

Abstract
A series of phosphoramidate-based prostate specific membrane antigen (PSMA) inhibitors of increasing lipophilicity were synthesized (4, 5, and 6), and their fluorine-18 analogs were evaluated for use as positron emission tomography (PET) imaging agents for prostate cancer. To gain insight into their modes of binding, they were also cocrystallized with the extracellular domain of PSMA. All analogs exhibited irreversible binding to PSMA with IC50 values ranging from 0.4 to 1.3 nM. In vitro assays showed binding and rapid internalization (80–95%, 2 h) of the radiolabeled ligands in PSMA(+) cells. In vivo distribution demonstrated significant uptake in CWR22Rv1 (PSMA(+)) tumor, with tumor to blood ratios of 25.6:1, 63.6:1, and 69.6:1 for [18F]4, [18F] 5, and [18F]6, respectively, at 2 h postinjection. Installation of aminohexanoic acid (AH) linkers in the phosphoramidate scaffold improved their PSMA binding and inhibition and was critical for achieving suitable in vivo imaging properties, positioning [18F]5 and [18F]6 as favorable candidates for future prostate cancer imaging clinical trials.
Graphical Abstract
INTRODUCTION
Prostate specific membrane antigen (PSMA) is an ideal cell surface biomarker and has been extensively pursued as a target in the development of imaging and therapeutic agents for prostate cancer (PCa).1–9 In the past several years, radiolabeled PSMA-targeted small molecules have successfully detected prostate tumor xenografts in mouse models and some have advanced to clinical studies.10 These small molecular scaffolds exhibit high affinity and specificity for PSMA, some of which rapidly internalize into the PSMA(+) tumor cells, overcoming the in vivo pharmacokinetic drawbacks presented by antibodies.1,11 Several urea-based inhibitors pioneered by Kozikowski and Pomper have been labeled with various isotopes such as 111In, 99mTc, 123I for SPECT12,13 and 68Ga, 18F, 124I for PET11,14,15 imaging. Some of these urea-based inhibitors (e.g., [18F]DCFBC and [18F]DCFPyL) have been translated to human imaging in clinical trials.16,17 While the pharmacokinetic profile and imaging with these urea-based agents appear superior to antibody-based diagnostics, the reversible mode of binding to PSMA may explain the observed tumor washout over several hours.18
In parallel, we developed a class of phosphoramidate compounds that exhibit irreversible or slow-reversible binding to PSMA, depending upon the structure, and that have shown similar promise for detection of PSMA(+) cells and tumors in mouse models.19–22 On the basis of their mode of binding to PSMA, we envisioned that this would result in reduced tumor washout. In PSMA(+) cells, we found that the mode of binding exhibited a significant effect upon cell uptake and internalization.23 Furthermore, when conjugated to either 18F-labeled pendent groups22 or chelators bearing 99mTc,21 these compounds displayed consistent uptake and retention in PSMA(+) tumors in mice with minimal washout over several hours.
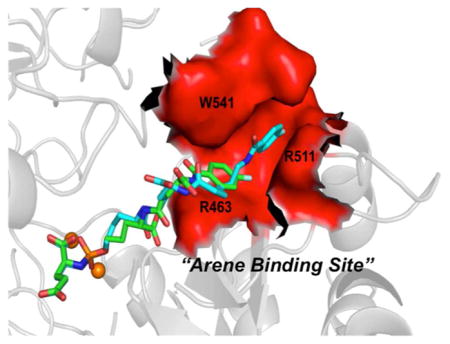
More recently, we reported a unique phosphoramidate-based PSMA inhibitor as a promising candidate for PET imaging of PCa (Figure 1).24 The 18F-labeled analogue [18F]5 displayed rapid uptake (2.35% ID/g at 1 h) and retention (2.35% ID/g at 4 h) in PSMA(+) CW22Rv1 tumor xenografts in mice with an exceptional tumor-to-blood ratio of 265:1 at 4 h postinjection. An important feature of 5, revealed by X-ray crystallography, was the π-stacking and π–cationic interaction of its fluorobenzamido (FB) ring with the Arg511/Try541 and Arg 463 residues, collectively known as the arene-binding site (ABS) located at the entrance PSMA’s active site. This additional unique interaction was thought to be responsible for enhanced PSMA affinity and favorable in vivo characteristics of this radiotracer compared to our previous reported analogues.
Figure 1.

Phosphoramidate-based PSMA inhibitors and radiolabling precursors.
In the current study, we modified the phosphoramidate scaffold of 5 by either omitting (4) or installing an extra (6) aminohexanoate (AH) linker to further understand the structure–activity relationship (SAR) of phosphoramidates with respect to the interactions with the PSMA arene-binding site and their corresponding in vivo pharmacokinetics and biodistribution (Figure 1). Herein, we report the synthesis, radiolabeling, and in vivo performance of phosphoramidates [18F]4 and [18F]6 in comparison to previously reported [18F]5. The in vitro cell uptake and internalization in PSMA(+) CWR22Rv1 and PSMA(−) PC3 cells were determined at 1 and 2 h postincubation. Additionally, in vivo biodistribution data at 1 and 2 h time points as well as microPET/CT imaging at 2 h postinjection were obtained in mice implanted with PSMA(+) CWR22Rv1 and PSMA(−) PC3 tumors.
RESULTS
Synthesis of Cold Standards and Radiolabeling Precursors
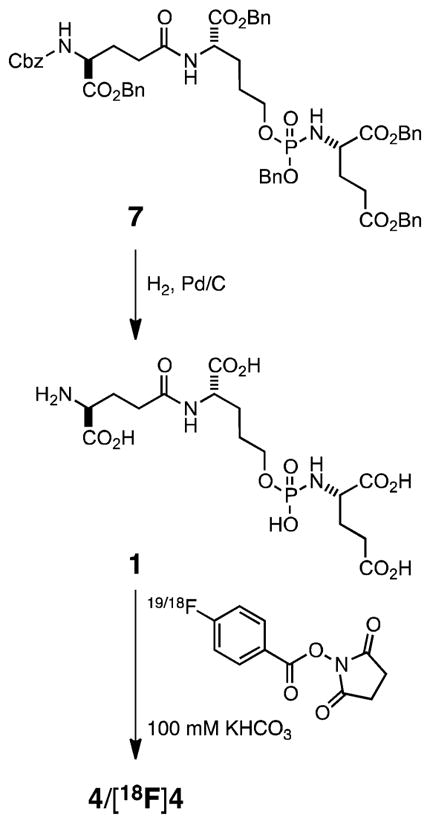
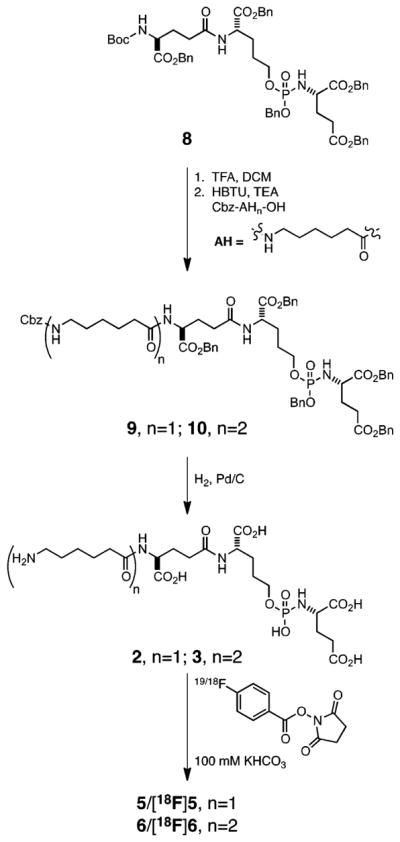
The phosphoramidate-based PSMA inhibitors and synthetic intermediates were prepared using the same general methodology as previously reported.24 Compound 1 was prepared in a final step by global deprotection of benzyl esters in precursor 7 (Scheme 1). Radiolabling precursor 1 represents a common structural core of the phosphoramidate analogues examined in the present study. The Boc-protected precursor 8 was N-deprotected and coupled to Cbz-AH-OH or Cbz-AH2-OH to generate 9 and 10, respectively. Subsequent global deprotection of 9 and 10 respectively yielded 2 and 3 (Scheme 2).
Scheme 1.
Synthetic Scheme for 4 and [18F]4
Scheme 2.
Synthetic sScheme for 2, 3, 5, 6, [18F]5, and [18F] 6
All the fluorobenzamide (FB and [18F]) derivatives were synthesized in quantitative yields via coupling of the phosphoramidate radiolabeling precursors 1, 2, and 3 with succinamidyl 4-fluorobenzoate (SFB) and [18F]SFB to generate 4 and [18F]4 (Scheme 1), 5 and[18F]5, and 6 and [18F]6 (Scheme 2), respectively. The nonradioactive analogues served as cold standards to determine IC50 values, mode of inhibition and as reference compounds to optimize the HPLC conditions for purification of their [18F]-labeled analogues.
IC50 and Mode of Inhibition
IC50 values and mode of inhibition (reversible, slowly reversible, or irreversible) were determined as described previously.25–28 As is common for small-molecule inhibitors of PSMA,19,22 derivatization of the N-terminal amine of the parent compounds 1 (IC50 = 27 nM), 2 (IC50 = 19 nM), and 3 (IC50 = 7.8 nM) with SFB led to enhanced inhibitory potency against PMSA (4, IC50 = 1.3 nM; 5, IC50 = 0.4 nM; 6, IC50 = 0.9 nM) (Figure 1). As observed previously for a number of phosphoramidate-based PSMA inhibitors, compounds 4, 5, and 6 exhibited an irreversible mode of binding to PSMA.
