

Abstract
Background and Purpose
cAMP is a central second messenger that broadly regulates cell function and can underpin pathophysiology. In chronic obstructive pulmonary disease, a lung disease primarily provoked by cigarette smoke (CS), the activation of cAMP‐dependent pathways, via inhibition of hydrolyzing PDEs, is a major therapeutic strategy. Mechanisms that disrupt cAMP signalling in airway cells, in particular regulation of endogenous PDEs, are poorly understood.
Experimental Approach
We used a novel Förster resonance energy transfer (FRET) based cAMP biosensor in mice in vivo, ex vivo precision cut lung slices (PCLS) and in human cell models, in vitro, to track the effects of CS exposure.
Key Results
Under fenoterol stimulation, FRET responses to cilostamide were significantly increased in in vivo, ex vivo PCLS exposed to CS and in human airway smooth muscle cells exposed to CS extract. FRET signals to rolipram were only increased in the in vivo CS model. Under basal conditions, FRET responses to cilostamide and rolipram were significantly increased in in vivo, ex vivo PCLS exposed to CS. Elevated FRET signals to rolipram correlated with a protein up‐regulation of PDE4 subtypes. In ex vivo PCLS exposed to CS extract, rolipram reversed down‐regulation of ciliary beating frequency, whereas only cilostamide significantly increased airway relaxation of methacholine pre‐contracted airways.
Conclusion and Implications
Exposure to CS, in vitro or in vivo, up‐regulated expression and activity of both PDE3 and PDE4, which affected real‐time cAMP dynamics. These mechanisms determine the availability of cAMP and can contribute to CS‐induced pulmonary pathophysiology.
Abbreviations
- CAG promoter
the hybrid CMV enhancer/chicken β‐actin promoter
- CFP
cyan fluorescence protein
- COPD
chronic obstructive pulmonary disease
- CS
cigarette smoke
- Epac1‐camps
exchange protein activated by cAMP type 1 based cAMP biosensor
- HASM
human airway smooth muscle cells
- PCLS
precision cut lung slices
- YFP
yellow fluorescence protein
Introduction
The ubiquitous second messenger http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2352 plays a key role in the signalling cascades that control a wide range of physiological and pathophysiological processes in the lung. Spatial and temporal localisation of intracellular cAMP concentrations are determined by the balance between generation by http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=257 and degradation by http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=260). At least two members of the PDE family, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=260#1299 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=260#1299, are pharmaco‐therapeutic targets for obstructive lung disease (Fan Chung, 2006) as they are highly expressed in airway smooth muscle and in airway epithelial cells (Rabe et al., 1993; Dent et al., 1998).
Cigarette smoke (CS) exposure is the principal risk factor for chronic obstructive pulmonary disease (COPD), which is characterized by progressive airflow limitation that is not fully reversible. COPD is associated with alterations in airway smooth muscle and epithelial cell function, a process paralleled by changes in activity and/or expression of cAMP signalling pathway effectors (Barnes, 2000; Qaseem et al., 2011). Though CS can reportedly reduce cAMP in association with activation of PDE4 in cultured human bronchial epithelial cells (Milara et al., 2014, 2012; Schmid et al., 2015), the full repertoire of mechanisms that modulate cAMP in response to CS in multi‐cellular airway are not known.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6962, an orally administered selective PDE4 inhibitor, is approved by the FDA and EMEA as a maintenance treatment for patients with severe COPD associated with bronchitis and a history of frequent exacerbations, as an add‐on to standard treatment (Abbott‐Banner and Page, 2014). However, oral PDE4 inhibitors have a limited therapeutic compliance, being associated with gastrointestinal side effects that are prohibitive for their development and have limited their wide use (Abbott‐Banner and Page, 2014). Compared to oral PDE4 inhibitors, inhaled dual PDE3/4 inhibitors are attracting considerably more interest for therapeutic use as they are more effective and well tolerated in COPD patients (Calzetta et al., 2013; Franciosi et al., 2013). Whether the efficacy of these inhibitors is linked to pathophysiological changes in PDE3 and PDE4 caused by CS exposure has not been investigated.
To elucidate the effects of CS on airway‐specific regulation of cAMP and associated signalling, we used a novel experimental system, combining precision cut lung slices (PCLS) that mimic the airway micro‐environment with Förster resonance energy transfer (FRET) that allows real‐time monitoring of cAMP in intact cells and tissues (Zaccolo and Pozzan, 2002; Sprenger and Nikolaev, 2013). We obtained PCLS from Epac1‐camps mice that express the cAMP FRET biosensor and monitored real‐time cAMP changes in response to CS in vivo and ex vivo. Human airway smooth muscle cells (HASM) and human epithelial cells (16HBE 14o−) exposed to CS served as in vitro models to assess the response to CS. To correlate cAMP changes with physiological responses, ciliary beating frequency (CBF) and airway smooth muscle tone in ex vivo PCLS exposed to CS extract were studied. We found that CS exposure in mouse tissue decreased airway cAMP levels by increasing the expression and activity of PDE3 and PDE4, thus diminishing the therapeutic efficacy of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=29 agonists. CS exposure modulates the PDE profile in a manner dependent on cell type, revealing differential regulation of PDE3 and PDE4. Inhibition of PDE4 reversed the reduction of CBF induced by exposure to CS extract. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=557, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5260 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5167 relaxed airways pre‐contracted by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7438. Only the relaxation induced by cilostamide was significantly increased in ex vivo PCLS exposed to CS extract. Our data offers a basis for more precise therapeutic strategies.
Methods
Transgenic mice
All animal care and experimental procedures complied with the animal protection and welfare guidelines and were approved by the University of Groningen Institutional Animal Care and Use Committee (Groningen, the Netherlands). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). Adult CAG‐Epac1‐camps FVB/N transgenic mice ubiquitously expressing the cAMP sensor Epac1‐camps under the control of a CAG promoter were used to monitor the global cAMP level in lung tissues (Figure 1). The generation of the CAG‐Epac1‐camps transgenic mice has been described by Calebiro et al. (2009). Mice were housed conventionally in rooms maintained at a 12‐h light/dark cycle and were provided ad libitum access to commercial diet and autoclaved water.
Figure 1.
