

Abstract
The opioid receptors are key targets in the treatment of acute and chronic pain, and the development of novel analgesics with reduced side effects is crucial in the search for more effective medications. The crystal structures of opioid receptors have provided a wealth of knowledge on many aspects of opioid receptor pharmacology and function, including ligand binding poses, location of the sodium allosteric binding site, conformational changes associated with activation and putative dimeric interfaces. These crystal structures also offer a starting point for molecular dynamics (MD) simulations to capture one aspect of drug design that static structures cannot resolve, namely protein dynamics. With the increase in computing power, MD simulations of crystal structures have become an influential tool in understanding the function of GPCRs in general. Here, we discuss lessons learned from MD simulations of opioid receptor crystal structures with reference to (i) the binding pathway of sodium to its crystallographic allosteric site, (ii) the dynamics of ligand–receptor and receptor–receptor interactions, both at the ligand‐ and G protein‐binding sites, (iii) the binding pathway and binding pose of novel ligands, and (iv) opioid receptor oligomerization.
Linked Articles
This article is part of a themed section on Emerging Areas of Opioid Pharmacology. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.14/issuetoc
Abbreviations
- CADD
computer‐aided drug design
- CG
coarse‐grained
- EL
extracellular loop
- MD
molecular dynamics
- NOP receptor
nociceptin opioid receptor
- PAM
positive allosteric modulator
- PDB
Protein Data Bank
- POPC
1‐palmitoyl,2‐oleoyl‐sn‐glycero‐3‐phosphocholine
- RMSD
root‐mean‐square deviation
- TM
transmembrane
Introduction
http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=50 are an important subfamily of GPCRs as they are among the main protein targets in the treatment of acute and chronic pain (Stein, 2016). Since classical opioid drugs are associated with a number of undesirable side effects, there is great interest in understanding fundamental aspects of opioid receptor function (e.g. allosteric modulation, functional selectivity and oligomerization), which may aid in the development of novel therapies.
The opioid receptor family is composed of three main subtypes: http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=317, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=318 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319. While a fourth opioid receptor subtype, the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=320 (NOP receptor), shares a high‐sequence identity with these three main opioid receptors, it does not bind the same ligands (Toll et al., 2016). Crystal structures of all opioid receptor subtypes bound to small‐molecule antagonists have been solved: μ receptor bound to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1631 (Protein Data Bank (PDB):4DKL (Manglik et al., 2012)), κ receptor bound to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7362 (PDB:4DJH (Wu et al., 2012)), δ receptor bound to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1641 (PDB:4EJ4 (Granier et al., 2012) and PDB:4N6H (Fenalti et al., 2014)), and NOP receptor bound to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7361 (PDB: 4EA3 (Thompson et al., 2012)), C‐35 (PDB: 5DHG (Miller et al., 2015)), or http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1693 (PDB: 5DHH (Miller et al., 2015)). At the time of writing, crystal structures are also available for the δ receptor bound to the tetrapeptide H‐Dmt‐Tic‐Phe‐Phe‐NH2 (DIPP‐NH2) (PDB:4RWA and PDB:4RWD (Fenalti et al., 2015)), which behaves as an antagonist at δ receptors but an agonist at μ receptors. Most recently, the activated structure of the μ receptor bound to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9363 and stabilized by a nanobody (PDB:5C1M (Huang et al., 2015)) was solved, providing an important contribution to our understanding of μ receptor activation (Figure 1).
Figure 1.
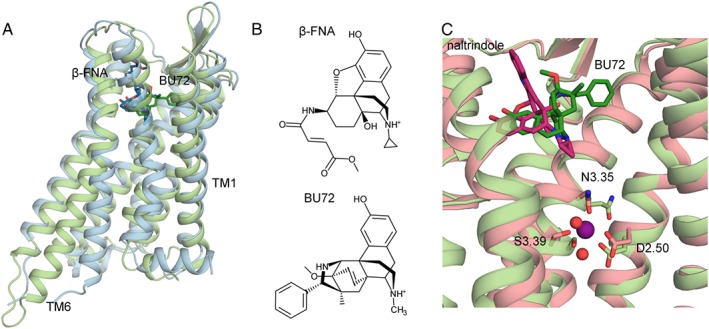
(A) Comparison of the inactive crystal structure of the μ receptor (PBD:4DKL, backbone: light blue, β‐FNA: dark blue) and the activated crystal structure of the μ receptor (PDB:5C1M backbone: light green, BU72: dark green); (B) 2D structure of β‐FNA and BU72, the ligands crystallized with the inactive and activated μ receptor; (C) sodium ion (purple sphere) coordination with D2.50, S3.39, N3.35 and two water molecules (red spheres) at the allosteric binding site of the inactive δ receptor crystal structure (PDB: 4N6H, backbone: pink, naltrindole: dark pink) compared with the collapsed sodium binding site of the activated μ receptor crystal structure (light green).
Although virtual screening and molecular docking using crystal structures have successfully been used as fast and inexpensive tools to discover novel opioid drugs, molecular dynamics (MD) simulations contribute crucial insights into protein dynamics in drug discovery that cannot be captured using most computer‐aided drug design (CADD) methods (e.g. see Ganesan et al., 2017, Miao and McCammon, 2016, and Tautermann et al., 2015, for recent reviews highlighting the importance of MD in drug design). While efforts have been made to incorporate dynamics into traditional CADD techniques (e.g. docking with flexible protein sidechains; Meiler and Baker, 2006; Morris et al., 2009), MD simulations are necessary to capture ligand‐induced conformational changes and to obtain a more realistic picture of the function of highly dynamic proteins such as opioid receptors and all other GPCRs. In addition to helping to understand how an antagonist or agonist binds to the orthosteric site of opioid receptors (i.e. the binding site of endogenous ligands), and either inactivates or activates the receptor, MD simulations provide important insights into the development of allosteric (i.e. molecules that bind at a secondary binding site) and functionally selective (i.e. biased signalling towards a specific pathway) modulators. In fact, a thorough understanding of the pharmacology of opioid receptors, as well as other GPCRs, requires more structural information than is available via crystal structures, as GPCRs naturally exist as an ensemble of different conformations in cells, and this conformational variability can be captured at an atomic resolution using MD simulations. Here, we discuss four areas in which MD simulations have provided unique insights into our understanding of the function of opioid receptors: (i) binding of sodium to the allosteric site; (ii) dynamics of ligand–receptor and receptor‐receptor interactions at both the ligand‐ and G protein‐binding sites; (iii) binding of novel ligands to opioid receptors; and (iv) oligomerization of opioid receptors.
