mice and their WT littermate controls of both sexes were used at 8–12 weeks of age to prepare heart mitochondria. Rats were killed by stunning followed by cervical dislocation. Mice were killed by cervical dislocation only. Hearts were removed into ice-cold STE buffer (250 mm sucrose, 5 mm Tris-HCl (pH 7.4, KOH), and 1 mm K-EGTA, supplemented with 0.1% (w/v) fatty acid–free BSA. Heart mitochondria were isolated by homogenization and differential centrifugation (700 × g for 3 min; 3 × 5,500 × g for 10 min) at 4 °C. Mitochondrial protein content was determined using the bicinchoninic acid assay with BSA as a standard.
Hydrogen peroxide efflux
H2O2 efflux from mitochondria was assayed using a plate reader fluorometer (SpectraMax GeminiXS, Molecular Devices; used at medium sensitivity). Resorufin (the product of Amplex Red oxidation) fluorescence was detected using λex = 570 nm and λem = 585 nm. Mitochondria (2 mg protein/ml) were incubated with 2.5 μm Amplex Red (Invitrogen), 5 units/ml HRP in STE with 10 mm potassium succinate at 37 °C. H2O2 production rates were linear over 10 min but were measured from 0 to 2 min to facilitate comparison with other measurements. The H2O2 response was calibrated using freshly prepared H2O2 standards (ϵ240 = 43.5 m−1 cm−1) that were added sequentially to mitochondrial incubations lacking only succinate to generate a linear calibration curve. Mitochondria-targeted test compounds were added 30 s before measurement.
Mitochondrial Δϵ
Mitochondrial Δϵ was measured by the uptake of radiolabeled [3H]TPMP as described (21). Mitochondria (2 mg protein/ml) were incubated at 37 °C with 10 mm succinate, 500 nm TPMP supplemented with [3H]TPMP (50 nCi/ml) with the test compounds for 2 min in 250 μl of medium in Eppendorf tubes. Mitochondria were pelleted by centrifugation (10,000 × g for 30 s). Supernatant (200 μl) was removed, and the pellets were dried with a rolled-up tissue and then solubilized in 40 μl of 20% (v/v) Triton X-100. Both the supernatant and pellets were then added to scintillant (Ultima-Gold liquid scintillant, PerkinElmer Life Sciences) and incubated for 1 h at room temperature and then vortexed, and [3H]TPMP content was assessed using a TriCarb LCS counter (PerkinElmer Life Sciences) counter with appropriate quench controls. To calculate the Δϵ, first the accumulation ratio was calculated assuming a mitochondrial matrix volume of 0.6 μl/mg protein, and the mitochondrial Δϵ was then calculated from the Nernst equation, assuming 40% binding of TPMP and that this was independent of mitochondrial Δϵ and consistent across all conditions (21).
CoQ extraction and detection
Mitochondria (2 mg of protein/ml) were incubated in 500–700 μl of STE with 10 mm potassium succinate at 37 °C on a shaking heat block for 2 min. At the end of the incubation, mitochondria were rapidly pelleted by centrifugation (10,000 × g for 30 s), the supernatant was removed, and pellets were snap-frozen in a dry ice/ethanol bath and stored at −80 °C until analysis. Immediately before HPLC analysis, the pellets were homogenized in 0.5 ml of ice-cold, nitrogen-purged 1-propanol in an ice-cold glass-on-glass homogenizer, 100 μl of ice-cold H2O was added, and the samples were centrifuged (16,000 × g at 4 °C for 5 min). Supernatants (200 μl) were immediately analyzed by HPLC on a 150 × 4.6-mm, 3 μ Hypersil ODS column (Thermo). Solvent A was MeOH, 50 mm NaClO4; solvent B was EtOH, 50 mm NaClO4. The gradient was 60% to 50% A over 15 min at a flow rate of 0.8 ml/min at 45 °C. Identity was established by retention time compared with authentic standards at the absorbance maxima for CoQ and CoQH2 (260 and 290 nm, respectively). CoQ9 redox state was determined from the peak areas of ubiquinone and ubiquinol at 292.5 nm, the isosbestic point for oxidized and reduced CoQ9. The percentage reduction of the CoQ9 pool was calculated as area of the reduced peak divided by the sum of both peak areas. Control incubations under conditions designed to maximally oxidize and reduce the CoQ pool demonstrated that this approach accurately reported the CoQ redox state, as the percentage reduction increased to ∼80% in the presence of cyanide or anoxia, whereas inhibition of the respiratory chain with malonate decreased the percentage reduction to ∼20%.