Cocrystallization of Phosphoramidate-Based PSMA Inhibitors with Glutamate Carboxypeptidase II and Structural Determination
The extracellular domain of PSMA, also known as human glutamate carboxypeptidase II (hGCPII; amino acids 44–750), was expressed in an S2 cell and purified using described protocols.29 To evaluate the effect of the AH chain length on PSMA (or hGCPII) binding, the phosphoramidate PSMA inhibitors 4 and 6 (the crystal structure of hGCPII/5 was recently reported24) were cocrystal-lized with hGCPII and the crystal structures of complexes hGCPII/4 and hGCPII/6 were determined at a resolution of 1.77 and 1.71 Å, respectively. The structural characteristics of hGCPII/4 (PDB code 4LQG, reported as hGCPII/CTT1056) and hGCPII/6 were compared to the previously reported hGCPII/524 (PDB code 4JYW, reported as hGCPII/CTT1057).
Structures were determined using difference Fourier methods, and the final models had well-defined crystallographic parameters (section 3, Table S1 of Supporting Information). The overall fold of all three structures is nearly identical with a maximum root-mean-square deviation of 0.33 Å for the 681 equivalent Cα pairs between hGCPII/5 and hGCPII/6 complex. The most pronounced difference in the loop arrangement is observed for the amino acids stretch spanning from Ser501 to Pro510.
Inhibitors were fitted into the Fo– Fc positive density peaks in the final stages of the refinement. In the case of 4, there is the strong Fo – Fc electron density for the inhibitor parts including the C-terminal glutamate, phosphoramidate group, and the linker up to the P1 carboxylate, and weaker, yet interpretable density for the remaining part of the molecule. As for 6, we built the model with the full confidence encompassing the C-terminal glutamate, phosphoramidate group, and the linker up to the P1 carboxylate and the adjacent peptide bond. However, the distal part of the inhibitor including the P2 residue, the lipophilic linker, and the fluorobenzoyl group is not seen in the Fo – Fc electron density; they are flexible and do not contribute to the interactions with GCPII (Figure 2A). In contrast to poorly defined distal parts of 4 and 6, positioning of 5 within the hGCPII binding pocket can be unambiguously assigned for the whole inhibitor (Figure 2B).
Figure 2.

(A) Individual inhibitors shown in stick representation: 4 (green; PBD code 4LQG), 5 (double inhibitor conformation; cyan; PDB code 4JYW), 6 (magenta). Corresponding Fo – Fc electron density map contoured at 3.0σ is shown in green. Note the completely missing electron density peaks for the distal part of 6, while there is very well-defined electron density for 5 and weaker, yet interpretable density for 4. (B) Comparison of binding modes of 4 (green), 5 (cyan, only a single conformation shown), and 6 (magenta). Zinc ions (orange spheres) and PSMA are shown in cartoon representation (gray). The arene-binding site (ABS) is highlighted in surface representation and colored red. (C, D) Details of interactions between 4 and 5, respectively, with PSMA. For the clarity, only residues having direct hydrogen-bonding interactions (shown as black broken lines) with the nonprime atoms of a given inhibitor are shown as lines. Canonical interactions within the S1′ site are not shown.
The core structural elements found to be common in all three inhibitors are the C-terminal glutamate, the phosphor-amidate group, the P1 hydroxypropylglycine residue, and the P2 glutamate linker. There is an overlapping of the C-terminal glutamate and the phosphoramidate moieties between the three inhibitors. The C-terminal glutamate occupies the S1′ pocket of the enzyme in the same mode as previously reported. This includes direct interactions between α- and γ-carboxylates of inhibitors and the side chains Arg210, Asn257, Tyr552, Lys699, and Tyr700.30,31 Free oxygen atoms from the phosphoramidate group chelate Zn2+ ions as well as interact with active-site residues including side chains of Tyr552, His553, Asp387, His377, Asp453, and Glu425. Additionally, the phosphorami-date amido group is involved in the hydrogen bonding with the Gly518 main-chain carbonyl and the Glu424 γ-carboxylate, whereas the oxygen atoms from the P–O bond and P1 hydroxypropylglycine form water-mediated contacts with the side chains of Asp453 and Arg536.
It is interesting to note that there are slight differences in the positioning between the AH linker and P1 carboxylate in 5 compared to that in 4 and 6. While the P1 carboxylate of 4 and 6 interact directly with both Arg534 and Arg536, this motif in 5 is shifted by approximately 1.1 Å (for the carbon atom of the P1 carboxylate), engaging NH1 of Arg536 only (3.1 Å).
The most important and prominent differences in the positioning of the inhibitor distal components are found in the lipophilic aminohexanoic linker and the fluorobenzoyl ring. For 4 (Figure 2C), the distance between the linker to the distal ring is approximately 13 Å. The distal fluorobenzoyl ring of 4 is positioned parallel to the guanidinium group of the Arg463 at a distance of approximately 4.0 Å with weak π–cation interactions in the arene-binding site. The terminal fluorobenzoyl functionality of 5 is wedged into the arene-binding cleft located at the “entrance lid” of the enzyme that is shaped by the side chains of Trp541 and Arg511 on sides and by the Arg463 side chain at the bottom. The plane of 5’s fluorobenzoyl ring is virtually parallel to both the indole and guanidinium groups of Trp541 and Arg511, respectively, and both these residues contribute to inhibitor binding (Figure 2D). In contrast to 4 and 5, the distal part of 6, possessing the longest linker (approximately 28 Å), is not observed in the electron density.
In Vitro Uptake and Internalization Study
Compounds [18F]4, [18F]5, and [18F]6 demonstrated specificity for PSMA showing uptake in PSMA(+) CWR22Rv1 cells but not in PSMA(−) PC3 cells. As early as 1 h postincubation, [18F]6 exhibited statistically significant higher uptake compared to that of [18F]4 and [18F]5 with P values of <0.0001 as determined by Student t-test; [18F]5 uptake was also statistically higher than [18F]4 with P value of 0.0012 (Table 1). The same trend was observed at 2 h. The activity measured within CWR22Rv1 cells, representing internalization for [18F]4, [18F]5, and [18F]6, at 1 h was 80.7%, 81.4%, and 84.9%, respectively, which increased to 94.2%, 84.2%, and 91.3%, respectively, at 2 h postincubation (Table 1).
Table 1.
Cell Uptake Data in PSMA(+) CWR22Rv1 and PSMA(−) PC3 Cells and Internalization Data in the PSMA(+) CWR22Rv1 Cell Line for [18F]4, [18F]5, and [18F]6
| % uptake | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| [18F]4 | [18F]5 | [18F]6 | ||||
|
|
|
|
||||
| 1 h | 2 h | 1 h | 2 h | 1 h | 2 h | |
| CW22Rv1 | 0.39 ± 0.06 | 0.70 ± 0.45 | 0.56 ± 0.05 | 2.28 ± 0.12 | 2.28 ± 0.14 | 4.23 ± 0.64 |
| PC3 | 0.18 ± 0.13 | 0.07 ± 0.02 | 0.04 ± 0.01 | 0.08 ± 0.02 | 0.14 ± 0.04 | 0.15 ± 0.01 |
|
| ||||||
| % internalization (CWR22Rv1) | ||||||
|
| ||||||
| [18F]4 | [18F]5 | [18F]6 | ||||
|
|
|
|
||||
| 1 h | 2 h | 1 h | 2 h | 1 h | 2 h | |
|
| ||||||
| internalized | 80.68 ± 1.76 | 94.15 ± 2.05 | 81.4 ± 2.7 | 84.2 ± 2.3 | 84.87 ± 3.88 | 91.31 ± 0.94 |
| bound | 19.32 ± 1.76 | 5.85 ± 2.05 | 18.6 ± 2.7 | 15.8 ± 2.3 | 15.13 ± 3.88 | 8.69 ± 0.94 |
In Vivo Imaging and Biodistribution Studies
As seen in the microPET/CT images in Figure 3, uptake of the tracers [18F]4, [18F]5, and [18F]6 was clearly observed in the PSMA(+) CWR22Rv1 tumors at 2 h postinjection but not in the PSMA(−) PC3 tumors. While all compounds showed uptake in the kidneys and clearance through the bladder, minimal uptake was seen in all other organs, including bone (Figure 3).
Figure 3.

3D MicroPET/CT images at 2 h postinjection of male nude mice bearing CWR22Rv1 and PC3 tumor xenografts respectively: (A) [18F]4; (B) [18F]5; (C) [18F]6. Arrows indicate tumor placement.