Binding pathway of sodium to its allosteric site
Over 40 years ago, sodium at physiological concentrations was found to inhibit the binding of agonists, but not antagonists, to μ receptors (Pert et al., 1973; Pasternak and Snyder, 1975). Later experiments in brain homogenates found that sodium similarly inhibits agonist binding to δ receptors and to a lesser extent, κ receptors (Werling et al., 1986). This so‐called ‘sodium effect’ was later identified in a number of other GPCRs (Katritch et al., 2014). While early mutagenesis experiments identified D2.50 as a crucial residue for sodium‐mediated inhibition, the high‐resolution crystal structure of the antagonist‐bound A2A adenosine receptor (PDB:4EIY) provided the first evidence of sodium binding at the so‐called ‘allosteric sodium binding site’ lined by two highly conserved residues in rhodopsin‐like GPCRs: D2.50 and S3.39 (Liu et al., 2012). To facilitate a direct comparison between different GPCRs, the Ballesteros‐Weinstein numbering scheme, where the first number indicates the transmembrane (TM) helix and the second number indicates the relative position to the most conserved residue in that helix, is used throughout (Ballesteros and Weinstein, 1995). Two high‐resolution, antagonist‐bound δ receptor crystal structures, specifically, naltrindole‐bound (PDB:4N6H at 1.80 Å resolution) and DIPP‐NH2‐bound (PDB:4RWD at 2.70 Å resolution), also revealed a sodium ion bound to the same site where the ion is stabilized by a salt‐bridge to the D2.50 sidechain and polar interactions to the sidechains of N3.35 and S3.39, as well as two water molecules (Figure 1C). While the resolution of the available crystal structures of inactive μ and κ receptors was insufficient to resolve a sodium ion at the allosteric binding site, the few available crystal structures of activated GPCRs, including the μ receptor, show that the sodium binding site is collapsed relative to that of inactive structures (Katritch et al., 2014).
The results of MD simulations of opioid receptor crystal structures at physiological sodium concentrations by Yuan et al. (2013, 2015) and ourselves (Shang et al., 2014) have provided molecular and dynamic details of sodium modulation of the receptor's physiological function that could not be obtained from other experimental studies. Since the MD simulations allowed free diffusion of sodium ions from the bulk solvent, not only did these studies further demonstrate that sodium ions enter the receptor from the extracellular side but they also provided details of sodium binding pathways, including preferred receptor locations visited by the ion before stably binding to the crystallographic sodium binding site in all three main opioid receptor subtypes, as well as molecular determinants contributing to the stabilization of inactive conformations of the receptor. Yuan et al. (2013) suggested that sodium interacted with E5.35 before entering the orthosteric binding site of the μ receptor and coordinating with D3.32. After 150 ns in the orthosteric ligand‐binding site, the ion moved into the crystallographic sodium allosteric site, where it was coordinated to D2.50 and S3.39. We compared the free binding of sodium to μ, κ and δ receptor crystal structures using a 1 μs and ten 100 ns simulations for each receptor subtype (Shang et al., 2014). Τhe ions in these simulations followed a similar path to that identified by Yuan et al. (2013). In all three cases, the sodium entered the receptor from the extracellular side, as evidenced by the presence of higher sodium density near the extracellular loops (ELs) in our simulations (Shang et al., 2014) and those of Yuan et al. (2013). The ion then proceeded to the orthosteric ligand‐binding site, where it formed a salt bridge with D3.32 and four polar interactions with water molecules. Notably, the ion density at D3.32 was lower than that observed at non‐conserved E6.58 and D204 (EL2) residues in κ receptor simulations, or EL3 D293 and D288 residues in δ receptor simulations, suggesting that the lack of these residues in the μ receptor was the reason for the ion's delayed access of the orthosteric binding site in μ receptor simulations, and perhaps also the different tendency of a second ion to enter the receptor from the extracellular side. These molecular differences were hypothesized to be the reason behind the experimentally observed differential increase in antagonist binding affinity for μ receptors, possibly due to a reduced level of competition between ion and ligand for the orthosteric site (Shang et al., 2014). Notably, once the ion reached the sodium allosteric site, the dominant ion‐receptor interactions were found to be identical to those revealed by the 1.8 Å naltrindole‐bound δ receptor crystal structure (Shang et al., 2014).
To determine the free energy barrier for unbinding of a sodium ion from the allosteric binding site of a δ receptor, Vickery et al. (2016) calculated the potential of mean force from umbrella sampling simulations. The free energy minimum corresponded to the position of the sodium in the high‐resolution naltrindole‐bound δ receptor crystal structure and had a 13 kJ·mol−1 barrier to move from the allosteric site to the orthosteric site. Applying membrane voltages of 250 to 1000 mV caused the free energy surface to tilt, such that the energy barrier for sodium to leave the allosteric site decreased, and eventually the orthosteric binding site became more energetically favourable than the allosteric site.
To examine the effect of the ligand on sodium binding, we also carried out simulations of a δ receptor starting with sodium at the allosteric site in the presence or absence of naltrindole at the orthosteric site (Shang et al., 2014). While sodium coordination remained identical to that of the high‐resolution crystal structure (Fenalti et al., 2014) in the simulations of naltrindole bound to the δ receptor, coordination immediately changed in the absence of naltrindole. In particular, coordination by N3.35 was quickly lost, probably because this residue is part of a hydrogen bond network connecting naltrindole to the sodium ion, which is likely to contribute to the stability of inactive structures of opioid receptors.
Dynamics of ligand‐receptor and receptor‐receptor interactions at the ligand binding site
While crystal structures capture a static picture of ligand‐protein interactions, MD simulations complement this picture by characterizing their dynamics. Many classical opioid receptor drugs form a salt bridge between the receptor's D3.32 and the amine group of these ligands (Figure 2A), which is present in the inactive crystal structures of δ, κ and μ receptors, as well as the activated μ receptor structure. This salt bridge remains formed in simulations of antagonists bound to inactive opioid receptors, including those of JDTic with the κ receptor (Cheng et al., 2016a), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9194 with the κ receptor (Cheng et al., 2016a), β‐FNA with the μ receptor (Shim et al., 2013), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1638 with the μ receptor (Shim et al., 2013), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1617 with the μ receptor (Shim et al., 2013), and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7209 with the μ receptor (Yuan et al., 2015). In the case of agonists bound to inactive receptor structures (e.g. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627 to the μ receptor (Yuan et al., 2015), a http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8868 derivative (Kothandan et al., 2014), and the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1681 peptide (NOP) to the NOP receptor (Kothandan et al., 2014; della Longa and Arcovito, 2016)), the salt bridge between the ligand and the receptor was stable or alternated between the two oxygens of D3.32 during simulations up to 3 μs, but broke completely for levallorphan with the κ receptor (Yuan et al., 2015). However, the possibility exists that this salt bridge broke because the agonist had been docked in the orthosteric site of the inactive crystal structure of the receptor. In fact, while the orthosteric sites of inactive and activated μ receptors are very similar, the Cα atom of D3.32 is shifted ~2 Å in the activated crystal structure relative to the inactive crystal structure (Huang et al., 2015).
Figure 2.
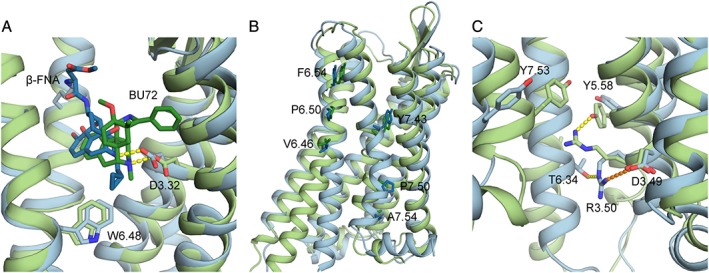
(A) Close‐up of the binding pockets of inactive and activated μ receptor crystal structures. Focus is on the salt bridges (yellow, dotted lines) formed by the amino group of β‐FNA (dark blue) and BU72 (dark green) and the side chain of D3.32 of the inactive (light blue) and activated (light green) crystal structures, respectively, as well as the hydrophobic interaction between W6.48 and β‐FNA; (B) residues of TM6 and TM7 used to calculate helix bending in (Cheng et al., 2016b); (C) residues involved in the ionic lock in inactive and activated μ receptor crystal structures. Shown as dotted lines are the salt bridge (orange) between D3.49 and R3.50 and the hydrogen bond (yellow) between T6.34 with R3.50 in the inactive μ receptor crystal structure, as well as the hydrogen bond (yellow) formed between Y5.58 and R3.50 in the activated crystal structure of the μ receptor.
