

Abstract
Background and Purpose
Opioid δ receptor agonists are potent antihyperalgesics in chronic pain models, but tolerance develops after prolonged use. Previous evidence indicates that distinct forms of tolerance occur depending on the internalization properties of δ receptor agonists. As arrestins are important in receptor internalization, we investigated the role of arrestin 2 (β‐arrestin 1) in mediating the development of tolerance induced by high‐ and low‐internalizing δ receptor agonists.
Experimental Approach
We evaluated the effect of two δ receptor agonists with similar analgesic potencies, but either high‐(SNC80) or low‐(ARM390) internalization properties in wild‐type (WT) and arrestin 2 knockout (KO) mice. We compared tolerance to the antihyperalgesic effects of these compounds in a model of inflammatory pain. We also examined tolerance to the convulsant effect of SNC80. Furthermore, effect of chronic treatment with SNC80 on δ agonist‐stimulated [35S]‐GTPγS binding was determined in WT and KO mice.
Key Results
Arrestin 2 KO resulted in increased drug potency, duration of action and decreased acute tolerance to the antihyperalgesic effects of SNC80. In contrast, ARM390 produced similar effects in both WT and KO animals. Following chronic treatment, we found a marked decrease in the extent of tolerance to SNC80‐induced antihyperalgesia and convulsions in arrestin 2 KO mice. Accordingly, δ receptors remained functionally coupled to G proteins in arrestin 2 KO mice chronically treated with SNC80.
Conclusions and Implications
Overall, these results suggest that δ receptor agonists interact with arrestins in a ligand‐specific manner, and tolerance to high‐ but not low‐internalizing agonists are preferentially regulated by arrestin 2.
Abbreviations
- ARM390
N,N‐diethyl‐4‐(phenyl‐piperidin‐4‐ylidenemethyl)‐benzamide
- CFA
complete Freund's adjuvant
- KO
knockout
- SNC80
(+)‐4‐[(αR)‐α‐((2S,5R)‐4‐Allyl‐2,5‐dimethyl‐1‐piperazinyl)‐3‐methoxybenzyl]‐N,N‐diethyl benzamide
- WT
wild‐type
Introduction
The opioid http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=317, also known as δOR, are GPCRs that play an important role in the regulation of pain processing and emotional responses (Gaveriaux‐Ruff et al., 2011; Pradhan et al., 2011). Recently, δ receptor agonists have attracted increasing research interest as a potential alternative to http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 agonists (Gendron et al., 2016). Compared to μ receptor agonists, δ receptor agonists are poor analgesics for acute pain (Gallantine and Meert, 2005), but they are highly effective in models of chronic pain. This difference in antinociceptive efficacy is likely due to increased functionality of δ receptors during chronic pain states (Fraser et al., 2000; Hurley and Hammond, 2000; Cahill et al., 2003; Kabli and Cahill, 2007; Gaveriaux‐Ruff et al., 2008; Pradhan et al., 2013). Unlike μ receptor agonists, δ receptor agonists do not show significant abuse liability (Negus et al., 1998; Brandt et al., 2001; Stevenson et al., 2005) and lack several side effects associated with μ receptor activation (constipation, respiratory depression, etc.) (Gallantine and Meert, 2005). However, some δ receptor agonists can cause convulsions, which has limited their clinical development (Comer et al., 1993; Broom et al., 2002a). In addition, prolonged exposure to δ or μ receptor agonists leads to the development of tolerance, which hinders their therapeutic use.
The extent and duration of GPCR signalling are governed by multiple factors, including receptor desensitization, internalization, down‐regulation and recycling. Arrestins are key regulators of GPCR desensitization and internalization (Luttrell and Lefkowitz, 2002; Reiter et al., 2012). In addition, arrestins act as multifunctional scaffolding proteins and signalling intermediates with effects on cytoskeletal remodelling (Mittal et al., 2013), protein ubiquitination (Shenoy, 2014), trafficking of ion channels (Nagi et al., 2015) and gene transcription (Ma and Pei, 2007). Similar to many GPCRs, ligand‐activated δ receptors are also modulated by arrestins (Lowe et al., 2002; Zhang et al., 2005; Qiu et al., 2007; Raehal and Bohn, 2011). In cellular models, both arrestin 2 (β‐arrestin 1) and arrestin 3 (β‐arrestin 2) can mediate δ receptor desensitization and internalization after receptor phosphorylation (Qiu et al., 2007; Hong et al., 2009). Interestingly, emerging evidence suggests that arrestins 2 and 3 play distinct roles in downstream functional outcomes upon GPCR activation (Taylor et al., 2016). In line with this functional specialization of arrestin isoforms, δ receptor export from the Golgi to the cell surface is selectively regulated by arrestin 2 through the RhoA/ROCK/LIMK pathway (Mittal et al., 2013). To add one more layer of complexity, several studies have demonstrated the existence of agonist‐specific recruitment of arrestin isoforms at many GPCRs, including the δ receptor. In mouse embryonic fibroblasts, DPDPE preferentially recruited arrestin 3 to induce phosphorylation‐dependent δ receptor internalization (Qiu et al., 2007). In contrast, arrestin 2 mediated etorphine‐activated δ receptor internalization in SK‐N‐BE cells, while facilitating receptor desensitization, but not internalization, in response to DPDPE or deltorphin I (Bowman et al., 2015). Recently, we reported that in vivo, the binding of the high‐internalizing agonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=1611 produces preferential interaction between δ receptors and arrestin 2, and low‐internalizing agonists like http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9005 or JNJ20788560 preferentially promote arrestin 3‐receptor interactions (Pradhan et al., 2016). An increased interaction between the δ receptor and arrestin 3 facilitated receptor resensitization and protected against the development of behavioural tolerance to low‐internalizing agonists (Pradhan et al., 2016).
In the present study, we explored the long‐term behavioural consequences of the ligand‐specific interaction between arrestin 2 and δ receptors using SNC80 and ARM390. We found that arrestin 2 mediates the development of tolerance to the antihyperalgesic and convulsive effects of SNC80, but not tolerance to the antihyperalgesic effects of ARM390. Furthermore, assays monitoring receptor function ex vivo after the establishment of tolerance to SNC80 revealed that δ receptors remain functionally coupled to G proteins in the absence of arrestin 2. These studies demonstrate that the ligand‐selective recruitment of arrestins by δ receptor agonists can have profound effects on tolerance to chronic drug administration.