Biodistribution data are provided for all three compounds in Table 2. Uptake of [18F]4, [18F]5, and [18F]6 at 1 h postinjection in the PSMA(+) CWR22Rv1 tumor was 1.54 ± 0.40, 3.16 ± 0.39, and 2.92 ± 0.30% ID/g with a tumor-to-blood ratio of 10, 20, and 24, respectively. At 2 h postinjection, the tumor accumulations were 1.57 ± 0.50, 1.65 ± 0.32, and 1.86 ± 0.14% ID/g with rapid clearance from blood, providing a tumor-to-blood ratio of 26, 64, and 70, respectively. Kidneys showed the expected high uptake and retention of all compounds. At 1 h postinjection, kidney uptake of [18F]4, [18F]5, and [18F]6 was 8.94 ± 2.93, 24.38 ± 3.72, and 5.87 ± 0.67% ID/g, respectively, and at 2 h was 9.97 ± 2.81, 21.54 ± 6.12, and 7.13 ± 1.45% ID/g, respectively. For the PSMA(−) PC3 xenografts, tumor uptake was similar to that in the background and nontarget organs while the uptake in kidneys for [18F]4, [18F]5, and [18F]6 at 2 h postinjection was 5.64 ± 2.41, 18.98 ± 4.75, and 4.44 ± 1.03% ID/g, respectively.
Table 2.
Biodistribution of [18F]4, [18F]5, and [18F]6 As Determined by Radioactivity Assays in PSMA(+) CWR22Rv1 Tumor-Bearing Mice (n = 4 in Each Group)a
| tissue | [18F]4 | [18F]5 | [18F]6 | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||
| CWR22Rv1 | PC3 | CWR22Rv1 | PC3 | CWR22Rv1 | PC3 | ||||
|
|
|
|
|
|
|
||||
| 1 h | 2 h | 2 h | 1 h | 2 h | 2 h | 1 h | 2 h | 2 h | |
| blood | 0.15 ± 0.07 | 0.07 ± 0.02 | 0.08 ± 0.04 | 0.17 ± 0.05 | 0.04 ± 0.02 | 0.05 ± 0.02 | 0.12 ± 0.03 | 0.03 ± 0.01 | 0.02 ± 0.01 |
| heart | 0.75 ± 0.32 | 0.50 ± 0.05 | 0.36 ± 0.21 | 0.34 ± 0.11 | 0.17 ± 0.06 | 0.21 ± 0.07 | 0.23 ± 0.05 | 0.06 ± 0.01 | 0.07 ± 0.03 |
| lung | 0.65 ± 0.34 | 0.43 ± 0.11 | 0.29 ± 0.09 | 0.43 ± 0.09 | 0.21 ± 0.10 | 0.27 ± 0.08 | 0.25 ± 0.07 | 0.11 ± 0.04 | 0.09 ± 0.02 |
| liver | 0.83 ± 0.23 | 0.5 ± 0.11 | 0.44 ± 0.27 | 0.49 ± 0.11 | 0.29 ± 0.08 | 0.28 ± 0.05 | 0.49 ± 0.07 | 0.25 ± 0.04 | 0.25 ± 0.04 |
| kidney | 8.94 ± 2.93 | 9.97 ± 2.81 | 5.46 ± 2.41 | 24.38 ± 3.72 | 21.54 ± 6.12 | 18.98 ± 4.75 | 5.87 ± 0.67 | 7.13 ± 1.45 | 4.44 ± 1.03 |
| spleen | 1.18 ± 0.08 | 0.87 ± 0.16 | 0.76 ± 0.35 | 1.02 ± 0.04 | 0.84 ± 0.30 | 1.38 ± 1.05 | 0.32 ± 0.12 | 0.19 ± 0.03 | 0.14 ± 0.03 |
| bone | 0.46 ± 0.04 | 0.5 ± 0.42 | 0.24 ± 0.06 | 0.38 ± 0.09 | 0.23 ± 0.15 | 0.17 ± 0.04 | 0.45 ± 0.12 | 0.21 ± 0.11 | 0.14 ± 0.04 |
| muscle | 0.29 ± 0.09 | 0.19 ± 0.01 | 0.17 ± 0.04 | 0.12 ± 0.04 | 0.10 ± 0.06 | 0.06 ± 0.01 | 0.15 ± 0.04 | 0.08 ± 0.02 | 0.03 ± 0.01 |
| tumor | 1.54 ± 0.40 | 1.57 ± 0.45 | 0.40 ± 0.17 | 3.16 ± 0.39 | 1.65 ± 0.32 | 0.38 ± 0.03 | 2.92 ± 0.30 | 1.86 ± 0.14 | 0.27 ± 0.07 |
| tumor/blood | 9.88 ± 5.21 | 25.61 ± 14.99 | 5.42 ± 1.43 | 20.01 ± 9.06 | 63.60 ± 18.08 | 9.15 ± 3.76 | 24.21 ± 3.21 | 69.60 ± 15.72 | 11.19 ± 1.31 |
Tissues were harvested at 1 and 2 h postinjection. Uptake values are expressed as % ID/g of tissue.
DISCUSSION
A series of FB ring-containing compounds was designed with or without a linker to separate it from the common phosphoramidate core 1. Compound 4, in which the FB group was directly attached to the inhibitor 1 via its terminal amine, was expected to behave like our first generation analogue II. The other new compound in the series was compound 6 with the FB ring separated from the core inhibitor by two AH units. Compound 5, where the inhibitor core was separated from the FB ring by one AH unit, has previously shown desirable PSMA targeting properties.24 [18F]5 retained high affinity for PSMA and showed consistent uptake and rapid internalization into CW22Rv1 (PSMA+) cells. The in vitro properties translated well to mouse models implanted with CW22Rv1 tumors xenografts. High uptake and retention of [18F]5 over 4 h was observed with exceptional blood-to-tumor ratios of 265:1 at 4 h postinjection, suggesting rapid clearance of the radiotracer from blood and all nontarget organs. Cocrystallization of PSMA with 5 revealed the binding of the fluorobenzoyl (FB) ring in the arene-binding site (ABS), initially identified by Zhang et al.32
It was previously revealed that an optimal distance between the targeting core and the FB group is necessary to achieve efficient binding of the FB ring with the ABS. On the basis of the results obtained from modeling studies, and literature precedent, it was anticipated that the FB ring of the [18F]4 would be too short to reach the ABS; the linker distance of >20 Å in [18F]6 was expected to allow for ABS binding as a contribution to a bimodal binding interaction. In addition to the pharmacodynamic consequences of linker length, we sought to assess the effect of varying the AH linker units on the in vivo pharmacokinetics of the targeting agents.
The syntheses of compounds 1, 2, 5, and [18F]5 were reported previously,22,24 while the syntheses of 4/[18F]4 and 6/[18F]6 are reported herein. Globally protected 3 was generated by acid deprotection of the Boc group of 8 followed by installation of Cbz-(AH)2-OH linker. Global deprotection afforded 3 in quantitative yield. Fluorobenzoylation of the N-terminal amines of 1 and 3 yielded fluorobenzamide (FB) compounds 4 and 6, respectively. As observed with 5 and other phosphoramidate analogues, these compounds maintained high affinity and irreversible modes of binding to PSMA upon derivatization of the N-terminal amine. Owing to the high affinity and the irreversible nature of the common scaffold, it was expected that compounds [18F]4 and [18F]6 would show uptake and retention in PSMA(+) tumors in vivo while rapidly clearing from all nontarget organs, as was observed in the case of [18F]5.24
To gather insight into the molecular interactions of these analogues with PSMA and draw a comparison with the previous analogue 5, compounds 4 and 6 were also cocrystallized with the extracellular domain of PSMA. While the P1′ carboxylate, the phosphoramidate binding group, and the P1 residue interact almost identically to PSMA, the distinct differences were the aminohexanoic acid linker(s) and the fluorobenzoyl ring. As anticipated, the linkage between the distal FB ring and the phosphoramidate core in 4 (approximately 13 Å) was insufficient to access the recently identified ABS.32 Rather, the distal FB group was found parallel to the guanidinium group of the Arg463 at a distance of approximately 4.0 Å (Figure 2C). Clearly, π interactions play a prominent role in the stabilization of this inhibitor’s positioning, although the strength of this interaction is likely much weaker compared to interactions in the ABS as observed for 5. The terminal fluorobenzoyl moiety of 5 was found to be parallel to both indole and guanidinium groups of Trp541 and Arg511 in the ABS residues contributing to enhanced inhibitor binding (Figure 2D). In the case of 6, possessing the longest linker (approximately 28 Å), the portion of the inhibitor distal to the active site was not observed in the electron density map, suggesting its positional flexibility and the absence of significant PSMA–inhibitor interactions (Figure 2A and Figure 2B).
The structural data are in a good agreement with the inhibitory properties toward PSMA contributed from the terminal FB group and the AH linker on the tested compounds. The coupling of the terminal FB group to the core structures 1, 2, and 3 led to an improvement of the IC50 values for 4, 5, and 6 by approximately 20-, 50-, and 9-fold, respectively. A mechanistic explanation for the increase in inhibition potency may be the result of additional binding interactions between the distal motifs of the inhibitors and the protein in addition to entropic contributions. The most prominent effect (50-fold improvement) was observed for 5, for which the terminal FB group is fully engaged with the ABS. Considerably less (20-fold) enhancement of inhibition was observed for 4, where there were less pronounced interactions between the SFB and the ABS. Interactions with the ABS were absent for 6, which exhibited the least enhancement of inhibitory potency compared to its inhibitor core 3. However, it should be noted that more complex effects (including entropic factors and solvation effects) may also play a role in defining the absolute values of inhibition constants for individual molecules. The simple bimodal binding as in 5 or unimodal binding in 6 cannot be the sole determining factor for the inhibition potencies.