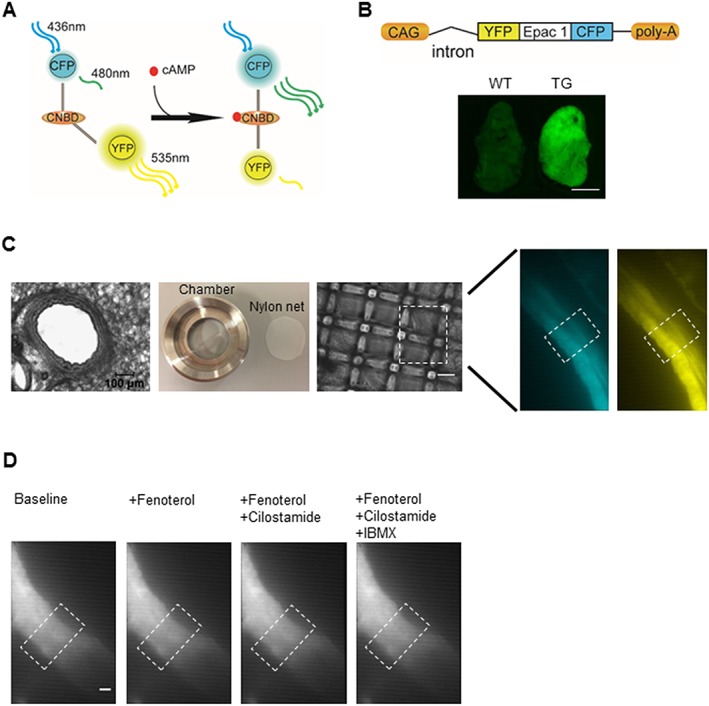
Monitoring cAMP FRET signals using lung slices of CAG‐Epac1‐camps transgenic mice. (A) An Epac1‐based FRET sensor for real‐time cAMP detection. This cAMP biosensor consists of CPF and YFP flanking a cyclic nucleotide binding domain (CNBD) from Epac1. Binding of cAMP leads to a conformational change, allowing an increase of CFP and a decrease of YFP fluorescence to be observed. (B) CAG‐Epac1‐camps transgenic mice express Epac1‐camps sensor ubiquitously, under the control of the CAG promoter. Fluorescent image of the superior lung lobe of a transgenic mouse compared with that of a wild type counterpart. Scale bar: 3 mm. (C) Slices of 200 μm thickness were cut using a vibratome. A lung slice was placed in the custom‐made image chamber, and 600 μL of FRET buffer was added into the chamber, and a home‐made net was used to fix the slice in the chamber in order to reduce airway movement artefacts. The fluorescent images (480 nm: left, 535 nm: right) of intrapulmonary airway are observed under inverted fluorescent microscope (×40). The region of interest is indicated in the white boxes. (D) Representative images showed no significant airway movement after treatment with fenoterol alone or in combination with cilostamide and IBMX. Similar images were observed with rolipram (not shown). Scale bar: 20 μm.
CS in vivo exposure
Adult CAG‐Epac1‐camps FVB/N transgenic mice (n = 6–8 per group, 18–23 weeks old) were exposed to mainstream smoke of filter‐free 3R4F research cigarettes (University of Kentucky, Lexington, USA) for four consecutive days by whole body exposure, as described previously (Kistemaker et al., 2013; Oldenburger et al., 2014). Each cigarette was smoked without a filter in 5 min at a rate of 5 L·h−1 in a ratio with 60 L·h−1 air using a peristaltic pump (45 rpm) (323 E/D; Watson Marlow, Rotterdam, the Netherlands). CS was directly distributed into a 6‐L perspex box. On day 1, mice were exposed to the smoke of one cigarette in the morning and three cigarettes in the afternoon. On days 2, 3 and 4, mice were exposed to the smoke of five cigarettes in the morning and five cigarettes in the afternoon. Control animals were handled in the same way but exposed to fresh air instead of CS. Sixteen hours after the last CS exposure, all animals were killed on day 5 by CO2, after which PCLS were prepared.
Precision cut lung slices (PCLS)
PCLS were prepared as previously described (Schlepütz et al., 2012). Briefly, adult CAG‐Epac1‐camps transgenic mice were killed by CO2, and tracheas were cannulated. Whole lungs were filled with low‐melting agarose through the cannula. As standard, we used 1.5% (w/v) low‐melting agarose for PCLS preparation. However, 3% (w/v) low‐melting agarose was used in the measurement of cAMP levels after methacholine treatment, as reported earlier (Delmotte and Sanderson, 2006). Lungs were then cooled on ice for 20 min to make sure that the agarose was solid for slicing. Afterwards, individual lung lobes were separated and fixed on the specimen holder. The cutting procedure was operated at a thickness of 200 μm with a vibratome (VT 1200S, Leica, Wetzlar, Germany) filled with 4°C cold slicing buffer (composition in mM, CaCl2·1.8, MgSO4·0.8, KCl 5.4, NaCl 116.4, NaH2PO4· 1.2, glucose 16.7, NaHCO3 26.1, HEPES 25.2, pH 7.2).
Lung slices were harvested and cultured in incubation buffer (in mM, CaCl2· 1.8, MgSO4·0.8, KCl 5.4, NaCl 116.4, NaH2PO4·1.2, glucose 16.7, NaHCO3 26.1, HEPES 25.2, sodium pyruvate 1.0, minimum essential medium (MEM) amino acids solution 1:50, MEM vitamin solution 1:100, glutamine 2.0, penicillin 50 U·mL−1, streptomycin 100 μg·mL−1, pH 7.2) in a humidified atmosphere at 37°C. In order to remove the agarose, the incubation medium was replaced every 30 min for 2 h.
FRET measurements
FRET measurements were performed using the method described by Börner et al. (2011), with minor modifications (Sprenger et al., 2015). A lung slice was placed in the Attofluor cell chamber (Invitrogen, Landsmeer, Netherlands). After washing once, 600 μL of FRET buffer (in mM, NaCl 144, KCl 5.4, MgCl2 1, CaCl2 1, HEPES 10, pH 7.3) was added into the chamber, and a home‐made net was used to fix the slice in the chamber in order to reduce airway movement artefacts. FRET response was monitored by an inverted fluorescent microscope (Nikon Ti) equipped with an oil immersion 40× objective and ImageJ software. Cyan fluorescence protein (CFP) was excited with 440 nm light from a CoolLED light source, and the images of CFP and yellow fluorescence protein (YFP) emission channels (after separation using DV2 DualView, Photometrics) were acquired by ORCA 03‐G camera (Hamamatsu, Hamamatsu, Japan) every 5 s. A total of 200 μL of different compounds, diluted with FRET buffer was added into the chamber as long as a stable baseline could be observed. All the raw data were corrected offline by the bleedthrough of the donor signal (CFP) into the acceptor signal (YFP) using the following equation: (YFP − 0.90 × CFP)/CFP.