While standard MD simulations are unable to capture the significant conformational changes expected upon opioid receptor activation, they can suggest testable hypotheses of interactions and motions involved in the activation process. After removing the agonist, BU72, from the μ receptorA, residues D3.32, N3.35 and I3.40 approached the positions observed in the inactive crystal structure during the simulation (Huang et al., 2015). Here, μ receptorA is used to denote the activated crystal structure to distinguish between simulations starting from the activated structure and those with agonists docked to the inactive structure. Notably, the Cα of N3.35, which is involved in coordination of the sodium ion at the allosteric site, underwent the largest change of the three residues during the simulation, moving 5.5 Å away from its position in the receptor activated structure.
Inactive crystal structures of opioid receptors bound to small molecules show interactions between hydrophobic groups (e.g. a cyclopropylmethyl group in β‐FNA and naltrindole or an isopropyl group in JDTic) and residue W6.48 (Figure 2A), the rotation of which has been implicated in GPCR activation (Deupi and Standfuss, 2011). MD simulations carried out on inactive and activated μ receptor crystal structures support earlier suggestions of a W6.48 rotamer toggle switch. In simulations of a ligand‐free, inactive μ receptor with sodium at its allosteric binding site (Shang et al., 2014), the W6.48 χ2 dihedral angle fluctuated around its crystallographic value (~70°). Similarly, in simulations of a ligand‐free inactive α2A adrenoceptor (Gutiérrez‐de‐Terán et al., 2013), the W6.48 dihedral angle remained at its crystal structure value if the sodium ion was bound, but rotated by 120° in two of three simulations in which sodium was not present. Notably, after removal of the agonist from the μ receptorA, the W6.48 χ2 angle remained stable at ~120° for 300 ns, but it changed by 180° afterwards (Huang et al., 2015). In contrast, the W6.48 dihedral angle maintained the value of the inactive crystal structure in simulations of the inactive μ receptor bound to the antagonist β‐FNA, or the value of the activated crystal structure in simulations of the μ receptorA bound to BU72 (Huang et al., 2015). After being placed in the inactive μ receptor structure, the agonist β‐FOA, which differs from the antagonist β‐FNA in that the cyclopropylmethyl group is replaced by a methyl, the W6.48 χ2 dihedral quickly moved away from its inactive value of ~70°, and finally fluctuated around ~100°, which is closer to the value assumed in the activated μ receptor crystal structure (Huang et al., 2015). While it was concluded that the exact orientation of W6.48 may be ligand‐dependent, based on the results of simulations of the inactive μ receptor bound to the agonist hydromorphine (W6.48 dihedral = 120°) or morphine (W6.48 dihedral = 0°)(Cong et al., 2015), the possibility that the differences between the agonists may have arisen due to docking of agonists in the μ receptor inactive crystal structure cannot be excluded.
One effect the motion of W6.48 could have on activation is the opening of the entryway to a water‐filled cavity on the intracellular part of the orthosteric binding pocket, which is connected to the orthosteric binding site in the activated μ receptor crystal structure (Huang et al., 2015), but not in the inactive crystal structure (Manglik et al., 2012). Yuan et al. (2015) found an average of four water molecules near D2.50 for antagonist‐bound opioid receptors (μ receptor with levallorphan and κ receptor with JDTic), significantly less than for agonist‐bound opioid receptors (μ receptor with morphine and κ receptor with levallorphan), which had an average of seven water molecules and as many as 11. An increase in internal hydration upon activation is also consistent with the results of MD simulations of rhodopsin (Leioatts et al., 2014) and the cannabinoid CB2 receptor (Hurst et al., 2010).
Dynamics of ligand‐receptor and receptor‐receptor interactions at the G protein‐binding site
Binding of agonists to GPCRs, including opioid receptors, leads to conformational changes on the intracellular side of the receptor upon activation to accommodate binding of intracellular proteins. Characterization of these changes as well as how signals propagate from the orthosteric ligand binding site to the intracellular side of the receptor can be obtained from MD simulations. A major conformational change exhibited by most GPCRs, including opioid receptors, is the outward motion of the intracellular end of TM6. While the orthosteric ligand‐binding sites of the inactive and activated crystal structures of opioid receptors share many similarities, the plasticity of the TM helices, which appears to be linked to the conformation of residues in the orthosteric site (Fossepre et al., 2014), leads to some notable differences. For instance, the fluctuations in angles formed by the Cα atoms of V6.46, P6.50, and F6.54 of TM6 and Y7.43, P7.50 and A7.54 of TM7 (Figure 2B) were significantly less pronounced in simulations where the antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1669 was bound to the κ receptor than when the agonist 6'GNTI was bound (Cheng et al., 2016b). TM6 also rotated 5° relative to its position in the κ receptor's inactive structure docked with 6'GNTI, due to a steric clash between 6'GNTI and I6.55 and an interaction formed between 6'GNTI and E6.58. Notably, when 5'GNTI was docked into the final structure from the 6'GNTI‐κ receptor complex simulation, TM6 returned to its orientation as in the 5'GNTI‐κ receptor complex. Increased bending of TM6 away from the z‐axis at residues close to P6.50 was also seen in simulations of the agonist levallorphan bound to the κ receptor and the agonist morphine bound to the μ receptor than for the antagonist JDTic bound to the κ receptor and the levallorphan μ receptor‐complex for which levallorphan acts as an antagonist (Yuan et al., 2015). Finally, the root‐mean‐square deviation (RMSD) of the Cα atoms of TM6 during the simulations of morphine with the μ receptor and hydromorphine with the μ receptor relative to the inactive crystal structure was larger than that of the β‐FNA‐μ receptor complex (Cong et al., 2015).
