

Abstract
In this study, we report a novel perpective of metabolic consequences for the m.8993T > G variant using fibroblasts from a proband with clinical symptoms compatible with Maternally Inherited Leigh Syndrome (MILS). Definitive diagnosis was corroborated by mitochondrial DNA testing for the pathogenic variant m.8993T > G in MT-ATP6 subunit by Sanger sequencing. The long-range PCR followed by massively parallel sequencing method detected the near homoplasmic m.8993T > G variant at 83% in the proband’s fibroblasts and at 0.4% in the mother’s fibroblasts. Our results are compatible with very low levels of germline heteroplasmy or an apparent de novo mutation. Our mitochondrial morphometric analysis reveals severe defects in mitochondrial cristae structure in the proband’s fibroblasts. Our live-cell mitochondrial respiratory analyses show impaired oxidative phosphorylation with decreased spare respiratory capacity in response to energy stress in the proband’s fibroblasts. We detected a diminished glycolysis with a lessened glycolytic capacity and reserve, revealing a stunted ability to switch to glycolysis upon full inhibition of OXPHOS activities. This dysregulated energy reprogramming results in a defective interplay between OXPHOS and glycolysis during an energy crisis. Our study sheds light on the potential pathophysiologic mechanism leading to chronic energy crisis in this MILS patient harboring the m.8993T > G variant.
Keywords: Mitochondrial encephalopathy, MT-ATP6 gene, de novo mutation, ATP synthase, Mitochondrial morphometric analysis, Extracellular flux analysis
1. Introduction
Maternally Inherited Leigh Syndrome (MILS, OMIM 516060) is a fatal neurodegenerative disease with an early childhood onset. It belongs to a subgroup of maternally inherited neurodegenerative disease, referred to as striatal necrosis syndromes, based on their characteristic bilateral lesions of the basal ganglia and brainstem with symmetrical foci of necrosis and capillary proliferation as shown by cranial magnetic resonance imaging (MRI) [1].
Patients with striatal necrosis syndromes show considerable clinical phenotypical variability and harbor maternally inherited mutations in the mitochondrial MT-ATP6 gene, which encodes the subunit 6 of the ATP synthase or complex V [2]. Among the five known pathogenic variants, the T > G point mutation mapping at position 8993 of the mitochondrial genome is the most frequent [3–7]. The m.8993T > G variant results in substitution of a highly conserved leucine residue to arginine (L156R), which affects a subset of the multi-copy mitochondrial genome, causing heteroplasmy characterized by the presence of both wild type (WT) and mutant mitochondrial DNAs (mtDNAs) within mitochondria. In general, the degree of heteroplasmy in combination with other factors, such as nuclear background, dictates not only the disease severity and age of onset, but also the clinical phenotype [2]. Patients carrying the m.8993T > G variant with a heteroplasmic load below 60% remain asymptomatic. Patients with a heteroplasmic load between 75% and 90% exhibit clinical manifestations of neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP), whereas patients with a heteroplasmic load > 90% are affected with the more severe disorder MILS [3,8]. MILS patients exhibit lactic acidosis and multisystemic manifestations with dominant neurological symptoms, which include developmental delay, psychomotor regression, ataxia, seizures, brainstem dysfunction, peripheral neuropathy, and optic atrophy [9].
Even though the molecular pathogenic mechanism of MILS remains to be fully elucidated, it has been firmly demonstrated that the m.8993T > G variant impairs ATP synthesis due to altered assembly of the ATP synthase [10–13]. ATP synthase is the last multisubunit complex of the mitochondrial oxidative phosphorylation (OXPHOS) pathway, where ATP synthesis occurs from ADP and inorganic phosphate using energy from the electrochemical proton gradient as a result of electron transport along the respiratory complexes I to IV. The efficiency by which the proton electrochemical gradient is converted to ATP is a key functional bioenergetic parameter for determining the capacity of a cell to generate the maximum ATP output [14]. All five OXPHOS complexes are embedded along the cristae membrane and modulate the cristae morphology to match the energy needs of the cell [15,16].
ATP synthase is composed of the F0 and F1 functional domains, with the F0 domain acting as a proton channel embedded in the mitochondrial inner membrane and the F1 domain protruding into the mitochondrial matrix and containing the catalytic site for ATP synthesis [17]. The mitochondrial-encoded ATPase 6 (MT-ATPase 6) subunit is part of the F0 domain and has four transmembrane helices that couple proton translocation with rotation of the F0 subunit c-ring to convert mechanical energy into chemical energy for ATP synthesis by the F1 domain [18]. The L156R substitution caused by the m.8993T > G variant prevents MT-ATPase 6 to induce rotation of the c-ring, resulting in decreased ATP synthesis.
In this study, we report a proband with a near homoplasmic m.8993T > G variant presenting clinical symptoms compatible with MILS. Our results from long-range PCR followed by deep sequencing are compatible with the conclusion of an apparent de novo m.8993T > G variant or very low level of germline heteroplasmy. Our functional metabolic studies provide the first evidence that the proband’s fibroblasts harboring the m.8993T > G variant not only exhibit impaired mitochondrial OXPHOS pathway, but also diminished glycolysis as a consequence of a reduced glycolytic capacity and reserve as well as a stunted compensatory glycolysis response. We provide evidence that the proband harboring the m.8993T > G variant dysregulate the metabolic switch from OXPHOS to glycolysis during an energy crisis.
2. Materials and methods
2.1. Subjects
This study was approved by the Institutional Review Board of the George Washington University and Children’s National Medical Center and was conducted in accordance with the ethical principles of the Declaration of Helsinki of 1975 (revised 1983). Patient skin biopsy was performed only after receiving written informed consent with permission to study the derived dermal fibroblasts.
2.2. Skin biopsy and fibroblast culture
Skin biopsy was performed on the 13-month-old proband and his 36-year-old mother. Dermal fibroblasts were derived from 3 mm skin biopsy in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 2 mM glutamine, 2.5 mM pyruvate, 0.2 mM uridine, FGF-2 (10 ng/mL) and 20% fetal bovine serum. Uridine has previously been indicated to preserve respiratory chain deficiencies in cultured fibroblasts [19]. Derived fibroblasts were frozen at passage 2 and never used beyond passage 10. Human primary dermal fibroblasts from healthy donors (GM00302C, 10-month-old; GM03349C; 10-year-old; GM03377E, 19-year-old) were obtained from the Coriell Cell Repositories (Camden, NJ).
2.3. DNA purification and determination of heteroplasmy
DNA was extracted from blood and dermal cultured fibroblast using the QIAamp DNA mini kit according to the manufacturer’s recommendations (Qiagen). Heteroplasmy was determined by Sanger for blood DNA or by a Long-Range PCR (LR-PCR)-based Next Generation Sequencing approach [20,21].
2.4. Transmission electron microscopy
Fibroblasts from the proband and his mother were fixed in 2.5% glutaraldehyde (Electron Microscopy Sciences), 1% paraformaldehyde in 0.12 M sodium cacodylate buffer (Electron Microscopy Sciences) for 20 min at room temperature followed by 40 min on ice. Cells were then fixed for one hour in 1% osmium tetroxide (Electron Microscopy Sciences) followed by en-bloc staining overnight in 1% aqueous uranyl acetate. The cells were dehydrated through a series of ethyl alcohol/deionized water solutions and propylene oxide before infiltration with Embed 812 epoxy resin. Blocks were cured for 48 h at 60 °C. Polymerized blocks were trimmed and 70 nm ultrathin sections were cut with a diamond knife on a Leica Ultramicut EM UC7 and transferred onto 200 mesh copper grids. Sections were counterstained with 1% ethanolic uranyl acetate for 10 min and lead citrate for 2 min. Samples were imaged with a FEI Talos F200X-transmission electron microscope (FEI Company) operating at an accelerating voltage of 80 kV equipped with a Ceta™ 16M camera.
