

Abstract
Background and Purpose
Insufficient prefrontal dopamine 1 (D1) receptor signalling has been linked to cognitive dysfunction in several psychiatric conditions. Because the PDE1 isoform B (PDE1B) is postulated to regulate D1 receptor‐dependent signal transduction, in this study we aimed to elucidate the role of PDE1 in cognitive processes reliant on D1 receptor function.
Experimental Approach
Cognitive performance of the D1 receptor agonist, SKF38393, was studied in the T‐maze continuous alternation task and 5‐choice serial reaction time task. D1 receptor/PDE1B double‐immunohistochemistry was performed using human and rat prefrontal brain sections. The pharmacological activity of the PDE1 inhibitor, ITI‐214, was assessed by measuring the increase in cAMP/cGMP in prefrontal brain tissue and its effect on working memory performance. Mechanistic studies on the modulation of prefrontal neuronal transmission by SKF38393 and ITI‐214 were performed using extracellular recordings in brain slices.
Key Results
SKF38393 improved working memory and attentional performance in rodents. D1 receptor/PDE1B co‐expression was verified in both human and rat prefrontal brain sections. The pharmacological activity of ITI‐214 on its target, PDE1, was demonstrated by its ability to increase prefrontal cAMP/cGMP. In addition, ITI‐214 improved working memory performance. Both SKF38393 and ITI‐214 facilitated neuronal transmission in prefrontal brain slices.
Conclusion and Implications
We hypothesize that PDE1 inhibition improves working memory performance by increasing prefrontal synaptic transmission and/or postsynaptic D1 receptor signalling, by modulating prefrontal downstream second messenger levels. These data, therefore, support the use of PDE1 inhibitors as a potential approach for the treatment of cognitive dysfunction.
Abbreviations
- 5‐CSRTT
5‐choice serial reaction time task
- AAALAC
Association for Assessment and Accreditation of Laboratory Animal Care
- ACSF
artificial CSF
- ITI
inter‐trial interval
- LH
limited hold
- SD
stimulus duration
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase
- TO
timeout
Introduction
The cognitive function of the prefrontal cortex (PFC) is known to be modulated by ascending monoaminergic neural systems. Lesions of the mesocortical dopaminergic projection have long been known to impair working memory performance, as previously shown in monkeys (Brozoski et al., 1979). http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=214, but not http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=215, antagonists impair delay‐dependent working memory performance when infused into substructures of the primate prefrontal brain (Sawaguchi and Goldman‐Rakic, 1991). These findings indicate that the D1 receptor has an important role in modulating cognitive performance under the control of the PFC and are consistent with human data showing beneficial effects of the mixed D1/D2 receptor agonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=48 (on longer, but not shorter, delays), in a delay‐dependent spatial working paradigm in humans (Muller et al., 1998). Post‐mortem tissue analysis of patients with schizophrenia indicates they have reduced dopaminergic innervation of the PFC (Akil et al., 1999), suggesting that an altered dopaminergic state in this brain area could be an important pathophysiological aspect of the disease and may contribute to the cognitive disturbances seen in this patient population. PET studies demonstrate increased D1 receptor availability in the PFC of drug‐naïve patients with schizophrenia, as well as patients with schizotypal personality disorder and those with schizophrenia and a medication history. However, this has an anticorrelation with working memory performance, indicating unsuccessful compensation of low dopaminergic input to the PFC (Abi‐Dargham et al., 2002; Thompson et al., 2014). In line with this idea, in a small exploratory study the D1 receptor agonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9267, was demonstrated to improve working memory in patients with schizotypal personality disorder (Rosell et al., 2015). Although working memory deficits in patients with schizophrenia are usually identified using the n‐back task, which has no delay, but depends on mental tracking of information with increasing span requirements (Gold et al., 2010), the data consistently point to a significant role of prefrontal D1 receptor functionality in cognitive performance. Restoring D1 receptor signalling in the PFC of patients with disorders characterized by prefrontal dopamine hypofunction, such as schizotypal spectrum disorders or schizophrenia, could therefore provide an important symptomatic treatment approach.
The D1 receptor is a GPCR that signals via Gαs, thus stimulating the conversion of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1713 into http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2352 by adenylate cyclase. Accordingly, inhibition of the cyclic nucleotide PDEs that degrade the second messenger molecule cAMP should have a D1 receptor agonist‐like effect. Because http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=260#1295 is postulated to regulate D1 receptor‐dependent signal transduction (Duinen et al., 2015; Snyder et al., 2016), this study aimed to elucidate the role of PDE1 in cognitive processes that are believed to depend on prefrontal D1 receptor function.
Methods
Subjects
Sixty‐four male Lister‐hooded rats (Charles River, Sulzfeld, Germany), weighing approximately 300 g at the start of training, were used in the 5‐choice serial reaction time task (5‐CSRTT). For the T‐maze, continuous alternation task studies, 145 male CD‐1 mice (Janvier, Le Genest St Isle, France), weighing approximately 28 g at start of training, were used and housed in reversed dark/light cycle conditions. For assessment of cAMP and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2347 levels, an additional 40 adult male CD‐1 mice were used. For acute slice experiments, 17 male Sprague Dawley rats (Janvier) aged 5–6 weeks were used. Animals were group‐housed under a 12 h light/dark cycle and controlled temperature (21 ± 1°C) with food and water initially available ad libitum. For the 5‐CSRTT training, the food was restricted shortly before the initiation of training and body weight was maintained at approximately 85% of free‐feeding weight. All experimental procedures were authorized by the Local Animal Care and Use Committee in accordance with local animal care guidelines and the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) regulations and the USDA Animal Welfare Act. All experimental studies were carried in an AAALAC certified facility. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Tissue specimens and immunohistochemistry
Rat brain tissue was fixed in 4% paraformaldehyde for 48 h and embedded in paraffin blocks. Human brain tissue from healthy donors (Folio Biosciences, Ohio, USA) was fixed in 4% paraformaldehyde, and samples containing PFC were identified, trimmed and processed into paraffin blocks. All procurement of human tissue was performed in accordance with protocols approved by the Institutional Review Board as well as all national and local regulatory guidelines; all human samples were obtained with informed consent. Tissue sections from rat and human brain were cut at 4 μm, adhered to glass slides and pretreated for 30 min at 95°C and pH 6 in a microwave oven. Double‐immunofluorescence was performed with primary antibodies against PDE1B (mouse monoclonal IgG2a, 1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and the D1 receptor (rabbit polyclonal, 1:100, Abcam, Cambridge, UK), followed by fluorochrome‐labelled secondary antibodies (goat anti‐mouse DyLight 488 and goat anti‐rabbit DyLight 633, respectively, both Thermo Fisher Scientific, Darmstadt, Germany). Stained slides were covered by using a DAPI mounting medium (ProLong™ Gold antifade, Invitrogen, Thermo Fisher Scientific) and imaged using a confocal microscope (LSM 700, Zeiss, Jena, Germany).
Determination of potency and selectivity
ITI‐214 (Li et al., 2016) was synthesized and its molecular potency determined by using a radiometric assay (Thompson and Appleman, 1971) with recombinant human PDE1A, PDE1B and PDE1C enzymes expressed in a baculoviral system. The selectivity of ITI‐214 against human PDE enzymes, expressed in insect Sf9 cells, was evaluated by using a fluorescence polarization assay (Lea and Simeonov, 2011).