The PSMA(+) LNCaP cell lines have been used most widely in the PSMA research by various research groups, including us.1,21,22 Although LNCaPs have a high PSMA concentration, their growth rate and proliferation as tumors in mice are unpredictable and often lead to tumor necrosis. In contrast, the CWR22Rv1 cells have a moderate PSMA expression (8-fold lower than LNCaPs) and a predictable growth pattern as tumor xenografts.24,33 In addition, the cellular PSMA concentration in the CRW22Rv1 cells is more akin to the expression levels expected in human prostate cancer metastases. Therefore, for the in vitro and in vivo studies in the present research, CWR22Rv1 has been the PSMA(+) cell line of choice.
The in vitro studies indicated uptake of all the radiotracers in PSMA(+) CW22Rv1 cells, where an increase in uptake was observed as the lipophilic linker length in these compounds increased from [18F]4 to [18F]6. While the increased uptake of [18F]5 can be attributed to its unique bimodal interactions with PSMA, no such binding interaction was observed with 6. Therefore, the enhanced uptake in PSMA(+) cells in vitro cannot be explained solely on the basis of PSMA affinity. This trend has also been reported previously in the literature, where high uptake in PSMA(+) cells in vitro and tumors in vivo has often been correlated with increased lipophilicity.12 As expected, all three compounds showed negligible uptake in PSMA(−) PC3 cell lines at all time points, due to lack of PSMA expression in this cell line. In addition, all compounds showed rapid internalization into PSMA(+) CW22Rv1 cells with more than 80% internalization at 1 h. We have previously reported that the mode of binding of PSMA inhibitors has a direct impact on the degree of internalization.28,33,34 The irreversible nature of the scaffolds and findings from in vitro uptake and internalization suggest that the radiotracers will likely lead to both uptake and retention in the PSMA(+) tumors in vivo over time, allowing clearance from the nontarget organs.
Tumor uptake was observed for [18F]4, [18F]5, and [18F]6 at 1 and 2 h postinjection in mice bearing PSMA(+) CWR22Rv1 tumor cells. At 1 h, the uptake of [18F]5 (3.16%) and [18F]6 (2.92%) was about 2-fold higher than [18F]4 (1.54%) (p < 0.05). However, there was no significant difference in tumor uptake between the three analogues at 2 h postinjection. At 2 h postinjection, all three tracers exhibited rapid clearance from blood and non-PSMA expressing organs, resulting in high tumor-to-blood ratios for [18F]5 and [18F]6, 64 and 70, respectively, that were significantly (p < 0.05) greater than [18F]4. The irreversible mode of binding of these compounds possibly contributes to its tumor retention while rapidly clearing from other nontarget organs, in contrast to the washout that is reported over time for most urea-based agents.35 As expected, kidney uptake was observed with all three analogues, due to the high expression of PSMA in mouse kidneys,36 with the highest uptake shown in the case of [18F]5, which can be considered as a secondary positive indicator of PSMA specificity. The kidney uptake of [18F]5 was significantly higher (p < 0.05) at both time points and in both tumor bearing animal models. It is unknown why the kidney uptake of [18F]5 was 2- to 2.5-fold higher, as the magnitude of the difference does not correlate to a significant chemical or in vitro characteristic. For compounds [18F]4 and [18F]6 there was no significant difference in kidney uptake at either time point, nor was there any difference for all three tracers between the kidney uptake in the PC3 versus the CWR22Rv1 animal model at 2 h.
Given the structural similarities, it was expected that [18F]4 would exhibit similar in vivo properties to our first-generation PET agent. In our previous work, [18F]II was evaluated in PSMA(+) LNCaP tumor xenografts in mice with imaging and biodistribution data collected at 2 h postinjection. PSMA expression in LNCaP cells is approximately 5- to 10-fold greater than that in CW22Rv1 cells.24 In comparison to the current work, [18F]4 had a modest uptake at 1.57% with a tumor-to-blood ratio of 64:1 in the lower PSMA expressing CW22Rv1 tumors vs 1.24% and tumor-to-blood ratio of 9:1 for [18F]II in LNCaP tumors. Though this may not be a head-to-head comparison because different tumor types were used, the in vivo uptake and clearance of [18F]4 suggest an overall trend toward higher affinity and more superior properties compared to [18F]II.
The most promising compounds of this study were [18F]5 and [18F]6, in which the installation of lipophilic AH linker units between the phosphoramidate targeting core and the FB ring improved the in vivo tumor uptake and clearance compared to that of [18F]4. While the excellent in vivo properties of [18F]5 can be attributed to its unique interactions with the ABS which resulted in higher PSMA affinity, the added lipophilicity from the additional AH spacer in [18F]6 may have contributed to reduced renal clearance and consequent tumor uptake. Although we observed no direct crystallographic evidence for the FB group of [18F]6 interacting with the ABS, the high degree of rotational freedom afforded by the two AH units may allow for the secondary binding at the ABS in solution.
The PET/CT images confirmed the observations of the biodistribution studies. All three tracers demonstrated significant uptake in the PSMA(+) CWR22Rv1 tumors but not in PSMA(−) PC3 tumors, confirming their specificity for PSMA. All these analogues exhibited high tumor-to-background ratios at 2 h postinjection with rapid blood clearance and minimal uptake and retention in nontarget organs. No evidence of metabolic defluorination and subsequent bone accumulation by any of the analogues was observed.
The tumor uptake and biodistribution patterns are in good agreement with the other known F-18 PSMA urea agents that have been translated into the clinical setting, although it is hard to make a direct head-to-head comparison between these compounds due to the different models used between the different studies and the variable expression levels of PSMA in the PSMA(+) cell lines used. However, the observed pharmacokinetics are indicative of an overall trend of the uptake and clearance of these agents. [18F]DCFBC, the first generation urea compound to be tested in human clinical trials, showed an uptake of 4.7% in PSMA(+) PC3-PIP cells and a tumor/blood ratio of 13:1.16 In comparison, compounds [18F]5 and [18F]6 show an uptake of 1.67% and 1.8%, with tumor/blood ratios of 64:1 and 69:1, respectively, at the 2 h time point. It has been shown that the concentration of PSMA in the PC3-PIP cells is significantly higher than that in CWR22Rv1 cells.37 Although the tumor uptake values are comparatively higher in the second generation urea agent, [18F]DCFPyL (39.4% at 2 h in PC3-PIP cells), the renal and liver uptake for this compound is also significantly higher than our phorphor-amidates.17 The renal and liver uptake values for [18F]DCFPyL at 2 h are 15.7% and 2.14%, respectively, as compared to 7.13% and 0.25% for [18F]6.
As both these reported urea compounds have been able to successfully detect primary and metastatic lesions in clinical trials,15,18 the comparable trends in tumor uptake and pharmacokinetics, and excellent clearance from nontarget organs, observed for [18F]5 and [18F]6 are positive indicators of their success in future clinical trials. In fact, after successful completion of preclinical, dosage, and toxicology studies, the first-in-human clinical trials for compound [18F]5 is currently underway.
CONCLUSION
We have successfully synthesized and evaluated a bracketed series of PSMA-targeted phosphoramidate analogues, differing in the lipophilicity compared to compound 5, and evaluated them as PET imaging agents for prostate cancer. While capitalizing the unique bimodal interaction with the “arene-binding site” in PSMA can be a key element for future designs of inhibitors with improved affinity, a balance between hydrophilic and lipophilic properties in the core structure may also be critical for favorable in vivo properties of the radiotracer. With their exceptional binding, tumor uptake and retention, and remarkable tumor-to-blood ratios, [18F]5 and [18F]6 are well-positioned as favorable candidates for translation to future prostate cancer imaging studies in human.
EXPERIMENTAL SECTION
Cell Lines, Reagents, and General Methods
CWR22Rv1 and PC-3 cells were obtained from the American Type Culture Collection (Manassas, VA). NCr-nu/nu mice (strain code 088) were purchased from Charles River (Hollister, CA). Z-6-Aminohexanoic acid (Cbz-AH-OH) was purchased from Sigma-Aldrich (St. Louis, MO). All chemicals and cell-culture reagents were purchased from Fisher Scientific (Sommerville, NJ) or Sigma-Aldrich. All solvents used in chemical reactions were anhydrous and obtained as such from commercial sources or distilled prior to use. All other reagents were used as supplied unless otherwise stated. Liquid flash chomatography (silica or C18) was carried out using a Flash Plus chromatography system (Biotage, Charlotte, NC). High-resolution mass spectrometry was performed using an ABS 4800 MALDI TOF/TOF analyzer (Applied Biosystems, Framingham, MA). 1H NMR chemical shifts were referenced to tetramethylsilane (δ = 0.00 ppm), CDCl3 (δ = 7.26 ppm), or D2O (δ = 4.87 ppm). 13C NMR chemical shifts were referenced to CDCl3 (δ = 77.23 ppm). 31P NMR chemical shifts in CDCl3 or D2O were externally referenced to 85% H3PO4 (δ = 0.00 ppm) in CDCl3 or D2O. Aqueous buffered solutions for in vitro experiments and HPLC chromatography were prepared with deionized distilled water (Milli-Q water system, Millipore, Billerica, MA).
The HPLC analysis and purification system for radioactive compounds were performed on a Waters model 600 multisolvent system pump equipped with a Shimudzu model SPD-10A UV detector and an in-line radioactivity detector (model 105s, Carroll and Ramsey Associates, Berkeley, CA) that was coupled to a data collection system (PeakSimple model 304, SRI, Torrance, CA).