2.5. Analysis of mitochondrial respiratory and glycolytic activities
Bioenergetic status was measured using the Seahorse Extracellular Flux XFp Analyzer (Seahorse Bioscience, Agilent Technologies). Optimal cell density (5000/well) and the uncoupler FCCP (fluoro 3-carbonyl cyanide-methoxyphenyl hydrazine; 2 μM) were determined using the Cell Energy Phenotype Test kit. Skin fibroblasts were seeded in triplicate on poly-D lysine-coated plates and incubated for 24 h at 37 °C in 5% CO2 atmosphere. Prior to the assay, the supplemented DMEM medium was changed to unbuffered Base Medium supplemented with 2 mM glutamine (Invitrogen), 2 mM pyruvate (Sigma), and/or 7.1 mM glucose (Sigma) depending on the assay and adjusted to at pH 7.4 with NaOH for 1 h at 37 °C. Using the XFp Mito Stress Test kit, OCR (oxygen consumption rate) and ECAR (extracellular acidification rate) were measured under basal conditions and after sequential injections of oligomycin (1 μM), FCCP (2 μM) and a mix of rotenone and antimycin A (1 μM) following the manufacturer’s recommendations. Using the XFp Glycolysis Stress Test kit, we measured the glycolytic functions under basal and stress conditions after sequential injections of glucose (10 mM), oligomycin (1 μM) and 2-DG (50 mM). Using the Glycolytic Rate Assay kit, we determined the total proton efflux and the glycolytic proton efflux by measuring the OCR and ECAR values. Prior to the assay, the supplemented DMEM medium was changed to the XF Base medium without phenol red supplemented with 2 mM glutamine, 10 mM glucose, 1 mM pyruvate, and 5.0 mM HEPES. OCR and ECAR were measured under basal conditions and after sequential injections of rotenone/antimycin A (0.5 μM final concentration) and 2-DG (50 mM final concentration). The data from three independent experiments were normalized to cell numbers quantified after the assay and plotted as OCR (pmol/min/cell ± S.D.) and ECAR (mpH/min/cell ± S.D.) as a function of time using the Seahorse Report Generator software. Statistical analyses were performed using the unpaired student t-test with p-value of < 0.05 considered statistically significant.
3. Results
3.1. Clinical manifestations
The proband was born full term via NSVD, weighing 3.9 kg. There were no prenatal nor postnatal complications. His mother received prenatal care. She was positive for group B strep and received in-trapartum antibiotics. There were no maternal or fetal infections noted. There were no maternal complications or bleeding noted. He developed normally up to four months of age when he received a set or vaccines (diphtheria, tetanus, and acellular pertussis vaccine; Haemophilus in-fluenza type b vaccine; inactivated poliovirus vaccine; pneumococcal conjugate vaccine; and rotavirus vaccine). He then appeared to be floppy with little to no energy. At six months of age, he had another set of vaccines and stopped breastfeeding and smiling. At seven months of age, he had regressed and was developmentally delayed. At nine months of age, his initial electroencephalogram (EEG) suggested an encephalopathy with generalized slowing. His first brain MRI scan revealed symmetric areas of T2/FLAIR signal change and diffusion restriction involving the bilateral basal ganglia, thalami and midbrain consistent with Leigh Syndrome (Fig. 1A). Laboratory testing performed during his initial visit demonstrated elevated lactate levels (54.4 mg/dL), increased plasma levels of the amino acid alanine (687.9 μmol/L), and elevated levels of L3 acylcarnitine (1.03 μmol/L) in keeping with a mitochondrial disease. He was started on riboflavin, thiamine, coenzyme Q10 and carnitine. His subsequent medical course was notable for development of infantile spasms requiring treatment with Clobazam, Topiramate, and Levetiracetam. He was also started on a Mitochondrial cocktail and Vitamin D.
Fig. 1.
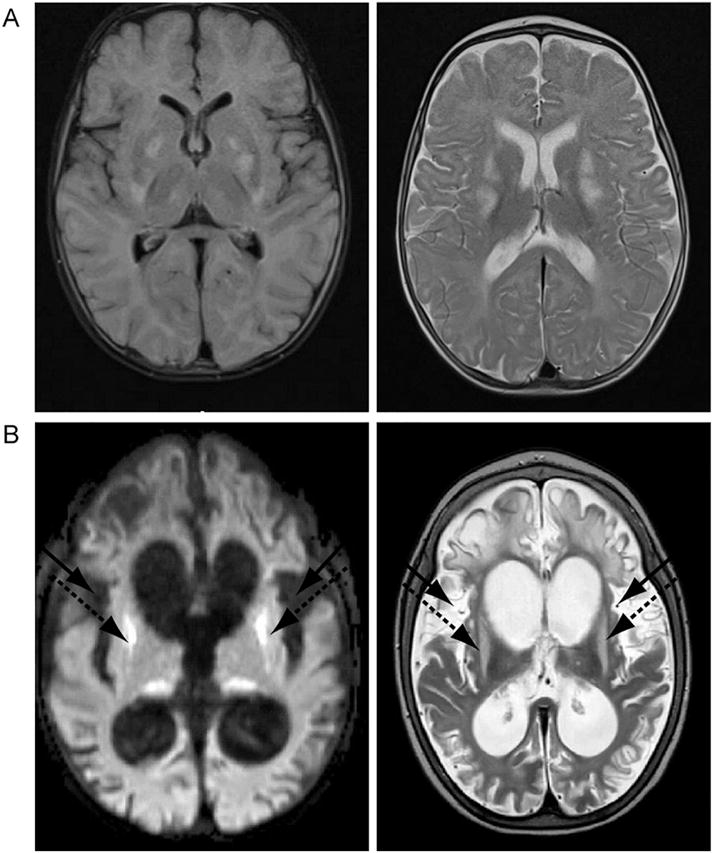
Brain magnetic resonance imaging of the proband. (A) The left panel illustrates the T2 weighted image performed at four months of age showing bilateral symmetric lesions in basal ganglia and thalami, while the right panel depicts the corresponding FLAIR imaging showing T2 hypersensitivities characteristic of Leigh Syndrome. (B) The left panel illustrates the T2 weighted image perfomed at nine months of age showing progressive cortical atrophy and resulting ventriculomegaly. In addition, there is increased T2 signal bilaterally in the putamen (dashed arrows) and insula (black arrows). The right panel shows the corresponding FLAIR imaging.
He also developed cortical blindness, apnea, and oral motor dysfunction requiring gastrostomy tube feeding. Following an inter-current illness with flu-like symptoms, his feeding abilities regressed and his seizures increased. Although he recovered, he remained slightly below his previous baseline. At two years of age, his development remained that of a newborn since he was unable to roll, sit or stand. He had no language and failed to use his hands in a purposeful manner. On examination, he had poor visual tracking, drooling and decreased gag response. He had both truncal and appendicular hypotonia. DTRs were 2+ without clonus.
Clinically, brief tonic extension of the upper extremities was noted. His subsequent EEG showed focal features, especially left occipital, and modified hypsarhythmia pattern. There were multifocal posterior dominant spike discharges and marked background discontinuity, dis-organization and slowing. A third EEG showed generalized and bi-occipital slowing frequent rhythmic high amplitude sharp discharges bilateral occipital regions (O1 and O2), at times near continuous, cluster of brief tonic seizures captured on this EEG. His EEG indicated voltage attenuation with superimposed faster frequency activity predominantly in the right hemisphere for about 4 s. These are consistent with a generalized and focal diffuse epileptic encephalopathy. His follow-up MRI taken a few months after his flu-like episode showed marked progressive cortical atrophy, resulting in ventriculomegaly (Fig. 1B). In addition, his T2 weighted axial image revealed increased T2 signal bilaterally in the putamen, consistent with mitochondrial encephalopathy (Fig. 1B).