Real‐time quantitative PCR
Total RNA was extracted from lung slices using the Maxwell 16 LEV simplyRNA Tissue Kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. The total RNA yield was determined by NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). Equal amounts of RNA were used to synthesize cDNA. An Illumina Eco Real‐Time PCR system was used to perform real‐time qPCR experiments. PCR cycling was performed with denaturation at 94°C for 30 s, annealing at 59°C for 30 s and extension at 72°C for 30 s for 45 cycles. RT‐qPCR data were analysed by LinRegPCR software 71. To analyse RT‐qPCR data, the amount of target gene was normalized to the reference genes 18S ribosomal RNA, B2M and RPL13A. Primer sequences are listed in Table 1.
Table 1.
Primer sequences used
| Species | Forward sequence 5′‐3′ | Reverse sequence 5′‐3′ | |
|---|---|---|---|
| 18S | Mus musculus | AAACGGCTACCACATCCAAG | CCTCCAATGGATCCTCGTTA |
| B2M | Mus musculus | ACCGTCTACTGGGATCGAGA | TGCTATTTCTTTCTGCGTGCA |
| RPL13A | Mus musculus | AGAAGCAGATCTTGAGGTTACGG | GTTCACACCAGGAGTCCGTT |
| PDE3A | Mus musculus | TGTTTGAAGACATGGGGCTCT | TAGAACATCGGTGGCATGGATT |
| PDE3B | Mus musculus | TGCATGCCACAGATGTCCTAC | CTGAATCTGCTTTGGTTTCCGT |
| PDE4A | Mus musculus | TATTCCAGGAGCGGGACTTAC | TGTGGACTGTAGCACATCGG |
| PDE4B | Mus musculus | GTCCAAACACATGAGCCTCCT | TACCATGTTGCGAAGAACCTGT |
| PDE4D | Mus musculus | GTTGCTCCAGGAAGAAAACTGT | CAGGTTCATGTGCTTCGACAT |
| 18S | Homo sapiens | CGCCGCTAGAGGTGAAATTC | TTGGCAAATGCTTTCGCTC |
| B2M | Homo sapiens | AAGCAGCATCATGGAGGTTTG | AAGCAAGCAAGCAGAATTTGGA |
| RPL13A | Homo sapiens | ACCGCCCTACGACAAGAAAA | GCTGTCACTGCCTGGTACTT |
| PDE3A | Homo sapiens | CAGCCTATTCCAGGCCTCTC | CCACATACAGCGCCATCAAC |
| PDE3B | Homo sapiens | GCCACAGATGTGCTACATGC | GACAGGCAGCCATAACTCTC |
| PDE4A | Homo sapiens | GGGGTGAAGACCGATCAAGA | TTATGGTAGGCCACGTCAGC |
| PDE4B | Homo sapiens | AATCTCACCAAGAAGCAGCG | AGGTCTGCACAGTGTACCAT |
| PDE4D | Homo sapiens | CACAGGTGGGCTTCATAGAC | TGACTGCCACTGTCCTTTTC |
Western blotting
Lung slices were lysed in RIPA buffer, and homogenized protein concentrations were measured by BCA protein assay (Pierce, Thermo Scientific, Landsmeer, Netherlands). Equal amounts of total protein were loaded into 8% SDS–polyacrylamide gel electrophoresis. After transferring to a nitrocellulose membrane, primary antibodies were incubated at 4°C overnight, followed by secondary antibody (anti‐mouse, 1:5000 or anti‐rabbit, 1:5000) incubation at room temperature for 1 h. Protein bands were developed on film using Western detection ECL‐plus kit (PerkinElmer, Waltman, MA). ImageJ software was used for band densitometry analysis.
CS extract preparation
CS extract was prepared as described in a previous report (Poppinga et al., 2015). Two 3R4F research cigarettes (University of Kentucky, Lexington, USA) without filter were combusted into 25 mL incubation buffer using a peristaltic pump (45 rpm, Watson Marlow 323E/D, Rotterdam, the Netherlands). After filtering through 0.22 μm filters (Millipore, Darmstadt, Germany) to remove large particles, the obtained solution was considered as 100% CS extract.
Cell culture and stimulation
HASM cells, immortalized by human telomerase reverse transcriptase were used for all the experiments (Gosens et al., 2006). Cells were maintained in DMEM (Life Technologies, Bleiswijk, Netherlands, 11965‐092) containing 10% FBS, 50 mg·mL−1 of penicillin, 50 U·mL−1 of streptomycin at 37°C, 5% CO2. HASM cells were seeded on coverslides (24 mm, Thermo Scientific) in six‐well plates at a density of 4 × 104 cells per well and incubated overnight. Adenovirus containing Epac1‐camps biosensor was used to infect HASM cells with 10 MOI for 33 h. After overnight serum‐free medium starvation, HASM cells were stimulated with or without 2.5% CS extract for 24 h until use.
The immortalised human bronchial epithelial cells (16HBE 14o−) were cultured in MEM (Life Technologies, Bleiswijk, Netherlands, 21090‐055), supplemented with 10% FBS, 50 mg·mL−1 of penicillin, 50 U·mL−1 of streptomycin and L‐glutamine (2 mM) in a humidified atmosphere of 37°C. For stimulation protocol, 16HBE 14o− cells were treated in the same way as HASM cells, but were plated at a density of 4 × 105 cells per well.
Exposure to CS extract ex vivo
After 2 h washing, slices were stimulated with medium containing CS extract (final concentration 2.5% , v/v) for 24 h, and the control group was incubated with control medium only.
Measurement of CBF with high‐speed digital microscopy
Measurement of CBF mainly followed previously described protocols (Delmotte and Sanderson, 2006; Francis and Lo, 2013). Briefly, the epithelial layers of lung slices were imaged with digital high‐speed imaging technique using a Leica DMI3000B (Leica Microsystems GmbH) inverted fluorescence microscope equipped with an oil immersion 63× objective. An optiMOS camera (Photometrics, Tucson, USA) at 200 frames s‐1 was used to capture the images. Video signals were digitized and processed using ImageJ (NIH, USA). Line tool was used to draw a raster line crossing the beating ciliary cells. After a ‘reslice’ of this line, a wave pattern could be used to measure the number of pixels (one pixel = one movie frame), from which the number of beats per minute (as Hz) can be calculated.