Redox state of mitochondrial NAD(P)H/NAD(P) pools
The redox state of the NAD(P)H/NAD(P)+ pool was determined by monitoring NAD(P)H fluorescence using a plate reader fluorimeter (SpectraMax GeminiXS; Molecular Devices) using λex = 365 nm and λem = 450 nm. Mitochondria were incubated as described for measuring H2O2 efflux. The signal was calibrated by subtraction of background fluorescence (mitochondria with no additions), and maximal reduction of the pool was set by incubating mitochondria with 5 mm malate and 5 μm rotenone for 5 min.
Combined respirometry and H2O2 production measurements
Combined respiration and H2O2 production by mitochondria was assessed using an Oxygraph2K (O2K) respirometer (Oroboros, Innsbruck, Austria) with a fluorescence LED module attachment. To assess the effect of O2 concentration on H2O2 production, mitochondria (200–250 μg of protein/ml) were suspended in 2 ml of KCl buffer (120 mm KCl, 10 mm Hepes, 1 mm EGTA, pH 7.2) supplemented with 50 units/ml SOD, 4 units/ml HRP, 0.2 mg/ml fatty acid–free BSA, 25 μm Amplex Red with stirring at 37 °C. Respiration was initiated by the addition of either 5 mm glutamate and 5 mm malate; 10 mm succinate; or 10 mm succinate with 5 μm rotenone. Where indicated, incubations were supplemented at the start with 5 μm rotenone, 500 nm FCCP, 1 μm antimycin A, or 1 μm nigericin. The concentration of O2 was adjusted by bubbling the buffer with N2. Amplex Red fluorescence was measured via the O2K fluorometer, and the corresponding voltage changes were calibrated via titration of known amounts of H2O2 (500 nm to 5 μm) in the presence of mitochondria, SOD, fatty acid–free BSA, HRP, and Amplex Red.
Western blotting
Mitochondrial pellets (∼250 μg of protein) were solubilized on ice in 50 μl of lysis buffer (100 mm Tris, 300 mm NaCl, 0.05% Nonidet P-40, pH 7.4, supplemented with protease and phosphatase inhibitors (Roche Applied Science). Protein was then quantified by the BCA assay and diluted in 4× loading buffer (Invitrogen), and 10 μg of protein was separated by SDS-PAGE on a 10% gel and transferred to polyvinylidene difluoride membrane. Membranes were incubated with a 1:20,000 dilution of rabbit serum raised against two AOX peptides (FKIETNDSTDEPNIEVENFPC and CVNHDLGSRKPDEQNPYPPGQ (49)) and a mouse monoclonal antibody against the voltage-dependent anion channel (1:1,000; Abcam ab14734)) and visualized using a LI-COR Odyssey flatbed scanner with anti-mouse and anti-rabbit secondary antibodies conjugated to IRDye 680RD and IRDye 800CW, respectively.