3.2. Identification of an apparent de novo mitochondrial m.8993T > G variant in the proband’s fibroblasts
Initial Sanger-based sequence analysis of the mitochondrial genome (mtDNA) from the blood samples revealed a homoplasmic m.8993T > G variant in the proband, but this variant was not detected in the mother, suggesting a possible de novo mutation that became near or homoplasmic in one generation. In order to determine its degree of heteroplasmy with great accuracy, we analyzed the mtDNA from dermal cultured fibroblasts using a LR-PCR-based Next Generation Sequencing (NGS) approach [20,21]. This analysis revealed near homoplasmic levels (83%) of the m.8993T > G variant in the proband (Fig. 2). In contrast, we detected 1 out of 250 reads in the mother’s fibroblasts, amounting to a very low level of heteroplasmy estimated at 0.4%. Such very low levels of heteroplasmy are within experimental error (Fig. 2) even though the LR-PCR-based NGS method can accurately calculate the level of heteroplasmy from as low as 1 read per 20,000 [20,21]. Thus, our results are compatible with either an apparent de novo mutation in the proband or very low levels of maternal germline mosaicism, two genetic scenarios that cannot be currently discriminated despite the increased sensitivity of the genomic technology.
Fig. 2.
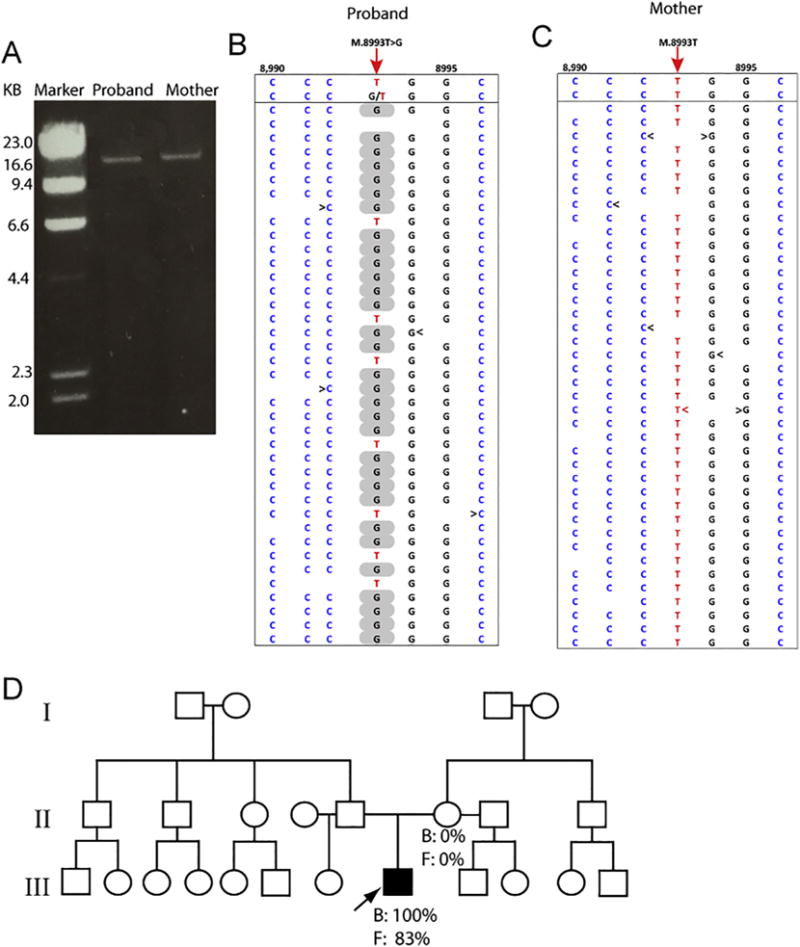
Detection of the m.8993T > G variant by LR-PCR-based next generation sequencing in fibroblasts of the proband and his mother. (A) Gel electrophoresis of the LR-PCR products from the proband and his mother. The size of the amplicon product is 16.6 kb. (B) The piled-up LR-PCR/massively parallel sequencing (MPS) result of the proband reveals that there are 83% T > G at the m.8993 position. (C) The piled-up LR-PCR/massively parallel sequencing (MPS) result of the proband’s mother indicates that all nucleotides at the m.8993 position are T’s. (D) Pedigree tree with the proband identified by an arrow. Percentages indicate the proportion of m.8993T > G variant in blood (B) and fibroblasts (F) of the proband and his mother.
3.3. Mitochondrial morphometric analysis of fibroblasts derived from the proband and his mother
Since the ultrastructure and morphology of cristae control the mitochondrial bioenergetic functions [22], we investigated by transmission electron microscopy whether mitochondria from the pro-band’s fibroblasts exhibited ultrastructural alterations of mitochondria when compared to those of his mother. The mother’s fibroblasts exhibited mitochondria with a normal ultrastructural morphology characterized with numerous and closely apposed cristae and a highly electron dense mitochondrial matrix (Fig. 3A). Furthermore, we observed mitochondrial fission-fusion events, indicative of normal mitochondrial dynamics (Fig. 3A). In contrast, most mitochondria from the proband’s fibroblasts have very few and short cristae that appear disorganized, while some mitochondria are devoid of cristae (Fig. 3B). Some mitochondria had enlarged cristae lumen width, a morphological characteristic associated with defective OXPHOS activity. In addition, some mitochondria exhibit abnormal “onion-like” concentric cristae, suggestive of abnormal cristae junctions between the inner boundary membrane and the cristae membrane (Fig. 3B). A similar morphological defect of mitochondrial cristae has been reported in muscle cells derived from patients affected with the MERRF mitochondrial disease [23]. Ultimately, mitochondrial identity needs to be confirmed by immune-gold electron microscopy [24]. Finally, the proband’s mitochondria consistently display a weak electron dense mitochondrial matrix (Fig. 3). Thus, our mitochondrial morphometric findings are consistent with the genetic diagnosis of a near homoplasmic m.8993T > G variant in the proband and absence of this variant in his mother.
Fig. 3.