Functional airway contraction and relaxation studies
Functional airway relaxation studies were performed, using slices from the same mouse, on control slices and slices exposed to 2.5% CS extract for 24 h. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7438 (1 μM) was used to induce about 50% airway narrowing followed by addition of fenoterol (1 μM), cilostamide (10 μM) or rolipram (10 μM) to dilate the airways. A nylon mesh with a hole in the middle and metal washer were used to fix the lung slice, but the airways were still able to contract and relax, as described previously (Chen and Sanderson, 2017). Lung slice images were captured in time lapse (1 frame per 2 s) using a microscope (Eclipse, TS100, Nikon) equipped with a 4× objective. To quantify airway luminal area, NIS‐Elements microscope imaging software 4.5 (Nikon, Amsterdam, Netherlands) was used. Luminal area is expressed as percent basal. To analyse relaxation induced by fenoterol, cilostamide or rolipram, we initially normalized the contractile state of an airway (measured the area after fenoterol/cilostamide/rolipram treatment) to its own initial contraction (measured the area after methacholine) by the formulae: % relaxation = (areafenoterol/cilostamide/rolipram − areamethacholine)/(100 − areamethacholine).
Data and statistical analyiss
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data were analysed by Origin Pro 8.5 software (OriginLab Corporation, Northampton, MA) and are expressed as mean ± SEM. In the in vivo CS exposure model, routinely more than six animals were used. In the ex vivo experiments, more than eight independent FRET measurements were made from at least three animals per treatment group. In the in vitro experiments, at least six independent experiments were analysed in each condition. The exact numbers of slices and animals and replicates are shown on the top of bars in the graphs and indicated in the Figure legends. One‐way ANOVA was used for simple two groups comparison, whereas for the ex vivo experiments, Student's paired t‐test was used to make comparisons between two groups. P < 0.05 was considered statistically significant.
Materials
Fenoterol was purchased from Boehringer Ingelheim (Ingelheim, Germany). Rolipram, cilostamide and BSA were from Sigma‐Aldrich (St‐Louis, MO, USA). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=388 was obtained from AppliChem (Darmstadt, Germany). Minimal essential medium, high‐glucose Dulbecco's modified Eagle's medium, L‐glutamine, fungizone, HEPE's, sodium pyruvate, MEM amino acids, MEM vitamin, gentamicin, penicillin–streptomycin and trypsin were obtained from Gibco Life Technologies (Paisely, UK). FBS was from Thermo Scientific. All other substances were from Sigma‐Aldrich.
In the present study, the following antibodies were used: anti‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1298 (antibody kindly provided by Chen Yan, Aab Cardiovascular Research Institute and Department of Microbiology and Immunology, University of Rochester School of Medicine and Dentistry, Rochester, NY, 14642, USA, 1:1000), anti‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1300 (61‐4, 1:1000), anti‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1301 (113‐4, 1:1000), anti‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1303 (ICOS 4D, 1:2000) and anti‐GAPDH (HyTest, 1:10 000). Anti‐PDE4A, anti‐PDE4B and anti‐PDE4D antibodies were kind gifts from Prof. Marco Conti (Department of Obstetrics, Gynecology and Reproductive Sciences, University of California, San Francisco, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
cAMP FRET measurement on lung slices
We used transgenic mice that ubiquitously express the Epac1‐camps cAMP sensor (Calebiro et al., 2009) to monitor cytosolic cAMP levels. The cAMP biosensor, termed Epac1‐camps (Nikolaev et al., 2004; Nikolaev and Lohse, 2006), includes the cAMP binding domain of Epac1 flanked by YFP and CFP. Binding of cAMP to the sensor induces a conformational change that results in an energy transfer between the two fluorophores (Figure 1A). In Epac1‐camps mice, the cAMP biosensor sequence was integrated under the control of the hybrid CMV enhancer/chicken β‐actin (CAG) promoter (Figure 1B). This supports robust cAMP biosensor expression in the lung, as demonstrated by fluorescent images of one lung lobe using fluorescence stereomicroscopy (Figure 1B). In order to explore real‐time dynamic changes of cAMP in the airways, we prepared PCLS, which included epithelial and smooth muscle layers, from Epac1‐camps transgenic mice, fixed with a home‐made nylon net (Figure 1C). In Figure 1D, white boxes indicate the region of interest studied in the FRET measurement.
Effect of CS on PDE3 and PDE4 signals in vivo
To address the hypothesis that CS exposure alters cAMP levels in airway cells, we monitored FRET responses in PCLS from mice exposed to CS in vivo (Kistemaker et al., 2013; Oldenburger et al., 2014). For this purpose, Epac1‐camps transgenic mice were exposed to CS for 4 days, then PCLS were prepared (Figure 2A). We observed that the baseline FRET ratio was significantly lower in PCLS from CS‐exposed mice compared to those from air‐exposed animals (Figure 2B). While calibrations of the FRET baseline are challenging and these results have to take this caveat into account, the data could indicate that CS exposure decreased the intracellular cAMP levels. Thus, we studied the impact of CS on the functional contribution of PDE3 and PDE4 to basal cAMP levels (in the absence of fenoterol), treating PCLS with the selective PDE3 inhibitor, cilostamide, and the PDE4 inhibitor, rolipram (Figure 2C–G). Compared to specimens from air‐exposed mice, in PCLS from CS‐exposed mice, we observed a significantly greater increase in cAMP‐dependent FRET in response to cilostamide or rolipram treatment (Figure 2C–G).
Figure 2.
