

Abstract
The decreased bone mineral density and compromised bone strength predispose individuals to skeletal osteoporosis. Both prostate cancer and bone metastasis caused by cancer invasion have remained a great challenge to researchers. With the advancement in the fields of biochemistry and biomechanics, the molecular mechanisms that make prostate cancer metastasize to bone have recently been identified, and they provide new molecular targets for drug development. Many biochemical by-products have been identified to help in understanding the interaction between the bone and the tumor. Enhanced clinical management of patients with bone metastases was reported during the past decade; however, the anticipated risk and the response to the therapy are still challenging to assess. In this review, the key players that play a dominant role in secondary osteoporosis are addressed. An attempt is made to provide the readers with a clear understanding of the communication pathways between each of the cell types involved in this vicious cycle. Furthermore, the role of Wnts, sclerostin, RANKL, PTHrP, and their respective clinical studies are addressed in this study.
Keywords: prostate cancer metastatic bone disease (PCa MBD), vicious cycle, macrophages, Wnts signaling
Introduction
As per 2016 statistics, the mortality rate associated with prostate cancer (PCa) stands fifth globally. Reports of surveillance research program of the National Institutes of Health unveiled that PCa is affecting male global population strongly. Accordingly, out of every 100,000 men, 124.9 new cases are registered, while 20.7 patients die annually. On the basis of 2011–2013 data, 12.9% male population has a PCa morbidity risk at any stage of life. In metastases, the final phase of PCa, neoplasmic cells proliferate in bones and cause fatal osteolytic and osteoblastic abrasions. Consequently, 90% of advanced-stage PCa patients have very low survival rate and they are more prone to complications such as skeletal fractures, intolerable bone pain, together with spinal cord compression. Unfortunately, 15%–35% of the patients develop metastases initiated by transition of primary PCa cells to mesenchymal entity. Modified cells disseminate from the primary source, attain bone-like morphology, and enter and multiply rapidly in the bone tissue causing bone fractures. However, the mechanism by which the PCa cells amend bone microenvironment to facilitate their survival and growth is ambiguous.
During the past decade, new therapeutic strategies have emerged to understand and cure bone metastasis in PCa.1 Homotypic interaction among cancerous cells in conjunction with heterotypic interaction among neoplasm cells, osteoclasts, and osteoblasts plays an important role in determining the course and complications of metastatic bone disease (MBD). Osteolysis releases many growth factors such as transforming growth factor (TGF), insulin-like growth factors (IGFs), and fibroblast growth factors (FGFs) into the tumor microenvironment that facilitate bone matrix remodeling. Consequently, the vicious cycle of metastasis is continuous by stimulating the growth of neoplasmic cells together with the growth factor-mediated phenotypic changes in cancerous cells. Bone loss from osteoclastic bone degradation is characteristic of solid tumor cells’ secondary metastases. This situation is further complicated in PCa due to inappropriate bone formation. Potential chemotherapeutic targets to restrain neoplasmic growth and subsequent bone metastasis include physical factors within the bone microenvironment, such as acidic pH, little oxygen, and high extracellular calcium levels.
In this article, keeping in view the need to explore the unrevealed biochemical mechanisms of PCa MBD, we address the key players of the vicious cycle, their communication pathways, and the preclinical/clinical trials and strategies to manipulate the role of such players, to minimize and/or reverse the predominant osteoblastic response and to halt the osteolytic activity.
Osteoblastic and osteolytic bone metastasis
Bone remodeling is a continuous mechanism through which the skeleton endures all over the life. Bone resorption and bone formation are two processes which occur in coupling during bone remodeling. Such a practice of bone proves it is a dynamic organ. A disparity among these processes gives rise to various bone diseases, including osteoporosis, arthritis, osteophytes (bone spur), osteolysis (bone degradation), craniosynostosis, knee effusion, and osteochondroma. The process of bone remodeling is based on a complex network comprising various growth factors and signaling pathways. The bone-resorbing cells (osteoclasts) and bone-forming cells (osteoblasts) are the key factors of targeted bone remodeling. Mesenchymal stem cells which are abundant in nature differentiate into preosteoblasts. Likewise, osteoclast progenitor cells differentiate into osteoclast precursors. A number of autocrine and paracrine factors are involved in such cell differentiation.2 The bone-forming connective tissue cells, named as active osteoblasts, originate at the surface of the bone from preosteoblasts. These cells fuse and do not cleave, and their main function includes the emission of the collagen matrix and calcium salts. Calcification of the extravasated matrix surrounding the osteoblastic cells traps the cells within it, due to which the cell changes its structure and is transformed into osteocyte; these osteocytes participate actively as bone-forming cells. The activity of the osteocytes and the substance they secrete are themselves influenced by a variety of factors including the mechanical stress on the bone, hormones, and calcium and phosphorous concentrations in the bloodstream.