Purity of compounds 4, 5, and 6 was verified to be >95% via 31P (formation of single product) and 1H NMR.
1. Syntheses of Phosphoramidate Compounds and Their Respective 18F Analogues
The synthetic sequence of compounds 4 and [18F]4 is shown in Scheme 1. Syntheses and characterization of 7 and its precursors are provided as Supporting Information (section 1). The general synthetic scheme for compounds 2, 3, 5, 6, [18F]5, and [18F]6 is shown in Scheme 2. Syntheses of compounds 2, 5, [18F]5, and 8 have been reported previously.24
1.1. Synthesis of 6-(6-(((Benzyloxy)carbonyl)amino)-hexanamido)hexanoic Acid, Cbz-AH2-OH
Synthesis of CBZ-AH-OH
6-Aminocaproic acid (8 g, 61 mmol, 1 equiv) and sodium carbonate (61 mmol, 1 equiv) were dissolved in water (18 mL) in a round-bottom flask (100 mL) and placed in an ice bath. Benzoyl chloroformate (67.1 mmol, 1.1 equiv) was added to the flask dropwise. Sodium hydroxide (2 N, 15 mL) was added in 5 mL portions over a 15 min period. The reaction was monitored by TLC and stirred until completion. The crude product was washed with diethyl ether (2 × 20 mL), and the aqueous phase was acidified to pH = 2 and placed in 5 °C. The white precipitate was filtered and washed with 10% HCl. TLC (silica, 4:1 DCM/EtOAc, 1% AcOH) showed the presence of CBZ-AH-OH (Rf = 0.27). CBZ-AH-OH was isolated as a white solid by column chromatography (silica, 4:1 DCM/EtOAc, 1% AcOH), yield 25.5%. Mp = 57–59 °C. 1H NMR (300 MHz, CDCl3): δ 1.36 (m, 2H), 1.50 (m, 2H), 1.65 (m, 2H), 2.35 (t, 2H), 2.35 (t, 2H), 3.19 (m, 4H), 4.78 (s, 1H), 5.09 (s, 2H), 5.64 (s, 1H), 7.35 (m, 5H). 13C NMR (100 MHz, CDCl3): δ 24.23, 26.08, 29.57, 33.82, 40.79, 66.64, 128.08, 128.48, 136.53, 156.44, 179.11. HR mass spectrometry: calculated 288.1212; found 288.12106 (M + Na+) for C14H19NO4.
Synthesis of CBZ-AH2-OH
CBZ-AH-OH (0.20 g, 0.754 mmol, 1 equiv) and HBTU (0.83 mmol, 1.0 equiv) were dissolved in distilled DCM (10 mL) and allowed to stir for 1 h under Ar(g) in a flame-dried flask (25 mL). A solution of NHS (0.83 mmol, 1.1 equiv) and TEA (0.75 mmol, 1 equiv) was prepared in distilled DCM (4 mL) and added to the reaction dropwise. This was allowed to stir overnight. The DCM was dried under vacuum. The crude product was extracted with ethyl acetate (25 mL) and washed with 10% HCl (3 × 25 mL), 10% NaHCO3 (3 × 25 mL), and brine (3 × 25 mL) and dried with Na2SO4. The presence of CBZ-AH-NHS was determined by 1H NMR (300 MHz, CDCl3): δ 1.43 (m, 2H), 1.51 (m, 2H), 1.73 (m, 2H), 2.59 (t, 2H), 2.35 (t, 2H), 2.76 (s, 4H), 3.19 (m, 4H), 4.98 (s, 1H), 5.07 (s, 2H), 5.64 (s, 1H), 7.35 (m, 5H). CBZ-AH-NHS was used as a crude mixture without further purification.
6-Aminocaproic acid (0.12 g, 0.90 mmol, 1.2 equiv) and sodium bicarbonate (1.35 mmol, 1.8 equiv) were dissolved in water (8 mL). CBZ-AH-NHS (0.752 mmol, 1 equiv) was dissolved in acetone (5.2 mL) and was added dropwise to the round-bottom flask and stirred overnight. The reaction mixture was acidified to pH 2, and the white precipitate was filtered and washed with 10% HCl. The product was isolated as a white solid without any further purification (68% yield, 0.61 mmol). TLC (silica, 49:1 EtOAc/AcOH) showed the presence of CBZ-AH2-OH (Rf = 0.17). Mp = 104–106 °C. 1H NMR (300 MHz, CDCl3): δ 1.36 (m, 4H), 1.50 (m, 4H), 1.61 (m, 4H), 2.16 (t, 2H), 2.35 (t, 2H), 3.16–3.34 (m, 4H), 4.94 (s, 1H), 5.09 (s, 2H), 5.69 (s, 1H), 7.34 (m, 5H). 13C NMR (300 MHz, CDCl3): δ 24.28, 25.28, 25.93, 26.06, 28.65, 29.15, 33.36, 35.57, 35.63, 38.73, 40.19, 65.85, 127.30, 127.48, 128.00, 137.05, 157.47, 174.60, 176.02. HR mass spectrometry: calculated 378.22, found 379.229 06 (M + H+) for C20H30N2O5.
1.2. 2-(((S)-((2R)-2-(4-Amino-4-carboxybutanamido)-2-carboxyethoxy)(hydroxy)phosphoryl)amino)pentanedioic Acid [1]
To a solution of a benzyl ester protected phosphoramidate (7) (0.100 g, 0.095 mmol) in THF (1 mL) were added 10% Pd/C (10 mg), K2CO3 (0.033 mg, 0.241 mmol), and H2O (1 mL). The mixture was stirred vigorously, purged with Ar(g), and then charged with H2(g) under balloon pressure overnight at room temperature. The solution was filtered through a 0.2 mm PTFE micropore filtration disk (Whatman). The solvent was removed in vacuo to yield a white solid, 1, in 96% yield. 1H NMR (300 MHz, D2O): δ 1.44–1.54 (m, 3H), 1.62–1.67 (m, 3H), 1.86–1.93 (m, 2H), 2.02–2.05 (m, 2H), 2.21 (m, 1H), 2.25 (m, 1H), 3.31 (m, 1H), 3.47 (m, 1h), 3.56 (t, 2H), 3.92 (m, 1H). 31P NMR (300 MHz, D2O): δ 8.46. HR mass spectrometry: calculated 471.3, found 470.20 (M – H) for C15H26N3O12P+.
1.3. 2-(((S)-((2R)-2-Carboxy-2-(4-carboxy-4-(4-fluorobenzamido)butanamido)ethoxy)(hydroxy)phosphoryl)-amino)pentanedioic Acid [4]
A solution of SFB (20.14 μmol, 1 equiv) in 400 μL of THF was added to a stirred solution of 1 (30.22 μmol, 1.5 equiv) in 600 μL of 0.1 M KHCO3. The reaction mixture was stirred for 6 h in the dark at room temperature. The unreacted 1 was scavenged by stirring with 25 mg of Si isocyanate resin (SiliCycle, Inc., Quebec, Canada) overnight at room temperature. The solution was subsequently centrifuged (7800 rcf, 10 min), and the supernatant was lyophilized in a 2 mL microcentrifuge tube. The unreacted materials and/or hydrolyzed SFB was removed by successively triturating the lyophilized solid with 1 mL portions of DMSO and centrifuging the mixture (16 200 rcf, 1 min) after each wash; this process was repeated 10 times. The resulting solid was dried in vacuo providing the desired 4-fluorobenzamidophosphoramidate 4 in quantitative yield. 1H NMR (300 MHz, D2O): δ 1.44–1.54 (m, 2H), 1.56–1.68 (m, 2H), 1.86–2.14 (m, 4H), 2.27–2.35 (m, 2H), 3.27–3.34 (m, 1H), 3.50 (t, 2H), 3.73–3.87 (m, 2H), 4.05–4.08 (t, 1H, 3.9 Hz), 4.15–4.19 (dd, 1H, 4.4 Hz, 9.15 Hz), 7.04–7.10 (t, 2H), 7.66–7.70 (dd, 2H). 31P NMR (300 MHz, D2O): δ 8.42. HR mass spectrometry: calculated 720.03, found 719.96 (M + 4Na + K) for C22H29FN3O13P+.
1.4. Synthesis of Dibenzyl 2-(((R)-(Benzyloxy)(((24R)-19,24-bis((benzyloxy)carbonyl)-3,10,17,22-tetraoxo-1-phenyl-2-oxa-4,11,18,23-tetraazaheptacosan-27-yl)oxy)phosphoryl)amino)-pentanedioate [10]
Cbz-AH2-acid (AH = aminohexanoic acid) (0.1 g, 0.264 mmol) was preactivated with HBTU (0.29 mmol, 1.1 equiv) and TEA (0.29 mmol, 1.1 equiv). 8 was treated with a mixture of dry TFA/DCM for deprotection of N-terminal Boc group and then added to the flask above with activated Cbz-AH2-acid. Purification was carried out using reversed phase C18 chromatography with 80% MeOH–water as the mobile phase. Pure 10 was isolated in 49% yield. 1H NMR (300 MHz, CDCl3): δ 1.28–1.30 (m, 4H), 1.41–1.46 (m, 4H), 1.56–1.61 (m, 6H), 1.86–1.89 (m, 2H), 2.09–2.27 (m, 10H), 2.37–2.39 (m, 2H), 3.12–3.18 (m, 4H), 3.74 (m, 1H), 3.89 (m, 2H), 4.51 (m, 2H), 4.91–4.96 (m, 2H), 5.05–5.11 (m, 10H), 5.95 (d, 1H, -NH), 6.98 (d, 1H, -NH), 7.03 (d, 1H, -NH), 7.27–7.31 (m, 27H). 31P NMR (300 MHz, CDCl3): δ 8.47. ESI mass spectrometry: calculated 1281.4, found 1282.4 (M + H), 1305.6 (M + Na) for C70H84N5O16P+.