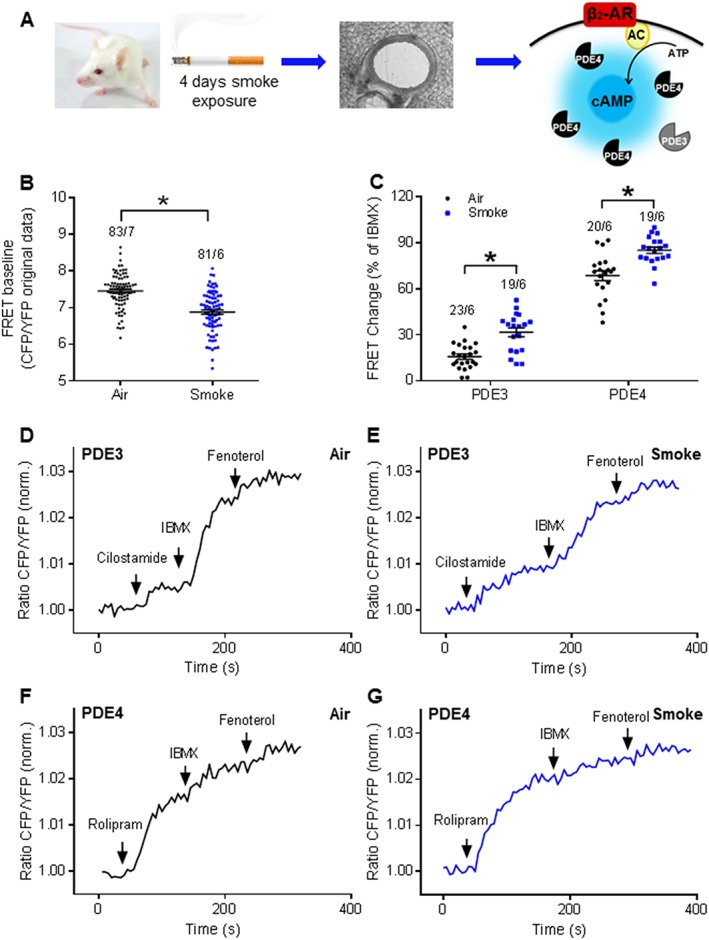
CS exposure affects PDE contributions to cAMP hydrolysis under basal conditions in the in vivo model. (A) Experimental design. Mice were exposed to the mainstream smoke of cigarettes for 4 days. Lung slices were prepared for FRET measurements with PDE3 and PDE4 selective inhibitors under basal conditions. β2‐AR, β2‐adrenoceptor; AC, adenylyl cyclase. (B) PCLS from smoke‐exposed mice showed a decreased baseline FRET ratio compared to those from air‐exposed animals, indicating decreased cAMP levels. (C) Quantification of FRET data indicated that clear effects of both PDE3 and PDE4 inhibitors on basal cAMP levels were detected after CS in vivo exposure. Representative FRET traces from slices of air‐exposed mice (D, F) or CS‐exposed mice (E, G). PDE3 and PDE4 were inhibited by cilostamide (10 μM; D, E) and rolipram (10 μM; F, G) respectively. Number of slices and mice is indicated above the bars. On average n = 19–23 independent slices from six to seven mice. Data are expressed as mean ± SEM. *P < 0.05; significantly different from air. One‐way ANOVA was used for simple two groups comparison; IBMX, 100 μM; fenoterol, 1 μM.
FRET recordings in the presence of the β2‐adrenoceptor agonist, fenoterol, and selective PDE inhibitors were made, comparing PCLS from CS‐ and air‐exposed mice (Figure 3A). The fenoterol concentration used was 1 μM (a fenoterol concentration–response curve is shown in Supporting Information Figure S1A) and IBMX was used to define the maximal response under basal conditions. Additional treatment with did not show further increase of cAMP signals (Supporting Information Figure S1K–L). FRET analysis revealed that, in response to fenoterol, cAMP concentration was significantly lower in PCLS from CS‐exposed mice (Figure 3B–F). However, inhibition of PDE3 with cilostamide increased cAMP FRET in lung preparations from CS‐exposed mice, a response that was significantly greater than a modest response observed in lungs from air‐exposed mice (Figure 3B–C, F). Similarly, PDE4 inhibition with rolipram resulted in significantly greater cAMP signals in PCLS from CS‐exposed mice (Figure 3D–G).
Figure 3.
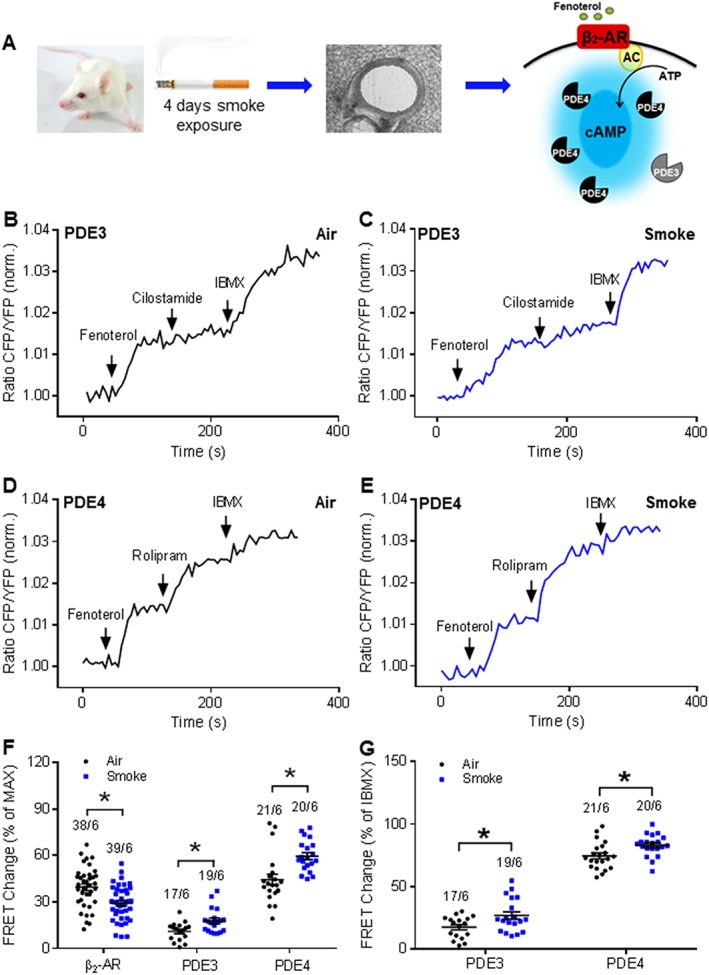
CS exposure changes PDE effects after β2‐adrenoceptor stimulation by fenoterol in the in vivo disease model. (A) Experimental design. Mice were exposed to mainstream smoke of cigarettes for 4 days. PCLS were prepared and used for FRET measurements with PDE3 and PDE4 selective inhibitors after β2‐adrenoceptor (β2‐AR) stimulation: AC, adenylyl cyclase. Representative FRET traces from slices of air‐exposed mice (B, D) or CS‐exposed mice (C, E). After treatment with fenoterol (1 μM), PDE3 and PDE4 were inhibited with 10 μM cilostamide (B, C) or 10 μM rolipram (D, E), respectively, and subsequently, the non‐selective PDE inhibitor, IBMX (100 μM), was used to define the maximal FRET response. After CS exposure, quantification of FRET data revealed an increase of both PDE3 and PDE4 contributions to cAMP hydrolysis in the presence of fenoterol. Decrease of fenoterol‐induced FRET response was also observed in the CS‐exposed group compared with the air‐exposed group. Maximal FRET response was considered as the response of fenoterol, cilostamide/rolipram and IBMX. IBMX FRET response was calculated as the response of cilostamide/rolipram and IBMX. Number of slices and mice is indicated above the bars. On average n = 17–39 independent slices from six mice have been used. Data are expressed as mean ± SEM. *P < 0.05; significantly different from air; One‐way ANOVA was used for simple two groups comparison.