Multifarious communication pathways among the osteoclast and osteoblast progenitors, at several phases of differentiation, participate in the bone remodeling cycle. Bone lining cells including the osteocytes, osteoblast progenitors, and osteoblast precursors monitor their own functioning along with the instigation of osteoclast synthesis, attachment, execution, and decay. In response, the osteoclasts govern the level of osteoblast functioning by releasing the factors (inside the milieu and membrane). Such a complex network consisting of various signaling pathways of inter-cellular interactions makes the bone capable to respond appropriately to hormonal, mechanical, and inflammatory stimuli to present a strong and versatile composition for human function.
Triphasic bone remodeling
In a healthy bone, remodeling may be described as the process where old or damaged tissue is resorbed by osteoclasts and the resorbed lacunae are filled to the original level by osteoblasts. The process is divided into three major phases: initiation, transition, and termination. These phases are depicted in Figure 1.
Figure 1.
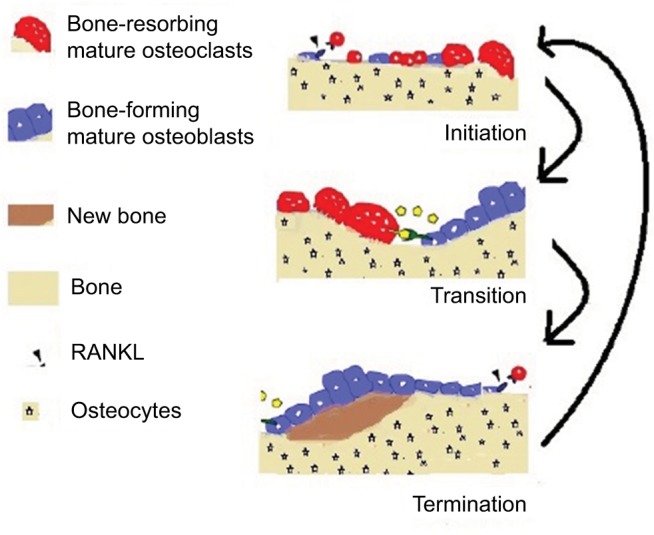
Overview of the bone dynamics.
At the initial stage of bone resorption, the mature osteoclast sticks tightly to the bone by creating a specialized isolated compartment. The membrane adjoining this part of the bone (ruffled membrane) contains proteins that acidify the compartment nearby the bone. The acid firstly liquefies the minerals in the bone, and then the digestive enzymes break down type I collagen and other proteins. Expiring of osteoclasts due to apoptosis ends up this procedure of bone resorption. The direction of major communication is opposite between the initiation and transition phases: from osteoblasts to osteoclast precursors in initiation, and from osteoclasts to osteoblast precursors (or bidirectionally) in transition. The initiation phase includes stimulation of osteoclast formation due to RANKL, which also directs the osteoclasts toward the sites of macrodamage. Duration of osteoclastic bone resorption is almost 21 days in human bone. Once the osteoclasts have removed the old tissue, they move forward and undergo apoptosis and the transition phase starts. This phase includes the recruitment and differentiation of osteoblasts and the grounding of resorbed surface for bone formation.3
The termination phase eventually mineralizes and forms osteoid (new bone), and the process of mass-neutral bone renewal is complete. The termination phase takes a longer period as compared to the initiation phase. Osteoprotegerin (OPG) is associated with the initial phase, in which it counteracts RANKL osteoclastogenic activity, but there are evidences which assure a possible role of OPG in the termination phase as well. OPG may, indeed, protect the osteoid from osteoclastic resorptive activity during bone formation. In Figure 1, the formation and resorption are presented. Bone mass and bone quality both are affected by osteoclast–osteoblast communications. Excessive remodeling decreases the bone mass, whereas restrained bone remodeling increases it. Suppressed bone remodeling reduces the bone quality due to accumulation of microfractures and reduced reformation of bone architecture.4 Deep understanding of osteoclast–osteoblast communication during bone remodeling is a prerequisite for designing interventions to avoid bone loss while retaining bone quality and for treating diseases such as osteoporosis, breast cancer and PCa, bone metastasis, and fracture repair.