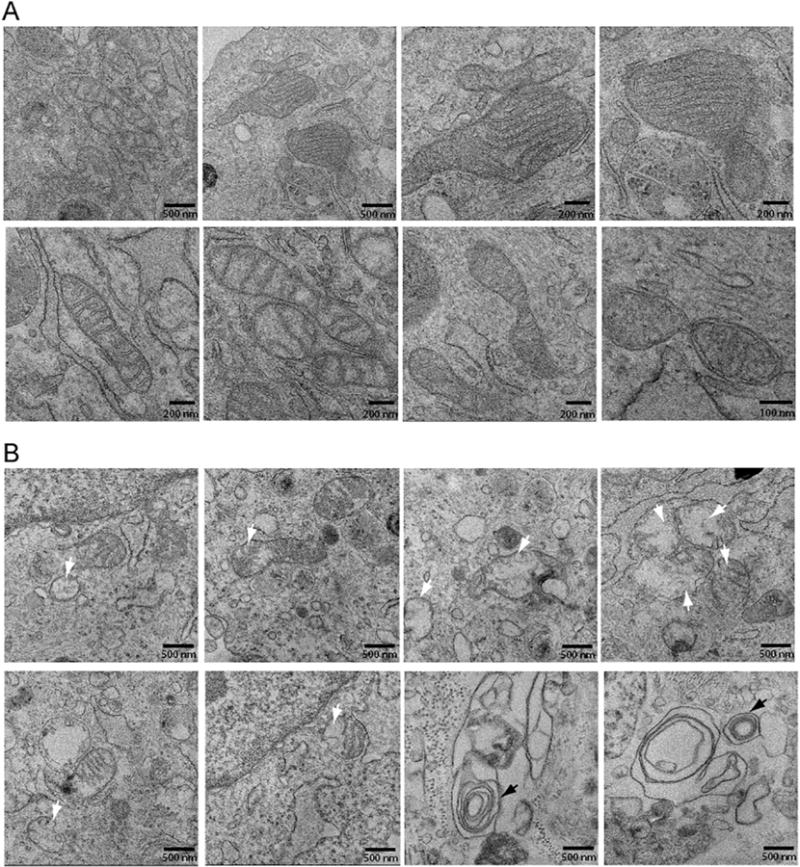
The m.8993T > G variant alters the morphology and abundance of cristae. (A) Mitochondrial morphometric analysis by transmission electron microscopy using dermal fibroblasts of the proband’s mother. The scale bar is indicated at the bottom right corner of each micrograph. (B) Mitochondrial morphometric analysis by transmission electron microscopy using dermal fibroblasts of the proband. The scale bar is indicated at the bottom right corner of each micrograph. The white arrows indicate mitochondria with abnormal cristae, while black arrows concentric cristae.
3.4. Functional outcome of the near homoplasmic m.8993T > G variant on mitochondrial respiration
Since the m.8993T > G variant affects the mitochondrial-encoded subunit ATPase 6 of the ATP synthase, we next investigated its functional impact on the OXPHOS metabolism in fibroblasts derived from the 13-month-old proband. We used the Seahorse Extracellular Flux XFp analyzer for live-cell measurement of the oxygen consumption rate (OCR), a key functional indicator of the mitochondrial ATP-coupled respiration. We initially determined the optimal cell seeding density and the concentration of the uncoupler compound FCCP using the cell energy phenotype kit (data not shown). We measured several key mitochondrial bioenergetics parameters of the OXPHOS pathway (Fig. 4A, B). As a control subject, we used dermal fibroblasts derived from a 10-month-old healthy infant. We also included additional control fibroblasts from two healthy subjects even though they were not age-matched, the GM00377E from a 19-year-old male subject and the GM03349C from a 10-year-old male child. We found that the mother’proband exhibited a similar mitochondrial respiration profile than the three controls, while the proband had a subtantially impaired OCR profile when compared to his mother and the three control subjects (Fig. 4B). We found a 40% decrease in the basal OCR of the MILS fibroblasts when compared to that of a 10-month-old healthy infant (Fig. 4C). By exposing the fibroblasts to oligomycin, an inhibitor of the ATP synthase, we detected a 36% decrease in ATP-linked respiration (Fig. 4C). The proband’s fibroblasts exhibited a similar response to saturated concentration of oligomycin (1 μM) than the controls, congruent with a previous study [25]. Next, we measured the maximal respiratory capacity evoked by FCCP, which disrupts the proton gradient across the inner mitochondrial membrane. We found a decrease in the maximal respiration capacity and spare respiratory capacity by 32% and 20%, respectively, indicating that the proband’s fibroblasts have a diminished energy capacity to respond to stress (Fig. 4C).
Fig. 4.
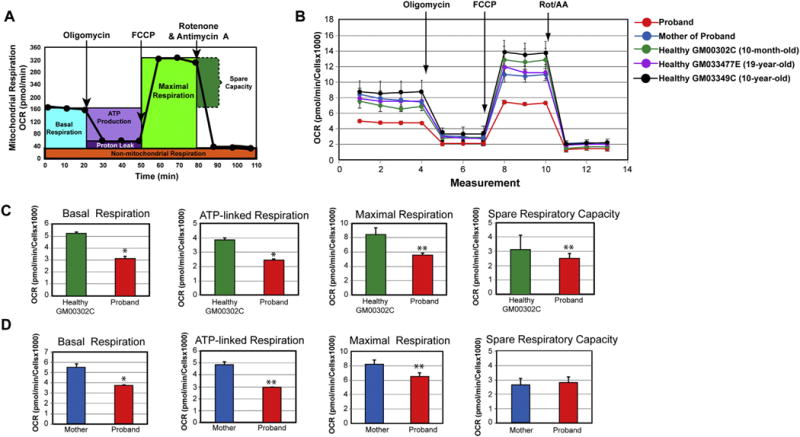
Impaired mitochondrial respiration functions in the fibroblasts of the proband. (A) Profile of the oxygen consumption rate (OCR) adapted from the Agilent Technologies brochure of the Mitochondrial Stress Test. (B) Compared OCR responses among the proband, his mother, and three healthy control subjects. (C) Quantitative data between the proband and the age-matched control (10-month-old infant) of basal respiration, ATP-linked respiration, maximal respiration, and spare respiratory capacity. Data are represented as means ± S.D., n = 3 of three independent experiments. * and ** indicate statistically significant differences with a p value of ≤0.005 and ≤0.0012, respectively, between the proband and healthy infant. (D) Quantitative data between the proband and his mother of basal respiration, ATP-linked respiration, maximal respiration, and spare respiratory capacity. Data are represented as means ± S.D., n = 3 of independent experiments. * and ** indicate statistically significant differences with a p value of 0.0003 and ≤0.0019, respectively, between the proband and his mother.
We compared the mitochondrial bioenergetics functions of the proband with those of his mother (Fig. 4D). The proband exhibited a decrease in basal OCR, maximum respiratory capacity, and ATP-linked respiration by 32%, 20%, and 40%, respectively (Fig. 4E). Interestingly, the spare respiratory capacity remained unaltered between the proband and mother (Fig. 4E). Collectively, our bioenergetics results reveal that the p.L156R MT-ATP6 subunit of ATP synthase significantly alters the overall OXPHOS pathway with respiratory defects related to respiratory complexes of the electron transfer chain that did not bear any mutations.
3.5. Functional outcome of the near homoplasmic m.8993T > G variant on the glycolysis pathway
We next assessed the contribution of the glycolysis pathway to meet the energy demand in the proband’s fibroblasts. We initiated our analysis by measuring basal extracellular acidification rate (ECAR) between the proband and a 10-month-old healthy infant using the glycolysis stress test assay (Fig. 5A and B). The cells were starved for glucose for 1 h prior to injecting glucose in order to measure bulk acidification as an indicator of glycolytic activity. The proband’s fibroblasts exhibited a non-statistically significant decrease in ECAR upon injection of glucose when compared to control healthy fibroblasts (Fig. 5C). We next evaluated the maximal glycolytic capacity by exposing the fibroblasts to oligomycin, which inhibits ATP synthase and therefore respiratory ATP production. We detected a 59% reduction in glycolytic capacity in the proband when compared to that of a healthy infant (Fig. 5C), indicating that the proband’s fibroblasts could not increase glycolysis sufficiently to compensate for the inhibition of mitochondrial ATP production, as healthy fibroblasts did. Finally, upon inhibition of the glucose metabolism via exposure to 2-deoxyglucose (2-DG), we observed a 79% reduction in glycolytic reserve in the proband’s fibroblasts, when compared to that of a healthy infant (Fig. 5C). We next compared the glycolytic activity between the mother and the proband and found that the proband’s fibroblasts exhibited lower glycolytic capacity and reserve by 57% and 78%, respectively, than the mother’s fibroblasts (Fig. 5D, E).
Fig. 5.