A highly conserved motif in rhodopsin‐like GPCRs, known as the D/ERY motif, is located on the intracellular end of TM3. Two residues of this motif, D/E3.49 and R3.50, have long been known to form an intra‐helical salt bridge in inactive GPCRs, and to participate quite substantially (e.g. see Li et al., 2001; Vogel et al., 2008) in the inter‐helical hydrogen‐bond network comprising the salt bridge between R3.50 and D/E6.30, which is commonly referred to in the literature as ‘the ionic lock’ for its role in stabilizing inactive receptor conformations (e.g. see Ballesteros et al., 2001). Notably, while opioid receptor inactive crystal structures confirmed the formation of the intra‐helical salt bridge between D3.49 and R3.50, the absence of an acidic residue at position 6.30 in opioid receptors impedes the formation of a salt bridge between residues 3.50 and 6.30. Yet, in line with earlier mutagenesis studies in which the T6.34K μ receptor mutant was found to be constitutively active while the T6.34D mutant was inactive (Huang et al., 2001; 2002), the crystal structures of inactive opioid receptors suggested a hydrogen bond formed between R3.50 and T6.34. Like the salt bridge between R3.50 and D3.49, this contact was broken in the activated crystal structure of the μ receptor. Notably, the results of MD simulations of inactive opioid receptors show that unlike the R3.50‐D3.49 salt bridge, the R3.50‐T6.34 hydrogen bond is very dynamic and is found to be broken at times, in line with earlier experimental observations that the inter‐helical TM3‐TM6 interaction in rhodopsin‐like GPCRs breaks more easily than the intra‐helical salt bridge between R3.50 and D3.49 (Vogel et al., 2008). For instance, the R3.50‐T6.34 hydrogen bond was never found to be formed in simulations of the ligand‐free inactive μ receptor or the receptor in complex with antagonists http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1639 or naloxone, while the R3.50‐D3.49 salt bridge remained formed during the entire simulation time (Yuan et al., 2013). In contrast, the R3.50‐T6.34 hydrogen bond distance stayed close to its value in the inactive μ receptor crystal structure in simulations of the μ receptor bound to the antagonist β‐FNA, and it only broke in simulations of agonists morphine and hydromorphine bound to the inactive μ receptor (Cong et al., 2015), although the latter could not be reproduced in a different set of simulations (Yuan et al., 2013). In a longer set of simulations, Yuan et al. (2015) did observe breaking of the R3.50‐D3.49 salt bridge for the μ receptor bound to the agonist morphine and the κ receptor bound to the agonist levallorphan, but the R3.50‐T6.34 hydrogen bond remained intact. Notably, Yuan et al. (2013) suggested that the R3.50‐T6.34 hydrogen bond could help break the R3.50‐D3.49 intra‐helical salt bridge, allowing R3.50 to finally interact with Y5.58, in line with the interaction seen in the activated crystal structure of the μ receptor (Huang et al., 2015). This R3.50‐Y5.58 interaction orients Y5.58, so it can form a water‐mediated interaction with Y7.53, which is part of the conserved NPxxY motif on the intracellular end of TM7 (Huang et al., 2015). Despite the Y5.58‐Y7.53 interaction also being present in crystal structures of activated rhodopsin and the β2 adrenoceptor, limited information about this interaction exists in the MD simulation studies published.
In the search for improved therapeutics to treat chronic pain, attention has recently shifted towards functionally selective ligands that only activate G‐protein signalling, since a few have recently been reported to exhibit fewer adverse effects (DeWire et al., 2013; Crowley et al., 2016; Manglik et al., 2016; Varadi et al., 2016). One such ligand, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7334 (oliceridine), is currently in phase 3 clinical trials (DeWire et al., 2013). To determine how TRV‐130 acts as a G protein‐biased ligand, we analysed the allosteric communication between the orthosteric and G‐protein binding sites using the NbIT method of LeVine and Weinstein (LeVine and Weinstein, 2014). This analysis of unbiased MD trajectories of TRV‐130‐μ receptorA (MORA) and morphine‐μ receptorA complexes showed clear differences in communication. While strong coupling was observed between the orthosteric site and intracellular ends of both TM3 and TM6 for the morphine‐μ receptorA, communication was only seen between the ligand binding pocket and the intracellular end of TM3 for the TRV‐130‐μ receptorA (Schneider et al., 2016). Notably, residues such as W7.35 acted as communicators between the ligand binding pocket and intracellular receptor regions only for the TRV‐130‐μ receptorA system. While Y2.42, W133 (EC1), Y7.43, F343 (H8), W6.48, Y7.53, F135 (EC1), D3.49 and I3.29 strongly contributed to the allosteric coupling in both the TRV‐130‐μ receptorA and morphine‐μ receptorA systems, residues F2.44, I2.43, N4.46 and R6.32 mostly contributed to allosteric communication in the morphine‐bound μ receptor, whereas W7.35, R3.50, Y3.34, F347 (H8) and Y1.55 mostly contributed to the transmission of information in the TRV‐130‐bound receptor.
Binding pathways and binding modes of novel ligands to opioid receptors
Simulations can play a key role in discerning ligand binding and unbinding pathways, which cannot be captured by crystal structures, as well as predicting the binding pose of a ligand for which no information from crystal structure exists. Standard all‐atom MD simulations of GPCRs are currently limited to timescales in the order of several microseconds, which is less than the timescale on which ligand binding occurs. One approach recently taken to overcome this problem is to use a high concentration of ligands and/or run extremely long MD simulations on specialized computer hardware (Dror et al., 2011; 2013) to increase the probability of effective binding to the receptor. We recently applied this approach to study the binding of TRV‐130, ultimately determining its binding pathway and energetically preferred binding pose (Schneider et al., 2016). Clustering the poses sampled during simulation based on ligand–receptor interaction fingerprints identified four highly populated clusters of the ligand along the binding pathway, in addition to a cluster at the orthosteric binding site. Consistent with previous simulations of the β2 adrenoceptor (Dror et al., 2011) and muscarinic M3 receptor (Kruse et al., 2012), a metastable state was identified in the so‐called vestibule delineated by TM2, TM3 and TM7. Notably, ligands from a cluster spatially close to this metastable state could not proceed to the orthosteric site without first unbinding. The representative TRV‐130 structures of the cluster at the orthosteric binding site (pink and orange in Figure 3B) partially overlapped with BU72, the agonist crystalized with the μ receptorA. The interaction of the TRV‐130 amine with D3.32 is not direct, but is mediated by two water molecules in both of the predicted binding poses of TRV‐130. Other residues that form contacts with TRV‐130 include M3.36, I6.51, H6.52, V6.55, W7.35 and I7.39.
Figure 3.
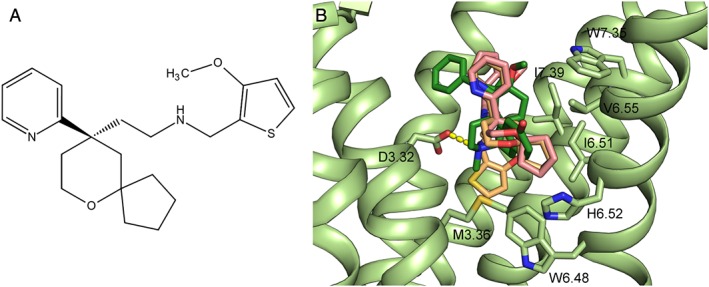
(A) Structure of TRV‐130, a biased agonist for the μ receptor, and (B) predicted binding poses of TRV‐130 (pink and light orange) in the μ receptor orthosteric binding site overlaid on the receptor activated crystal structure (light green) bound to BU72 (dark green). The salt‐bridge between the amino group of BU72 and D3.32 is shown as a yellow dotted line. The residues that interact with TRV‐130 within a 4 Å distance cut‐off are labelled.
More computationally efficient strategies to study ligand binding to GPCRs use enhanced MD techniques (Schneider et al., 2015; Miao and McCammon, 2016). One such method is metadynamics (Laio and Parrinello, 2002), which was more recently used to characterize JDTic and LY2456302 unbinding from the orthosteric site of the κ receptor (Cheng et al., 2016a) as well as the binding of a positive allosteric modulator (PAM), BMS‐986187, to the δ receptor (Shang et al., 2016). Metadynamics alleviates the conformation sampling issue seen in standard MD simulations by biasing the potential along reaction coordinates chosen by the user to represent the slowest degrees of freedom relevant to the process studied. Cheng and co‐authors (Cheng et al., 2016a) used the RMSD of the ligand with respect to the initial pose and the distance between atoms in the ligand and protein to perform 100 ns of well‐tempered metadynamics. While these simulations identified possible metastable states in the binding of JDTic and LY2456302, the relevance of these states is unclear since the simulations were too short to see more than one unbinding event.