Vicious cycle and its mechanics
PCa cells release factors responsible for bone remodeling. A schematic of the vicious cycle is presented in Figure 2. A number of factors contributing to bone remodeling are secreted by PCa cells. These numerous factors include the bone morphogenetic proteins (BMPs; they are multifunctional growth factors that belong to the TGF-β superfamily), TGF-β, (PTHrP: this is a protein secreted by the cancer cells and acts as an endocrine, autocrine, paracrine, and intracrine hormone), adrenomedullin, OPG, RANK, and RANKL. This process is called osteomimicry. Osteoclast differentiation into more mature stages gets faster because of these secreted factors. Such an effect occurs directly or indirectly through osteoblastic cells. In turn, during bone resorption, the osteoclasts secrete a number of growth factors that are accumulated in the impregnated bone milieu and proficient to stimulate tumor development. The factors involved in the vicious cycle are remarkably diverse and are responsible for the histologic and phenotypic heterogeneity of bone metastases patients. PCa patients have a high affinity to encounter osteoblastic bone metastasis. This phenotype is found to be the most prevailing malady at present and the main reason for cancer patients’ fatality and infirmity. The vicious cycle is shown in Figure 2. The means regulating the intensification of this phenotype are not well identified yet. Generally, the characteristic response linked with PCa bone metastasis is found to be osteoblastic, but there are some adequate facts proving that osteolytic events are also in progress.5 A part of the osteolysis may be ascribed to androgen dearth, while the osteolytic features expressed by the PCa cells may take a part as well. Despite the instigating phenomena, osteolysis is extremely expected to result in extricate of persuasive growth factors from the bone matrix that positively affect the development of cancer cells. Several studies have been conducted to address this issue directly, in which the effects of osteolysis-hindering agents on PCa bone metastases have been analyzed. Such agents include the use of bisphosphonates to target the active osteoclasts or exploiting the OPG/RANK/RANKL signaling pathway to hinder the differentiation of preosteoclasts.
Figure 2.
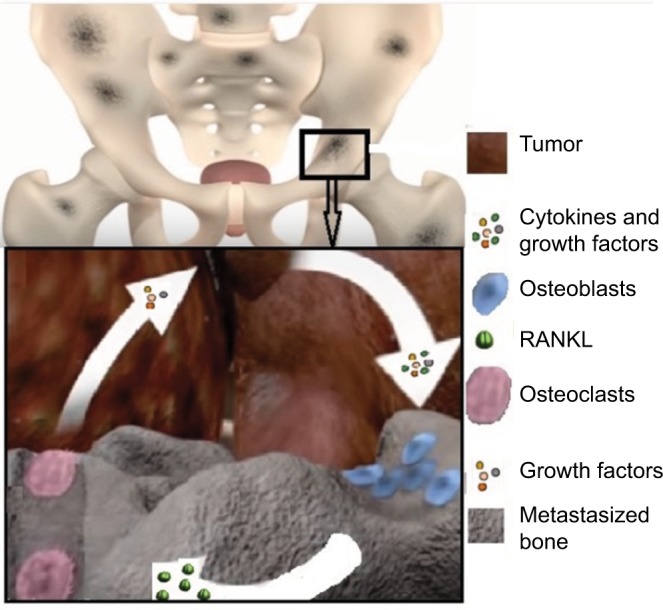
The schematic of vicious cycle.
Several studies are available in literature to demonstrate bone remodeling. Komarova and her research group6 discussed a model in which the key factors considered were the cell population dynamics of osteoclasts and osteoblasts. The chemical quantities were not elaborated as functions of time or location. However, the biochemical mechanisms were integrated in an abstract way. The inclusion of osteocytes, their biochemical processes, and their interactions with other bone remodeling cells in a mathematical model may play a vital role to give a significant advancement to this field. In 2013, Graham et al7 developed an improved mathematical model for the biochemical systems in bone remodeling. The derivation of this model was motivated by the fact that in most of the clinical trials, explicit chemical data for this mechanism are not available. The model focused on the RANK/RANKL/OPG pathway and its consequences. The actions of growth factors such as TGF-β and other cytokines on bone remodeling cells were also incorporated in this model.
Sclerostin: an osteocyte-enhanced local regulator of bone remodeling
Osteocytes express growth factors that encourage bone formation. They secrete sclerostin, which is a cysteine-knot-containing protein that inhibits bone formation and affects both osteoblast and osteoclast activity. Sclerostin inhibits the Wnt signaling pathway by binding to LRP5 coreceptor, ensuing mutations in gene encoding. Gkotzamanidou et al8 summarized the function of sclerostin in bone metastases of cancer. They analyzed that in most of the cancers, high sclerostin levels are found in patients’ plasma and serum, whereas the situation is reverse in PCa. Low levels of sclerostin along with morphogenic protein-6 may be considered a biomarker of PCa metastatic progression. In breast cancer, in vitro data suggest that within the metastatic bone region, the osteoblastic activity is inhibited by high expression of sclerostin. On the basis of this observation, it was concluded that sclerostin might be involved in inhibition of bone formation in breast cancer and malignant neoplasm-related bone diseases.