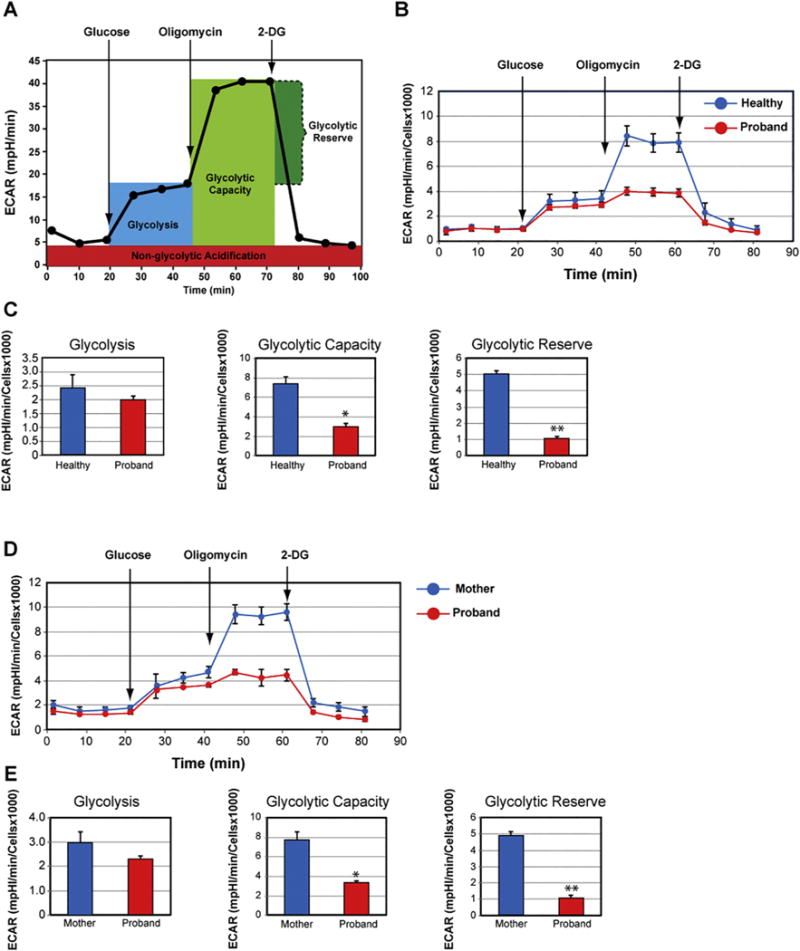
Impaired glycolytic activity in the proband’s fibroblasts. (A) Profile of the extracellular acidification rate (ECAR) adapted from the Agilent Technologies brochure of the Glycolysis Stress Test. (B) Compared ECAR responses between the proband and a healthy infant. (C) Quantitative data of glycolysis, glycolytic capacity, and glycolytic reserve. Data are represented as means ± S.D., n = 3 of independent experiments. * and ** indicate statistically significant differences with a p value of 0.0005 and 0.0001, respectively, between the proband and healthy infant. (D) Compared ECAR responses between the proband and his mother. (E) Quantitative data of glycolysis, glycolytic capacity, and glycolytic reserve. Data are represented as means ± S.D., n = 3 of independent experiments. * and ** indicate statistically significant differences with a p value of 0.0026 and 0.0005, respectively, between the proband and his mother.
Since bulk acidification measured by the Glycolysis Stress Test assay ECAR does not discriminate between mitochondrial acidification as a by-product of the tricarboxylic acid cycle (TCA) cycle and acidification from glycolysis-derived lactate, the glycolytic rate is overestimated. During mitochondrial respiration, TCA produces CO2 that is exported outside the cells to give rise by hydration to six carbonate ions and protons, thereby substantially contributing to the acidification of the medium [26]. Thus, we performed the complementary Glycolysis Rate assay, which measures the glycolytic proton production rate (Gly-coPER) instead of bulk acidification (ECAR). The GlycoPER correlates one-on-one with lactate accumulation, a precise and direct measurement of glycolytic activity. The Glycolytic Rate assay was performed under physiological conditions in the absence of the stressor, glucose starvation, and upon blocking the mitoxondrial OXPHOS pathway at the complexes I and III via injection of rotenone and antimycin A, thereby precluding mitochondrial acidification [27]. We found that the glycolytic proton efflux rate of the MILS fibroblasts decreases by 57% when compared to that of healthy fibroblasts, indicating a significant decrease in basal glycolysis (Fig. 6C). We next assessed the compensatory glycolysis response upon blocking the mitochondrial OXPHOS, which indicates the ability of the cells to switch to glycolysis to produce ATP. We found that MILS fibroblasts exhibited a significantly diminished compensatory glycolysis by 63% when compared to that of healthy fibroblasts (Fig. 6C). This diminished glycolytic response is consistent with the 79% reduction in glycolytic reserve quantified using the glycolysis stress test assay (Fig. 5C). Finally, we found that the MILS fibroblasts had half of the amount of mitochondrial acidification than healthy fibroblasts (Fig. 6B, C), consistent with the observed 40% decrease in mitochondrial OXPHOS metabolism (Fig. 4C). We next compared the glycolytic response between the mother and the proband and detected a decrease in basal glycolysis and compensatory glycolysis by 32% and 35%, respectively, in the proband (Fig. 6D, E). Additionally, we detected a significant decrease by 33% decrease in mitochondrial acidification in the proband (Fig. 6E), congruent with the observed reduction in mitochondrial OXPHOS functions (Fig. 4 E). Thus, our collective metabolic results show that the proband’s fibroblasts harboring the m.8993T > G variant exhibit a stunted ability to switch to glycolysis to compensate for defective OXPHOS activities.
Fig. 6.
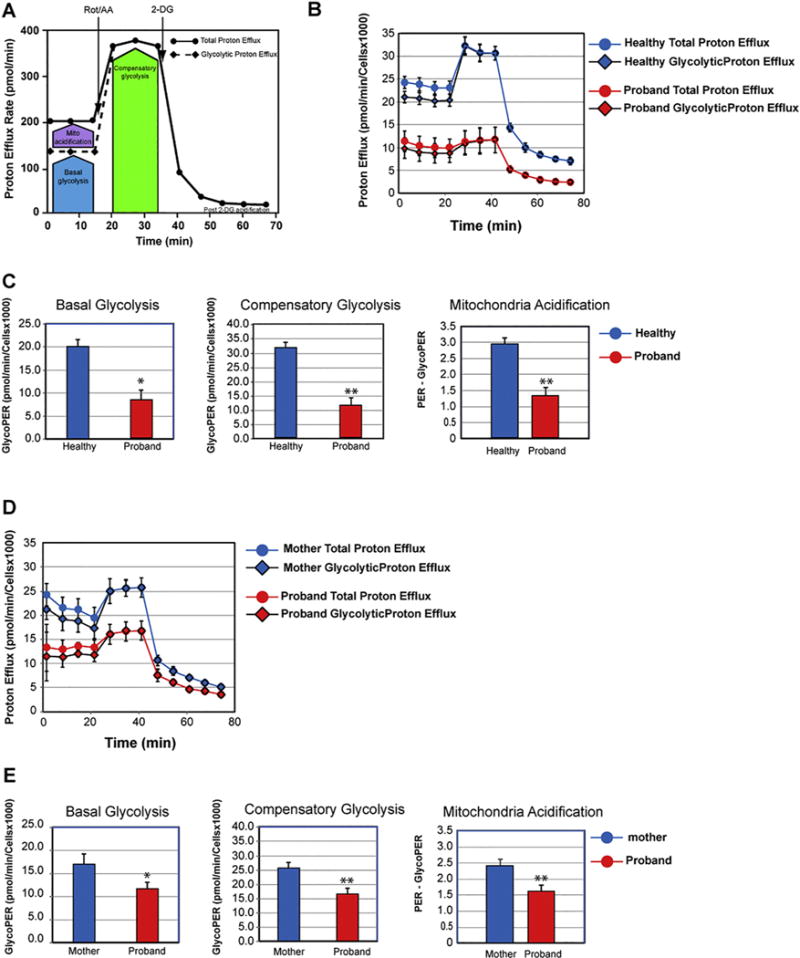
Altered glycolytic adaptation in the fibroblast’s proband. (A) Schematic representation of proton efflux adapted from the Agilent Technologies brochure of the Glycolytic Rate Test. (B) Compared proton efflux rate between the proband and healthy infant. (C). Quantitative data of basal glycolysis, compensatory glycolysis, and mitochondrial acidification. Data are represented as means ± S.D., n = 3 of independent experiments. * and ** indicate statistically significant differences with a p value of 0.001 and ≤0.0005, respectively, between the proband and healthy infant. (D) Compared proton efflux rate between the proband and his mother. (E). Quantitative data of basal glycolysis, compensatory glycolysis, and mitochondrial acidification. Data are represented as means ± S.D., n = 3 of independent experiments. * and ** indicate statistically significant differences with a p value of 0.002 and ≤0.0006, respectively, between the proband and his mother.