Extracellular field potential recordings
Rats were anaesthetized with isoflurane, decapitated and brains quickly removed and immersed in ice‐cold (4°C) ACSF containing 124 mM sodium chloride, 2.45 mM potassium chloride, 8.8 mM magnesium sulfate, 1.2 mM monopotassium phosphate, 25.6 mM sodium bicarbonate, 2.25 mM calcium chloride and 10 mM D‐glucose, pH 7.4, saturated with 95% oxygen and 5% carbon dioxide; 400 μm coronal slices were cut and left to rest at room temperature for at least 90 min before the recordings were obtained in a holding chamber containing ACSF. The slices were transferred to integrated brain slice chambers as described previously (Kroker et al., 2011) and continuously superfused (2.5 mL·min−1, room temperature) with the same ACSF used for the recovery phase, except with 1.2 mM of magnesium sulfate (regular ACSF). fEPSPs were recorded from layer V of the prelimbic and infralimbic PFC and evoked by single electrical stimulus pulses (100 μs in duration, every 30 s) of the superficial layers II–III, delivered through a monopolar stimulating electrode. The PFC has a modular architecture based on the aggregation of neurons arranged in minicolumns. The supragranular layers II–III, which are the major source of corticocortical projections, also receive sensory information, whereas the infragranular layer V is the main output to subcortical structures. Following the concept of such columnar microcircuits in the PFC, and in accordance with the physiological information flow, we recorded fEPSPs from layer V in response to paired stimulus pulses in the superficial layers II–III. The stimulation and field‐recording electrodes (2–6 MΩ) were filled with regular ACSF. The amplitude of the fEPSPs was measured for 30 min at baseline, followed by a 30–60 min period of drug administration. Between 3 and 14 slices were recorded per group. Changes in the fEPSP amplitude were calculated in relation to the baseline fEPSP responses during the last 10 min before drug administration (100%). A modular electrophysiology system (npi electronic, Tamm, Germany), conducted the low‐noise recordings of extracellular signals. Signals were amplified 1000 times and subsequently filtered with a low‐pass (5 kHz) and a high‐pass (3 Hz) filter.
Determination of cGMP and cAMP in mouse brain
Cyclic nucleotide levels were determined as described previously (Kroker et al., 2014). Briefly, mice were killed by focused microwave irradiation of the brain, 30 min after p.o. administration of vehicle (0.5% Natrosol in water containing 0.01% Tween‐80) or various doses of ITI‐214 (n = 8 animals per group, for all groups). The PFC was isolated, homogenized and centrifuged in 0.5 N of hydrogen chloride. The cyclic nucleotide concentrations of the supernatants were then measured using cGMP/cAMP elisa kits according to manufacturer's protocol (Enzo Life Science GmbH, Lörrach, Germany). The detection limit for cGMP was 0.02 pmol·mg−1 brain tissue and for cAMP was 0.03 pmol·mg−1 brain tissue.
T‐maze continuous alternation task in mice
The T‐maze continuous alternation was performed as described previously (Delotterie et al., 2015). This behavioural task is based on the natural propensity of the animal to explore new environments, referred as to as neophilia, or novelty preference. A normal mouse, with intact cognitive abilities, would present a high degree of spontaneous alternation in this task, basing its alternated turning on the memory of the previously visited arm and choosing the unvisited one. The percentage of spontaneous alternation is considered to be an index of working memory competence. Testing was performed in the active period of the mice, that is, the dark period, in mice housed under a reversed light cycle. Briefly, the apparatus consisted of an enclosed T‐maze made of grey polyvinyl chloride, which was elevated 1 m above the floor in a dimmed (10 lux) testing room. Extra maze cues were present on each arm side and directly illuminated by a low ceiling lighting (6–10 lux). The start arm measured 54 × 8.5 × 20 cm, against 30 × 8.5 × 20 cm for the two horizontal arms facing each other (left and right goal arms). Two removable guillotine doors permitted manual control of access to these goal arms during the experiment. A third guillotine door, located at the bottom part of the start arm, permitted restriction of the mouse to a specific start area (14 × 8.5 × 20 cm) for 5 s before starting the behavioural testing. Once the guillotine door of the start arm was opened, the evaluation began. During the first trial, a forced choice was imposed by closing the door of one goal arm; left and right goal arms were balanced across treatment groups. Criteria for arm entry was set as ‘full body including the tail’. Subsequent trials were ‘free‐choice’, whereby mice, once returned to the start arm, were free to choose between left and right goal arms. After the mouse had completely entered one of the two goal arms, access to the other goal arm was immediately blocked by closing the corresponding door. The mouse had to go back to the terminal part of the start arm to trigger the opening of the guillotine door and allow the onset of a new choice phase. Mice were allowed 14 trials in 14 min. Mice not completing at least 50% of the trials in 14 min were excluded from the study. After each evaluation, partitions of the T‐maze were carefully cleaned with ethanol (70%) and a paper towel to eliminate the smell and traces of the previous animal. Administration of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2403 (dizocilpine) has previously been shown to impair performance in the spontaneous alternation task (van der Staay et al., 2011). Mice were pretreated with MK‐801 (0.075 or 0.1 mg·kg−1, s.c.) 30 min before the start of testing and received http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=935 (2, 4 or 8 mg·kg−1, s.c.) 15 min or ITI‐214 (0.1–75 mg·kg−1, p.o.) immediately after MK‐801 administration. The percentage of alternation over the 14 free‐choice trials was determined for each mouse and was used as an index of working memory performance.
5‐CSRTT in rats
Thirty‐two operant chambers (Med Associates Inc., St. Albans, VT, USA) were used for 5‐CSRTT training and drug testing as described previously (Isherwood et al., 2015, 2017). Briefly, each chamber consisted of five evenly spaced apertures (2.5 × 2.5 × 4 cm) containing an LED light set into a curved wall at the rear of the chamber. A centrally located food depot was situated on the opposite wall of the chamber, into which 45 mg reward pellets could be delivered (Sandown Scientific, Middlesex, UK). Infrared beams located at the entrance of each aperture and the food depot allowed detection of nose pokes. Task parameters and data collection were controlled by Med Associates Inc. software. Each training session consisted of 100 self‐paced trials and lasted no longer than 30 min. At later training stages, 100 trials were normally completed within 20 min. Training sessions started with the illumination of the house light and depot light and by the delivery of a 45 mg reward pellet (Sandown Scientific). Collection of the reward initiated the first trial. A single trial consisted of an inter‐trial interval (ITI), followed by the pseudo‐random illumination of one of the five apertures for a fixed duration [stimulus duration (SD)]. Following stimulus detection, a nose poke into the corresponding aperture, within a fixed time interval [limited hold (LH)] was required for reward delivery. Premature responses made during the ITI, incorrect responses and responses made outside the LH (an omission) resulted in a timeout (TO) period, during which time no food was delivered and the house light was extinguished for 5 s. Percentage accuracy was defined as the number of correct responses divided by the sum of correct and incorrect responses. Animals were deemed to be trained when they completed ≥50 correct trials with ≥70% accuracy and ≤ 20% omissions (SD 0.7 s; ITI 5 s; LH 5 s). At this stage, perseverative responses (additional responses made to the same aperture following a correct response) resulted in a 5 s TO and loss of food reward. Screening for high‐ and low‐performing rats consisted of 10 challenge sessions where the SD was reduced from 700 to 500 ms. The mean accuracy for each rat across the challenge sessions was calculated. All rats were ranked, based on the mean accuracy, from high to low performance. The upper and lower 15% of rats were deemed high‐performing (n = 9) and low‐performing (n = 9), respectively, and used for subsequent drug testing. SKF38393 (3 or 6 mg·kg−1, administered 10 min before the start of the experiment) testing was performed in a Latin square design with William randomization.
Experimental design and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data were analysed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). Behavioural data from the mouse T‐maze continuous alternation task were analysed using the Kruskal–Wallis non‐parametric one‐way ANOVA followed by Dunn's multiple comparisons test. One sample t‐test versus 50% chance level was also performed. The drug‐induced reversal of MK‐801‐induced memory deficit was calculated by setting the respective response of the saline/vehicle as 100% and that of the group MK‐801/vehicle as 0% reversion. Repeated‐measures ANOVA with a between‐subjects factor was used to compare data derived from the high‐ and low‐performing rats. Where significant main effects were indicated, post hoc analysis using Dunnett's test was performed. For data analysis of the slice recordings, NOTOCORD‐hem software (Croissy‐sur‐Seine, France) was used. Data from slice recordings are shown as mean ± SEM of the percentage of the baseline amplitude. In each experiment, n represents the number of animals. Student's paired t‐test was used for data analysis. Data derived from the second messenger concentration‐determination experiments were analysed using a one‐way ANOVA followed by Dunnett's post hoc test. Statistical significance was set at α = 0.05. All data are shown as mean ± SEM, and statistical group differences are indicated in the figure legends and tables.