Osteocytes not only secrete the growth factors that stimulate bone formation, but also secrete sclerostin which is a cysteine-knot-containing protein that inhibits bone formation and affects both osteoblast and osteoclast activity. Sclerostin is a bone-specific molecule that binds LRP5, and thus prevents binding of Wnt proteins, leading to inhibition of the pathway loss-of-function mutations of gene encoding.
There are preclinical trials9 and clinical trials10 available in literature, emphasizing on the role of sclerostin in bone remodeling. In 2012, Yavropoulou et al11 performed a clinical trial to investigate the effect of serum sclerostin levels in Paget’s disease and PCa with bone metastases. A significant positive relationship between sclerostin levels and bone turnover markers in metastatic PCa was detected. They highlighted a number of factors that can stimulate sclerostin production. Cytokines produced by PCa cells could also contribute to the increased circulating levels of sclerostin observed in PCa metastasis to the skeleton. This increased sclerostin production may, in turn, stimulate RANKL and bone resorption. Sclerostin is encoded as SOST gene product that reduces osteoblastic bone formation by inhibiting canonical Wnt/β-catenin signaling (discussed in the “Wnts induced activities in PCa bone metastases” section). Sclerostin was recently shown to upregulate the expression of RANKL by osteocyte-like cells and promotes osteoclastogenesis,12 supporting the view that osteocytes are targets for the action of sclerostin.
Later, in 2013, Larson et al10 performed for the first time a detailed analysis of bone remodeling proteins in human specimens of PCa bone metastases. Based on the study findings, they acknowledged the findings in Yavropoulou et al11 and revealed that sclerostin was elevated in PCa bone metastases and was found to inhibit bone formation, promote osteoclast formation, and promote bone degradation. In studying the regulation of bone mass, the measurement of serum sclerostin level is a new approach and may serve as an effective tool in monitoring the efficiency of several therapies and detecting bone disorders. In multiple myeloma, sclerostin is produced by the myeloma cells, and is elevated in the serum or plasma of the patients and correlates with extensive bone disease and adverse myeloma features. Sclerostin expression is reduced in case of PCa, and in combination with BMP-6 and noggin expression, may work as prognostic predictor for metastatic progression. In case of breast cancer, in vitro data indicate that malignant cell induces the expression of sclerostin to inhibit osteoblasts in the metastatic bone area. In PCa patients, the role of sclerostin in bone metabolism as well as its relation with sex steroids is not explicitly known. In order to answer these mysteries, Garcia-Fontana et al13 conducted a cross-sectional study that included healthy controls and PCa patients. They gave a brief statistical analysis and concluded that the sclerostin levels were increased significantly in PCa patients, particularly in those who had androgen-deprivation therapy. They also concluded that there is an inverse relation between testosterone and sclerostin levels.
Wnts induced activities in PCa bone metastases
The Wnt gene family was discovered in 1982 by Nusse and Varmus. Currently having 19 members, the Wnt proteins are a big family of cysteine-rich glycoproteins. They primarily work during development in order to control branching morphogenesis and body axis symmetry.14 Wnts exert their biological effect through three signaling pathways, which we can separate by their ability to stabilize catenin. Non-canonical Wnts bind to a variety of receptors, including receptor tyrosine kinases, and mediate a lot of actions, some of which oppose canonical Wnt signaling. It has been proven that Wnt signaling plays some very important roles during organ and tissue homeostasis, bone biology, and embryonic development. The Wnt signaling pathways are defined as a group of signal transduction pathways which consist of proteins that pass signals into cell surface receptors. Wnt signaling also regulates bone mass with the help of multiple mechanisms such as proliferation, coupling to osteoclast function and osteoblast differentiation.15
Several proteins involved in Wnt signaling are involved in prostate tumor-induced osteoblastic activity,15 indicating that small-molecular targeting Wnt signaling may be an effective intervention for PCa metastasis. Very recently, Tseng et al16 revealed through preclinical trials that caffeic acid phenethyl ester (CAPE), a main bioactive component extracted from honeybee hive propolis, suppresses migration and invasion of PCa cells via activation of non-canonical Wnt signaling. Therefore, CAPE treatment can prove to be a potential intervention for PCa metastasis.