Next, we measured mRNA and protein abundance of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1298, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1300, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1301 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1303, and mRNA of http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=260#1299, in PCLS from CS‐ and air‐exposed mice. The specificity of antibodies for the PDEs of interest was verified by studies in knockout mice (Supporting Information Figure S1F–I). Quantitative real‐time PCR revealed PDE4D was the predominant isoform in the lung. We found that CS exposure resulted in a significant increase in PDE4D and PDE4B, but not β2‐adrenoceptor mRNA (Figure 4A) and their protein (Figure 4D–E). Interestingly, mRNA of PDE3A and PDE3B and protein abundance of PDE3A were not altered by CS exposure (Figure 4A–C).
Figure 4.
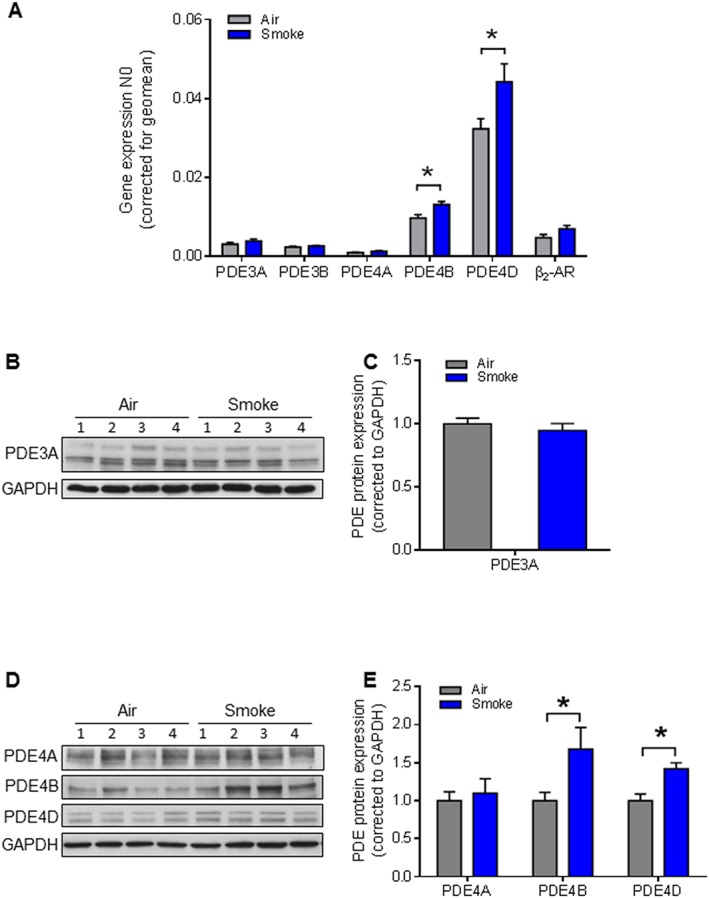
Effects of CS exposure on gene and protein expression of PDE3 and PDE4 in the in vivo model. (A) Gene expression of PDE3 (PDE3A and PDE3B), PDE4 subfamilies (PDE4A, PDE4B and PDE4D) and β2‐adrenoceptor) were analysed by real‐time quantitative PCR in lung slice lysates. CS increased the gene expressions of both PDE4B and PDE4D. Data presented are from at least seven animals. (B‐E) Protein expression of PDE3A and PDE4 subfamilies (PDE4A, PDE4B and PDE4D) was determined by Western blot analysis in lung homogenates. Quantification and representative blots are shown. PDE3A: air n = 6, smoke n = 6; PDE4A, PDE4B and PDE4D: air n = 6, smoke n = 6. Data are expressed as mean ± SEM, *P < 0.05; significantly different from air; One‐way ANOVA was used for simple two groups comparison.
Effect of CS extract on PDE3 and PDE4 signals in HASM cells and 16HBE 14o − cells
To more directly investigate the effects of CS on PDE signals in structural cells from lung, we used Epac1‐camps‐transduced HASM cells exposed to CS extract for 24 h. We measured FRET responses in HASM cells in the absence of fenoterol and found that CS extract exposure led to significantly higher cAMP FRET in responses to cilostamide and rolipram compared with control HASM cultures (Figure 5A, B). The fenoterol concentration used in HASM cells was 100 nM (fenoterol concentration–response curve, see Supporting Information Figure S1B). We next measured the effects of PDE3 and PDE4 inhibition on fenoterol‐induced cAMP‐dependent FRET responses. Interestingly, with PDE3 inhibition (cilostamide), CS extract exposure was sufficient to significantly increase cAMP‐dependent FRET, normalized to maximal and to IBMX‐induced FRET responses (Figure 5C–D). Although we observed a similar increase cAMP with PDE4 inhibition (rolipram) in HASM exposed to CS extract, this was only evident when normalized as percent IBMX‐induced FRET (Figure 5C–D). Quantitative real‐time PCR further revealed that exposure to CS extract resulted in significantly increased mRNA for PDE3B and PDE4D (Figure 5E), and immunoblotting revealed that PDE3A and PDE4D protein abundance was significantly increased in HASM after exposure to CS extract (Figure 5F–G).
Figure 5.