Drugs
The http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=75 antagonist, (+)‐MK‐801 hydrogen maleate (Sigma, Germany), was dissolved in saline and stored in daily aliquots at −20°C. On each experimental day, MK‐801 aliquots were defrosted at room temperature and were administered s.c. to mice 30 min prior to testing (mouse T‐maze continuous alternation task) at a dose of 0.075 or 0.1 mg·kg−1. The D1 receptor agonist, (±)‐SKF38393 (Sigma), was freshly dissolved in saline and administered s.c. to mice 15 min prior to the test, or i.p. to rats 10 min prior to the test. The PDE1 inhibitor, ITI‐214 (Li et al., 2016), was synthesized and dissolved in a standard Natrosol solution (0.5% Natrosol in water containing 0.01% Tween‐80) and administered p.o. to mice 30 min before the start of testing. Administration times and routes were based on an internal pharmacokinetic investigation and previous internal studies.
For ex vivo slice recordings, all drugs were initially dissolved in DMSO and diluted further by regular artificial CSF (ACSF) to a final DMSO concentration of 0.05%.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Results
The D1 receptor agonist, SKF38393, reverses MK‐801‐induced memory impairment in the mouse T‐maze continuous alternation task
As shown in Figure 1A, MK‐801 treatment was associated with a significant reduction of spontaneous alternation compared with the performance of vehicle‐injected mice (P < 0.05); moreover, mice showed a mean per cent alternation significantly below chance level (P < 0.05). SKF38393 produced an increase in the alternation performance of MK‐801‐treated mice in the T‐maze continuous alternation task. The recovery in the cognitive performance was 57%, 67% and 43% for SKF38393 2, 4 and 8 mg·kg−1, respectively, suggesting a decline at higher doses. Statistical analysis confirmed that 2 and 4 mg·kg−1 doses of SKF38393 improved the MK‐801‐induced deficits in spontaneous alternation (P < 0.05 value vs. MK‐801; P > 0.05 for both doses vs. vehicle, for SKF38393 2 and 4 mg·kg−1, respectively). By contrast, the highest dose of 8 mg·kg−1 was ineffective in improving MK‐801‐induced deficits in spontaneous alternation (P > 0.05 vs. both MK‐801 and vehicle). Spontaneous alternation was significantly above chance level in mice treated with the dose of 4 mg·kg−1 (P < 0.05 vs. 50%), but not for the doses of 2 and 8 mg·kg−1 SKF38393 (P > 0.05), suggesting not only an amelioration of MK‐801‐induced deficits but also a full restoration of normal working memory. SKF38393 treatment did not change the time spent in the T‐maze when compared with the MK‐801/vehicle‐treated group (P > 0.05, Supporting Information Figure S1A). In summary, these results indicate that SKF38393 reversed MK‐801‐induced cognitive disruption in the mouse T‐maze continuous alternation task with a maximal effect of approximately 67% recovery in the cognitive function of mice.
Figure 1.
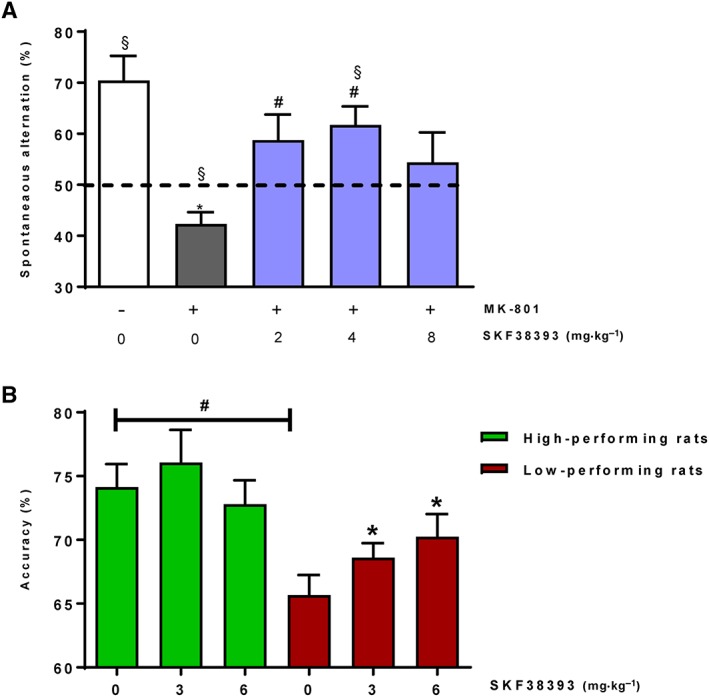
Effects of s.c. administered SKF38393 on reversal of MK‐801‐induced memory impairment in mouse T‐maze continuous alternation task (A) and effects of SKF38393 on accuracy in the 5‐CSRTT in low‐ and high‐performing rats (B). (A) SKF38393 showed a significant reversal of memory impairment induced by the NMDA receptor antagonist MK‐801 (0.075 mg·kg−1). Data are presented as mean ± SEM (all groups n = 13, except the MK801/4 mg·kg−1 SKF38393‐treated group: n = 14); * P < 0.05 versus control group; # P < 0.05 versus MK‐801 treated group; § P < 0.05 versus 50% chance level. For drug effect: Kruskal–Wallis one‐way ANOVA followed by Dunnett's test; for significance versus chance level (indicated by a dashed line), one sample t‐test. (B) SKF38393 dose‐dependently improved accuracy in low‐performing rats. Data are presented as mean ± SEM (n = 9 all groups); # P < 0.05 (high‐ vs. low‐performing rats); * P < 0.05 (vs. vehicle treatment); repeated‐measures ANOVA followed by Dunnett's post hoc testing.
D1 receptor activation by SKF38393 improves attentional performance in low‐performing rats
High‐ and low‐performing rats were selected based on the mean accuracy measured in the 5‐CSRTT. Two‐way repeated‐measures ANOVA with Bonferroni correction indicated that accuracy of vehicle‐treated high‐ and low‐performing rats differed significantly (mean ± SEM accuracy 73.3 ± 1.78 vs. 66.4 ± 1.57, respectively; P < 0.05, Figure 1B). In contrast, there were no significant differences in other task parameters between vehicle‐treated high‐ and low‐performing rats (Table 1). SKF38393 treatment produced a significant dose‐dependent enhancement of accuracy in low‐performing (F (1,13) = 5.51, P < 0.05, Figure 1B) but not high‐performing rats (F (2,15) = 0.96, P > 0.05, Figure 1B). Post hoc testing indicated that SKF38393 treatment significantly increased accuracy of the low‐performing rats at 3 and 6 mg·kg−1 (mean ± SEM accuracy 69.4 ± 0.91 vs. 70.89 ± 1.89, respectively; P < 0.05). SKF38393 slightly increased reward and correct response latencies and reduced premature responses but had no effect on perseverative responses (Table 1). In contrast to the accuracy performance, in which SKF38393 only increased accuracy in low‐performing rats, these effects were largely consistent between both groups of rats (Table 1).
Table 1.
Effect of the D1 receptor agonist, SKF38393, on multiple task parameters of the 5‐CSRTT in high‐ and low‐performing rats
| Dose (mg·kg−1) | High‐performing rats | Low‐performing rats | ||||
|---|---|---|---|---|---|---|
| 0 | 3 | 6 | 0 | 3 | 6 | |
| Correct latency (s) | 0.69 ± 0.02 | 0.79 ± 0.02* | 0.83 ± 0.03* | 0.72 ± 0.03 | 0.77 ± 0.02 | 0.85 ± 0.03* |
| Reward latency (s) | 1.06 ± 0.05 | 1.24 ± 0.06* | 1.27 ± 0.06* | 1.07 ± 0.04 | 1.21 ± 0.04* | 1.31 ± 0.06* |
| Omissions (n) | 4.11 ± 1.01 | 11.24 ± 2.64* | 21.65 ± 2.98* | 5.11 ± 1.10 | 10.61 ± 1.70* | 17.74 ± 2.58* |
| Premature responses (%) | 7.28 ± 1.85 | 3.17 ± 0.72 | 1.97 ± 0.69* | 10 ± 1.59 | 3.06 ± 0.27* | 2.15 ± 0.49* |
| Perseverative responses (n) | 0.78 ± 0.28 | 0.94 ± 0.26 | 1.06 ± 0.27 | 1.22 ± 0.34 | 0.83 ± 0.26 | 0.67 ± 0.20 |
Data are presented as mean ± SEM (n = 9 all groups).