For a complex process such as ligand unbinding from a GPCR, there is no guarantee that the first unbinding event captures all aspects of the pathway. Multiple unbinding/binding events are necessary to ensure sampling of all metastable states, which can be accomplished using a method like multiple‐walker metadynamics. For instance, we used multiple‐walker metadynamics to enhance the sampling of the binding of the BMS‐986187 PAM to the δ receptor with the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1611 agonist bound to the orthosteric site (Shang et al., 2016). Clustering of the sampled binding poses was performed using interaction fingerprints, which considered both direct and water‐mediated interactions between the receptor and ligand. Two poses with essentially the same free energy (poses 1 and 2 in Figure 4) were thus identified as possible modes of binding of BMS‐986187 at an allosteric site delineated by TM helices TM1, TM2, TM6 and TM7. Both pose 1 (red in Figure 4) and pose 2 (blue in Figure 4) exhibited similar interactions between the receptor and the 2‐methyl‐benzyl group of BMS‐986187 but different orientations of the fused tricyclic moiety. The two predicted BMS‐986187 poses, which are hypothesized to be equally populated at room temperature, differed mainly in interactions with residues L1.29, L1.31 and L2.65 seen in pose 1 but not 2, as well as interactions with residues W6.58, L7.35, Q2.60 and K2.63 seen in pose 2 but not 1. To experimentally validate the predicted poses, mutagenesis experiments were performed on selected residues of the proposed allosteric site, specifically: Y56(1.39)A, Q105(2.60)A, K108(2.63)A, K108(2.63)N, Y109(2.64)A, Y109(2.64)I, W284(6.58)K, L300(7.35)W, H301(7.36)R and H301(7.36)Y. While some of these mutants (i.e. Q105(2.60)A, W284(6.58)K, and H301(7.36)Y) only affected the BMS‐986187 binding affinity, the Y109(2.64)A and L300(7.35)W mutants appeared to affect both the allosteric binding affinity and its cooperativity, whereas H301(7.36)R and K108(2.63)N only affected allosteric cooperativity (Shang et al., 2016). In spite of a higher support for pose 2 as the preferred binding pose of BMS‐986187 (Shang et al., 2016), these computational predictions and experimental validations were deemed to be insufficient to discriminate unambiguously between the two predicted binding poses.
Figure 4.
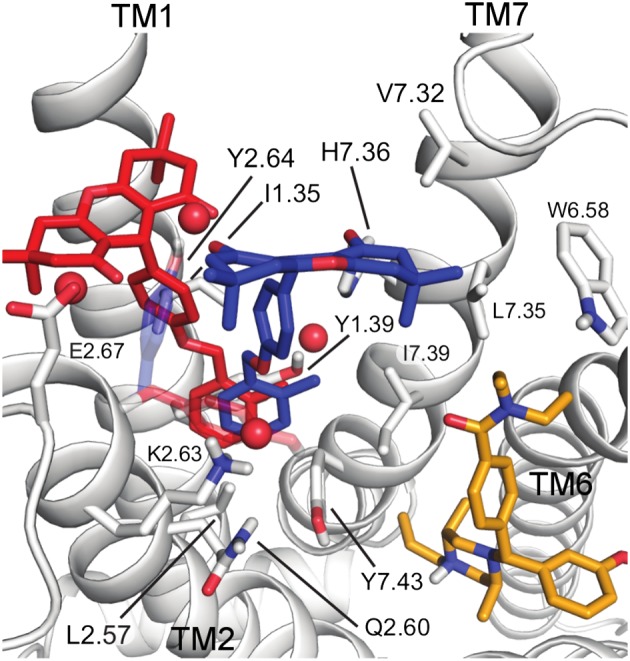
An extracellular view of the two lowest energy binding poses of BMS‐986187 in red (pose 1) and blue (pose 2) bound to the δ receptor. The agonist, SNC‐80, is in orange. Water molecules which participate in water‐mediated hydrogen bonds between BMS‐986187 and the receptor are shown as red spheres. Residues that interact with both pose 1 and pose 2 are labelled. Adapted with permission from Shang et al. (2016) Proposed mode of binding and action of positive allosteric modulators at opioid receptors. ACS Chemical Biology, 11: 1220‐9. Copyright 2016 American Chemical Society.
Oligomerization of opioid receptors
A fundamental question about the function of opioid receptors and other rhodopsin‐like GPCRs is whether or not their oligomerization in the cell membrane plays a significant role in signalling. While glutamate‐like GPCRs form constitutive dimers, there is still a debate on the functional role of oligomerization for rhodopsin‐like GPCRs (Ferre et al., 2014). Targeting opioid receptor heteromers may represent a possible route for new treatments of acute and chronic pain as some of them, for example, μ receptor‐δ receptor (Gomes et al., 2013) and μ receptor‐mGlu5 receptor (Akgün et al., 2013) heteromers, have been suggested to be involved in antinociceptive activity.
Implicit in the elucidation of the function of oligomerization in opioid receptor signalling is a clear picture of which dimer interfaces form. The crystal structures of inactive and active μ receptors and inactive κ receptors showed parallel crystallographic interfaces, although the evidence is inconclusive as to whether they represent physiological interfaces or are just artefacts of crystallization. While all three structures had a TM1,2,H8/TM1,2,H8 interface, the inactive μ receptor also had a TM5,6/TM5,6 interface, which could not be formed by the active μ receptor due to the outward swing of TM6 upon activation. Figure 5 shows the three putative dimer interfaces seen in the crystal structures of inactive opioid receptors currently available. Capturing the oligomerization of GPCRs currently requires a coarse‐grained (CG) model, that is, a reduced representation with one bead describing multiple heavy atoms, since all‐atom MD simulations would be too computationally expensive. The MARTINI CG model has been used to study self‐assembly of a number of GPCRs, including opioid receptors (Provasi et al., 2015; Marino et al., 2016), rhodopsin (Periole et al., 2007; 2012), β2 adrenoceptors (Prasanna et al., 2014) and the chemokine receptor CCR4 (Pluhackova et al., 2016). All‐atom MD has thus far only been applied to the preformed crystallographic dimers of the μ receptor and not their free association (Shim et al., 2013; Huang et al., 2016a). To obtain an estimate of the relative stability of the opioid receptor crystallographic interfaces in a lipid bilayer, enhanced sampling techniques are preferred over standard MD simulation methods, because the latter have a limited ability to sample multiple associated and dissociated states of the receptors during the currently affordable simulation time scales of microseconds. Among these enhanced sampling techniques is umbrella sampling, a powerful method that improves the efficiency of MD simulations by first splitting the reaction coordinate into subintervals, and then performing MD simulations in each subinterval, from which the free‐energy profile of the system under study can be derived. We performed umbrella sampling simulations of crystal dimers of the inactive μ receptor and κ receptor in a 1‐palmitoyl,2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC)/10% cholesterol membrane and calculated the free energy profile of the two opioid receptor protomers using the distance between their centres of mass as a reaction coordinate (Johnston and Filizola, 2014). The TM5,6/TM5,6 interface of the μ receptor was found to have a free energy minimum at a slightly shorter distance than its crystallographic value, probably due to the absence of the T4L fusion protein, which was part of the interface in the crystal structure. In contrast to the TM5,6/TM5,6 interface, the free energy of the alternative TM1,2,H8/TM1,2,H8 crystallographic interface of the μ receptor was found to have two free energy minima separated by a very small energy barrier, one of which was very close to the crystallographic value. Notably, the structurally equivalent TM,1,2,H8/TM1,2,H8 interface of the κ receptor was found to have a much deeper free energy minimum with respect to the TM,1,2,H8/TM1,2,H8 interface of the μ receptor, at a distance shorter than the crystal minimum. These differences in free energy were attributed to specific residue differences between κ and μ receptors and hence contact variations at the interface in both the crystallographic structures and the dimeric minima from the simulations such as different residues at positions 1.29, 1.32, 1.36 and 2.62, as well as contacts formed in the κ receptor but not in the μ receptor (e.g. involving residue 1.43), or the μ receptor but not the κ receptor (e.g. involving residue 1.48).