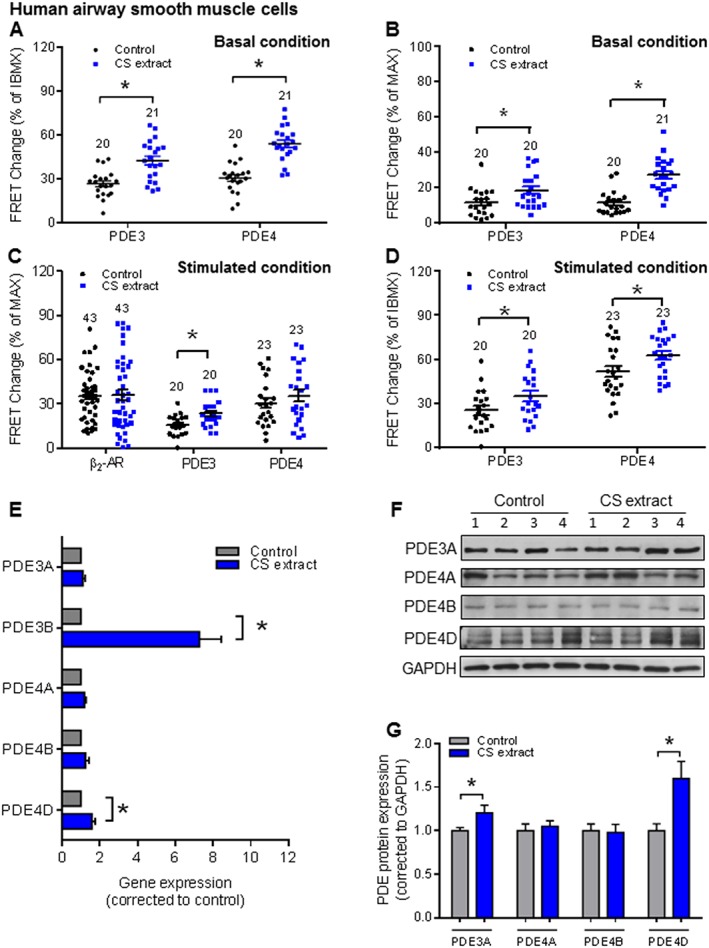
CS extract induces PDE3 and PDE4 changes in HASM. HASM cells were incubated with control medium or medium containing CS extract (2.5% v/v; final concentration) for 24 h. FRET data analysis of HASM (A–D) under basal conditions (A, B) and fenoterol stimulated conditions (C, D). Quantification of basal PDE inhibitor responses revealed that both PDE3 and PDE4 were significantly up‐regulated by CS extract (A, B). We observed a significant increase of PDE3 contributions induced by CS extract after β2‐adrenoceptor stimulation, while fenoterol‐induced and rolipram‐induced FRET responses were not affected (C). In HASM cells, CS extract significantly increased both PDE3 and PDE4 profiles in total PDEs (D). Number of independent experiments is indicated above the bars. (E) Gene expression of PDE3 (PDE3A and PDE3B) and PDE4 subfamilies (PDE4A, PDE4B and PDE4D) was analysed by real‐time quantitative PCR in lung slice lysates. CS extract increased the gene expressions of both PDE3B and PDE4D. (F, G) Protein expression of PDE3A and PDE4 subfamilies (PDE4A, PDE4B and PDE4D) were determined by Western blot analysis in lung homogenates. Quantification and representative blots are shown. Data are from nine independent experiments. Data are expressed as mean ± SEM, *P < 0.05; significantly different from control; One‐way ANOVA was used for simple two groups comparison. Rolipram, 10 μM; cilostamide, 10 μM; IBMX, 100 μM; fenoterol, 100 nM for HASM cells.
We also used 16HBE 14o− cells transduced with the Epac1‐camps cAMP sensor to measure effects of exposure to CS extract . In the absence of fenoterol, after exposure to CS extract, we did observe a significantly increased cAMP‐dependent FRET in response to PDE4 inhibition, but not to PDE3 inhibition (Figure 6A, B). the fenoterol concentration used in 16HBE 14o− cells was 1 nM (fenoterol concentration–response curve, see Supporting Information Figure S1C). In contrast to HASM, for epithelial cells, we found that pretreatment with CS extract had no effect on the FRET response to PDE3 or PDE4 inhibitors in the presence of fenoterol (Figure 6C–D). Furthermore, although exposure to CS extract led to accumulation of PDE4D mRNA (Figure 6E–G), the mRNA and protein for PDE3 and protein for PDE4 were not altered by such exposure of epithelial cells (Figure 6E–G).
Figure 6.
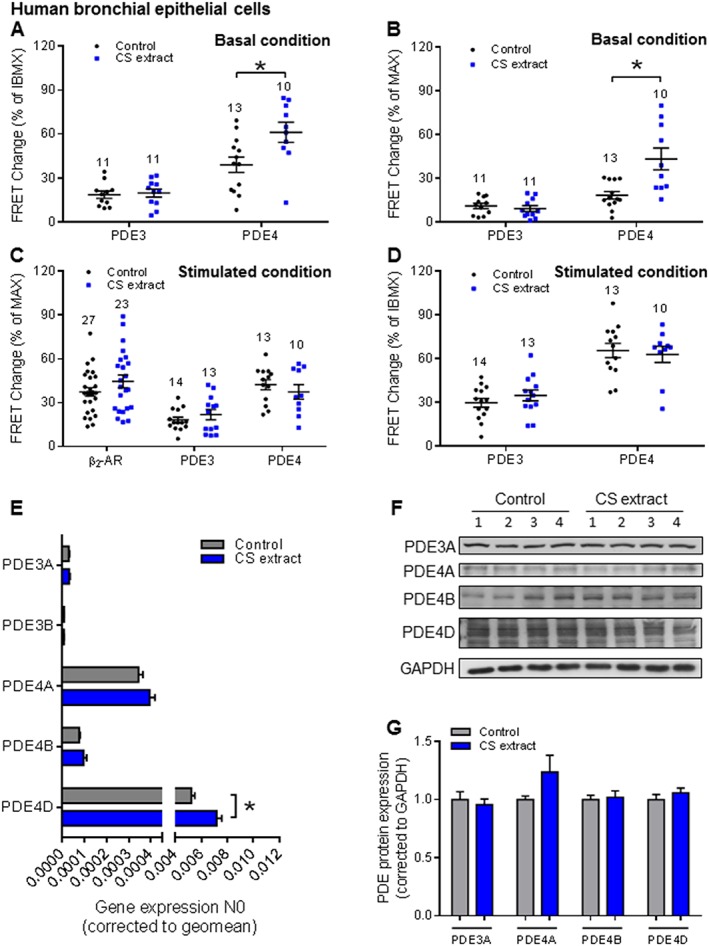
CS extract induces PDE3 and PDE4 changes in 16HBE 14o− cells. 16HBE 14o− cells were incubated with control medium or medium containing CS extract (2.5% v/v; final concentration), for 24 h. FRET data analysis of 16HBE 14o− (A–D) under basal conditions (A, B) and fenoterol stimulated conditions (C, D). Under the basal condition, clear increases of the PDE4, but not PDE3, on basal cAMP levels were detected after CS extract treatment (A, B). FRET experiments stimulated with fenoterol and different PDE inhibitors showed that after β2‐adrenoceptor stimulation, none of the different treatments with cilostamide or rolipram was affected by CS extract (C, D). Number of independent experiments is indicated above the bars. (E) Gene expression of PDE3 (PDE3A and PDE3B) and PDE4 subfamilies (PDE4A, PDE4B and PDE4D) was analysed by real‐time quantitative PCR in lung slice lysates. CS increased the gene expression PDE4D. (F, G) Protein expression of PDE3A and PDE4 subfamilies (PDE4A, PDE4B and PDE4D) was determined by Western blot analysis in lung homogenates. Quantification and representative blots are shown. Data shown are from six independent experiments. Data are expressed as mean ± SEM, *P < 0.05; significantly different from control. One‐way ANOVA was used for simple two groups comparison; Rolipram, 10 μM; cilostamide, 10 μM; IBMX, 100 μM; fenoterol, 1 nM for 16HBE 14o− cells.