P < 0.05 (vs. vehicle treatment); repeated‐measures ANOVA followed by Dunnett's post hoc testing. Note that vehicle‐treated high‐ and low‐performing rats did not differ in any parameter.
PDE1B and D1 receptor are co‐expressed in rat and human prefrontal cortex
To verify cellular co‐expression of PDE1B with the D1 receptor in brain tissue, double immunohistochemistry was used. D1 receptor expression in the rat PFC was evenly distributed across the different subregions and across all layers (Figure 2). Strong D1 receptor expression could also be found in the striatum (not shown). PDE1B proved to have a similar corticostriatal expression profile, and thorough analysis of the PFC indicated that the majority of PDE1B‐positive neurons co‐expressed the D1 receptor (Figure 2). In human prefrontal brain sections, evaluation of D1 receptor expression indicated a comparable expression profile to that determined in the rat, with most PDE1B‐positive neurons also being positive for the D1 receptor expression (Figure 2).
Figure 2.
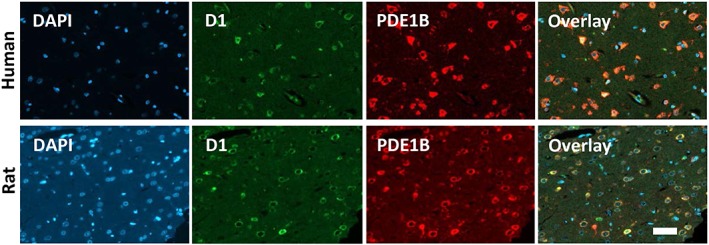
Double fluorescence labelling of the D1 receptor (D1) and PDE1B in the human (upper panel; 20× magnification) and rat PFC (lower panel, 20× magnification, border zone between infralimbic and prelimbic cortex). In both species, the prefrontal cortical layers showed a comparable staining pattern for both D1 receptors (green) and PDE1B (red). Merged images (overlay) indicate that the minority of neurons stained for only one marker. The vast majority of neurons of human and rat PFC co‐expressed D1 receptors and PDE1B (yellow). Blue colour indicates the DAPI staining of the cell nuclei. Representative images from processed brains of rats (n = 3) and human donors (n = 5). Bar graph = 50 μm.
PDE1 inhibition by ITI‐214 leads to increases in cGMP and cAMP concentrations in the mouse prefrontal cortex
ITI‐214 proved to be a very potent inhibitor of all three PDE1 isoforms (i.e. picomolar potency) with a negligible preference for PDE1A over PDE1B and PDE1C, and a high selectivity for PDE1 over other major PDE isoforms (Table 2), confirming previous reports (Snyder et al., 2016). ITI‐214 administration in mice rapidly increased PFC cGMP (F (3,27) = 6.54, P < 0.05; Figure 3A) and cAMP (F (3,27)= 5.35, P < 0.05; Figure 3B). Post hoc analysis indicated a significant increase at the highest dose tested (P < 0.05 for both, cGMP and cAMP), confirming the pharmacological activity in the brain area of interest.
Table 2.
Inhibition of PDE1A, 1B, 1C, 4A, 4B, 5A, 6, 7A and 10A by ITI‐214 as determined using human enzymes with radiometric assay for measuring cGMP or cAMP
| PDE | cXMP | IC50 |
|---|---|---|
| 1A | cGMP | 31 pM |
| 1B | cGMP | 75 pM |
| 1C | cGMP | 53 pM |
| 4A | cAMP | >100 nM |
| 4B | cAMP | >100 nM |
| 5A | cGMP | >100 nM |
| 6 | cGMP | >100 nM |
| 7 | cAMP | >100 nM |
| 10A | cAMP | >100 nM |
Figure 3.
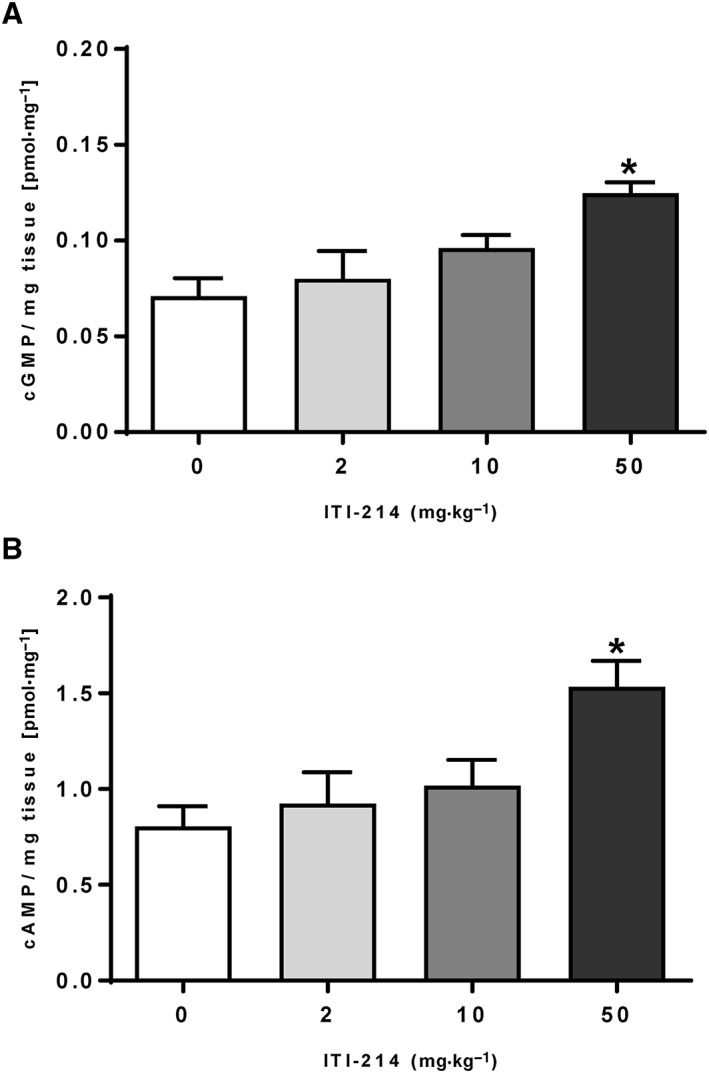
Inhibition of PDE1 by ITI‐214 significantly increases (A) cGMP and (B) cAMP in the PFC of mice. Data are presented as mean ± SEM (n = 8 all groups); * P < 0.05versus vehicle, one‐way ANOVA followed by Dunnett's post hoc test.
PDE1 inhibition by ITI‐214 reverses MK‐801‐induced memory impairment in the mouse T‐maze continuous alternation task
As shown in Figure 4, MK‐801 was associated with significant reduction of spontaneous alternation compared with the performance of vehicle‐injected mice (approximate 23% reduction, P < 0.05). ITI‐214 produced an increase in the alternation performance of MK‐801‐treated mice in the T‐maze continuous alternation task. The change in alternation of MK‐801‐treated mice was 0%, 13%, 12%, 10%, 7% and 2% for the doses of 0.1, 1, 3, 10, 30 and 75 mg·kg−1 of ITI‐214 respectively. The recovery in cognitive performance was 1%, 57%, 51%, 45%, 32% and 11% for the same doses of ITI‐214 respectively. The effect was statistically significant for the 1 mg·kg−1 dose (P < 0.05vs. MK‐801; P > 0.05 vs. vehicle), and there was a trend towards a significant effect at 3 mg·kg−1 (P = 0.059 vs. MK‐801; P > 0.05 vs. vehicle) of ITI‐214. Notably, both groups of mice treated with 1 and 3 mg·kg−1 doses showed a spontaneous alternation significantly above chance level (P < 0.05 vs. 50% chance), suggesting a full restoration of normal cognitive abilities. There was a progressive decline in the effect of ITI‐214 as the doses increased from 10 mg·kg−1; the effect was significantly reduced, if not abolished, at 75 mg·kg−1 compared with that observed at 1 and 3 mg·kg−1. ITI‐214 treatment did not change time spent in the T‐maze when compared with the MK‐801/vehicle‐treated group (P > 0.05, Supporting Information Figure S1B). In summary, the results indicate that ITI‐214 reversed MK‐801‐induced cognitive disruption in the mouse T‐maze continuous alternation task with a maximal effect of approximately 57% recovery in the cognitive function of mice.