Studies are available in literature to investigate the contribution of the Wnt signaling pathway in PCa growth, incidence of bone metastases, and osteoblastic phenotype of bone metastases. Hall et al17 demonstrated elevated Dickkopf-1 (DKK-1, a secreted glycoprotein encoded by the DKK1 gene) expression as an early event in PCa and declined DKK-1 expression as a later event in progressed PCa (advanced bone metastases). The decline of DKK-1 in bone metastases is interlinked with Wnts’ osteoblastic activity and supports a model in which DKK-1 is a molecular switch that transitions the phenotype of PCa osseous lesions from osteolytic to osteoblastic. Thudi et al18 adopted a strategy of overexpressing the Wnt antagonist, DKK-1, in the mixed osteoblastic and osteolytic Ace-1 PCa cells. They concluded that DKK-1 proved to be oncogenic and inhibited Wnt signaling and, thus, the bone formation (osteoinduction).
Wnt mRNAs are expressed in PCa cell lines and as compared with non-neoplastic prostate tissues, Wnt mRNA expression increases in primary PCa. In PCa bone metastases, Wnts induce osteoblastic activity, although it is not yet clear if Wnt mediates this activity due to a direct action on the osteoblasts or by activating some other pro-osteoblastic factors, for instance, BMP. Cancer models have confirmed that mutations or altered expression of components of Wnt pathways are strongly related with prostate tumors. Wnt pathway and the downstream effector, catenin, play a vital role in the development and progression of PCa skeletal metastasis. Wnts influence tumor growth and survival (autocrine effects) and modulate bone cell growth as well as differentiation (paracrine effects). Canonical Wnts stimulate osteoblast mineralization and differentiation. In 2006, Hall et al15 identified that the Wnts produced by PCa induce osteoblastic activity in bone metastases and Wnt signaling could work both in paracrine and autocrine manner to help tumor cell growth and osteoblast differentiation. The preclinical and clinical studies to control the osteoblastic activities require much attention, especially to understand the pathways and the interference with endothelin-1 and BMPs.
In 2008, Robinson et al19 speculated on targeted studies of Wnt pathways for therapeutic treatment, and Dai et al20 conducted clinical trials to explore Wnt-induced BMP mechanism. It was observed that PCa mediates pro-osteoblast activity through both canonical and non-canonical Wnts, which, in turn, contributes to osteoblast activity through BMP-dependent and BMP-independent pathways.
In 2009, Bisson and Prowse21 reported that Wnt signaling regulates the differentiation and self-renewal of PCa cells with the characteristics of stem cells. Their studies revealed that treatment with Wnt inhibitors may reduce both self-renewal and prostasphere size, while addition of Wnt 3a caused increases self-renewal and the prostasphere size. Tai et al22 reported a new molecular therapeutic target in PCa bone metastasis and concluded that by downregulating miR 126, osteoblast-derived, Wnt-induced secreted protein 1 strengthens PCa. Various proteins involved in Wnt signaling have a role in prostate tumor-induced osteoblastic activity, which indicates that small-molecular targeting Wnt signaling might be an efficient intervention for PCa metastasis. Very recently, Tseng et al16 revealed through preclinical trials that via activation of non-canonical Wnt signaling, CAPE suppresses the migration and invasion of PCa cells. Therefore, CAPE treatment can prove to be a possible intervention for PCa metastasis.
Klipp and Liebermeister23 meticulously reviewed the signaling pathways and discovered their hidden role in the stimulation and development of oncogenesis. It was revealed that the cross talk between ERK signaling proteins and Wnt pathway generates positive feedback oncogenic loop that promotes different types of neoplasms. They were able to demonstrate the existent ERK/Wnt pathway cross talk mechanism via a persuasive model through amalgamation of the inclusive pathways’ mathematical models and experimental results. Based on the accepted assumption that mutations in signaling pathways are a conductive factor for cancer, it was found that mutations could activate both the pathways simultaneously resulting in permanent changes in the structure and function of both the pathways via cross talk-embedded feedback loop.