Calculations
This section describes calculations required to determine the thermodynamic driving force for RET from the data in Figs. 2 and 5 to generate the graphs shown in Fig. 7. The thermodynamic driving force for RET is derived from Equation 5. In mV, it is as follows.
| (6) |
In the presence of nigericin, Δp is unchanged, whereas ΔpH = 0. Hence, ΔpH = Δψ (+nigericin) − Δψ (−nigericin). From Fig. 2C, 61.5ΔpH = −18.1 mV and the matrix pH = 7.7.
| (7) |
As the matrix pH is 7.7, Eh for the NAD+/NADH couple in mV is as follows.
| (8) |
In the presence of nigericin, the matrix pH will be 7.4, and under those conditions, the following will be true.
| (9) |
As the matrix pH is 7.7, Eh for the CoQ/CoQH2 couple in mV is as follows.
| (10) |
In the presence of nigericin, the matrix pH will be 7.4, and under those conditions, the following will be true.
| (11) |
Statistical analysis and experimental design
Data were expressed as mean ± S.E., with p values calculated using a two-tailed Student's t test for pairwise comparisons whereas one-way analysis of variance (ANOVA) followed by Tukey's post hoc test was used for multiple comparisons. Statistical analyses were performed using GraphPad Prism version 7 software.
Author contributions
E. L. R. and A. R. H. designed and carried out the experiments, with assistance from T. A. P. S. E. carried out the CoQ assays. M. S. and C. V. provided the AOX mice. A. M. J. provided advice and helped with data interpretation. M. P. M. directed the project and wrote the manuscript, with assistance from all other authors.
Acknowledgment
We are grateful to Judy Hirst for helpful discussions and suggestions.
This work was supported by the Medical Research Council UK (Grants MC_U105663142 (to M. P. M.) and MC_UU_00015/5 (to C. V.)) and by a Wellcome Trust Investigator award (110159/Z/15/Z) (to M. P. M). The authors declare that they have no conflicts of interest with the contents of this article.
- O2˙̄
- superoxide
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- RET
- reverse electron transport
- CoQ
- coenzyme Q
- Δp
- proton-motive force
- Δψ
- membrane potential
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- AOX
- alternative oxidase
- TPMP
- methyltriphenylphosphonium
- HRP
- horseradish peroxidase
- TPP
- triphenylphosphonium
- ANOVA
- analysis of variance
- CN-POBS
- N-cyclohexyl-4-(4-nitrophenoxy)benzenesulfonamide.
References
- 1. Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong H. S., Dighe P. A., Mezera V., Monternier P. A., and Brand M. D. (2017) Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 292, 16804–16809 10.1074/jbc.R117.789271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Collins Y., Chouchani E. T., James A. M., Menger K. E., Cochemé H. M., and Murphy M. P. (2012) Mitochondrial redox signalling at a glance. J. Cell Sci. 125, 801–806 10.1242/jcs.098475 [DOI] [PubMed] [Google Scholar]
- 4. Holmström K. M., and Finkel T. (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell. Biol. 15, 411–421 10.1038/nrm3801 [DOI] [PubMed] [Google Scholar]
- 5. Finkel T. (2011) Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15 10.1083/jcb.201102095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Janssen-Heininger Y. M., Mossman B. T., Heintz N. H., Forman H. J., Kalyanaraman B., Finkel T., Stamler J. S., Rhee S. G., and van der Vliet A. (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic. Biol. Med. 45, 1–17 10.1016/j.freeradbiomed.2008.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goncalves R. L., Quinlan C. L., Perevoshchikova I. V., Hey-Mogensen M., and Brand M. D. (2015) Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 290, 209–227 10.1074/jbc.M114.619072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. St-Pierre J., Buckingham J. A., Roebuck S. J., and Brand M. D. (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 277, 44784–44790 10.1074/jbc.M207217200 [DOI] [PubMed] [Google Scholar]