Figure 5.
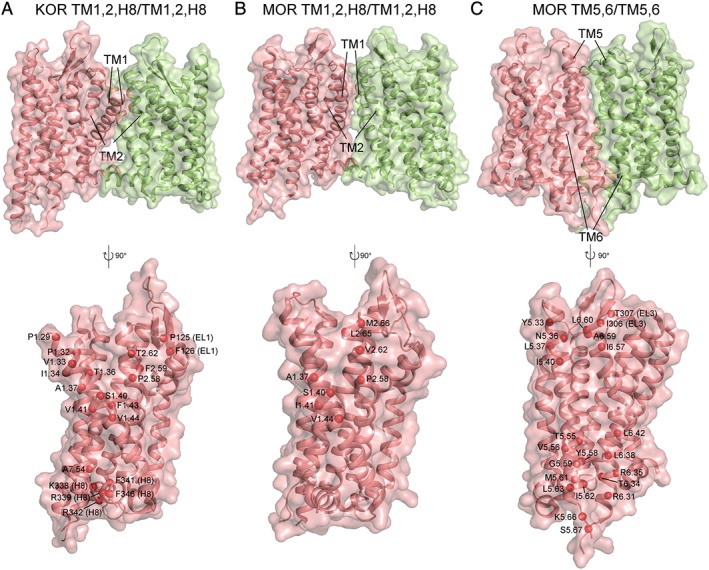
The three putative dimeric interfaces seen in opioid receptor inactive crystal structures (A) κ receptor (KOR) TM1,2,H8/TM1,2,H8, (B) μ receptor (MOR) TM1,2,H8/TM1,2,H8 and (C) μ receptor (MOR) TM5,6/TM5,6. The top row shows the interface with protomer A in pink and protomer B in green. The bottom row shows a cross section of the interface. The red spheres represent the Cα atoms of residues involved in cross‐interface contacts, where a contact is considered to be formed between two residues if their Cα atoms are within 8 Å of each other.
While the aforementioned umbrella sampling simulations were able to confirm the stability of the crystallographic interfaces in a lipid bilayer, they could not determine if these interfaces were the most favoured. To characterize other probable interfaces, we recently performed unbiased MD simulations of arrays of 16 inactive, CG protomers, which were able to freely move in a POPC/10% cholesterol membrane (Provasi et al., 2015). Five sets of simulations were performed to characterize the formation of both homo‐ and heterodimers and higher order opioid receptor oligomers: 16 δ receptors, 16 κ receptors, 16 μ receptors, 8 δ/8 μ receptors and 8 δ/8 κ receptors. All three simulated opioid receptors formed similar homodimer interfaces albeit with different probabilities. The predominant interfaces formed by two δ receptors were the TM1,2/TM4,5 (41%) and the TM1,2/TM5,6 (32%); κ receptor homodimers with TM1,2,H8/TM1,2,H8, TM1,2/TM4,5, TM1,2/TM5,6 and TM5/TM5 interfaces formed with similar probabilities (~25% each). The most frequently observed interfaces formed by the μ receptor were TM5/TM5 (43%) and TM1,2/TM5,6 (37%). While the crystallographic TM1,2,H8/TM1,2H8 interfaces were seen for both κ (RMSD of 4.1 Å from 4DJH) and μ receptors (RMSD of 6.7 Å from 4DKL), the TM5,6/TM5,6 interface did not form during these simulations. Since the TM5/TM5 interface formed for both the μ and κ receptor, the TM5,6/TM5,6 interface was concluded to be kinetically unable to form on the timescale of these simulations. Notably, the TM4,5/TM5,6 interface formed during simulations of all opioid receptors and the TM4,5/TM4,5 interface formed for the δ receptor homodimer, which is in line with earlier cross‐linking experiments that provided evidence for the involvement of TM4 and TM5 in the formation of δ receptor dimers (Johnston et al., 2011). Although the asymmetric interfaces that formed during simulation (e.g. TM1,2,H8/TM4,5 and TM1,2,H8/TM5,6) have not been observed crystallographically for opioid receptors, asymmetric interfaces were indeed seen in crystal structures of closely related GPCRs such as chemokine receptors CXCR4 (PDB: 3OE8 (Wu et al., 2010)) and CCR5 (PDB: 4MBS (Tan et al., 2013)) albeit these interfaces are not exactly the same as those from the aforementioned simulations.
In interpreting the results of dimerization studies of opioid receptors, it is worth emphasizing that the lipid environment plays a key role in the function of these receptors through direct protein‐lipid interactions and/or indirect modulation of membrane properties, similar to other GPCRs and membrane proteins (Phillips et al., 2009; Oates and Watts, 2011). For instance, experiments have clearly shown that μ receptor oligomerization is sensitive to cholesterol levels (Zheng et al., 2012) and modulating serum cholesterol levels has been shown to affect the analgesic profile of opioids (Huang et al., 2016b). To examine the effect of the lipids on opioid receptor oligomerization, we (Marino et al., 2016) recently examined the supramolecular organization of the inactive and/or activated conformations of the μ receptor in a 63‐component idealized plasma membrane model (Ingólfsson et al., 2014). These simulations suggested a specific mechanism by which lipid molecules appear to regulate the dimerization process of opioid receptors. The lipid order was found to be helix dependent with the lipids near TM1, TM5 and TM6 showing more order than the average, while the lipids near TM4 were less ordered. The ordered regions correlated with regions rich in sphingomyelin lipids, which have less flexible hydrophobic tails than other lipid types. Because ordered regions tended to coalesce with other ordered regions, lipid order acted as a long range sorting mechanism aligning protomer faces involving TM1 or TM5/TM6 prior to interface formation. Thus, interfaces like TM1,2,H8/TM1,2,H8 and TM1,2,H8/TM5 were much more likely to form than a TM4/TM4 dimer interface. While lipid order influenced dimer formation on a long‐range, the final interfaces that formed were dictated by the protomer conformation. TM1,2,H8/TM4 and TM1,2,H8/TM4,5 interfaces were those most frequently formed by two inactive protomers of the μ receptor, while two active protomers preferentially formed the TM1,2,H8/TM1,2,H8, TM1,2,H8/TM5 and TM1,2,H8/TM5,6 interfaces.