4. Discussion
In this study, we report the comprehensive metabolic signature of a proband harboring the m.8993T > G variant in the MT-ATPase 6 gene responsible to cause the NARP/MILS syndrome. Despite the well-established consensus that the m.8993T > G variant alters the rate of ATP synthesis, little is known about its global impact on the mitochondrial and glycolytic metabolism in patient-derived cells. Previously, the bioenergetic impact of the m.8993T > G variant was evaluated in the absence of the input from the nuclear genome since these studies were performed using the m8993. T > G cybrid system, the yeast model, or sub-mitochondrial particles isolated from platelets of patients [10,13,28–33]. Herein, we provide the first comprehensive functional analysis of mitochondrial respiratory and glycolytic functions in patient-derived fibroblasts harboring a near homoplasmic m.8993T > G variant. Our mitochondrial morphometric analysis combined with our live-cell mitochondrial respiratory analysis demonstrate a functional link among disrupted mitochondrial ultrastructure, impaired mitochondrial OXPHOS efficiency, and diminished glycolytic response, preventing an efficient switch from OXPHOS to glycolysis in the pro-band carrying a near homoplasmic m.8993T > G variant.
The subtle discrepancy in the degree of m.8993T > G heteroplasmy between blood and fibroblasts may be owing to: (a) Sanger analysis is not quantitative, which usually has a background of 20% removed, qualifying a heteroplasmy level of 83% to be near homoplasmic; (b) although we used cultured fibroblasts below 10 passages, repetitive culturing of fibroblasts has been shown to reduce the heteroplasmic level of mtDNA mutant; and (c) there may be a difference in the degree of heteroplasmy between blood and skin fibroblasts. Our LR-PCR-NGS results are consistent with an apparent de novo variant or very low levels of germline heteroplasmy. The frequency of de novo variants is low with only four cases of de novo m.8993T > G variant among a total of 105 patients [34]. It is worth emphasizing that the de novo status of these variants may results from the technique used to quantify heteroplasmy. Until recently, heteroplasmy was assessed by Sanger sequencing, which lacks sensitivity to detect low levels of heteroplasmy, resulting in variants being labeled as de novo with a one-generation jump from zero to near 100%. With the advent of deep sequencing, detecting very low levels of heteroplasmy is now feasible with the highly sensitive and accurate long-range PCR followed by massively parallel sequencing, thereby bringing into question the previously reported de novo variants. These variants may result from very low levels of germline heteroplasmy, thereby affecting their heritability frequency. Thus, the highly sensitive LR-PCR-NGS method allows for a comprehensive and accurate assessment of heteroplasmy, thereby clarifying the low frequency of apparent de novo mutations in future studies and making it possible to perform longitudinal studies using multiple tissues from patients affected with a mitochondrial disease due to a mitochondrial DNA mutation.
The near homoplasmic pathogenic m.8993T > G variant disrupts the shape and number of mitochondrial cristae with mitochondria being devoid of cristae or with altered cristae morphology, such as short cristae/or concentric-lamellar cristae. Furthermore, the proband’s fibroblasts contain small mitochondria when compared to those of the mother’s fibroblasts, consistent with dysregulated mitochondrial dynamics and abnormal cristae ultrastructural morphology. An earlier electron microscopy study reported the presence of unusual cristae in mitochondria from a patient carrying the m.8993T > G variant [35]. Ultrastructural defects of mitochondrial cristae have also been observed in a Drosophila ATP6 mutant model harboring mitochondria with numerous short, round cristae and a honeycomb appearance [36]. The nuclear-mitochondrial crosstalk may lend a potential explanation for the difference in cristae morphology between the human MILS fibroblasts and the Drosophila mutant model. The dynamin-related GTPase Optic Atrophy 1 (OPA1) located in the mitochondrial inner membrane also plays a critical role in both cristae architecture and mitochondrial fusion [22,37,38].
The structural role of ATP synthase as one of the main regulators of cristae morphology has been well-established in several non-pathological mammalian cellular and yeast systems [15,16,39,40]. Deletion or knockdown of the subunit e or g, both involved in the formation of ATP synthase dimerization and oligomerization, promotes onion-like concentric mitochondrial cristae [41–43]. Yeast cells treated with the in vivo cross-linker DsRed, which affects ATP synthase oligomerization, harbor mitochondria either devoid of cristae or with concentric lamellar cristae, thereby lending further support to the role of ATP synthase as a key determinant of the cristae topology [44]. Furthermore, electron cryotomography-based studies have unraveled the intertwined functional and morphological roles of ATP synthase dimerization by demonstrating that rows of ATP synthase dimers are concentrated at the tip of the mitochondrial cristae, resulting in membrane curvature where a local gradient of protons is created to optimize mitochondrial ATP synthesis [45,46].
Our live-cell bioenergetic functional results demonstrate that the near homoplasmic m.8993 T > G variant induces a profound decline in ATP-linked respiration supporting our morphometric findings of altered mitochondrial cristae architecture and the functional role of dimerized ATP synthase as proton trap to generate a local proton sink for effective ATP synthesis [46]. Increased pH in the mitochondrial matrix has previously been reported in T8993G mutant cybrids, which may be explained by altered topography of cristae membranes and local proton gradient in the cristae space [31]. In the case of the m.8993T > G variant, the proton trap is affected given the key role of the subunit ATPase 6 for dimerization of ATP synthase [47]. Several studies have demonstrated that defect in ATP synthesis associated with the m.8993T > G variant is the result of incomplete assembly rather than degradation of Complex V [18,25,26]. The F1 sub-complex is a stable assembly intermediate waiting to bind the mitochondrial encoded AT-Pase 6 subunit to conclude complex V assembly [17]. Furthermore, the substitution of leucine with arginine at the position 156 of the ATP synthase does not prevent competent proton translocation through F0, but rather affects its coupling with ATP synthesis in lymphocytes from patients harboring the m.8993T > G variant [18].