- 9. Pryde K. R., and Hirst J. (2011) Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 286, 18056–18065 10.1074/jbc.M110.186841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hinkle P. C., Butow R. A., Racker E., and Chance B. (1967) Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavin-cytochrome β region of the respiratory chain of beef heart submitochondrial particles. J. Biol. Chem. 242, 5169–5173 [PubMed] [Google Scholar]
- 11. Fernández-Aguera M. C., Gao L., González-Rodríguez P., Pintado C. O., Arias-Mayenco I., García-Flores P., García-Pergañeda A., Pascual A., Ortega-Sáenz P., and López-Barneo J. (2015) Oxygen sensing by arterial chemoreceptors depends on mitochondrial complex I signaling. Cell Metab. 22, 825–837 10.1016/j.cmet.2015.09.004 [DOI] [PubMed] [Google Scholar]
- 12. Orr A. L., Ashok D., Sarantos M. R., Shi T., Hughes R. E., and Brand M. D. (2013) Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic. Biol. Med. 65, 1047–1059 10.1016/j.freeradbiomed.2013.08.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mills E. L., Kelly B., Logan A., Costa A. S. H., Varma M., Bryant C. E., Tourlomousis P., Däbritz J. H., Gottlieb E., Latorre I., Corr S. C., McManus G., Ryan D., Jacobs H. T., Szibor M., Xavier R. J., Braun T., Frezza C., Murphy M. P., and O'Neill L. A. (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e13 10.1016/j.cell.2016.08.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chouchani E. T., Pell V. R., James A. M., Work L. M., Saeb-Parsy K., Frezza C., Krieg T., and Murphy M. P. (2016) A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263 10.1016/j.cmet.2015.12.009 [DOI] [PubMed] [Google Scholar]
- 15. Chouchani E. T., Methner C., Nadtochiy S. M., Logan A., Pell V. R., Ding S., James A. M., Cochemé H. M., Reinhold J., Lilley K. S., Partridge L., Fearnley I. M., Robinson A. J., Hartley R. C., Smith R. A., et al. (2013) Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 19, 753–759 10.1038/nm.3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szibor M., Dhandapani P. K., Dufour E., Holmström K. M., Zhuang Y., Salwig I., Wittig I., Heidler J., Gizatullina Z., Gainutdinov T., Fuchs H., Gailus-Durner V., de Angelis M. H., Nandania J., Velagapudi V., et al. (2017) Broad AOX expression in a genetically tractable mouse model does not disturb normal physiology. Dis. Model. Mech. 10, 163–171 10.1242/dmm.027839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bienert G. P., Møller A. L., Kristiansen K. A., Schulz A., Møller I. M., Schjoerring J. K., and Jahn T. P. (2007) Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 282, 1183–1192 10.1074/jbc.M603761200 [DOI] [PubMed] [Google Scholar]
- 18. Bienert G. P., Schjoerring J. K., and Jahn T. P. (2006) Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 1758, 994–1003 10.1016/j.bbamem.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 19. Miller E. W., Dickinson B. C., and Chang C. J. (2010) Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 15681–15686 10.1073/pnas.1005776107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang B., Zhao D., and Verkman A. S. (2006) Evidence against functionally significant aquaporin expression in mitochondria. J. Biol. Chem. 281, 16202–16206 10.1074/jbc.M601864200 [DOI] [PubMed] [Google Scholar]
- 21. Ross M. F., Prime T. A., Abakumova I., James A. M., Porteous C. M., Smith R. A. J., and Murphy M. P. (2008) Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem. J. 411, 633–645 10.1042/BJ20080063 [DOI] [PubMed] [Google Scholar]
- 22. Murphy M. P. (2012) Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 16, 476–495 10.1089/ars.2011.4289 [DOI] [PubMed] [Google Scholar]