Summary and future challenges
As a complement to experiments, MD simulations have added a great deal of knowledge on the function of opioid receptors and will continue to advance our understanding in the future. However, a number of challenges still limit their use. Among the main limitations is the timescale problem, which remains the key issue in the successful application of all‐atom MD simulations to lengthy processes such as opioid receptor ligand binding and unbinding, the conformational transitions due to activation, and receptor oligomerization. Additionally, continued improvements in force‐field parameterization will be necessary to more realistically describe atomic interactions.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by National Institutes of Health grants DA038882, DA026434, and DA034049. Computations in the Filizola lab are run on resources available through the Scientific Computing Facility at the Icahn School of Medicine at Mount Sinai, and the Extreme Science and Engineering Discovery Environment under MCB080077, which is supported by National Science Foundation grant number ACI‐1053575.
Marino K. A., Shang Y., and Filizola M. (2018) Insights into the function of opioid receptors from molecular dynamics simulations of available crystal structures, British Journal of Pharmacology, 175:2834–2845, https://doi.org/10.1111/bph.13774.
References
- Akgün E, Javed MI, Lunzer MM, Smeester BA, Beitz AJ, Portogese PS (2013). Ligands that interact with putative MOR‐mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc Natl Acad Sci U S A 110: 11595–11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SGF, Shi L, Gether U et al. (2001). Activation of the β2‐adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem 276: 29171–29177. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H (1995). Integrated methods for the construction of three‐dimensional models and computational probing of structure‐function relations in G protein‐coupled receptors In: Stuart CS. (ed). Methods in Neurosciences. Academic Press: San Diego, pp. 366–428. [Google Scholar]
- Cheng J, Li W, Liu G, Zhu W, Tang Y (2016a). Computational insights into different inhibition modes of the κ‐opioid receptor with antagonists LY2456302 and JDTic. RSC Adv 6: 13626–13635. [Google Scholar]
- Cheng J, Sun X, Li W, Liu G, Tu Y, Tang Y (2016b). Molecular switches of the kappa opioid receptor triggered by 6′‐GNTI and 5′‐GNTI. Sci Rep 6: 18913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong X, Campomanes P, Kless A, Schapitz I, Wagener M, Koch T et al. (2015). Structural determinants for the binding of morphinan agonists to the mu‐opioid receptor. PLoS One 10 e0135998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley RS, Riley AP, Sherwood AM, Groer CE, Shivaperumal N, Biscaia M et al. (2016). Synthetic studies of neoclerodane diterpenes from Salvia divinorum: Identification of a potent and centrally acting μ opioid analgesic with reduced abuse liability. J Med Chem 59: 11027–11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- della Longa S, Arcovito A (2016). A dynamic picture of the early events in nociceptin binding to the NOP receptor by metadynamics. Biophys J 111: 1203–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deupi X, Standfuss J (2011). Structural insights into agonist‐induced activation of G‐protein‐coupled receptors. Curr Opin Struct Biol 21: 541–551. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM et al. (2013). A G protein‐biased ligand at the μ‐opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfuncation compared with morphine. J Pharmacol Exp Ther 344: 708–717. [DOI] [PubMed] [Google Scholar]
- Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC et al. (2013). Structural basis for modulation of a G‐protein‐coupled receptor by allosteric drugs. Nature 503: 295–299. [DOI] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y et al. (2011). Pathway and mechanism of drug binding to G‐protein‐coupled receptors. Proc Natl Acad Sci U S A 108: 13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V et al. (2014). Molecular control of delta‐opioid receptor signalling. Nature 506: 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G, Zatsepin NA, Betti C, Giguere P, Han GW, Ishchenko A et al. (2015). Structural basis for bifunctional peptide recognition at human delta‐opioid receptor. Nat Struct Mol Biol 22: 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre S, Casado V, Devi LA, Filizola M, Jockers R, Lohse MJ et al. (2014). G protein‐coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev 66: 413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossepre M, Leherte L, Laaksonen A, Vercauteren DP (2014). On the modularity of the intrinsic flexibility of the μ opioid receptor: a computational study. PLoS One 9: e115856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A, Coote ML, Barakat K (2017). Molecular dynamics‐driven drug discovery: leaping forward with confidence. Drug Discov Today 22: 249–269. [DOI] [PubMed] [Google Scholar]
- Gomes I, Fujita W, Gupta A, Saldanha SA, Negri A, Pinello CE et al. (2013). Identification of a μ‐δ opioid receptor heteromerbiased agonist with antinociceptive activity. Proc Natl Acad Sci U S A 110: 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI et al. (2012). Structure of the delta‐opioid receptor bound to naltrindole. Nature 485: 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez‐de‐Terán H, Massink A, Rodríguez D, Liu W, Han Gye W, Joseph Jeremiah S et al. (2013). The role of a sodium ion binding site in the allosteric modulation of the A2A adenosine G protein‐coupled receptor. Structure 21: 2175–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kaushik S, Coop A, MacKerell AD (2016a). Conformational heterogeneity of intracellular loop 3 of the μ‐opioid G ‐protein coupled receptor. J Phys Chem B 120: 11897–11904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Li J, Chen C, Visiers I, Weinstein H, Liu‐Chen L‐Y (2001). Functional role of a conserved motif in TM6 of the rat μ opioid receptor: Constitutively active and inactive receptors result from substitutions of Thr6.34(279) with Lys and Asp. Biochemistry 40: 13501–13509. [DOI] [PubMed] [Google Scholar]
- Huang P, Visiers I, Weinstein H, Liu‐Chen L‐Y (2002). The local environment at the cytoplasmic end of TM6 of the μ opioid receptor differs from those of rhodopsin and monoamine receptors: introduction of an ionic lock between the cytoplasmic ends of helices 3 and 6 by a L6.30(275)E mutation inactivates the μ opioid receptor and reduces the constitutive activity of its T6.34(279)K mutant. Biochemistry 41: 11972–11980. [DOI] [PubMed] [Google Scholar]
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL et al. (2015). Structural insights into μ‐opioid receptor activation. Nature 524: 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Liang L, Li L, Xu M, Li X, Sun H et al. (2016b). Opioid doses required for pain management in lung cancer patients with different cholesterol levels: negative correlation between opioid doses and cholesterol levels. Lipids Health Dis 15: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, Gawrisch K et al. (2010). A lipid pathway for ligand binding is necessary for a cannabinoid G protein‐coupled receptor. J Biol Chem 285: 17954–17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingólfsson HI, Melo MN, van Eerden FJ, Arnarez C, Lopez CA, Wassenaar TA et al. (2014). Lipid organization of the plasma membrane. J Am Chem Soc 136: 14554–14559. [DOI] [PubMed] [Google Scholar]
- Johnston JM, Aburi M, Provasi D, Bortolato A, Urizar E, Lambert NA et al. (2011). Making structural sense of dimerization interfaces of delta opioid receptor homodimers. Biochemistry 50: 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JM, Filizola M (2014). Differential stability of the crystallographic interfaces of mu‐ and kappa‐opioid receptors. PLoS One 9: e90694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC (2014). Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39: 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothandan G, Gadhe CG, Balupuri A, Ganapathy J, Cho SJ (2014). The nociceptin receptor (NOPR) and its interaction with clinically important agonist molecules: a membrane molecular dynamics simulation study. Mol Biosyst 10: 3188–3198. [DOI] [PubMed] [Google Scholar]
- Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E et al. (2012). Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482: 552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laio A, Parrinello M (2002). Escaping free‐energy minima. Proc Natl Acad Sci U S A 99: 12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leioatts N, Mertz B, Martinez‐Mayorga K, Romo TD, Pitman MC, Feller SE et al. (2014). Retinal ligand mobility explains internal hydration and reconciles active rhodopsin structures. Biochemistry 53: 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine MV, Weinstein H (2014). NbIT – A new information theory‐based analysis of allosteric mechanisms reveals residues that underlie function in the leucine transporter LeuT. PLoS Comput Biol 10 e1003603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Huang P, Chen C, de Riel JK, Weinstein H, Liu‐Chen L‐Y (2001). Constitutive activation of the μ opioid receptor by mutation of D3.49(164), but not D3.32(147): D3.49(164) is critical for stabilization of the inactive form of the receptor and for its expression. Biochemistry 40: 12039–12050. [DOI] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V et al. (2012). Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK et al. (2012). Crystal structure of the μ‐opioid receptor bound to a morphinan antagonist. Nature 485: 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G et al. (2016). Structure‐based discovery of opioid analgesics with reduced side effects. Nature 537: 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino KA, Prada‐Gracia D, Provasi D, Filizola M (2016). Impact of lipid composition and receptor conformation on the spatio‐temporal organization of mu‐opioid receptors in a multi‐component plasma membrane model. PLoS Comput Biol 12 e1005240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler J, Baker D (2006). ROSETTALIGAND: protein–small molecule docking with full side‐chain flexibility. Proteins 65: 538–548. [DOI] [PubMed] [Google Scholar]
- Miao Y, McCammon JA (2016). G‐protein coupled receptors: advances in simulation and drug discovery. Curr Opin Struct Biol 41: 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RL, Thompson AA, Trapella C, Guerrini R, Malfacini D, Patel N et al. (2015). The importance of ligand‐receptor conformational pairs in stabilization: spotlight on the N/OFQ G Protein‐coupled receptor. Structure 23: 2291–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS et al. (2009). AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30: 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates J, Watts A (2011). Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr Opin Struct Biol 21: 802–807. [DOI] [PubMed] [Google Scholar]
- Pasternak GW, Snyder SH (1975). Identification of novel high affinity opiate receptor binding in rat brain. Nature 253: 563–565. [DOI] [PubMed] [Google Scholar]
- Periole X, Huber T, Marrink S‐J, Sakmar TP (2007). G protein‐coupled receptos self‐assemble in dynamics simulations of model bilayers. J Am Chem Soc 129: 10126–10132. [DOI] [PubMed] [Google Scholar]
- Periole X, Knepp AM, Sakmar TP, Marrink SJ, Huber T (2012). Structural determinants of the supramolecular organization of G protein‐coupled receptors in bilayers. J Am Chem Soc 134: 10959–10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pert CB, Pasternak G, Snyder SH (1973). Opiate agonists and antagonists discriminated by receptor binding in brain. Science 182: 1359–1361. [DOI] [PubMed] [Google Scholar]
- Phillips R, Ursell T, Wiggins P, Sens P (2009). Emerging roles for lipids in shaping membrane‐protein function. Nature 459: 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluhackova K, Gahbauer S, Kranz F, Wassenaar TA, Böckmann RA (2016). Dynamic cholesterol‐conditioned dimerization of the G protein coupled chemokine receptor type 4. PLoS Comput Biol 12 e1005169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna X, Chattopadhyay A, Sengupta D (2014). Cholesterol modulates the dimer interface of the β2‐adrenergic receptor via cholesterol occupancy sites. Biophys J 106: 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi D, Boz MB, Johnston JM, Filizola M (2015). Preferred supramolecular organization and dimer interfaces of opioid receptors from simulated self‐association. PLoS Comput Biol 11 e1004148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Provasi D, Filizola M (2015). The dynamic process of drug‐GPCR binding at either orthosteric or allosteric sites evaluated by metadynamics. Methods Mol Biol 1335: 277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider S, Provasi D, Filizola M (2016). How oliceridine (TRV‐130) binds and stabilizes a mu‐opioid receptor conformational state that selectively triggers G protein signaling pathways. Biochemistry 55: 6456–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, LeRouzic V, Schneider S, Bisignano P, Pasternak GW, Filizola M (2014). Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions. Biochemistry 53: 5140–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Yeatman HR, Provasi D, Alt A, Christopoulos A, Canals M et al. (2016). Proposed mode of binding and action of positive allosteric modulators at opioid receptors. ACS Chem Biol 11: 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J, Coop A, MacKerell AD Jr (2013). Molecular details of the activation of the mu opioid receptor. J Phys Chem B 117: 7907–7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C (2016). Opioid receptors. Annu Rev Med 67: 433–451. [DOI] [PubMed] [Google Scholar]
- Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I et al. (2013). Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science 341: 1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautermann CS, Seeliger D, Kriegl JM (2015). What can we learn from molecular dynamics simulations for GPCR drug design? Comput Struct Biotechnol J 13: 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E et al. (2012). Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485: 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Bruchas MR, Calo G, Cox BM, Zaveri NT (2016). Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacol Rev 68: 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi A, Marrone GF, Palmer TC, Narayan A, Szabo MR, Le Rouzic V et al. (2016). Mitragynine/Corynantheidine pseudoindoxyls as opioid analgesics with mu agonism and delta antagonism, which do not recruit beta‐arrestin‐2. J Med Chem 59: 8381–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery ON, Machtens J‐P, Tamburrino G, Seeliger D, Zachariae U (2016). Structural mechanisms of voltage sensing in G protein‐coupled receptors. Structure 24: 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel R, Mahalingam M, Lüdeke S, Huber T, Siebert F, Sakmar TP (2008). Functional role of the “ionic lock”—An interhelical hydrogen‐bond network in family A heptahelical receptors. J Mol Biol 380: 648–655. [DOI] [PubMed] [Google Scholar]
- Werling LL, Brown SR, Puttfarcken P, Cox BM (1986). Sodium regulation of agonist binding at opioid receptors. II. Effects of sodium replacement on opioid binding in guinea pig cortical membranes. Mol Pharmacol 30: 90–95. [PubMed] [Google Scholar]
- Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V et al. (2010). Structures of the CXCR4 chemokine GPCR with small‐molecule and cyclic peptide antagonists. Science 330: 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E et al. (2012). Structure of the human κ‐opioid receptor in complex with JDTic. Nature 485: 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Palczewski K, Peng Q, Kolinski M, Vogel H, Filipek S (2015). The mechanism of ligand‐induced activation or inhibition of mu‐ and kappa‐opioid receptors. Angew Chem Int Ed Engl 54: 7560–7563. [DOI] [PubMed] [Google Scholar]
- Yuan S, Vogel H, Filipek S (2013). The role of water and sodium ions in the activation of the mu‐opioid receptor. Angew Chem Int Ed Engl 52: 10112–10115. [DOI] [PubMed] [Google Scholar]
- Zheng H, Pearsall EA, Hurst DP, Zhang Y, Chu J, Zhou Y et al. (2012). Palmitoylation and membrane cholesterol stabilize μ‐opioid receptor homodimerization and G protein coupling. BMC Cell Biol 13: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]