The proband’s fibroblasts also exhibit a decreased basal respiration as a result of decline in substrate oxidation congruent with the drop in mitochondrial acidification due to decreased CO2 production from the mitochondrial tricarboxylic acid cycle. Consistent with mitochondrial dysfunction is the decrease in maximum respiration detected upon exposure to the protonophore FCCP. Thus, the functional bioenergetic consequences of the m.8993T > G variant are not confined to ATP synthase, but rather expand to the other upstream respiratory complexes. Our results are in keeping with the reported decreased complex I, III, and IV biochemical activities in muscle biopsies from NARP/MILS patients with a high heteroplasmy load of the m.8993T > G variant [48,49]. Compromised biochemical activities of the complexes I, III, and IV have also been detected in 8993T > G cybrids, suggesting that impaired respiratory activities of those complexes devoid of any pathogenic mutations are not dictated by the nuclear genome [13,30]. In the presence of the m.8993T > G variant, the spare respiratory capacity is decreased, indicating that the proband’s fibroblasts lack the ability to respond to a sudden increase in energy demand, substantiating the severe lactic acidosis measured in the proband and his clinical symptoms associated with chronic energy deficit.
Until now, the bioenergetics consequences of the p.L156R of ATP6 on the glycolytic pathway have not been investigated. Our study provides a novel perpective of metabolic consequences of the m.8993T > G variant not previously examined by showing that this pathogenic variant greatly impacts the glycolytic pathway by lessening the glycolytic capacity and reserve. Most importantly, our novel and unexpected metabolic results reveal that the proband’s fibroblasts harboring the m.8993T > G variant had a diminished ability to switch to glycolysis upon full inhibition of the mitochondrial OXPHOS pathway. Thus, this variant in the proband prevents an efficient metabolic switch from OXPHOS to glycolysis, which is critical to maintain ATP production under conditions simulating an acute ATP crisis. Similarly, we observed a stunted glycolytic response during glucose starvation, which has clinical relevance in light of the feeding difficulties linked to the MILS disease. Therefore, the overall decreased glycolytic activities combined with a deficient metabolic switch most likely lead to aggravated clinical manifestations, significant disability and premature death.
In conclusion, the near homoplasmic m.8993T > G variant provokes severe defects in the mitochondrial ultrastructure of the cristae membranes with detrimental functional bioenergetic consequences on the mitochondrial OXPHOS pathway. Moreover, the proband’s fibroblasts display a defective interplay between the two energy metabolic pathways, OXPHOS and glycolysis. Collectively, our results provide insight on the potential pathogenic mechanism of the m.8993T > G variant leading to chronic energy crisis in this MILS patient and set the stage for future studies to confirm this metabolic disturbance in a cohort of patients harboring the same variant and to ultimately design novel therapeutic avenues.
Acknowledgments
Funding
This work was funded by the NIH National Institute of Neurological Disorders and Stroke [NS085282 to AC], from the National Institute of Child Health and Development [1U54HD090257] and the NIH National Center for Advancing Translational Sciences [UL1TR00075].
Footnotes
Conflict of interest
There are no conflict of interest to disclose.
References
- 1.Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. J Bioenerg Biomembr. 1997;29:131–149. doi: 10.1023/a:1022685929755. [DOI] [PubMed] [Google Scholar]
- 2.Schon EA, Santra S, Palloti F, Girvin ME. Pathogenesis of primary defects in mitochondrial ATP synthesis, Semin. Cell Dev Biol. 2001;12:441–448. doi: 10.1006/scdb.2001.0281. [DOI] [PubMed] [Google Scholar]
- 3.Tatuch Y, Christodoulou J, Feigenbaum A, Clarke JTR, Wherret J, Smith C, Rudd N, Petrova-Benedict R, Robinson BH. Heteroplasmic mtDNA mutation (T > G) at 8993 can cause Leigh disease when the percentage of abnormal mtDNA is high. Am J Hum Genet. 1992;50:852–858. [PMC free article] [PubMed] [Google Scholar]
- 4.Ciafaloni E, Santorelli FM, Shanske S, Deonna T, Roulet E, Janzer C, Pescia G, DiMauro S. Maternally inherited Leigh syndrome. J Pediatr. 1993;122:419–422. doi: 10.1016/s0022-3476(05)83431-6. [DOI] [PubMed] [Google Scholar]
- 5.Santorelli FM, Shanske S, Macaya A, DeVivo DC, DiMauro S. The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh’s syndrome. Ann Neurol. 1993;34:827–834. doi: 10.1002/ana.410340612. [DOI] [PubMed] [Google Scholar]
- 6.Pastores GM, Santorelli FM, Shanske S, Gelb B, Fyfe B, Wolfe D, Willner JP. Leigh syndrome and hypertrophic cardiomyopathy in an infant with a mitochondrial DNA point mutation (T8993G) Am J Hum Genet. 1994;50:265–271. doi: 10.1002/ajmg.1320500310. [DOI] [PubMed] [Google Scholar]
- 7.Uziel G, Moroni I, Lamantea E, Fratta GM, Ciceri E, Carrara F, Zeviani M. Mitochondrial disease associated with the T8993G mutation of the mitochondrial ATPase 6 gene: a clinical, biochemical, and molecular study in six families. J Neurol Neurosurg Psychiatry. 1997;63:16–22. doi: 10.1136/jnnp.63.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holt IJ, Harding AE, Petty RKH, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428–433. [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman S, Blok RB, Dahl HHM, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- 10.Baracca A, Barogi S, Carelli V, Lenaz G, Solaini G. Catalytic activities of mitochondrial ATP synthase in patients with mitochondrial DNA T8993G mutation in the ATPase 6 gene encoding subunit a. J Biol Chem. 2000;275:4177–4182. doi: 10.1074/jbc.275.6.4177. [DOI] [PubMed] [Google Scholar]
- 11.Nijtmans LGJ, Henderson NS, Attardi G, Holt IJ. Impaired ATP synthase assembly associated with a mutation in the human ATP synthase subunit 6 gene. J Biol Chem. 2001;276:6755–6762. doi: 10.1074/jbc.M008114200. [DOI] [PubMed] [Google Scholar]
- 12.Pallotti F, Baracca A, Hernandez-Rosa E, Walker WF, Solaini G, Lenaz G, D’Eril GVMelzi, DiMauro S, Davidson MM. Biochemical analysis of respiratory function in hybrid cell lines harbouring mitochondrial DNA mutations. Biochem J. 2004;384:287–293. doi: 10.1042/BJ20040561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Aurelio M, Vives-Bauza C, Davidson MM, Manfredi G. Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Nucleic Acids Res. 2010;19:374–386. doi: 10.1093/hmg/ddp503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cogliati S, Enriquez JA, Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci. 2016;41:261–273. doi: 10.1016/j.tibs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Quintana-Cabrera R, Mehrotra A, Rigoni G, Soriano ME. Who and how in the regulation of mitochondrial cristae shape andfunction. Biochem Biophys Res Commun. 2017 doi: 10.1016/j.bbrc.2017.04.088. http://dx.doi.org/10.1016/j.bbrc (2017.04.088 [published Oline First: 21 April 2017]) [DOI] [PubMed]
- 17.Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP synthase: architecture, function and pathology. J Inherit Metab Dis. 2012;35:211–225. doi: 10.1007/s10545-011-9382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scarbi G, Baracca A, Lenaz G, Valentino LM, Carelli V, Solaini G. Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA. Biochem J. 2006;395:493–500. doi: 10.1042/BJ20051748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bourgeron T, Chretien D, Rötig A, Munnich A, Rustin P. Fate and expression of the deleted mitochondrial DNA differ between human heteroplasmic skin fibroblast and Epstein-Barr virus-transformed lymphocyte cultures. J Biol Chem. 1993;268:19369–19376. [PubMed] [Google Scholar]