Role of PTH/PTHrP in bone metastasis
Besides structure, parathyroid hormone (PTH) and parathyroid hormone-related protein (PTHrP) share similarity in biological action and are important mediators of bone remodeling through osteoclastogenesis. Under normal physiological conditions, bone remodeling process consists of a series of highly coordinated and closely regulated steps. Together with regulating calcium concentration, both aforementioned peptide mediators encourage osteolysis by binding to PTH-1R receptor on osteoblasts. The aftermath of this action is the upregulation of RANKL, coupled with the suppression of its decoy receptor, osteoprotegrin, by osteoblasts. RANKL activates preosteoclasts’ differentiation, migration, and fusion to form multinucleate mature osteoclasts to initiate bone resorption. Release of lysosomal enzymes such as cathepsin K by the osteoclasts results in irregular scalloped cavities, Haversian canals, on the bone surface. Afterward, the macrophage cells replace the osteoclasts to degrade collagen, deposit proteoglycans for bone cement lining, and release growth factors to stimulate osteoblasts for the formation phase. Osteoblasts complete the bone remodeling cycle by filling the cavities along with deposition of mineralizable matrix. Clinical analysis of various tumors from breast, prostate, thyroid, lung, and kidney identified PTHrP as a neoplasm-derived humoral factor.24 Upregulation of PTHrP and PTH-1R receptor in tumor cells contributes to their survival, proliferation, and adhesion via intracrine, paracrine, and autocrine signaling, hence reinforcing the metastatic potential of the tumor. It has been found that PTHrP plays a key role in cyclin D1 regulation leading to cell proliferation in other cell systems. PTHrP coupled with ectopic receptor expression augmented DNA synthesis in MCF-7 breast cancer cell line.25 Proliferation of human SV40-immortalized breast epithelial cells and colon cancer cells was also triggered by PTHrP. PTHrP enhanced the PC-3 PCa cell adhesion to fibronectin, collagen type 1, and laminin.26 Antiapoptotic effect of PTHrP was also observed in lung and breast cancer cells through intracrine mechanism.27
PTHrP not only facilitates tumor localization and growth in the bone, but also is closely associated with tumor-born skeletal lesions and complications. It was suggested that overexpression of PTHrP provides survival advantages to tumor cells in the bone matrix, most probably by stimulating osteoclastic destruction. Tumor cells release mediators to either stimulate differentiation of hematopoietic stem cells into osteoclasts or activate mature osteoclasts or do both.28 It was observed that in breast carcinoma, in particular, PTHrP produced by the tumor cells is primarily responsible for osteolytic bone destruction at the site of tumor metastasis to bone. After invading the hard calcified bone matrix, the tumor increases the expression of PTHrP together with other factors such as IGF-1, IGF-II, FGF1, FGF2, TGFs (TGF-α, TGF-β) and platelet-derived growth factors to dramatically modify the bone microenvironment and gain growth advantages leading to bone lesion and complications.29 Only a small cell population of the primary tumor, endowed with specific phenotypic features, has the capacity for successful bone metastases. These cells invade the basement membrane, evoke angiogenesis, adhere to bony skeleton, and ultimately extravasate into its microenvironment to proliferate exponentially, thus instigating macrometastases. A large number of factors in the bone tissue, such as interleukins, PTHrP, and TGF-β, create a friendly environment for rapid proliferation of tumor cells. Clinical evidences indicate the strong involvement of PTHrP in the pathophysiology of metastases to bone as >90% cases of breast carcinoma show predilection to bone metastases and are associated with overexpression of PTHrP.30 Bone resorption results in elevated local calcium concentration and releases TGF-β growth factor, which is stored abundantly in the bone microenvironment. High calcium level facilitates cancer cell proliferation through calcium sensing receptor and also induces PTHrP synthesis from the tumor, providing a cyclic feedback maintenance system for the cancer cells’ localization and growth in bone.31 A large body of studies indicate that the interaction of activated osteoclasts and cytokines released by the cancer cells results in enhanced bone lysis leading to loss of its architectural integrity.
Conclusion and open questions
PCa is not a truly localized disease and to control this fact, the recommended treatment is “androgen ablation”, a strategy that takes advantage of the tumor’s dependence on androgens as a growth factor. The majority of PCa patients receiving androgen ablation therapy will ultimately develop castration-resistant PCa within 13 years with a median survival time of 12 years. Median time to prostate-specific antigen progression was recorded to be significantly shorter with bone metastases.1
In tumor cell dormancy in PCa, the dormant state confers resistance to conventional chemotherapeutic agents and prevents the elimination of disseminated tumor cells from the bone using current drug therapies.32 Expansion of limited understanding of the molecular mechanisms underpinning disseminated prostate tumor cell dormancy is critical for the future development of novel drug therapies aimed at the prevention of bone metastasis.
Successful therapeutic strategies for the treatment of osteolytic MBD include the administration of intravenous bisphosphonates or subcutaneous inhibitors of receptor activator of RANKL. Inhibitors of SRC and cABL kinases and cathepsin K are under clinical investigation as potential antiosteolytics. Sclerostin is encoded as a SOST gene product that reduces osteoblastic bone formation by inhibiting canonical Wnt/β-catenin signaling. It has proved to upregulate the expression of RANKL by osteocyte-like cells and promotes osteoclastogenesis.12 Similarly, CAPE may prove to be a potential intervention for PCa metastasis, since preclinical trials verified that it suppresses the migration and invasion of PCa cells via activation of non-canonical Wnt signaling.