Figure 4.
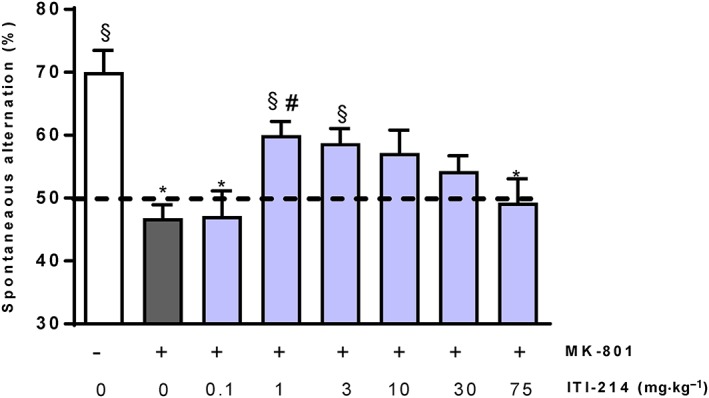
Effects of p.o. ITI‐214 on the reversal of MK‐801‐induced memory impairments in mouse T‐maze continuous alternation task. ITI‐214 significantly reversed the memory impairments induced by the NMDA receptor antagonist MK‐801 (0.1 mg·kg−1). Data are presented as mean ± SEM (n = 10 all groups, except MK‐801/vehicle‐treated group and MK‐801/3 mg·kg−1 ITI‐214‐treated group: n = 9); * P < 0.05 versus control group; # P < 0.05 and versus MK‐801 treated group; § P < 0.05 versus 50% chance level. For drug effect: Kruskal–Wallis one‐way ANOVA followed by Dunnett's test; for significance versus chance level, one sample t‐test.
PDE1 inhibition by ITI‐214 modulates synaptic transmission in the rat prefrontal cortex
Mechanistic studies aiming to improve the understanding of the mode of action of ITI‐214 were conducted by studying synaptic transmission in acute slices of the rat PFC. Because the D1 receptor is coupled to Gαs, the adenylyl cyclase activator http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5190 was used and demonstrated an increase in fEPSP amplitude in a concentration‐dependent manner (Figure 5A). PDE1 inhibition by ITI‐214 did not have an effect on fEPSP amplitude on its own (Figure 5B). The Ca2+/calmodulin complex was activated by increasing the Ca2+ in the cytosol through administration of the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=159) inhibitor, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5351, which is required to activate PDE1 (Bender and Beavo, 2006). Activation of PDE1 by the Ca2+/calmodulin complex reduced the forskolin‐induced fEPSP amplitude (Figure 5C). ITI‐214 then restored this forskolin‐induced fEPSP amplitude in the presence of activated PDE1 (Figure 5C). In comparison, the D1 receptor agonist SKF38393 increased synaptic transmission, as demonstrated by increased fEPSP amplitude (Figure 5D), as expected.
Figure 5.
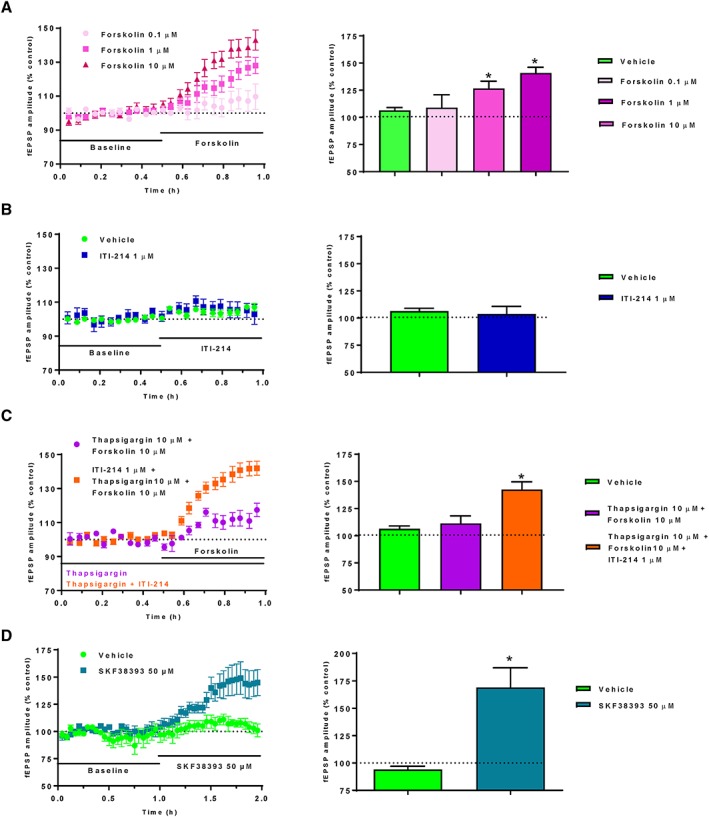
Effect of cAMP‐modulating compounds on synaptic transmission in acute slices of the rat PFC. (A) The adenylyl cyclase activator forskolin increased fEPSPs concentration‐dependently (vehicle group n = 4; 0.1 μM group: n = 3; 1 μM group: n = 4; 10 μM group: n = 14). (B) The PDE1 inhibitor ITI‐214 (n = 5) did not show any effects per se (vehicle: n = 4), but (C) rescued the reduced fEPSPs amplitudes elicited by forskolin when PDE1 was activated by the Ca2+/calmodulin complex (via a Ca2+ increase in the cytosol mediated by the SERCA inhibitor thapsigargin) (vehicle group: n = 4; thapsigargin group: n = 3; thapsigargin + ITI‐214 group: n = 4). (D) The D1 receptor agonist SKF38393 increased synaptic transmission, presumably via intracellular cAMP increase through D1 receptor activation (vehicle group: n = 5; SKF38393 group: n = 7). Bar charts represent average drug‐induced changes in the fEPSP amplitude in relation to the baseline fEPSP responses of the last 10 min of drug administration.
Discussion
Insufficient prefrontal D1 receptor activation caused by dopamine hypofunction is hypothesized to underlie deficits in working memory and attention. The current findings support our hypothesis that the PDE1 family may potentially be key enzymes in the intracellular D1 receptor pathway and could possibly be involved in terminating D1 receptor‐linked downstream signalling events. Moreover, our data may indicate a mechanistic link between the D1 receptor and PDE1 in cognitive processes, which in turn may support the use of PDE1 inhibitors as a potential therapeutic approach for the treatment of cognitive dysfunction in psychiatric diseases characterized by prefrontal dopamine hypofunction. We also hypothesize, based on our localization studies, that PDE1B may be the PDE1 isoform predominantly responsible for these effects.
The importance of dopamine for cognition was discovered almost 30 years ago in primates, whilst subsequent work indicated that insufficient prefrontal D1 (but not D2) receptor function may be linked to working memory impairments (reviewed in Goldman‐Rakic et al., 2000). Since then, reproduction and extension of these results has led to the hypothesis of an inverted U‐shaped function relating cognitive performance to D1 receptor stimulation. Consequently, it was shown that moderate fostering of prefrontal D1 receptor function can improve cognitive processes in aged animals or in pharmacologically induced (cholinergic) models of cognitive impairment (Amico et al., 2007; Mizoguchi et al., 2009). We further extend these findings and demonstrate the efficacy of SKF38393 in reversing working memory impairments induced by the NMDA receptor antagonist, MK‐801, in the mouse T‐maze continuous alternation task. This is an important finding, as glutamatergic dysfunction is more closely related to schizophrenia than cholinergic dysfunction. Thus, the current data are compatible with a hypothesized mechanism of dopamine‐related augmentation of depolarizing synaptic inputs to prefrontal pyramidal cells, including those mediated by the NMDA receptor.