- 20.Zhang W, Cui H, Wong LJC. Comprehensive one-step molecular analyses of mitochondrial genome by massively parallel sequencing. Clin Chem. 2012;58:1322–1331. doi: 10.1373/clinchem.2011.181438. [DOI] [PubMed] [Google Scholar]
- 21.Cui H, Li F, Chen D, Wang G, Truong CK, Enns GM, Graham B, Milone M, Landsverk ML, Wang J, Zhang W, Wong LJ. Comprehensive next-generation sequence analyses of the entire mitochondrial genoe reveal new insights into the molecular diagnosis of mitochondrial disorders. Genet Med. 2013;5:388–394. doi: 10.1038/gim.2012.144. [DOI] [PubMed] [Google Scholar]
- 22.Cogliati S, Frezza C, Soriano ME, Varnita T, Quintana-Cabrea R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernadez-Silva P, Enriquez JA, Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vincent AE, Ng YS, White K, Davey T, Manella C, Falkous G, Feeney C, Schafer AM, McFarland R, Gorman GS, Taylor RW, Turnbull DM, Picard M. The spectrum of mitochondrial ultrastructure defects in mitochondrial myopathy. Sci Rep. 2016;6:30610. doi: 10.1038/srep30610. http://dx.doi.org/10.1038/srep30610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eskelinen EV, Kovács A. Double membranes vs. lipid bilayers, and their significance for correct identification of macroautophagic structures. Autophagy. 2011;9:931–932. doi: 10.4161/auto.7.9.16679. [DOI] [PubMed] [Google Scholar]
- 25.Cortéz-Hernàndez P, Vázquez-Memije ME, García JJ. ATP6 homoplasmic mutations inhibit and destabilize the human F1F0-ATP synthase without preventing enzyme assembly and oligomerization. J Biol Chem. 2007;282:1051–1058. doi: 10.1074/jbc.M606828200. [DOI] [PubMed] [Google Scholar]
- 26.Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta. 2015;1847:171–181. doi: 10.1016/j.bbabio.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Mookerjee SA, Nicholls DG, Brand MD. Determining maximum glycolytic capacity using extracellular flux measurements. PLoS One. 2016;11(3):e0152016. doi: 10.1371/journal.pone.0152016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.García JJ, Ogilvie I, Robinson BH, Capaldi RA. Structure, functioning, and assembly of the ATP synthase in cells from patients with the T8993G mitochondrial DNA mutation. J Biol Chem. 2000;275:11075–11081. doi: 10.1074/jbc.275.15.11075. [DOI] [PubMed] [Google Scholar]
- 29.Carelli V, Baracca A, Barogni S, Pallotti F, Valentino ML, Montagna P, Zeviani M, Pini A, Lenaz G, Baruzzi A, Solaini G. Biochemical-clinical correlation in patients with different loads of the mitochondrial DNA T8993G mutation. Arch Neurol. 2002;59:264–270. doi: 10.1001/archneur.59.2.264. [DOI] [PubMed] [Google Scholar]
- 30.Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, Schon EA. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002;30:394–399. doi: 10.1038/ng851. [DOI] [PubMed] [Google Scholar]
- 31.Mattiazzi M, Vijayvergiya C, Gajewski CD, DeVivo DC, Lenaz G, Wiedmann M, Manfredi G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum Mol Genet. 2004;13:869–879. doi: 10.1093/hmg/ddh103. [DOI] [PubMed] [Google Scholar]
- 32.Kucharczyk R, Rak M, di Rago JP. Biochemical consequences in yeast of the human mitochondrial DNA 8993T > C mutation in the ATPase6 gene found in NARP/MILS patients. Biochim Biophys Acta. 2009;1793:817–824. doi: 10.1016/j.bbamcr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 33.López-Gallardo E, Emperador S, Solano A, Llobet L, Martín-Navarro A, López-Pérez MJ, Briones P, Pineda M, Artuch R, Barraquer E, Jericó I, Ruiz-Pesini E, Montoya J. Expanding the clinical phenotypes of MT-ATP6 mutations. Hum Mol Genet. 2014;23:6191–6200. doi: 10.1093/hmg/ddu339. [DOI] [PubMed] [Google Scholar]
- 34.Sallevet SC, de Die-Smulders CE, Hendrickx AT, Hellebrekers DM, de Coo IF, Alston CL, Knowles C, Taylor RW, McFarland R, Smeets HJ. De novo mtDNA point mutations are common and have a low recurrence risk. J Med Genet. 2017;54:73–83. doi: 10.1136/jmedgenet-2016-103876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brantová O, Tesarová M, Hansíková H, Elleder M, Zeman J, Sládková J. Ultrastructural changes of mitochondria in the cultivated skin fibroblasts of patients with point mutations in mitochondrial DNA. Ultrastruct Pathol. 2006;30:239–245. doi: 10.1080/01913120600820112. [DOI] [PubMed] [Google Scholar]
- 36.Cellotto AM, Frank AC, McGrath SW, Fergestad T, Van Voorhies WA, Buttle KF, Mannella CA, Palladino MJ. Mitochondrial encephalopathy in Drosophila. J Neurosci. 2006;26:810–820. doi: 10.1523/JNEUROSCI.4162-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, Strooper BDe, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 38.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 39.Velours J, Dautant A, Salin B, Sagot I, Brèthes D. Mitochondrial F1F0-ATP synthase and organellar internal architecture. Int J Biochem Cell Biol. 2009;41:1783–1789. doi: 10.1016/j.biocel.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Zick M, Rabl R, Reichert AS. Cristae formation-linking ultrastructure and function of mitochondria. Biochem Biophys Acta. 2009;1793:5–19. doi: 10.1016/j.bbamcr.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 41.Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brèthes D, di Rago JP, Velours J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arselin G, Vaillier J, Salin B, Schaeffer J, Giraud MF, Dautant A, Brèthes D, Velours J. The modulation in subunits e and g amounts of yeast ATP synthase modifies mitochondrial cristae morphology. J Biol Chem. 2004;279:40392–40399. doi: 10.1074/jbc.M404316200. [DOI] [PubMed] [Google Scholar]
- 43.Habersetzer J, Larrieu I, Priault M, Salin B, Rossignol R, Brèthes D, Paumard P. Human F1F0 ATP synthase, mitochondrial ultrastructure and OXPHOS impairment: a super-complex matter. PLoS One. 2013;8(10):e75429. doi: 10.1371/journal.pone.0075429. http://dx.doi.org/10.1371/journal.pone.0065429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gavin PD, Prescott M, Luff SE, Devenish RJ. Cross-linking ATP synthase complexes in vivo eliminates mitochondrial cristae. J Cell Sci. 2004;117:2333–2343. doi: 10.1242/jcs.01074. [DOI] [PubMed] [Google Scholar]
- 45.Strauss M, Hofhaus G, Schröder RR, Kühlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008;27:1154–1160. doi: 10.1038/emboj.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davies KM, Strauss M, Daum B, Kief JH, Osiewacz HD, Rycovska A, Zickermann V, Kühlbrandt W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Natl Acad Sci U S A. 2001;108:14121–14126. doi: 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wittig I, Schägger H. Structural organization of mitochondrial ATP synthase. Biochem Biophys Acta. 2008;1777:592–598. doi: 10.1016/j.bbabio.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 48.Parfait B, de Lonlay P, von Kleist-Retzow JC, Cormier-Daire V, Chretien D, Rötig A, Rabier D, Saudubray JM, Rustin P, Munnich A. The neurogenic weakness, ataxia, and retinitis pigmentosa (NARP) syndrome mtDNA mutation (T8993G) triggers muscle ATPase deficiency and hypocitrullinaemia. Eur J Pediatr. 1999;158:55–58. doi: 10.1007/s004310051009. [DOI] [PubMed] [Google Scholar]
- 49.Morava E, Rodenburg RJ, Hol F, de Vries M, Janssen A, van den Heuvel L, Nijtmans L, Smeitink J. Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am J Med Genet A. 2006;140(8):863. doi: 10.1002/ajmg.a.31194. [DOI] [PubMed] [Google Scholar]