1.5. Synthesis of 2-(((S)-((4R)-4-(4-(6-(6-Aminohexanamido)-hexanamido)-4-carboxybutanamido)-4-carboxybutoxy)-(hydroxy)phosphoryl)amino)pentanedioic Acid [3]
To a solution of benzyl ester protected phosphoramidate (10) (0.160 g, 0.124 mmol) in THF (1 mL) were added 10% Pd/C (16 mg), K2CO3 (0.044 mg, 0.318 mmol), and H2O (1 mL). The mixture was stirred vigorously, purged with Ar(g), and then charged with H2(g) under balloon pressure overnight at room temperature. The solution was filtered through a 0.2 mm PTFE micropore filtration disk (Whatman). The solvent was removed in vacuo to yield a white solid, 3, in 94% yield. 1H NMR (300 MHz, D2O): δ 1.14–1.19 (m, 2H), 1.36 (m, 4H), 1.38–1.50 (m, 10H), 1.59–1.68 (m, 2H), 1.89 (m, 2H), 1.99–2.19 (m, 8H), 2.86 (t, 2H), 3.34 (m, 1H), 3.56 (dd, 1H), 3.94 (m, 3H). 31P NMR (300 MHz, D2O): δ 8.43. HR mass spectrometry: calculated 698.30, found 698.35 (M + H) for C27H49N5O14P+.
1.6. Synthesis of 2-(((S)-(((22R)-17,22-Dicarboxy-1-(4-fluoro-phenyl)-1,8,15,20-tetraoxo-2,9,16,21-tetraazapentacosan-25-yl)oxy)(hydroxy)phosphoryl)amino)pentanedioic Acid [6]
A solution of 3 (0.028g, 0.003 mmol, 1.5 equiv) was made in 500 μL of 100 mmol of KHCO3, and SFB (0.005g, 1 equiv) in 400 μL of THF was added and stirred for 5 h. The unreacted 3 was scavenged by stirring with 5 mg of Si isocyanate resin (SiliCycle, Inc., Quebec, Canada) overnight at room temperature. The solution was subsequently centrifuged (7800 rcf, 10 min), and the supernatant was lyophilized in a 2 mL microcentrifuge tube. The unreacted materials and/or hydrolyzed SFB was removed by successively triturating the lyophilized solid with 1 mL portions of DMSO and centrifuging the mixture (16 200 rcf, 1 min) after each wash; this process was repeated 10 times. The resulting solid was dried in vacuo providing the desired 4-fluorobenzamidophosphoramidate 6 in quantitative yield. 1H NMR (300 MHz, D2O): δ 1.09–1.19 (m, 2H), 1.16–1.24 (m, 6H), 1.30–1.35 (m, 2H), 1.41–1.45 (m, 5H), 1.61–1.68 (m, 5H),1.99–2.07 (m, 6H), 2.14–2.21 (m, 2H), 2.91–2.95 (m, 2H), 3.16–3.21(m, 2H), 3.25–3.33 (m, 2H), 3.54–3.56 (m, 2H), 3.87–3.96 (m, 2H), 7.02–7.08 (m, 2H), 7.56–7.61 (m, 2H). 31P NMR (300 MHz, D2O): δ 8.43. HR mass spectrometry: calculated 820.38, found 820.43 (M + H) and 858.40 (M + K) for C34H51N5FO15P+.
1.7. Synthesis of [18F]4, [18F]5, or [18F]6
Succinimidyl [18F]fluorobenzoate ([18F]SFB) was synthesized in a Neptis synthesizer (ORA—Optimized Radiochemical Applications, Belgium) equipped with commercially available kits and cassettes (ABX GmbH, Germany) that was then coupled to 1, 2, and 3 as was previously described with modifications.22 The PSMA analogues (2 mg) were dissolved in 100 mL of H2O, 20 mL of 0.1 M K2CO3, and an amount of 100 mL of [18F]SFB in acetonitrile was added to a 1 dram vial charged with a stir bar (pH 9.5–10). The coupling reaction took place at 40 °C for 15 min. Prior to in vitro and in vivo studies, the radioconjugate was purified on a semipreparative RP-HPLC using a Phenomenex C18(2) 100 Å, 250 mm × 10 mm, 5 μm column, linear gradient (20–90% over 22 min) of solvent B in solvent A (A, 0.1% formic acid in water; B, 0.1% formic acid in acetonitirile) at a flow rate of 5 mL/min and UV detection at 254 nm. [18F]4, [18F]5, or [18F]6 peak was collected and concentrated via QMA light SPE method using 0.5% NaCl for elution and diluted with 1× PBS for in vitro and in vivo studies. Radiochemical yields ranged between 50% and 60% decay-corrected from [18F]SFB. Analytical RP-HPLC of the purified peak confirmed >95% purity of all three compounds via coinjection with the respective nonradioactive analogs.
2.1. General Method of Determining IC50 Values
Inhibition studies were performed as previously described with minor modifications.25,34 Description is provided in Supporting Information (section 2).
2.2. Mode of Inhibition Study
The mode of inhibition studies followed the procedure described in our previous work.19 A description is provided in Supporting Information (section 2).
3.1. GCPII Expression, Purification, Crystallization, and Data Collection
The extracellular part of human GCPII (hGCPII; amino acids 44–750) was expressed in S2 cell and purified according to procedures described previously.29 The final protein preparation in 20 mM Tris-HCl, 150 mM NaCl, pH 8.0, was concentrated to 9 mg/mL and stored at −80 °C until further use.
Complexes of hGCPII/4 and hGCPII/6 were prepared by mixing stock solutions of hGCPII (9 mg/mL) and a given inhibitor (20 mM in water, pH adjusted to 8.0 by the addition of NaOH) at the 9:1 ratio (v/v). Crystals were grown from 2 μL droplets made by mixing equal volumes of GCPII/inhibitor and reservoir solutions (33% pentaery-thritol propoxylate (Sigma), 1.5% polyethylene glycol 3350 (Sigma), and 100 mM Tris-HCl, pH 8.0) using the hanging-drop vapor diffusion setup at 293 K. Diffraction quality crystals were flash-frozen in liquid nitrogen directly from the crystallization droplet. The diffraction data for both complexes were collected at 100 K using synchrotron radiation at the MX 14.2 beamline (BESSYII, Helmholtz-Zentrum Berlin, Germany; 0.918 Å). The complete data set was collected from a single crystal, and data were processed with the XDSAPP package.38
3.2. Structure Determination and Refinement
Structures were determined by the difference Fourier methods using the structure of the hGCPII/NAAG complex (PDB entry 3BXM39) as the template model. Model building was accomplished using Coot,40 and calculation steps were performed using Refmac 5.1.41 The restrains library and the coordinate files for individual inhibitors were prepared using the PRODRG server,42 and the inhibitors/substrates were fitted into the positive electron density map in the final stages of the refinement.
The stereochemical quality of final models was evaluated using MolProbity,43 and the final model, together with experimental amplitudes, was deposited in the RCSB Protein Data Bank under the entry code 4LQG (hGCPII/4; deposited as hGCPII/CTT1056). Data collection and structure refinement statistics are provided in the Supporting Information, section 3, Table S1.
4. Cell Lines and Cell Culture
CWR22Rv1 and PC-3 cells were incubated in T-75 flasks with complete growth medium (RPMI 1640 containing 10% heat-inactivated fetal calf serum (FBS), 100 units of penicillin, and 100 μg/mL streptomycin) in a humidified incubator at 37 °C and 5% CO2.
4.1. In Vitro Cell Uptake Studies
Confluent CWR22Rv1 and PC-3 cells were detached with 0.25% trypsin–0.53 mM EDTA solution. The cells were washed three times with 1% FBS 1X-RPMI 1640 phosphate-free medium. Cells were subdivided into micro-centrifuge tubes to contain approximately 500 000 cells each in 250 μL of 1% FBS 1X-RPMI 1640 phosphate-free medium and 6.25 μL of ethanol. A solution of [18F]4, [18F]5, or [18F]6 (2 μCi in 3 μL) was added to microcentrifuge tubes containing one of the following: CWR22Rv1 cells (n = 5) in 1% FBS 1X-RPMI 1640 phosphate-free medium; PC-3 cells (n = 5) in 1% FBS 1X-RPMI 1640 phosphate-free medium; 1 mL of 1% FBS 1X-RPMI 1640 phosphate-free medium (n = 3) which served as reference for the total activity incubated with the cells. All samples above were incubated at 37 °C and 5% CO2 for 1 and 2 h. At each time point, the cell pellets were repeatedly washed with PBS (1 mL), centrifuged (2 min at 14 700g) and the medium was removed. The radioactivity of the cell pellets was counted and compared to the total activity references.