- 23. Lambert A. J., and Brand M. D. (2004) Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Biol. Chem. 279, 39414–39420 10.1074/jbc.M406576200 [DOI] [PubMed] [Google Scholar]
- 24. Lambert A. J., and Brand M. D. (2004) Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 382, 511–517 10.1042/BJ20040485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. El-Khoury R., Dufour E., Rak M., Ramanantsoa N., Grandchamp N., Csaba Z., Duvillié B., Bénit P., Gallego J., Gressens P., Sarkis C., Jacobs H. T., and Rustin P. (2013) Alternative oxidase expression in the mouse enables bypassing cytochrome c oxidase blockade and limits mitochondrial ROS overproduction. PLoS Genet. 9, e1003182 10.1371/journal.pgen.1003182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El-Khoury R., Kemppainen K. K., Dufour E., Szibor M., Jacobs H. T., and Rustin P. (2014) Engineering the alternative oxidase gene to better understand and counteract mitochondrial defects: state of the art and perspectives. Br. J. Pharmacol. 171, 2243–2249 10.1111/bph.12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dry I. B., Moore A. L., Day D. A., and Wiskich J. T. (1989) Regulation of alternative pathway activity in plant mitochondria: nonlinear relationship between electron flux and the redox poise of the quinone pool. Arch. Biochem. Biophys. 273, 148–157 10.1016/0003-9861(89)90173-2 [DOI] [PubMed] [Google Scholar]
- 28. Wikstrom M., Krab K., and Saraste M. (1981) Cytochrome Oxidase: A Synthesis, Academic Press, London [Google Scholar]
- 29. Kelso G. F., Porteous C. M., Coulter C. V., Hughes G., Porteous W. K., Ledgerwood E. C., Smith R. A., and Murphy M. P. (2001) Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596 10.1074/jbc.M009093200 [DOI] [PubMed] [Google Scholar]
- 30. Szeto H. H., and Schiller P. W. (2011) Novel therapies targeting inner mitochondrial membrane–from discovery to clinical development. Pharm. Res. 28, 2669–2679 10.1007/s11095-011-0476-8 [DOI] [PubMed] [Google Scholar]
- 31. Bridges H. R., Jones A. J., Pollak M. N., and Hirst J. (2014) Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 462, 475–487 10.1042/BJ20140620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Owen M. R., Doran E., and Halestrap A. P. (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348, 607–614 10.1042/bj3480607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nicholls D. G., and Ferguson S. J. (2013) Bioenergetics 4, Academic Press, London [Google Scholar]
- 34. Hirst J., King M. S., and Pryde K. R. (2008) The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 36, 976–980 10.1042/BST0360976 [DOI] [PubMed] [Google Scholar]
- 35. Kussmaul L., and Hirst J. (2006) The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7607–7612 10.1073/pnas.0510977103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lambert A. J., Buckingham J. A., and Brand M. D. (2008) Dissociation of superoxide production by mitochondrial complex I from NAD(P)H redox state. FEBS Lett. 582, 1711–1714 10.1016/j.febslet.2008.04.030 [DOI] [PubMed] [Google Scholar]
- 37. Lambert A. J., Buckingham J. A., Boysen H. M., and Brand M. D. (2008) Diphenyleneiodonium acutely inhibits reactive oxygen species production by mitochondrial complex I during reverse, but not forward electron transport. Biochim. Biophys. Acta 1777, 397–403 10.1016/j.bbabio.2008.03.005 [DOI] [PubMed] [Google Scholar]
- 38. Efremov R. G., Baradaran R., and Sazanov L. A. (2010) The architecture of respiratory complex I. Nature 465, 441–445 10.1038/nature09066 [DOI] [PubMed] [Google Scholar]