Methods
Animals
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015) and the US National Research Council's Guide for the Care and Use of Laboratory Animals. Mice were group‐housed with a maximum of five animals per cage in clear polypropylene cages with corn cob bedding and nestlets as enrichment. Arrestin 2 knockout (KO) mice were generously provided by R. Lefkowitz (Duke University). Both male and female mice were used. Mice, aged 8–20 weeks, were housed in a temperature‐ and humidity‐controlled animal colony on a 12 h light/dark cycle. Mice had free access to food and water at all times. All animal experiments were performed according to Association for Assessment and Accreditation of Laboratory Animal Care guidelines as administered by the University of Illinois at Chicago and University of Michigan Animal Care Committees. A total of 242 mice were used in these experiments – 118 wild‐type (WT) and 124 arrestin 2 KO.
Drugs
All drugs were administered in a volume of 10 mL·kg−1. SNC80 (Tocris Bioscience, Pittsburgh, PA, USA) was dissolved in acidic 0.9% saline, pH 5.5, and injected i.p. ARM390 (AstraZeneca, R&D, Wilmington, DE, USA) was dissolved in dH2O and administered by oral gavage. Unless otherwise stated, mechanical sensitivity was tested 45 min after treatment with SNC80 or ARM390. All other drugs were purchased from Sigma‐Aldrich (St. Louis, MO, USA), unless noted otherwise.
Inflammatory pain model
The complete Freund's adjuvant (CFA)‐induced inflammatory pain model was performed as described previously (Pradhan et al., 2016; Vicente‐Sanchez et al., 2016). Briefly, inflammation was induced by injecting 13 μL of CFA (Sigma‐Aldrich) into the plantar surface of the hindpaw, and animals were tested 72 h after injection. Mice were habituated to the testing area for 20 min daily for 2 days before baseline testing. The threshold for mechanical responses of the hindpaw to punctate mechanical stimuli (mechanical hyperalgesia) was assessed according to the up‐and‐down method (Chaplan et al., 1994). A series of eight von Frey filaments with bending force ranging from 0.01 to 2 g was used. A response was defined as a lifting or shaking of the paw upon stimulation. Mechanical responses were determined prior to CFA injection to establish baseline sensitivity. Animals were randomly assigned to treatment groups, and the experimenter was blinded to the drug treatment and/or genotype when testing.
Observation of SNC80‐induced convulsions
Mice were randomized to receive a s.c. injection of SNC80 (3.2, 10, 32 mg·kg−1) every 24 ± 1 h for 5 days. Following SNC80 administration, mice were observed continuously in individual cages for 30 min for convulsions. Convulsions were comprised of a tonic phase characterized by sudden tensing of the musculature and extension of the forepaws followed by clonic contractions that extended the length of the body. The severity of each convulsion was quantified using the following modified Racine scale (Racine, 1972) adapted from Jutkiewicz et al. (2006): scale 1 – teeth chattering or face twitching; 2 – head bobbing or twitching; 3 – tonic extension or clonic convulsion lasting less than 3 s; 4 – tonic extension or clonic convulsion lasting longer than 3 s; 5 – tonic extension or clonic convulsion lasting more than 3 s with loss of balance. Post‐convulsion catalepsy‐like behaviour was assessed by a placing response in which a horizontal rod was placed under the forearms of the mouse, and a positive catalepsy score was assigned if the mouse did not move its forepaws onto the rod or cage floor within 30 s.
GTPγS assay
Brain membrane preparations were carried out as described previously (Befort et al., 2001). Animals that were treated and tested chronically with vehicle or SNC80 for 5 days were anaesthetised with isoflurane gas and killed by decapitation 24 h after the final treatment day. Whole brains from WT and KO mice chronically treated and tested with agonist or vehicle were removed, immediately frozen in isopentane on dry ice and stored at −80°C prior to use. Whole brain membranes were prepared by homogenizing the brain in ice‐cold 0.25 M sucrose solution 10 vol (mL·g−1 wet weight of tissue). Samples were then centrifuged at 1100× g for 10 min. Supernatants were collected and diluted five times in buffer containing 50 mM Tris‐HCl (pH 7.4) and 1 mM EDTA, following which they were centrifuged at 25000× g for 30 min. The pellets were homogenized in 2 mL ice‐cold sucrose solution (0.32 M), aliquoted and kept at −80°C until further use.
For the [35S]‐GTPγS binding assay, 5 μg of protein was used per well. Samples were incubated with varying concentrations of SNC80 (10−5 to 10−12 M) for 1 h at 25°C in assay buffer containing 50 mM Tris HCl (pH 7.4), 3 mM MgCl2, 100 mM NaCl, 0.2 mM EGTA, 30 μM GDP and 0.1 nM [35S]‐GTPγS (Perkin Elmer USA). Incubation was terminated by rapid filtration and washing in ice‐cold buffer (50 mM Tris HCl, 5 mM MgCl2, 50 mM NaCl, pH 7.4). Bound radioactivity was quantified using a liquid scintillation counter. Non‐specific binding was defined as binding in the presence of 10 μM GTPγS, and basal binding indicates binding in the absence of any agonist. Samples were run in triplicate. Plates were counterbalanced so that different combinations of groups were represented on a plate, and the experimenter was blinded to the groups.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are reported as mean ± SEM. All graphs were produced in GraphPad Prism version 6 or 7, and all statistical analysis was performed in SigmaStat software. Experiments were designed to have an equal n per group. However, animals were excluded from analysis if there was an error in drug injection, genotyping or if they were outliers. Outliers were defined using the Grubb's test, and based on this analysis, one data point was removed from the dataset shown in Figure 3A. In addition, in the convulsion experiments, arrestin 2 KO mice died or were killed following 32 mg·kg−1 SNC80; and one arrestin 2 KO mouse died due to a misplaced injection that damaged the liver for the 10 day SNC80 experiment outlined in Figure 3C. For pain and convulsion experiments, two‐way repeated‐measures ANOVAs were performed or where cited two‐way ANOVA was used. For all tests, the level of significance α was set to 0.05. Post hoc analysis was conducted using a Holm–Sidak post hoc analysis. Post hoc analysis was only performed when F values achieved P < 0.05. For convulsion data, a Tukey's post hoc analysis was performed. For the GTPγS assays, curve fitting was performed using GraphPad Prism. SNC80 was fit with a nonlinear fit, one‐site model. R 2 values were used to assess goodness of fit. EC50 values were determined from pooled, fitted data (n = 3–4 mice per group). Each data point for each mouse was the average of a triplicate, and this average was considered as n = 1.