We have emphasized on Wnt signaling pathways due to its complex role in tumor-induced osteoblastic activity in advanced-stage bone metastasis. Wnts and their downstream effectors such as BMP, PTHrP, and OPG are important in tumor growth and metastasis leading to osteoblastic phenotype transition. PCa is distinct from other cancers in that the early stage of bone metastasis is marked by bone lysis, while the advanced stage is associated with osteoblastic lesions, that is, bone formation. Constitutive activation of Wnt signaling pathway contributes toward bone metastasis together with its osteoblastic phenotype. Studies have established the oncogenic activity of Wnts, and it is found that both canonical and non-canonical Wnt signaling pathways activate BMP-dependent as well as BMP-independent osteoblast differentiation leading to osteoblastic lesions. Thus, Wnt is a potential target from the therapeutic development standpoint that interferes with PCa bone metastasis. In this regard, new agents have been discovered and successively tested in preclinical models to mend Wnt signaling.
Applications of these therapeutic strategies could change the status of “hormone-refractory PCa” metastatic to bone from irremediable to manageable clinical pathosis. This discussion provides an overview of the main putative molecular mechanisms in PCa bone metastasis reported from multiple experimental in vitro and in vivo models, and may help to discover new therapeutic strategies to control tumor invasion and suppress the PCa metastatic bone lesions.
Acknowledgments
This work is partially supported by the National Natural Foundation of China under Grant No. 61603285 and FDCT of Macau under Grant No. 122/2017/A3.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Lee GT, Kang DI, Ha YS, et al. Prostate cancer bone metastases acquire resistance to androgen deprivation via WNT5Aa-mediated BMP-6 induction. BrJ Cancer. 2014;110(6):1634–1644. doi: 10.1038/bjc.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kular J, Tickner J, Chim SM, Xu J. An overview of the regulation of bone remodelling at the cellular level. Clin Biochem. 2012;45(12):863–873. doi: 10.1016/j.clinbiochem.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 3.Tran Van P, Vignery A, Baron R. An electron-microscopic study of the bone-remodeling sequence in the rat. Cell Tissue Res. 1982;225(2):283–292. doi: 10.1007/BF00214682. [DOI] [PubMed] [Google Scholar]
- 4.Mashiba T, Turner CH, Hirano T, et al. Effects of high-dose etidronate treatment on microdamage accumulation and biomechanical properties in beagle bone before occurrence of spontaneous fractures. Bone. 2001;29(3):271–278. doi: 10.1016/s8756-3282(01)00575-0. [DOI] [PubMed] [Google Scholar]
- 5.Camacho DE, Pienta KJ. A multi-targeted approach to treating bone metastases. Cancer Metastasis Rev. 2014;33(2–3):545–553. doi: 10.1007/s10555-013-9476-y. [DOI] [PubMed] [Google Scholar]
- 6.Komarova SV. Mathematical model of paracrine interactions between osteoclasts and osteoblasts predicts anabolic action of parathyroid hormone on bone. Endocrinology. 2005;146(8):3589–3595. doi: 10.1210/en.2004-1642. [DOI] [PubMed] [Google Scholar]
- 7.Graham JM, Ayati BP, Holstein SA, Martin JA. The role of osteocytes in targeted bone remodeling: a mathematical model. PLoS One. 2013;8(5):e63884. doi: 10.1371/journal.pone.0063884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gkotzamanidou M, Dimopoulos MA, Kastritis E, Christoulas D, Moulopoulos LA, Terpos E. Sclerostin: a possible target for the management of cancer-induced bone disease. Expert Opin Ther Targets. 2012;16(8):761–769. doi: 10.1517/14728222.2012.697154. [DOI] [PubMed] [Google Scholar]
- 9.Sinder BP, Salemi JD, Ominsky MS, Caird MS, Marini JC, Kozloff KM. Rapidly growing BRTL/+ mouse model of osteogenesis imperfecta improves bone mass and strength with sclerostin antibody treatment. Bone. 2015;71:115–123. doi: 10.1016/j.bone.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larson SR, Zhang X, Dumpit R, et al. Characterization of osteoblastic and osteolytic proteins in prostate cancer bone metastases. Prostate. 2013;73(9):932–940. doi: 10.1002/pros.22639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yavropoulou MP, van Lierop AH, Hamdy NA, Rizzoli R, Papapoulos SE. Serum sclerostin levels in Paget’s disease and prostate cancer with bone metastases with a wide range of bone turnover. Bone. 2012;51(1):153–157. doi: 10.1016/j.bone.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 12.Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One. 2011;6(10):e25900. doi: 10.1371/journal.pone.0025900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Fontana B, Morales-Santana S, Varsavsky M, et al. Sclerostin serum levels in prostate cancer patients and their relationship with sex steroids. Osteoporosis Int. 2014;25(2):645–651. doi: 10.1007/s00198-013-2462-y. [DOI] [PubMed] [Google Scholar]