Attentional performance is also known to be modulated by ascending monoaminergic systems, especially dopamine (Robbins et al., 1993). Attentional deficits contribute significantly to functional disability of patients with schizophrenia among other patient populations (Green et al., 2004; Marder and Fenton, 2004). In a previous investigation, pro‐attentive properties of SKF38393 were determined in rats with phencyclidine‐altered attentional performance (Barnes et al., 2016). More akin to our study design and attentional behavioural assay, a previous study determined the effect of local infusions of SKF38393 on attentional performance in rats selected for high and low 5‐CSRTT performance (Granon et al., 2000). Interestingly, SKF38393 effects were baseline‐dependent; an improvement in attentional performance was seen in low‐ but not high‐performing rats, an effect largely replicated in our study following systemic administration of SKF38393. It is conceivable that no further improvement in attentional performance could be detected in high‐performing rats owing to a ceiling effect, or potentially D1 receptor engagement is already close to optimum. Regardless, the data suggest that D1 receptor activation was insufficient in low‐performing animals.
The role of prefrontal D1 receptors in cognitive processes is further supported by data from patients with schizophrenia and aged subjects. Post‐mortem tissue analysis from the former has previously indicated reduced prefrontal dopaminergic innervation (Akil et al., 1999). Furthermore, aged subjects with working memory deficits showed reduced prefrontal dopaminergic neurotransmitter tonus (Ota et al., 2006). A recently described genetic link between a functional D1 receptor gene polymorphism and cognition indicates that reduced D1 receptor expression impairs cognition in both patients with psychiatric disorders and healthy volunteers (Tsang et al., 2015). Furthermore, a study of schizotypal personality disorder indicated that the D1 receptor agonist dihydrexidine improved working memory (Rosell et al., 2015). However, the poor pharmacokinetic characteristics (reviewed by Arnsten et al., 2017) and the known cardiovascular effects of D1 receptor agonists (e.g. severe hypotension) limit their clinical use. Therefore, approaches targeting psychiatric conditions characterized by dopaminergic hypofrontality whilst avoiding pitfalls of direct D1 receptor agonism are highly attractive.
D1 receptors are Gαs‐coupled and thus activate adenylyl cyclase, increasing cAMP synthesis. PDE1 enzymes hydrolyse cAMP and cGMP, and PDE1B is highly expressed in corticostriatal areas, where a role in D1 receptor signalling is suggested (Duinen et al., 2015; Li et al., 2016; Snyder et al., 2016). Among the PDE1 subtypes, PDE1C shows the strongest gene expression in the cerebellum and striatum but is absent in cortical areas (Yan et al., 1996), whereas PDE1A shows a discrete cortical expression pattern (Sonnenburg et al., 1993). However, gene and protein expression of PDE1B occurs in brain structures known to be D1 receptor‐enriched, such as the striatum and PFC (Polli and Kincaid, 1994; Yan et al., 1994). Furthermore, PDE1A has a much higher affinity for cGMP than cAMP (Ramirez and Smith, 2014), and we therefore concluded that PDE1B might be linked to D1 receptor function. Indeed, immunohistochemistry suggested both a corticostriatal PDE1B expression profile in rat brains and PDE1‐D1 receptor co‐localization in human and rat prefrontal neurons, suggesting the putative involvement of PDE1B in D1 receptor‐dependent downstream signalling. Within the PFC, D1 receptors have been shown to be expressed on distal dendrites and spines of pyramidal cells and GABAergic interneurons, preferentially parvalbumin‐containing basket and chandelier cells (Goldman‐Rakic et al., 2000). Given the almost complete overlap between PDE1B in D1 receptor expression in the PFC seen in this study, the data might indicate that PDE1B is expressed by both pyramidal cells as well as interneurons. However, in‐depth ultrastructural analyses and additional light microscopy studies need to determine the cellular identity and localization of neuronal PDE1B expression within the PFC. Further evidence for a putative involvement of PDE1B in D1 receptor‐dependent downstream signalling comes also from a study showing overshooting phosphorylation of downstream substrates after D1 receptor agonism in brain slices from PDE1B‐knockout mice versus wild‐type mice (Reed et al., 2002). Although a role for PDE1A in D1 receptor‐mediated downstream signalling is not excluded, it supports the hypothesis that PDE1B has a role in terminating intracellular downstream effects of D1 receptor activation. Using the potent and selective PDE1 inhibitor, ITI‐214, (Li et al., 2016, Snyder et al., 2016) in naïve mice resulted in a dose‐dependent increase in prefrontal cAMP levels, confirming the relevance of PDE1 enzymes for the regulation of prefrontal cAMP. It is noteworthy that this kind of in vivo and/or ex vivo assay is not suitable for predicting effective doses in behavioural cognition tasks, due to issues such as dilution effects during tissue homogenization and assay processing. Therefore, it is perhaps not surprising that higher doses of ITI‐214 were required to increase second messenger levels in prefrontal tissue than those found to be efficacious in the behavioural assay. This is consistent with data on other PDE inhibitors, as previously reported by us and others (Verhoest et al., 2009; Kroker et al., 2014). Nevertheless, the assay used can demonstrate pharmacological activity of PDE inhibitors (Kleiman et al., 2012; Kroker et al., 2014) such as ITI‐214, in relevant brain areas.
One main finding of this study is the pro‐cognitive effect of PDE1 inhibition, that is, that ITI‐214 reverses MK‐801‐induced working memory impairments in the T‐maze continuous alternation task, in line with a recent report demonstrating that ITI‐214 enhances acquisition, consolidation and retrieval memory processes in the novel object recognition task (Snyder et al., 2016; Dyck et al., 2017). In the present study, the response to different doses of ITI‐214 suggests an inverted U‐shaped dose‐response, an observation also made by Snyder et al. (2016), consistent with the idea that optimal cognitive performance requires optimal engagement of prefrontal dopamine signal transduction (Goldman‐Rakic et al., 2000). Together, these data support PDE1‐linked degradation of cAMP which can be formed upon D1 receptor activation and, thus, a potential D1 receptor agonistic‐like effect of PDE1 inhibitors. It is perhaps therefore not surprising that ITI‐214 and SKF38393 produced qualitatively comparable effects in improving working memory impairments. However, given that PDE1s are dual‐specific, it cannot be excluded that cGMP elevation contributes to its efficacy. In fact, cGMP is an important intracellular messenger for synaptic plasticity and memory function in rodents, and we previously demonstrated that cGMP elevation by inhibition of the cGMP‐specific isoform PDE9 increased hippocampal long‐term potentiation and exhibited pro‐cognitive effects in rodents (Kroker et al., 2014). Therefore, both cGMP and cAMP elevation by PDE1 inhibition may contribute to restoring MK‐801‐mediated working memory impairments.
The distinguishing feature of the PDE1 family versus the other 11 PDE families is regulation by Ca2+/calmodulin, binding of which stimulates cyclic nucleotide hydrolysis (Bender and Beavo, 2006; Ramirez and Smith, 2014). Considering the Ca2+/calmodulin sensitivity of PDE1, it was expected that ITI‐214 would only have an effect after increasing cytosolic Ca2+ through the SERCA inhibitor thapsigargin. In this setting, Ca2+‐activated PDE1 reduced forskolin‐induced cAMP levels and thus decreased synaptic potential amplitude, whilst PDE1 inhibition with ITI‐214 rescued the forskolin‐induced fEPSP amplitude. The effect of ITI‐214 on fEPSP amplitude was comparable to the increase achieved by SKF38393, mimicking effects previously reported by Huang and Kandel (1995). The results suggest that the increase in fEPSP amplitude by ITI‐214 is promoted via a D1 receptor/cAMP‐mediated pathway, dependent on elevated intracellular Ca2+. This highlights a unique, activity‐dependent quality of the PDE1 enzyme that may provide tight coupling of this substrate to neuronal activity, setting this PDE family apart from other enzyme families implicated in enhancing cognitive performance (e.g. PDE4 and PDE10). However, due to methodological limitations, we were not able to directly test the proposed link between PDE1 and the D1 receptor/cAMP pathway, since even low concentrations of a D1 receptor antagonist induced strong disinhibition and epileptiform discharges in the slices, which made the recording of fEPSPs impossible.