Figure 3.
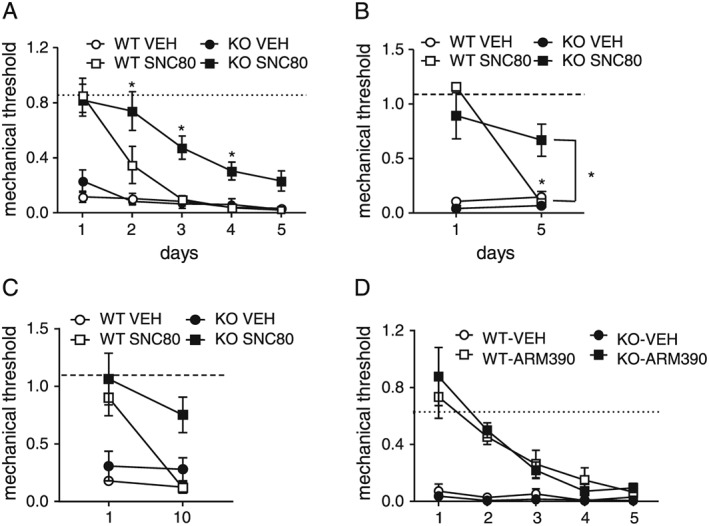
The extent of chronic analgesic tolerance after repeated exposure to SNC80 is attenuated in arrestin 2 KO mice. Arrestin 2 WT and KO mice were injected with equipotent doses of SNC80 (10 mg·kg−1 for WT, and 3 mg·kg−1 for KO) daily for 5 days, and tested daily, 45 min following each injection (A, n = 10 per WT group, n = 12 per KO group; two‐way RM ANOVA P < 0.05 time, genotype and interaction; * P < 0.05 as compared to WT‐SNC80 group). We also treated mice daily but only tested on the first and the fifth day of treatment (B, n = 8 per group; two‐way RM ANOVA, P < 0.05 genotype × time interaction, * P < 0.05 as compared to day 1, and as compared to WT‐SNC80 on day 5); or on the first and tenth day of treatment (C, n = 5–6 per group; two‐way RM ANOVA, P < 0.05 genotype and time). Unlike SNC80, tolerance to repeated ARM390 treatment was unaltered in arrestin 2 KO mice (D). WT and KO mice were injected with ARM390 (10 mg·kg−1, p.o.) daily for 5 days, and tested 45 min following each injection. n = 6 per group, two‐way RM ANOVA P < 0.05 for time only.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Effect of arrestin 2 deletion on the antihyperalgesic response to SNC80 and ARM390
To evaluate the role of arrestin 2 in δ receptor‐mediated antihyperalgesia, CFA was injected into the hindpaw, and 72 h later, mice were injected with different doses of SNC80 or ARM390. To assess potential differences in the duration of drug action, mice were tested 45 min (Figure 1A, C) and 3 h (Figure 1B, D) following injection. In WT mice, SNC80 and ARM390 produced dose‐dependent increases in mechanical threshold 45 min after administration (Figure 1A, C). After 3 h, SNC80 was no longer effective in WT animals potentially due to drug clearance. However, in arrestin 2 KO mice SNC80, but not ARM390, was enhanced at this time point; and SNC80 continued to produce significant antihyperalgesic effects (Figure 1B). ARM390 failed to produce antihyperalgesic effects in either genotype 3 h post‐injection (Figure 1D). These data show that SNC80, but not ARM390, has increased efficacy and duration of action in arrestin 2 KO mice.
Figure 1.
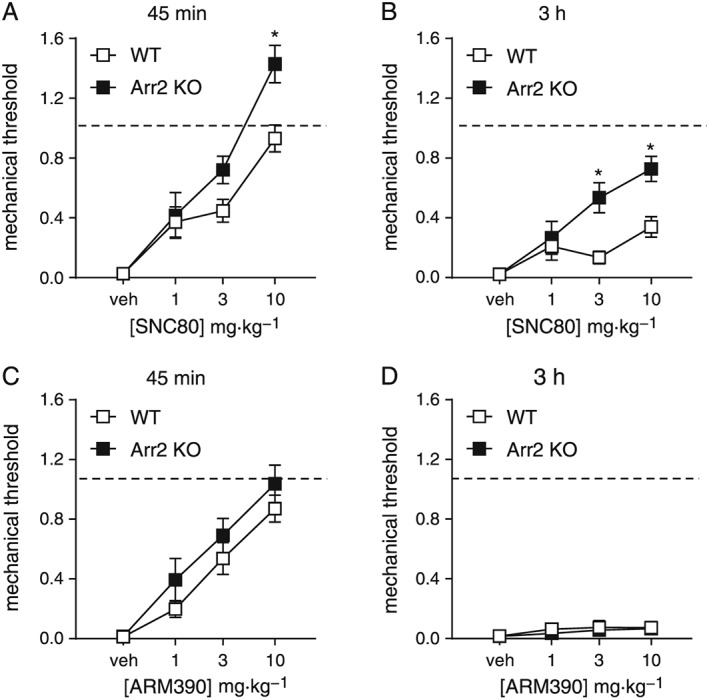
Potency and duration of SNC80‐ or ARM390‐induced antihyperalgesia in WT and arrestin 2 KO mice. In a CFA model of inflammatory pain, we performed a dose‐ and time‐response to SNC80 (i.p.) and ARM390 (p.o.). (A) The antihyperalgesic effect of SNC80 was significantly enhanced in arrestin 2 KO mice, and the effect of SNC80 was longer lasting in these animals (B), n = 8 per group. Two‐way ANOVA with Holm–Sidak post hoc analysis at both time points, P < 0.05 genotype, dose and interaction; * P < 0.05. (C) ARM390 showed similar effects in arrestin 2 KO and WT mice (two‐way ANOVA, P < 0.05 effect of dose), and regardless of genotype, this antihyperalgesic effect was lost 3 h post‐administration (D), n = 7; vehicle and 10 mg·kg−1; n = 9; 1 and 3 mg·kg−1. Dashed lines represent baseline mechanical responses assessed pre‐CFA injection.