- 14.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 15.Hall CL, Kang S, MacDougald OA, Keller ET. Role of Wnts in prostate cancer bone metastases. J Cell Biochem. 2006;97(4):661–672. doi: 10.1002/jcb.20735. [DOI] [PubMed] [Google Scholar]
- 16.Tseng JC, Lin CY, Su LC, Fu HH, Yang SD, Chuu CP. Cape suppresses migration and invasion of prostate cancer cells via activation of non-canonical Wnt signalling. Oncotarget. 2016;7(25):38010–38024. doi: 10.18632/oncotarget.9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall CL, Daignault SD, Shah RB, Pienta KJ, Keller ET. DICKKOPF-1 expression increases early in prostate cancer development and decreases during progression from primary tumor to metastasis. Prostate. 2008;68(13):1396–1404. doi: 10.1002/pros.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thudi NK, Martin CK, Murahari S, et al. Dickkopf-1 (DKK-1) stimulated prostate cancer growth and metastasis and inhibited bone formation in osteoblastic bone metastases. Prostate. 2011;71(6):615–625. doi: 10.1002/pros.21277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson DR, Zylstra CR, Williams BO. Wnt signaling and prostate cancer. Curr Drug Targets. 2008;9(7):571–580. doi: 10.2174/138945008784911831. [DOI] [PubMed] [Google Scholar]
- 20.Dai J, Hall CL, Escara-Wilke J, Mizokami A, Keller JM, Keller ET. Prostate cancer induces bone metastasis through Wnt-induced bone morphogenetic protein-dependent and independent mechanisms. Cancer Res. 2008;68(14):5785–5794. doi: 10.1158/0008-5472.CAN-07-6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bisson I, Prowse DM. Wnt signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 2009;19(6):683–697. doi: 10.1038/cr.2009.43. [DOI] [PubMed] [Google Scholar]
- 22.Tai HC, Chang AC, Yu HJ, et al. Osteoblast-derived WNT-induced secreted protein 1 increases VCAM-1 expression and enhances prostate cancer metastasis by down-regulating miR-126. Oncotarget. 2014;5(17):7589–7598. doi: 10.18632/oncotarget.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klipp E, Liebermeister W. Mathematical modeling of intracellular signaling pathways. BMC Neurosci. 2006;7(Suppl 1):S10. doi: 10.1186/1471-2202-7-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coleman RE. Skeletal complications of malignancy. Cancer. 1997;80(8 Suppl):1588–1594. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1588::aid-cncr9>3.3.co;2-z. [DOI] [PubMed] [Google Scholar]
- 25.Hoey RP, Sanderson C, Iddon J, Brady G, Bundred NJ, Anderson NG. The parathyroid hormone-related protein receptor is expressed in breast cancer bone metastases and promotes autocrine proliferation in breast carcinoma cells. Br J Cancer. 2003;88(4):567–573. doi: 10.1038/sj.bjc.6600757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen X, Falzon M. PTH-related protein modulates PC-3 prostate cancer cell adhesion and integrin subunit profile. Mol Cell Endocrinol. 2003;199(1):165–177. doi: 10.1016/s0303-7207(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 27.Hastings RH, Araiza F, Burton DW, Zhang L, Bedley M, Deftos LJ. Parathyroid hormone-related protein ameliorates death receptor-mediated apoptosis in lung cancer cells. Am J Physiol Cell Physiol. 2003;285(6):C1429–C1436. doi: 10.1152/ajpcell.00269.2003. [DOI] [PubMed] [Google Scholar]
- 28.Taichman RS. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood. 2005;105(7):2631–2639. doi: 10.1182/blood-2004-06-2480. [DOI] [PubMed] [Google Scholar]
- 29.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2(8):584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 30.Powell GJ, Southby J, Danks JA, et al. Localization of parathyroid hormone-related protein in breast cancer metastases: increased incidence in bone compared with other sites. Cancer Res. 1991;51(11):3059–3061. [PubMed] [Google Scholar]
- 31.Liao J, Schneider A, Datta NS, McCauley LK. Extracellular calcium as a candidate mediator of prostate cancer skeletal metastasis. Cancer Res. 2006;66(18):9065–9073. doi: 10.1158/0008-5472.CAN-06-0317. [DOI] [PubMed] [Google Scholar]
- 32.Quayle L, Ottewell PD, Holen I. Bone metastasis: molecular mechanisms implicated in tumour cell dormancy in breast and prostate cancer. Curr Cancer Drug Targets. 2015;15(6):469–480. doi: 10.2174/1568009615666150506092443. [DOI] [PubMed] [Google Scholar]