A number of previous behavioural and neurochemical studies probed the efficacy of SKF38393 and other D1 receptor agonists following behavioural alteration induced by the D1 receptor antagonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=943 (Imperato et al., 1987; Granon et al., 2000; Bueno‐Nava et al., 2012; Avila‐Luna et al., 2018a,b), thereby strengthening the D1 receptor dependency of SKF38393's effects. Future studies should use such interaction studies and employ D1 receptor antagonist‐induced working memory or attentional deficits as a paradigm to probe the efficacy of both D1 receptor agonists and PDE1 inhibitors to confirm the mechanistic PDE1‐D1 receptor link and the D1 receptor agonistic‐like pharmacological profile of PDE1 inhibitors. Additional mechanistic studies will be required to further elucidate the extent ITI‐214 treatment can restore NMDA versus D1 receptor associated cognitive impairments and to further dissect the relative contribution of prefrontal cGMP versus cAMP elevation to its efficacy. Looking forward, selective PDE1B inhibitors will be required to confirm the specific relevance of PDE1B for D1 receptor‐linked cognitive processes.
We demonstrated that SKF38393 improves working memory and attentional performance in rodents and increases synaptic transmission in the PFC. Immunohistochemistry revealed potential PDE1‐D1 receptor co‐localization in rodent and human PFC. ITI‐214 proved to be a potent PDE1 inhibitor, which increased prefrontal cAMP/cGMP and reversed MK‐801‐induced working memory, potentially by increasing prefrontal synaptic plasticity. In summary, our data suggest that a mechanistic PDE1‐D1 receptor link may be conceivable and also indicate that PDE1 could potentially regulate D1 receptor signalling by modulating cAMP levels, highlighting the role of a putative PDE1‐D1 receptor pathway in cognitive processes and indicating that PDE1 inhibitors have a D1 receptor agonistic‐like pharmacological profile. This supports the use of PDE1 inhibitors as a potential approach for the treatment of cognitive dysfunction in psychiatric conditions characterized by prefrontal dopamine hypofunction.
Author contributions
A.P., H.R. and N.S. designed the studies and/or conducted most of the experiments, analysed and interpreted the data. A.P. wrote in addition the manuscript. C.D.C. contributed to writing by critical revision. B.S. was responsible for the immunohistochemistry and image acquisition, and S.D. performed the T‐maze studies with SKF38393, analysed and interpreted the relative data and contributed to writing of respective manuscript sections. All authors read and approved the manuscript.
Conflict of interests
All authors are employees of Boehringer Ingelheim Pharma GmbH & Co. KG.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effect of SKF38393 (a) and ITI‐214 (b) on time spent in T‐Maze. (a) MK‐801 treatment reduced the overall time spent to complete the T‐maze task significantly versus control group, but SKF38393 treatment at any dose tested did not change this measure further. (b) MK‐801 treatment reduced the overall time spent to complete the T‐maze task significantly versus control group, but ITI‐214 treatment at any dose tested did not change this measure further. Data are presented as mean ± SEM, n‐size per group as for Figure 1a and Figure 5. * P < 0.05 mean effect of MK‐801 versus control group.
Acknowledgements
We thank Dr. G. Birk and Dr. E. Adriambeloson for support of confocal microscopy and T‐maze studies, respectively, as well as N. Kötteritzsch, A. Blasius, M.‐T. Trinz and P. Schorn for excellent technical assistance. Language revision and editing provided by Heather Shawcross, PhD, and Michelle Marvel of Fishawack Communications, UK, which was funded by Boehringer Ingelheim.
This research did not receive any specific grant from funding agencies in the public, or not‐for‐profit sectors, but was supported in full by Boehringer Ingelheim Pharma GmbH & Co. KG, Div. Research Germany, Birkendorf Strasse 65, 88397, Biberach an der Riss, Germany.
Pekcec A., Schülert N., Stierstorfer B., Deiana S., Dorner‐Ciossek C., and Rosenbrock H. (2018) Targeting the dopamine D1 receptor or its downstream signalling by inhibiting phosphodiesterase‐1 improves cognitive performance, British Journal of Pharmacology, 175, 3021–3033, https://doi.org/10.1111/bph.14350.
References
- Abi‐Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y et al (2002). Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci 22: 3708–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR et al (1999). Lamina‐specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry 156: 1580–1589. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The concise guide to PHARMACOLOGY 2017/2018: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The concise guide to PHARMACOLOGY 2017/2018: enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E (2017d). The concise guide to PHARMACOLOGY 2017/2018: voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amico F, Spowart‐Manning L, Anwyl R, Rowan MJ (2007). Performance‐ and task‐dependent effects of the dopamine D1/D5 receptor agonist SKF 38393 on learning and memory in the rat. Eur J Pharmacol 577: 71–77. [DOI] [PubMed] [Google Scholar]
- Arnsten AF, Girgis RR, Gray DL, Mailman RB (2017). Novel dopamine therapeutics for cognitive deficits in schizophrenia. Biol Psychiatry 81: 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila‐Luna A, Galvez‐Rosas A, Alfaro‐Rodriguez A, Reyes‐Legorreta C, Garza‐Montano P, Gonzalez‐Pina R et al (2018a). Dopamine D1 receptor activation maintains motor coordination in injured rats but does not accelerate the recovery of the motor coordination deficit. Behav Brain Res 336: 145–150. [DOI] [PubMed] [Google Scholar]
- Avila‐Luna A, Galvez‐Rosas A, Durand‐Rivera A, Ramos‐Languren LE, Rios C, Arias‐Montano JA et al (2018b). Dopamine D1 receptor activation maintains motor coordination and balance in rats. Metab Brain Dis 33: 99–105. [DOI] [PubMed] [Google Scholar]
- Barnes SA, Young JW, Bate ST, Neill JC (2016). Dopamine D1 receptor activation improves PCP‐induced performance disruption in the 5C‐CPT by reducing inappropriate responding. Behav Brain Res 300: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AT, Beavo JA (2006). Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58: 488–520. [DOI] [PubMed] [Google Scholar]
- Brozoski TJ, Brown RM, Rosvold HE, Goldman PS (1979). Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science 205: 929–932. [DOI] [PubMed] [Google Scholar]
- Bueno‐Nava A, Gonzalez‐Pina R, Alfaro‐Rodriguez A, Avila‐Luna A, Arch‐Tirado E, Alonso‐Spilsbury M (2012). The selective inhibition of the D(1) dopamine receptor results in an increase of metabolized dopamine in the rat striatum. Neurochem Res 37: 1783–1789. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delotterie DF, Mathis C, Cassel JC, Rosenbrock H, Dorner‐Ciossek C, Marti A (2015). Touchscreen tasks in mice to demonstrate differences between hippocampal and striatal functions. Neurobiol Learn Mem 120: 16–27. [DOI] [PubMed] [Google Scholar]
- Duinen MV, Reneerkens OA, Lambrecht L, Sambeth A, Rutten BP, Os JV et al (2015). Treatment of cognitive impairment in schizophrenia: potential value of phosphodiesterase inhibitors in prefrontal dysfunction. Curr Pharm Des 21: 3813–3828. [DOI] [PubMed] [Google Scholar]
- Dyck B, Branstetter B, Gharbaoui T, Hudson AR, Breitenbucher JG, Gomez L et al (2017). Discovery of selective phosphodiesterase 1 inhibitors with memory enhancing properties. J Med Chem 60: 3472–3483. [DOI] [PubMed] [Google Scholar]
- Gold JM, Hahn B, Zhang WW, Robinson BM, Kappenman ES, Beck VM et al (2010). Reduced capacity but spared precision and maintenance of working memory representations in schizophrenia. Arch Gen Psychiatry 67: 570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman‐Rakic PS, Muly EC 3rd, Williams GV (2000). D(1) receptors in prefrontal cells and circuits. Brain Res Brain Res Rev 31: 295–301. [DOI] [PubMed] [Google Scholar]
- Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW (2000). Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into rat prefrontal cortex. J Neurosci 20: 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MF, Kern RS, Heaton RK (2004). Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr Res 72: 41–51. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (1995). D1/D5 receptor agonists induce a protein synthesis‐dependent late potentiation in the CA1 region of the hippocampus. Proc Natl Acad Sci U S A 92: 2446–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperato A, Mulas A, Di Chiara G (1987). The D‐1 antagonist SCH 23390 stimulates while the D‐1 agonist SKF 38393 fails to affect dopamine release in the dorsal caudate of freely moving rats. Eur J Pharmacol 142: 177–181. [DOI] [PubMed] [Google Scholar]
- Isherwood SN, Pekcec A, Nicholson JR, Robbins TW, Dalley JW (2015). Dissociable effects of mGluR5 allosteric modulation on distinct forms of impulsivity in rats: interaction with NMDA receptor antagonism. Psychopharmacology (Berl) 232: 3327–3344. [DOI] [PubMed] [Google Scholar]
- Isherwood SN, Robbins TW, Nicholson JR, Dalley JW, Pekcec A (2017). Selective and interactive effects of D2 receptor antagonism and positive allosteric mGluR4 modulation on waiting impulsivity. Neuropharmacology 123: 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010). Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman RJ, Chapin DS, Christoffersen C, Freeman J, Fonseca KR, Geoghegan KF et al (2012). Phosphodiesterase 9A regulates central cGMP and modulates responses to cholinergic and monoaminergic perturbation in vivo. J Pharmacol Exp Ther 341: 396–409. [DOI] [PubMed] [Google Scholar]
- Kroker KS, Rosenbrock H, Rast G (2011). A multi‐slice recording system for stable late phase hippocampal long‐term potentiation experiments. J Neurosci Methods 194: 394–401. [DOI] [PubMed] [Google Scholar]
- Kroker KS, Mathis C, Marti A, Cassel JC, Rosenbrock H, Dorner‐Ciossek C (2014). PDE9A inhibition rescues amyloid beta‐induced deficits in synaptic plasticity and cognition. Neurobiol Aging 35: 2072–2078. [DOI] [PubMed] [Google Scholar]
- Lea WA, Simeonov A (2011). Fluorescence polarization assays in small molecule screening. Expert Opin Drug Discov 6: 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Zheng H, Zhao J, Zhang L, Yao W, Zhu H et al (2016). Discovery of potent and selective inhibitors of phosphodiesterase 1 for the treatment of cognitive impairment associated with neurodegenerative and neuropsychiatric diseases. J Med Chem 59: 1149–1164. [DOI] [PubMed] [Google Scholar]
- Marder SR, Fenton W (2004). Measurement and treatment research to improve cognition in schizophrenia: NIMH MATRICS initiative to support the development of agents for improving cognition in schizophrenia. Schizophr Res 72: 5–9. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi K, Shoji H, Tanaka Y, Maruyama W, Tabira T (2009). Age‐related spatial working memory impairment is caused by prefrontal cortical dopaminergic dysfunction in rats. Neuroscience 162: 1192–1201. [DOI] [PubMed] [Google Scholar]
- Muller U, Von Cramon DY, Pollmann S (1998). D1‐ versus D2‐receptor modulation of visuospatial working memory in humans. J Neurosci 18: 2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota M, Yasuno F, Ito H, Seki C, Nozaki S, Asada T et al (2006). Age‐related decline of dopamine synthesis in the living human brain measured by positron emission tomography with L‐[beta‐11C]DOPA. Life Sci 79: 730–736. [DOI] [PubMed] [Google Scholar]
- Polli JW, Kincaid RL (1994). Expression of a calmodulin‐dependent phosphodiesterase isoform (PDE1B1) correlates with brain regions having extensive dopaminergic innervation. J Neurosci 14: 1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez AD, Smith SM (2014). Regulation of dopamine signaling in the striatum by phosphodiesterase inhibitors: novel therapeutics to treat neurological and psychiatric disorders. Cent Nerv Syst Agents Med Chem 14: 72–82. [DOI] [PubMed] [Google Scholar]
- Reed TM, Repaske DR, Snyder GL, Greengard P, Vorhees CV (2002). Phosphodiesterase 1B knock‐out mice exhibit exaggerated locomotor hyperactivity and DARPP‐32 phosphorylation in response to dopamine agonists and display impaired spatial learning. J Neurosci 22: 5188–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW, Roberts AC, Owen AM, Sahakian BJ, Everitt BJ, Wilkinson LS et al (1993). Monoaminergic dependent cognitive functions of the prefrontal cortex in monkey and man In: Christen Y. (ed). Prefrontal Cortext. Springer: Heidelberg. [Google Scholar]
- Rosell DR, Zaluda LC, Mcclure MM, Perez‐Rodriguez MM, Strike KS, Barch DM et al (2015). Effects of the D1 dopamine receptor agonist dihydrexidine (DAR‐0100A) on working memory in schizotypal personality disorder. Neuropsychopharmacology 40: 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaguchi T, Goldman‐Rakic PS (1991). D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science 251: 947–950. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Prickaerts J, Wadenberg ML, Zhang L, Zheng H, Yao W et al (2016). Preclinical profile of ITI‐214, an inhibitor of phosphodiesterase 1, for enhancement of memory performance in rats. Psychopharmacology (Berl) 233: 3113–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg WK, Seger D, Beavo JA (1993). Molecular cloning of a cDNA encoding the “61‐kDa” calmodulin‐stimulated cyclic nucleotide phosphodiesterase. Tissue‐specific expression of structurally related isoforms. J Biol Chem 268: 645–652. [PubMed] [Google Scholar]
- Thompson WJ, Appleman MM (1971). Multiple cyclic nucleotide phosphodiesterase activities from rat brain. Biochemistry 10: 311–316. [PubMed] [Google Scholar]
- Thompson JL, Rosell DR, Slifstein M, Girgis RR, Xu X, Ehrlich Y et al (2014). Prefrontal dopamine D1 receptors and working memory in schizotypal personality disorder: a PET study with [(1)(1)C]NNC112. Psychopharmacology (Berl) 231: 4231–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang J, Fullard JF, Giakoumaki SG, Katsel P, Katsel P, Karagiorga VE et al (2015). The relationship between dopamine receptor D1 and cognitive performance. NPJ Schizophr 1: 14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Staay FJ, Rutten K, Erb C, Blokland A (2011). Effects of the cognition impairer MK‐801 on learning and memory in mice and rats. Behav Brain Res 220: 215–229. [DOI] [PubMed] [Google Scholar]
- Verhoest PR, Proulx‐Lafrance C, Corman M, Chenard L, Helal CJ, Hou X et al (2009). Identification of a brain penetrant PDE9A inhibitor utilizing prospective design and chemical enablement as a rapid lead optimization strategy. J Med Chem 52: 7946–7949. [DOI] [PubMed] [Google Scholar]
- Yan C, Bentley JK, Sonnenburg WK, Beavo JA (1994). Differential expression of the 61 kDa and 63 kDa calmodulin‐dependent phosphodiesterases in the mouse brain. J Neurosci 14: 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Zhao AZ, Bentley JK, Beavo JA (1996). The calmodulin‐dependent phosphodiesterase gene PDE1C encodes several functionally different splice variants in a tissue‐specific manner. J Biol Chem 271: 25699–25706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of SKF38393 (a) and ITI‐214 (b) on time spent in T‐Maze. (a) MK‐801 treatment reduced the overall time spent to complete the T‐maze task significantly versus control group, but SKF38393 treatment at any dose tested did not change this measure further. (b) MK‐801 treatment reduced the overall time spent to complete the T‐maze task significantly versus control group, but ITI‐214 treatment at any dose tested did not change this measure further. Data are presented as mean ± SEM, n‐size per group as for Figure 1a and Figure 5. * P < 0.05 mean effect of MK‐801 versus control group.