Effect of arrestin 2 deletion on acute analgesic tolerance to SNC80 and ARM390
We have previously shown that a single injection of the high‐internalizing δ receptor agonist SNC80 produced acute analgesic tolerance while the low‐internalizing agonist ARM390 did not (Pradhan et al., 2009). In those experiments, we tested Emax doses of drug (10 mg·kg−1). Because SNC80 is more effective in arrestin 2 KO mice than WT mice, we tested equipotent doses of the drug between the two genotypes. Thus, KOs received a 3 mg·kg−1 dose of SNC80 and WTs a 10 mg·kg−1 dose. For ARM390, both genotypes were administered a 10 mg·kg−1 dose. CFA was injected into the mouse paw, and severe mechanical hypersensitivity was observed 72 h later (Figure 2, dashed line vs. vehicle controls). In WT and arrestin 2 KO mice, an initial injection of SNC80 and ARM390 attenuated CFA‐induced hyperalgesia (Figure 2, Injection 1). Consistent with previous findings, a second injection of SNC80 given 4 h later was ineffective in WT animals (Figure 2A, Injection 2). However, this acute behavioural desensitization was not observed in arrestin 2 KO mice (Figure 2B, Injection 2). A second injection of ARM390 was equally effective in both WT and KO mice. These results imply that acute tolerance to SNC80, but not ARM390, is preferentially mediated by arrestin 2.
Figure 2.

Acute behavioural tolerance to SNC80 is not observed in arrestin 2 knockout mice. Mechanical responses were determined in the CFA model of inflammatory pain. Equipotent doses of SNC80 and ARM390 were compared. Injection 1, mechanical responses in (A) WT and (B) arrestin 2 KO mice treated with vehicle (control, n = 10 per genotype), SNC80 (3 mg·kg−1 for KO mice, 13 per group, and 10 mg·kg−1 for WT mice, 12 per group i.p.), or ARM390 (10 mg·kg−1, p.o., 12 per genotype). Injection 2, animals re‐challenged with the same drug and dose 4 h following Injection 1. Dashed lines represent baseline mechanical responses pre‐CFA injection. For consistency, all animals were injected i.p. and p.o. Two‐way RM ANOVA for each genotype, P < 0.05 drug, time and interaction for WT; P < 0.05 for drug in KO. * P < 0.05 as compared to Injection 1, Holm–Sidak post hoc analysis. In addition, P < 0.05 when Injection 2 of SNC80 was compared across genotypes; t‐test.
Effect of arrestin 2 deletion on the development of chronic analgesic tolerance to SNC80 and ARM390
To confirm the role of arrestin 2 in SNC80‐induced behavioural tolerance, we examined the consequences of repeated SNC80 or ARM390 treatment in arrestin 2 WT and KO animals. WT mice treated daily with SNC80 showed full tolerance to the antihyperalgesic effects of SNC80 by the third day of treatment (Figure 3A). The development of tolerance to SNC80 was significantly delayed in arrestin 2 KO mice (Figure 3A). To rule out the possibility that these effects were due to WT and KO mice developing behavioural/associative tolerance to the assay at different rates, we repeated the same experiment but tested only on the first and fifth days of treatment (Figure 3B). Again, SNC80 failed to produce antihyperalgesic effects in WT animals after repeated treatment, indicating pharmacological tolerance. However, in arrestin 2 KO mice, the last injection of SNC80 still produced significant antihyperalgesic effects (Figure 3B). In order to determine if an even longer term treatment would induce pharmacological tolerance in arrestin 2 KOs, we treated animals with vehicle or SNC80 daily for 10 days. Even after this prolonged treatment, SNC80 continued to be effective in arrestin 2 KO mice as compared to WT controls (Figure 3C). In contrast to SNC80, WT and arrestin 2 KO mice developed chronic tolerance to the antihyperalgesic effects of ARM390 at similar rates (Figure 3D). Arrestin 2 appears to be necessary for the development of tolerance to SNC80, but not to the low‐internalizing agonist, ARM390.
The effect of arrestin 2 deletion on tolerance to SNC80‐induced convulsions
To explore the role of arrestin 2 in the development of tolerance to other δ receptor‐mediated behaviours, tolerance to SNC80‐induced convulsions was evaluated in WT and arrestin 2 KO mice (Figure 4). ARM390 does not produce convulsions, even at high doses, and therefore was not included to study the development of tolerance to this phenomenon. Mice were treated with SNC80 daily for 5 days, and the severity of convulsions was evaluated using a modified Racine scale. In WT mice, the dose of 32 mg·kg−1 SNC80 was tested as 10 mg·kg−1 does not reliably induce convulsions. SNC80 produced severe convulsive effects on day 1 of treatment, but failed to produce significant convulsive effects on subsequent days in WTs. In contrast, arrestin 2 KO mice exhibited significant convulsive effects in response to lower doses of SNC80 (3.2 or 10 mg·kg−1), and this effect was observed on all 5 days. A group of four arrestin 2 KO mice was also tested with 32 mg·kg−1 SNC80. However, on day 1, this dose of SNC80 produced fatal convulsions in two mice and a sustained convulsion in a third mouse that prompted the experimenters to kill it immediately by pentobarbital overdose. Therefore, this dose of SNC80 was not further evaluated in arrestin 2 KO animals. Overall, mice lacking arrestin 2 showed increased sensitivity and attenuated tolerance to SNC80‐induced convulsive effects.
Figure 4.
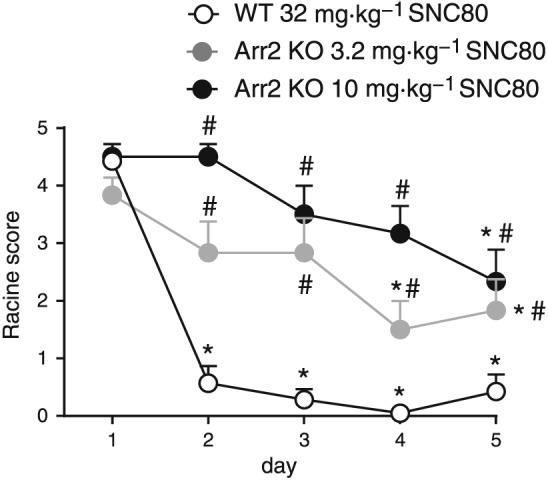
Development of tolerance to the convulsive effects of SNC80 is attenuated in arrestin 2 KO mice. The severity of convulsions produced by acute administration of SNC80 in WT mice (32 mg·kg−1) or arrestin 2 KO mice (3.2 or 10 mg·kg−1) did not differ significantly. In WT mice, SNC80 failed to produce significant convulsive effects after the first day of treatment. Repeated daily administration of SNC80 produced significant convulsive effects in arrestin 2 KO mice on all test days. For all groups n = 6, two‐way RM ANOVA, P < 0.05 time, group and interaction. * P < 0.05 compared to same treatment group on day 1, # P < 0.05 compared to the WT group on the same day.