4.2. Internalization Studies
Internalization studies followed the method describe for the in vitro cell uptake studies with a minor modification. Following the incubation periods (1 and 2 h) the cells were washed as described in the uptake studies with PBS cooled to 4 °C. The resulting cells were then treated with a solution of 50 mM glycine and 100 mM NaCl at pH 3 for 2 min at 37 °C and 5% CO2. Cells were then centrifuged (2 min at 12 000g), and the supernatants were collected. This treatment was repeated two additional times, and the combined supernatants were counted for external binding, while the cell pellet was counted for internalization.44,45
5. In Vivo PET Imaging Studies
All animal experiments were conducted in accordance with the UCSF IACUC approved protocol. Approximately 106 CWR22Rv1 PSMA(+) or PC3 PSMA(−) cells in 50:50 mixture of complete medium and matrigel matrix were implanted in the right shoulder of athymic NCr-nu/nu male mice (approximately 8 weeks old) from Charles River (Hollister, CA). Approximately 4 weeks after implantation, animals with tumors reaching 150–300 mm3 were anesthetized by isoflurane inhalation and were administered with [18F]4, [18F]5, or [18F]6 at 200–250 μCi in 250 μL of saline through tail vein injection. The resulting animals were imaged with 10 min acquisition by a microPET/CT imaging system (Inveon, Siemens, Germany) at 2 h postinjection. PET imaging data were acquired in list mode and reconstructed with the iterative OSEM 2-D reconstruction algorithm provided by the manufacturer.
6. Biodistribution Studies
Four to five weeks after the implantation of CWR22Rv1 PSMA(+) cells or three to four weeks after the implantation of PC3 PSMA(−) cells, tumor bearing mice were anesthetized by isoflurane inhalation and administered with 50 μCi of [18F]4, [18F]5, or [18F]6 in 150 μL of saline through tail vein injection. These mice (CWR22Rv1 at 1 and 2 h postinjection; PC3 at 2 h post injection; n = 4 for each time point) were euthanized for biodistribution analysis. Blood was collected by cardiac puncture. Major organs (heart, lung, liver, spleen, kidney, muscle, bone, and tumor xenografts) were harvested, weighed, and counted in an automated γ counter (Wizard 2, PerkinElmer, Waltham, MA). The percent injected dose per gram (% ID/g) of tissue was calculated by comparison with standards of known radioactivity. Statistical analysis was performed using two-tailed Student t-test (Microsoft Excel Prism software). A p value of <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health (Grant R01CA140617) and the Department of Defense (Grant W81XWH-11-1-0464). The authors extend their gratitude for technical assistance to G. Helms and W. Hiscox at the WSU Center for NMR Spectroscopy, G. Munske at the WSU Laboratory for Bioanalysis and Biotechnology for mass spectrometry analysis, P. Daniel for the help with X-ray data collection, Dr. B. Hann for assistance of animal models, and Dr. Y. Seo for the 3D microPET/CT images. We thank Helmholtz-Zentrum Berlin for the allocation of synchrotron radiation beamtime that received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under BioStruct-X (Grant Agreement 283570). C.B. acknowledges the support from the Czech Science Foundation (Grant 301/12/1513). This publication is supported by Project “BIOCEV” (CZ.1.05/1.1.00/02.0109), from the ERDF.
ABBREVIATIONS USED
- PSMA
prostate-specific membrane antigen
- PCa
prostate cancer
- ABS
arene-binding site
- AH
aminohexanoic acid
- CT
computed tomography
- SFB
succinamidyl 4-fluoroben-zoate
- FB
fluorobenzoate
- hGCPII
human glutamate carbox-ypeptidase II
- nM
nanomolar
- 18F
fluorine-18
- 99mTc
technitium-99m
- ID/g
injected dose/gram
- [18F]DCFBC
N[N-[(S)-1,3-dicarboxypropyl]carbamoyl]-4-[18F] fluorobenzyl-L-cysteine
- [18F]DCFPyL
2-(3-(1-carboxy-5-[(6-[18F]-fluoropyridine-3-carbonyl)amino]pentyl)ureido)pentanedioic acid
Footnotes
Accession Codes
PDB codes are the following: 4LQG (hGCPII/4; deposited as hGCPII/CTT1056) and 4JYW (PSMA/5; deposited as hGCPII/CTT1057). Authors will release the atomic coordinates and experimental data upon manuscript publication.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.5b01850.
Synthesis and characterization of compound 7 and its precursors; complete experimental details of IC50 and mode of inhibition studies; table of crystallographic data and refinement statistics of the compounds (PDF) Molecular formula strings (CSV)
References
- 1.Foss CA, Mease RC, Fan H, Wang Y, Ravert HT, Dannals RF, Olszewski RT, Heston WD, Kozikowski AP, Pomper MG. Radiolabeled small-molecule ligands for prostate-specific membrane antigen: in vivo imaging in experimental models of prostate cancer. Clin Cancer Res. 2005;11:4022–4028. doi: 10.1158/1078-0432.CCR-04-2690. [DOI] [PubMed] [Google Scholar]
- 2.Gao X, Cui Y, Levenson RM, Chung LW, Nie S. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 3.Humblet V, Lapidus R, Williams LR, Tsukamoto T, Rojas C, Majer P, Hin B, Ohnishi S, De Grand AM, Zaheer A, Renze JT, Nakayama A, Slusher BS, Frangioni JV. High-affinity near-infrared fluorescent small-molecule contrast agents for in vivo imaging of prostate-specific membrane antigen. Mol Imaging. 2005;4:448–462. doi: 10.2310/7290.2005.05163. [DOI] [PubMed] [Google Scholar]
- 4.Pomper MG, Musachio JL, Zhang J, Scheffel U, Zhou Y, Hilton J, Maini A, Dannals RF, Wong DF, Kozikowski AP. 11C-MCG: synthesis, uptake selectivity, and primate PET of a probe for glutamate carboxypeptidase II (NAALADase) Mol Imaging. 2002;1:96–101. doi: 10.1162/15353500200202109. [DOI] [PubMed] [Google Scholar]
- 5.Smith-Jones PM, Vallabhajosula S, Navarro V, Bastidas D, Goldsmith SJ, Bander NH. Radiolabeled monoclonal antibodies specific to the extracellular domain of prostate-specific membrane antigen: preclinical studies in nude mice bearing LNCaP human prostate tumor. J Nucl Med. 2003;44:610–617. [PubMed] [Google Scholar]
- 6.Tasch J, Gong M, Sadelain M, Heston WD. A unique folate hydrolase, prostate-specific membrane antigen (PSMA): a target for immunotherapy? Crit Rev Immunol. 2001;21:249–261. [PubMed] [Google Scholar]
- 7.Liu T, Wu LY, Choi JK, Berkman CE. Targeted photodynamic therapy for prostate cancer: inducing apoptosis via activation of the caspase-8/-3 cascade pathway. Int J Oncol. 2010;36:777–784. doi: 10.3892/ijo_00000553. [DOI] [PubMed] [Google Scholar]
- 8.Fracasso G, Bellisola G, Cingarlini S, Castelletti D, Prayer-Galetti T, Pagano F, Tridente G, Colombatti M. Anti-tumor effects of toxins targeted to the prostate specific membrane antigen. Prostate. 2002;53:9–23. doi: 10.1002/pros.10117. [DOI] [PubMed] [Google Scholar]
- 9.Lu J, Celis E. Recognition of prostate tumor cells by cytotoxic T lymphocytes specific for prostate-specific membrane antigen. Cancer Res. 2002;62:5807–5812. [PubMed] [Google Scholar]
- 10.Mease RC, Foss CA, Pomper MG. PET imaging in prostate cancer: focus on prostate-specific membrane antigen. Curr Top Med Chem. 2013;13:951–962. doi: 10.2174/1568026611313080008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Foss CA, Byun Y, Nimmagadda S, Pullambhatla M, Fox JJ, Castanares M, Lupold SE, Babich JW, Mease RC, Pomper MG. Radiohalogenated prostate-specific membrane antigen (PSMA)-based ureas as imaging agents for prostate cancer. J Med Chem. 2008;51:7933–7943. doi: 10.1021/jm801055h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banerjee SR, Foss CA, Castanares M, Mease RC, Byun Y, Fox JJ, Hilton J, Lupold SE, Kozikowski AP, Pomper MG. Synthesis and evaluation of technetium-99m- and rhenium-labeled inhibitors of the prostate-specific membrane antigen (PSMA) J Med Chem. 2008;51:4504–4517. doi: 10.1021/jm800111u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banerjee SR, Pullambhatla M, Shallal H, Lisok A, Mease RC, Pomper MG. A modular strategy to prepare multivalent inhibitors of prostate-specific membrane antigen (PSMA) Oncotarget. 2011;2:1244–1253. doi: 10.18632/oncotarget.