- 39. Zhu J., Vinothkumar K. R., and Hirst J. (2016) Structure of mammalian respiratory complex I. Nature 536, 354–358 10.1038/nature19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Birrell J. A., Yakovlev G., and Hirst J. (2009) Reactions of the flavin mononucleotide in complex I: a combined mechanism describes NADH oxidation coupled to the reduction of APAD+, ferricyanide, or molecular oxygen. Biochemistry 48, 12005–12013 10.1021/bi901706w [DOI] [PubMed] [Google Scholar]
- 41. Sled V. D., Rudnitzky N. I., Hatefi Y., and Ohnishi T. (1994) Thermodynamic analysis of flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I). Biochemistry 33, 10069–10075 10.1021/bi00199a034 [DOI] [PubMed] [Google Scholar]
- 42. Lapuente-Brun E., Moreno-Loshuertos R., Acin-Pérez R., Latorre-Pellicer A., Colás C., Balsa E., Perales-Clemente E., Quirós P. M., Calvo E., Rodríguez-Hernández M. A., Navas P., Cruz R., Carracedo Á., López-Otín C., Pérez-Martos A., et al. (2013) Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570 10.1126/science.1230381 [DOI] [PubMed] [Google Scholar]
- 43. Smith R. A., and Murphy M. P. (2010) Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. NY Acad. Sci. 1201, 96–103 10.1111/j.1749-6632.2010.05627.x [DOI] [PubMed] [Google Scholar]
- 44. Reily C., Mitchell T., Chacko B. K., Benavides G., Murphy M. P., and Darley-Usmar V. (2013) Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 1, 86–93 10.1016/j.redox.2012.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. James A. M., Sharpley M. S., Manas A. R., Frerman F. E., Hirst J., Smith R. A., and Murphy M. P. (2007) Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 282, 14708–14718 10.1074/jbc.M611463200 [DOI] [PubMed] [Google Scholar]
- 46. Wheaton W. W., Weinberg S. E., Hamanaka R. B., Soberanes S., Sullivan L. B., Anso E., Glasauer A., Dufour E., Mutlu G. M., Budigner G. S., and Chandel N. S. (2014) Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 3, e02242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Barzilai N., Crandall J. P., Kritchevsky S. B., and Espeland M. A. (2016) Metformin as a tool to target aging. Cell Metab. 23, 1060–1065 10.1016/j.cmet.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chouchani E. T., Pell V. R., Gaude E., Aksentijević D., Sundier S. Y., Robb E. L., Logan A., Nadtochiy S. M., Ord E. N. J., Smith A. C., Eyassu F., Shirley R., Hu C. H., Dare A. J., James A. M., et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scialò F., Sriram A., Fernández-Ayala D., Gubina N., Lõhmus M., Nelson G., Logan A., Cooper H. M., Navas P., Enríquez J. A., Murphy M. P., and Sanz A. (2016) Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 23, 725–734 10.1016/j.cmet.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fernandez-Ayala D. J., Sanz A., Vartiainen S., Kemppainen K. K., Babusiak M., Mustalahti E., Costa R., Tuomela T., Zeviani M., Chung J., O'Dell K. M., Rustin P., and Jacobs H. T. (2009) Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab. 9, 449–460 10.1016/j.cmet.2009.03.004 [DOI] [PubMed] [Google Scholar]
- 51. Vinogradov A. D., and Grivennikova V. G. (2005) Generation of superoxide-radical by the NADH:ubiquinone oxidoreductase of heart mitochondria. Biochemistry 70, 120–127 [DOI] [PubMed] [Google Scholar]
- 52. Stepanova A., Konrad C., Guerrero-Castillo S., Manfredi G., Vannucci S., Arnold S., and Galkin A. (2018) Deactivation of mitochondrial complex I after hypoxia–ischemia in the immature brain. J. Cereb. Blood Flow Metab., in press 10.1177/0271678X18770331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guarás A., Perales-Clemente E., Calvo E., Acín-Pérez R., Loureiro-Lopez M., Pujol C., Martínez-Carrascoso I., Nuñez E., García-Marqués F., Rodríguez-Hernández M. A., Cortés A., Diaz F., Pérez-Martos A., Moraes C. T., Fernández-Silva P., et al. (2016) The CoQH2/CoQ ratio serves as a sensor of respiratory chain efficiency. Cell Rep. 15, 197–209 10.1016/j.celrep.2016.03.009 [DOI] [PubMed] [Google Scholar]