The effect of arrestin 2 deletion on G protein‐coupling to δ receptors after chronic exposure to SNC80
We then investigated receptor functionality of δ receptors after the establishment of tolerance to SNC80. Arrestin 2 WT and KO animals were injected and tested with vehicle or equipotent doses of SNC80 (10 mg·kg−1 WT, 3 mg·kg−1 KO) once daily for 5 days, and tissue was collected 24 h after the final treatment day. SNC80‐induced G protein‐coupling to δ receptors was examined in brain membrane preparations. [35S]GTPγS binding was severely attenuated in the brains of WT animals treated repeatedly with SNC80, in line with the observed chronic tolerance observed in these animals. In contrast, SNC80‐treated arrestin 2 KO mice showed [35S]GTPγS binding that was comparable to SNC80‐naïve controls (Figure 5A, B). These results indicate that in arrestin 2 KO mice δ receptor‐G protein coupling is preserved, even after chronic treatment with SNC80.
Figure 5.
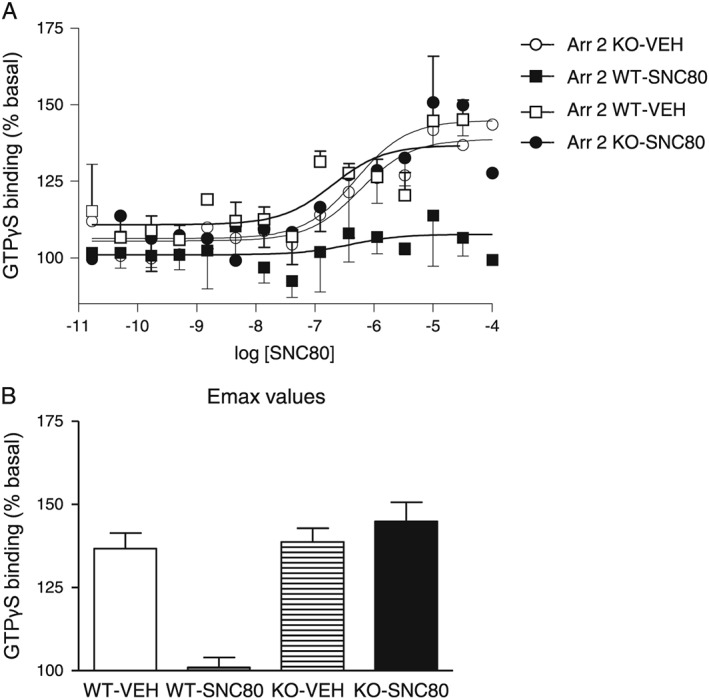
Arrestin 2 KO mice showed intact G protein‐δ receptor coupling after chronic SNC80 treatment. Graphs show (A) concentration–response curves for SNC80‐induced [35S]‐GTPγS binding and (B) Emax values calculated from these responses. The y‐axis shows mean ± SEM specific [35S]‐GTPγS binding expressed as percentage basal binding; n = 3 mice per group.
Discussion
In this study, we examined the role of arrestin 2 in the development of tolerance to the behavioural effects of a high‐ (SNC80) or a low‐ (ARM390) internalizing δ receptor agonist. We found that KO of arrestin 2 results in increased efficacy and duration of action of the antihyperalgesic effects of SNC80 and increased the potency of SNC80 to produce convulsions. Loss of arrestin 2 also attenuated the development of tolerance to both the antihyperalgesic and convulsive effects of chronic SNC80 treatment. Although some chronic tolerance to the antihyperalgesic effects of SNC80 was observed, it was considerably diminished and was significantly dependent on repeated testing (behavioural/associative tolerance). In accordance with these findings, δ receptors in KO mice treated chronically with SNC80 remained functionally coupled to G proteins, an effect not observed in the WTs. In contrast, the low‐internalizing agonist ARM390 produced similar analgesic effects and tolerance in both WT and KO arrestin 2 animals. Overall, these results suggest a ligand‐selective recruitment of arrestins by different δ receptor agonists and reveal that SNC80 may preferentially recruit arrestin 2 to promote tolerance whereas ARM390 does not.
We have previously shown that loss of arrestin 2 enhances the antihyperalgesic effects of SNC80 and inhibits the development of acute tolerance (Pradhan et al., 2016). This study reproduce those results in another group of animals and also demonstrate a potential role for arrestin 2 in regulating the development of tolerance to chronic daily administration of SNC80. After repeated administration, a marked reduction in tolerance to the antihyperalgesic effects of SNC80 was clear in arrestin 2 KO animals tested only twice, at the beginning and end of a 5‐ or 10‐day treatment, but not in animals tested every day. A previous study showed that repeated testing of mice treated with vehicle for 5 days resulted in a 50% decrease of the analgesic effects of a subsequent challenge with SNC80 or ARM390 (Pradhan et al., 2010). It is known that environmental cues and processes related to memory and learning can facilitate tolerance development during repeated exposure to opioids (Gamble and Milne, 1989; Mitchell et al., 2000). Thus, our data demonstrate that the tolerance that develops to SNC80‐induced antihyperalgesia in arrestin 2 KOs is significantly dependent on associative learning. Our data also show that the development of tolerance to the convulsive effects of SNC80 was attenuated in arrestin 2 KO mice, suggesting that arrestin 2 is also an important regulator of this behaviour. Although seizure activity can have profound effects on behaviour, it is unlikely to have affected antihyperalgesic responses to SNC80, as convulsive effects are lost within 10–15 min post‐administration (Broom et al., 2002a). In addition, the arrestin 2 KOs developed behavioural/associative tolerance similar to WTs, suggesting that their learning and memory was not affected. Loss of δ receptor in GABAergic forebrain neurons was sufficient to block all SNC80‐induced seizurogenic activity (Chung et al., 2015). It remains to be seen whether arrestin 2 acts as a negative regulator of δ receptor‐mediated convulsions within these neurons. Furthermore, the role these neurons play in the development of tolerance to SNC80‐induced convulsions should be examined. Taken together, these behavioural results also imply that both central (convulsions) and peripheral (antihyperalgesia) effects of SNC80 are regulated by arrestin 2.