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banerjee SR, Pullambhatla M, Byun Y, Nimmagadda S, Green G, Fox JJ, Horti A, Mease RC, Pomper MG. 68Ga-labeled inhibitors of prostate-specific membrane antigen (PSMA) for imaging prostate cancer. J Med Chem. 2010;53:5333–5341. doi: 10.1021/jm100623e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Pullambhatla M, Foss CA, Byun Y, Nimmagadda S, Senthamizhchelvan S, Sgouros G, Mease RC, Pomper MG. 2-(3-{1-Carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pen tanedioic acid, [18F]DCFPyL, a PSMA-based PET imaging agent for prostate cancer. Clin Cancer Res. 2011;17:7645–7653. doi: 10.1158/1078-0432.CCR-11-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho SY, Gage KL, Mease RC, Senthamizhchelvan S, Holt DP, Jeffrey-Kwanisai A, Endres CJ, Dannals RF, Sgouros G, Lodge M, Eisenberger MA, Rodriguez R, Carducci MA, Rojas C, Slusher BS, Kozikowski AP, Pomper MG. Biodistribution, tumor detection, and radiation dosimetry of 18F-DCFBC, a low-molecular-weight inhibitor of prostate-specific membrane antigen, in patients with metastatic prostate cancer. J Nucl Med. 2012;53:1883–1891. doi: 10.2967/jnumed.112.104661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szabo Z, Mena E, Rowe SP, Plyku D, Nidal R, Eisenberger MA, Antonarakis ES, Fan H, Dannals RF, Chen Y, Mease RC, Vranesic M, Bhatnagar A, Sgouros G, Cho SY, Pomper MG. Initial Evaluation of [(18)F]DCFPyL for Prostate-Specific Membrane Antigen (PSMA)-Targeted PET Imaging of Prostate Cancer. Mol Imaging Biol. 2015;17:565–574. doi: 10.1007/s11307-015-0850-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mease RC, Dusich CL, Foss CA, Ravert HT, Dannals RF, Seidel J, Prideaux A, Fox JJ, Sgouros G, Kozikowski AP, Pomper MG. N-[N-[(S)-1,3-Dicarboxypropyl]carbamoyl]-4-[18F]-fluorobenzyl-L-cysteine, [18F]DCFBC: a new imaging probe for prostate cancer. Clin Cancer Res. 2008;14:3036–3043. doi: 10.1158/1078-0432.CCR-07-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu T, Toriyabe Y, Kazak M, Berkman CE. Pseudoirreversible inhibition of prostate-specific membrane antigen by phosphoramidate peptidomimetics. Biochemistry. 2008;47:12658–12660. doi: 10.1021/bi801883v. [DOI] [PubMed] [Google Scholar]
- 20.Liu T, Nedrow-Byers JR, Hopkins MR, Berkman CE. Spacer length effects on in vitro imaging and surface accessibility of fluorescent inhibitors of prostate specific membrane antigen. Bioorg Med Chem Lett. 2011;21:7013–7016. doi: 10.1016/j.bmcl.2011.09.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nedrow-Byers JR, Jabbes M, Jewett C, Ganguly T, He H, Liu T, Benny P, Bryan JN, Berkman CE. A phosphoramidate-based prostate-specific membrane antigen-targeted SPECT agent. Prostate. 2012;72:904–912. doi: 10.1002/pros.21493. [DOI] [PubMed] [Google Scholar]
- 22.Lapi SE, Wahnishe H, Pham D, Wu LY, Nedrow-Byers JR, Liu T, Vejdani K, VanBrocklin HF, Berkman CE, Jones EF. Assessment of an 18F-labeled phosphoramidate peptidomimetic as a new prostate-specific membrane antigen-targeted imaging agent for prostate cancer. J Nucl Med. 2009;50:2042–2048. doi: 10.2967/jnumed.109.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nedrow-Byers JR, Moore AL, Ganguly T, Hopkins MR, Fulton MD, Benny PD, Berkman CE. PSMA-targeted SPECT agents: mode of binding effect on in vitro performance. Prostate. 2013;73:355–362. doi: 10.1002/pros.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganguly T, Dannoon S, Hopkins MR, Murphy S, Cahaya H, Blecha JE, Jivan S, Drake CR, Barinka C, Jones EF, VanBrocklin HF, Berkman CE. A high-affinity [(18)F]-labeled phosphoramidate peptidomimetic PSMA-targeted inhibitor for PET imaging of prostate cancer. Nucl Med Biol. 2015;42:780–787. doi: 10.1016/j.nucmedbio.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu LY, Anderson MO, Toriyabe Y, Maung J, Campbell TY, Tajon C, Kazak M, Moser J, Berkman CE. The molecular pruning of a phosphoramidate peptidomimetic inhibitor of prostate-specific membrane antigen. Bioorg Med Chem. 2007;15:7434–7443. doi: 10.1016/j.bmc.2007.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maung J, Mallari JP, Girtsman TA, Wu LY, Rowley JA, Santiago NM, Brunelle AN, Berkman CE. Probing for a hydrophobic a binding register in prostate-specific membrane antigen with phenylalkylphosphonamidates. Bioorg Med Chem. 2004;12:4969–4979. doi: 10.1016/j.bmc.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 27.Anderson MO, Wu LY, Santiago NM, Moser JM, Rowley JA, Bolstad ES, Berkman CE. Substrate specificity of prostate-specific membrane antigen. Bioorg Med Chem. 2007;15:6678–6686. doi: 10.1016/j.bmc.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu T, Toriyabe Y, Kazak M, Berkman CE. Pseudoirreversible inhibition of prostate-specific membrane antigen by phosphoramidate peptidomimetics. Biochemistry. 2008;47:12658–12660. doi: 10.1021/bi801883v. [DOI] [PubMed] [Google Scholar]
- 29.Barinka C, Mlcochova P, Sacha P, Hilgert I, Majer P, Slusher BS, Horejsi V, Konvalinka J. Amino acids at the N- and C-termini of human glutamate carboxypeptidase II are required for enzymatic activity and proper folding. Eur J Biochem. 2004;271:2782–2790. doi: 10.1111/j.1432-1033.2004.04209.x. [DOI] [PubMed] [Google Scholar]
- 30.Barinka C, Rovenska M, Mlcochova P, Hlouchova K, Plechanovova A, Majer P, Tsukamoto T, Slusher BS, Konvalinka J, Lubkowski J. Structural insight into the pharmacophore pocket of human glutamate carboxypeptidase II. J Med Chem. 2007;50:3267–3273. doi: 10.1021/jm070133w. [DOI] [PubMed] [Google Scholar]
- 31.Pavlicek J, Ptacek J, Barinka C. Glutamate carboxypeptidase II: an overview of structural studies and their importance for structure-based drug design and deciphering the reaction mechanism of the enzyme. Curr Med Chem. 2012;19:1300–1309. doi: 10.2174/092986712799462667. [DOI] [PubMed] [Google Scholar]
- 32.Zhang AX, Murelli RP, Barinka C, Michel J, Cocleaza A, Jorgensen WL, Lubkowski J, Spiegel DA. A remote arene-binding site on prostate specific membrane antigen revealed by antibody-recruiting small molecules. J Am Chem Soc. 2010;132:12711–12716. doi: 10.1021/ja104591m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nedrow JR, Latoche JD, Day KE, Modi J, Ganguly T, Zeng D, Kurland BF, Berkman CE, Anderson CJ. Targeting PSMA with a Cu-64 labeled phosphoramidate inhibitor for PET/CT imaging of variant PSMA-expressing xenografts in mouse models of prostate cancer. Mol Imaging Biol. 2016;18:402–410. doi: 10.1007/s11307-015-0908-7. [DOI] [PubMed] [Google Scholar]
- 34.Liu T, Wu LY, Kazak M, Berkman CE. Cell-Surface labeling and internalization by a fluorescent inhibitor of prostate-specific membrane antigen. Prostate. 2008;68:955–964. doi: 10.1002/pros.20753. [DOI] [PubMed] [Google Scholar]
- 35.Hillier SM, Maresca KP, Femia FJ, Marquis JC, Foss CA, Nguyen N, Zimmerman CN, Barrett JA, Eckelman WC, Pomper MG, Joyal JL, Babich JW. Preclinical evaluation of novel glutamate-urea-lysine analogues that target prostate-specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res. 2009;69:6932–6940. doi: 10.1158/0008-5472.CAN-09-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bacich DJ, Pinto JT, Tong WP, Heston WD. Cloning, expression, genomic localization, and enzymatic activities of the mouse homolog of prostate-specific membrane antigen/NAALADase/folate hydrolase. Mamm Genome. 2001;12:117–123. doi: 10.1007/s003350010240. [DOI] [PubMed] [Google Scholar]
- 37.Ghosh A, Wang X, Klein E, Heston WDW. Novel role of prostate-specific membrane antigen in suppressing prostate cancer invasiveness. Cancer Res. 2005;65:727–731. [PubMed] [Google Scholar]
- 38.Krug M, Weiss MS, Heinemann U, Mueller U. XDSAPP: a graphical user interface for the convenient processing of diffraction data using XDS. J Appl Crystallogr. 2012;45:568–572. [Google Scholar]
- 39.Klusak V, Barinka C, Plechanovova A, Mlcochova P, Konvalinka J, Rulisek L, Lubkowski J. Reaction mechanism of glutamate carboxypeptidase II revealed by mutagenesis, X-ray crystallography, and computational methods. Biochemistry. 2009;48:4126–4138. doi: 10.1021/bi900220s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 43.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nedrow-Byers JR, Jabbes M, Jewett C, Ganguly T, He H, Liu T, Benny P, Bryan JN, Berkman CE. A phosphoramidate-based prostate-specific membrane antigen-targeted SPECT agent. Prostate. 2012;72:904–912. doi: 10.1002/pros.21493. [DOI] [PubMed] [Google Scholar]
- 45.Kuppuswamy D, Pike LJ. Ligand-induced desensitization of 125I-epidermal growth factor internalization. J Biol Chem. 1989;264:3357–3363. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.