Tolerance to the convulsive effects of SNC80 developed rapidly relative to antihyperalgesic tolerance, and almost no convulsions were observed in WT mice in the second day of SNC80 treatment. These results are in keeping with a previous report that showed that tolerance to the convulsive effects of SNC80 developed faster than tolerance to its antidepressant‐like effects and that this rapid tolerance corresponded with differential tolerance rates in δ receptor signalling (Jutkiewicz et al., 2005). One hypothesis is that δ receptor agonists have a low efficacy requirement to produce convulsive effects, as compared to their pain‐relieving properties (Broom et al., 2002b).
Consistent with the in vivo data, our GTPγS assays revealed that δ receptors remain functionally coupled to G proteins in arrestin 2 KO mice repeatedly treated with SNC80. SNC80 has been shown to induce robust δ receptor internalization in vitro and in vivo (Lecoq et al., 2004; Scherrer et al., 2006; Pradhan et al., 2009, 2010; Charfi et al., 2014). This loss of surface δ receptor expression likely accounts for the decreased δ receptor function observed after repeated administration of SNC80 to WT mice. Following prolonged agonist exposure, internalized δ receptors are targeted to lysosomes and degraded (Ko et al., 1999; Tsao and von Zastrow, 2000; Whistler et al., 2002), a process that is also induced after chronic treatment with SNC80 (Lecoq et al., 2004; Pradhan et al., 2010). This receptor down‐regulation results in generalized tolerance to all the behavioural effects triggered by SNC80 (Pradhan et al., 2010). Because arrestin 2 is involved in δ receptor internalization (Zhang et al., 2005, 2008; Qiu et al., 2007; Mittal et al., 2013), the increased analgesic effects of SNC80 and decreased tolerance observed in the absence of arrestin 2 could be due to diminished receptor internalization. Therefore, more δ receptors would be accessible to SNC80 resulting in increased receptor activation. Mittal et al. (2013) found that loss of arrestin 2 increased the export of δ receptors to the cell membrane in response to SNC80 via a deregulation of the ROCK‐LIMK pathway. This enhancement of agonist‐induced externalization could also account for the differences observed in the behavioural effects of SNC80 in arrestin 2 KOs. We hypothesize that our findings could result from a combination of both phenomena, a decrease in receptor internalization along with an increase in δ receptors externalization, thus resulting in a net enhancement of SNC80 effects.
Unlike SNC80, the antihyperalgesic effects of ARM390 were not altered in arrestin 2 KO mice. Previous BRET studies demonstrated that the low‐internalizing agonist ARM390 recruits arrestin 2 at the δ receptor; however, no changes in the antihyperalgesic effects of ARM390 were observed in KOs of arrestin 2 (Pradhan et al., 2016). Moreover, no significant differences in the development of acute and chronic tolerance to ARM390 were detected between arrestin 2 KOs and WT animals. Importantly, the mechanisms involved in the development of tolerance to SNC80 and ARM390 are different. ARM390 does not promote robust receptor endocytosis, and animals treated with ARM390 do not develop acute analgesic tolerance (Pradhan et al., 2010; Pradhan et al., 2016). In addition, chronic tolerance to ARM390 is independent of receptor internalization and relies on cellular adaptations occurring down‐stream of the receptor (Pradhan et al., 2010). Thus, contrary to SNC80, recruitment of arrestin 2 at ARM390‐activated δ receptors may serve a different function to the traditional role of arrestins as attenuators of GPCR signalling.
Numerous lines of evidence in vitro indicate that SNC80‐activated δ receptors recruit both arrestin 2 and arrestin 3 (Cen et al., 2001; Audet et al., 2012; Mittal et al., 2013; Chiang et al., 2016; Pradhan et al., 2016). However, our results suggest functional specialization in vivo for arrestin 2 in modulating the antihyperalgesic effects of SNC80 in the CFA model of chronic inflammatory pain. Furthermore, KO of arrestin 3 had no effect on SNC80‐induced analgesia and acute tolerance (Pradhan et al., 2016). Notwithstanding, a recent study has shown a direct correlation between the efficacy of SNC80 and other δ receptor agonists to induce recruitment of arrestin 3 and increased alcohol consumption (Chiang et al., 2016). Thus, it is a possibility that our results are specific to regions controlling pain processing and convulsions and that arrestin 3 may play an important role in regulation of SNC80‐activated δ receptor in other CNS areas. Further work will be needed to explain potential regional differences in agonist‐specific receptor‐arrestin interactions.
These results suggest that arrestin 2 differentially regulates δ receptor effects in an agonist‐dependent manner. The ligand‐specific recruitment of arrestins observed in our studies is likely due to distinct conformational changes induced by the binding of different δ receptor agonists. The δ receptor agonists induce specific receptor conformations that possess different affinities for arrestins, resulting in distinct receptor desensitization, internalization and signalling profiles that ultimately dictate physiological outcomes (Aguila et al., 2012; Audet et al., 2012). In BRET studies, it has been shown that agonists like SNC80 stabilize a δ receptor conformation in which its C‐terminal tail becomes closer to Gβγ subunits. This spatial rearrangement of the receptor increased the stability of its interaction with arrestin 3 and caused poor recycling and marked receptor desensitization (Audet et al., 2012). Differentially, the δ receptor agonist DPDPE moved the C‐terminal tail of the δ receptor away from Gβγ subunits and promoted a transient interaction with arrestin 3 that resulted in receptor recycling and sustained analgesia (Audet et al., 2012). These studies support the notion that agonists for the same receptor can promote distinct receptor–arrestin complexes.
Our study indicates that arrestin 2 may be selectively recruited in vivo by high‐internalizing δ agonists to modulate chronic pain. The specific deletion of arrestin 2 produced enhanced δ receptor function to the high‐internalizing agonist SNC80 even after chronic treatment but did not modify the behavioural effects to the low‐internalizing agonist ARM390. Collectively, our results demonstrate the behavioural significance that ligand‐specific interactions between arrestin 2 and the δ receptor may have and reveal a potential role of arrestin 2 as a mediator of the analgesic effects and tolerance to high‐internalizing δ receptor agonist.
Author contributions
A.V.S., I.J.D., A.F.T., H.A. and A.A. performed the experiments. A.V.S., I.J.D., E.M.J. and A.A.P. analysed the data. A.V.S., I.J.D., E.M.J. and A.A.P. planned the experiments, wrote and edited the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This research was supported by NIH‐NIDA Grants DA031243 and DA040688 (A.A.P.), and in part by a PhRMA Foundation Research Starter Grant (E.M.J.). A.A.P. has previously received funding from Trevena Inc. The authors would like to thank Zachariah Bertels for help with animal injections.
Vicente‐Sanchez A., Dripps I. J., Tipton A. F., Akbari H., Akbari A., Jutkiewicz E. M., and Pradhan A. A. (2018) Tolerance to high‐internalizing δ opioid receptor agonist is critically mediated by arrestin 2, British Journal of Pharmacology, 175, 3050–3059, https://doi.org/10.1111/bph.14353.
References
- Aguila B, Coulbault L, Davis A, Marie N, Hasbi A, Le Bras F et al (2012). βarrestin1‐biased agonism at human δ‐opioid receptor by peptidic and alkaloid ligands. Cell Signal 24: 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audet N, Charfi I, Mnie‐Filali O, Amraei M, Chabot‐Dore AJ, Millecamps M et al (2012). Differential association of receptor‐Gbetagamma complexes with beta‐arrestin2 determines recycling bias and potential for tolerance of delta opioid receptor agonists. J Neurosci 32: 4827–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Befort K, Filliol D, Decaillot FM, Gaveriaux‐Ruff C, Hoehe MR, Kieffer BL (2001). A single nucleotide polymorphic mutation in the human mu‐opioid receptor severely impairs receptor signaling. J Biol Chem 276: 3130–3137. [DOI] [PubMed] [Google Scholar]
- Bowman SL, Soohoo AL, Shiwarski DJ, Schulz S, Pradhan AA, Puthenveedu MA (2015). Cell‐autonomous regulation of Mu‐opioid receptor recycling by substance P. Cell Rep 10: 1925–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt MR, Furness MS, Rice KC, Fischer BD, Negus SS (2001). Studies of tolerance and dependence with the delta‐opioid agonist SNC80 in rhesus monkeys responding under a schedule of food presentation. J Pharmacol Exp Ther 299: 629–637. [PubMed] [Google Scholar]
- Broom DC, Jutkiewicz EM, Folk JE, Traynor JR, Rice KC, Woods JH (2002a). Convulsant activity of a non‐peptidic delta‐opioid receptor agonist is not required for its antidepressant‐like effects in Sprague‐Dawley rats. Psychopharmacology (Berl) 164: 42–48. [DOI] [PubMed] [Google Scholar]
- Broom DC, Nitsche JF, Pintar JE, Rice KC, Woods JH, Traynor JR (2002b). Comparison of receptor mechanisms and efficacy requirements for delta‐agonist‐induced convulsive activity and antinociception in mice. J Pharmacol Exp Ther 303: 723–729. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Hoffert C, O'Donnell D, Beaudet A (2003). Up‐regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain 101: 199–208. [DOI] [PubMed] [Google Scholar]
- Cen B, Xiong Y, Ma L, Pei G (2001). Direct and differential interaction of beta‐arrestins with the intracellular domains of different opioid receptors. Mol Pharmacol 59: 758–764. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Meth 53: 55–63. [DOI] [PubMed] [Google Scholar]
- Charfi I, Nagi K, Mnie‐Filali O, Thibault D, Balboni G, Schiller PW et al (2014). Ligand‐ and cell‐dependent determinants of internalization and cAMP modulation by delta opioid receptor (DOR) agonists. Cell Mol Life Sci 71: 1529–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang T, Sansuk K, van Rijn RM (2016). β‐arrestin 2 dependence of δ opioid receptor agonists is correlated with alcohol intake. Br J Pharmacol 173: 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung PC, Boehrer A, Stephan A, Matifas A, Scherrer G, Darcq E et al (2015). Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80‐induced seizures. Behav Brain Res 278: 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer SD, Hoenicke EM, Sable AI, McNutt RW, Chang KJ, De Costa BR et al (1993). Convulsive effects of systemic administration of the delta opioid agonist BW373U86 in mice. J Pharmacol Exp Ther 267: 888–895. [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser GL, Gaudreau GA, Clarke PB, Menard DP, Perkins MN (2000). Antihyperalgesic effects of delta opioid agonists in a rat model of chronic inflammation. Br J Pharmacol 129: 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallantine EL, Meert TF (2005). A comparison of the antinociceptive and adverse effects of the mu‐opioid agonist morphine and the delta‐opioid agonist SNC80. Basic Clin Pharmacol Toxicol 97: 39–51. [DOI] [PubMed] [Google Scholar]
- Gamble GD, Milne RJ (1989). Repeated exposure to sham testing procedures reduces reflex withdrawal and hot‐plate latencies: attenuation of tonic descending inhibition? Neurosci Lett 96: 312–317. [DOI] [PubMed] [Google Scholar]
- Gaveriaux‐Ruff C, Karchewski LA, Hever X, Matifas A, Kieffer BL (2008). Inflammatory pain is enhanced in delta opioid receptor‐knockout mice. Eur J Neurosci 27: 2558–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaveriaux‐Ruff C, Kieffer BL (2011). Delta opioid receptor analgesia: recent contributions from pharmacology and molecular approaches. Behav Pharmacol 22: 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron L, Cahill CM, von Zastrow M, Schiller PW, Pineyro G (2016). Molecular Pharmacology of delta‐Opioid Receptors. Pharmacol Rev 68: 631–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong MH, Xu C, Wang YJ, Ji JL, Tao YM, Xu XJ et al (2009). Role of Src in ligand‐specific regulation of delta‐opioid receptor desensitization and internalization. J Neurochem 108: 102–114. [DOI] [PubMed] [Google Scholar]
- Hurley RW, Hammond DL (2000). The analgesic effects of supraspinal mu and delta opioid receptor agonists are potentiated during persistent inflammation. J Neurosci 20: 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutkiewicz EM, Baladi MG, Folk JE, Rice KC, Woods JH (2006). The convulsive and electroencephalographic changes produced by nonpeptidic delta‐opioid agonists in rats: comparison with pentylenetetrazol. J Pharmacol Exp Ther 317: 1337–1348. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz EM, Kaminsky ST, Rice KC, Traynor JR, Woods JH (2005). Differential behavioral tolerance to the delta‐opioid agonist SNC80 ([(+)‐4‐[(alphaR)‐alpha‐[(2S,5R)‐2,5‐dimethyl‐4‐(2‐propenyl)‐1‐piperazinyl]‐(3‐methoxyphenyl)methyl]‐N,N‐diethylbenzamide) in Sprague‐Dawley rats. J Pharmacol Exp Ther 315: 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabli N, Cahill CM (2007). Anti‐allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain 127: 84–93. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JL, Arvidsson U, Williams FG, Law PY, Elde R, Loh HH (1999). Visualization of time‐dependent redistribution of delta‐opioid receptors in neuronal cells during prolonged agonist exposure. Brain Res Mol Brain Res 69: 171–185. [DOI] [PubMed] [Google Scholar]
- Lecoq I, Marie N, Jauzac P, Allouche S (2004). Different regulation of human delta‐opioid receptors by SNC‐80 [(+)‐4‐[(alphaR)‐alpha‐((2S,5R)‐4‐allyl‐2,5‐dimethyl‐1‐piperazinyl)‐3‐methoxybenz yl]‐N,N‐diethylbenzamide] and endogenous enkephalins. J Pharmacol Exp Ther 310: 666–677. [DOI] [PubMed] [Google Scholar]
- Lowe JD, Celver JP, Gurevich VV, Chavkin C (2002). mu‐Opioid receptors desensitize less rapidly than delta‐opioid receptors due to less efficient activation of arrestin. J Biol Chem 277: 15729–15735. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ (2002). The role of beta‐arrestins in the termination and transduction of G‐protein‐coupled receptor signals. J Cell Sci 115 (Pt 3): 455–465. [DOI] [PubMed] [Google Scholar]
- Ma L, Pei G (2007). Beta‐arrestin signaling and regulation of transcription. J Cell Sci 120 (Pt 2): 213–218. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JM, Basbaum AI, Fields HL (2000). A locus and mechanism of action for associative morphine tolerance. Nat Neurosci 3: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal N, Roberts K, Pal K, Bentolila LA, Fultz E, Minasyan A et al (2013). Select G‐protein‐coupled receptors modulate agonist‐induced signaling via a ROCK, LIMK, and beta‐arrestin 1 pathway. Cell Rep 5: 1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagi K, Charfi I, Pineyro G (2015). Kir3 channels undergo arrestin‐dependant internalization following delta opioid receptor activation. Cell Mol Life Sci 72: 3543–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Gatch MB, Mello NK, Zhang X, Rice K (1998). Behavioral effects of the delta‐selective opioid agonist SNC80 and related compounds in rhesus monkeys. J Pharmacol Exp Ther 286: 362–375. [PubMed] [Google Scholar]
- Pradhan A, Smith M, McGuire B, Evans C, Walwyn W (2013). Chronic inflammatory injury results in increased coupling of delta opioid receptors to voltage‐gated Ca2+ channels. Mol Pain 9: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Becker JA, Scherrer G, Tryoen‐Toth P, Filliol D, Matifas A et al (2009). In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoSOne 4: e5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Befort K, Nozaki C, Gaveriaux‐Ruff C, Kieffer BL (2011). The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci 32: 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Perroy J, Walwyn WM, Smith ML, Vicente‐Sanchez A, Segura L et al (2016). Agonist‐specific recruitment of arrestin isoforms differentially modify delta opioid receptor function. J Neurosci 36: 3541–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Walwyn W, Nozaki C, Filliol D, Erbs E, Matifas A et al (2010). Ligand‐directed trafficking of the delta‐opioid receptor in vivo: two paths toward analgesic tolerance. J Neurosci 30: 16459–16468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Loh HH, Law PY (2007). Phosphorylation of the delta‐opioid receptor regulates its beta‐arrestins selectivity and subsequent receptor internalization and adenylyl cyclase desensitization. J Biol Chem 282: 22315–22323. [DOI] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32: 281–294. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM (2011). The role of beta‐arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 60: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, Lefkowitz RJ (2012). Molecular mechanism of beta‐arrestin‐biased agonism at seven‐transmembrane receptors. Annu Rev Pharmacol Toxicol 52: 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer G, Tryoen‐Toth P, Filliol D, Matifas A, Laustriat D, Cao YQ et al (2006). Knockin mice expressing fluorescent delta‐opioid receptors uncover G protein‐coupled receptor dynamics in vivo. Proc Natl Acad Sci USA 103: 9691–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK (2014). Arrestin interaction with E3 ubiquitin ligases and deubiquitinases: functional and therapeutic implications. Handb Exp Pharmacol 219: 187–203. [DOI] [PubMed] [Google Scholar]
- Stevenson GW, Folk JE, Rice KC, Negus SS (2005). Interactions between delta and mu opioid agonists in assays of schedule‐controlled responding, thermal nociception, drug self‐administration, and drug versus food choice in rhesus monkeys: studies with SNC80 [(+)‐4‐[(alphaR)‐alpha‐((2S,5R)‐4‐allyl‐2,5‐dimethyl‐1‐piperazinyl)‐3‐methoxybenz yl]‐N,N‐diethylbenzamide] and heroin. J Pharmacol Exp Ther 314: 221–231. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Castonguay A, Ghogha A, Vayssiere P, Pradhan AA, Xue L et al (2016). Neuroimmune regulation of GABAergic neurons within the ventral tegmental area during withdrawal from chronic morphine. Neuropsychopharmacology 41: 949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao PI, von Zastrow M (2000). Type‐specific sorting of G protein‐coupled receptors after endocytosis. J Biol Chem 275: 11130–11140. [DOI] [PubMed] [Google Scholar]
- Vicente‐Sanchez A, Segura L, Pradhan AA (2016). The delta opioid receptor tool box. Neuroscience 338: 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Enquist J, Marley A, Fong J, Gladher F, Tsuruda P et al (2002). Modulation of postendocytic sorting of G protein‐coupled receptors. Science (New York, NY) 297: 615–620. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wang F, Chen X, Chen Y, Ma L (2008). Post‐endocytic fates of delta‐opioid receptor are regulated by GRK2‐mediated receptor phosphorylation and distinct beta‐arrestin isoforms. J Neurochem 106: 781–792. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wang F, Chen X, Li J, Xiang B, Zhang YQ et al (2005). Beta‐arrestin1 and beta‐arrestin2 are differentially required for phosphorylation‐dependent and ‐independent internalization of delta‐opioid receptors. J Neurochem 95: 169–178. [DOI] [PubMed] [Google Scholar]