Effect of CS extract on PDE3 and PDE4 signals in ex vivo
To further elucidate our findings that exposure to CS directly induces PDE changes in airway structural cells, we next performed studies to assess effects of 24 h exposure to CS extract ex vivo using PCLS from Epac1‐camps mice (Figure 7A). Notably, the basal cAMP dependent FRET response to cilostamide was significantly increased after this exposure (Figure 7B, C, F), a finding that is consistent with results using both PCLS from CS‐exposed mice (Figure 2C) and CS extract challenged HASM cells (Figure 5A, B). We also observed a clear increase in PDE4 inhibitor‐induced cAMP FRET after exposing PLCS to CS extract (Figure 7D–F), an observation that is in line with results from CS‐exposed mice and HASM and 6HBE 14o− cells exposed to CS extract (Figures 2C; 5A, B and 6A, B).
Figure 7.
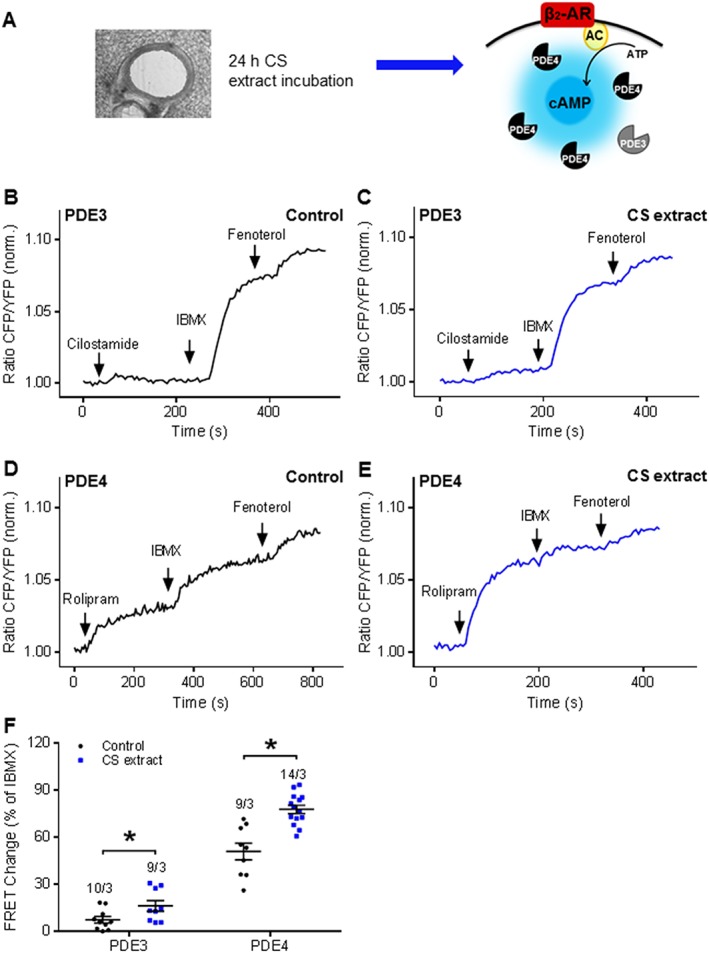
CS extract alters PDE contributions to cAMP hydrolysis under basal conditions in the ex vivo model. (A) Experimental design. Lung slices were treated with CS extract ex vivo for 24 h, and PDE3 and PDE4 inhibitor effects were monitored under the basal conditions. β2‐AR, β2‐adrenoceptor; AC, adenylyl cyclase. (B, D) Representative FRET traces from slices after 24 h incubation with control medium or (C, E) with medium containing CS extract (2.5% v/v; final concentration). After basal inhibition of PDE3 and PDE4 by using selective inhibitors, cilostamide (B, C) and rolipram (D, E) respectively. (F) Quantification of FRET data demonstrated a significant increase of PDE4 and PDE3 contributions to basal cAMP hydrolysis after treatment with medium containing CS extract (2.5%). Number of slices and mice is indicated above the bars. On average n = 9–14 independent slices from three mice have been used. Data are expressed as mean ± SEM, *P < 0.05; significantly different from control. One‐way ANOVA was used for simple two groups comparison; Cilostamide, 10 μM; rolipram, 10 μM; IBMX, 100 μM; fenoterol, 1 μM.
We also explored the contribution of PDE3 and PDE4 to fenoterol‐induced cAMP levels and the effects of exposure to CS extract. Consistent with our results using lung preparations from CS‐exposed mice (Figure 3F, G), in PLCS exposed to CS extract and stimulated with fenoterol, only the PDE3 inhibitor cilostamide induced a significant increase in total cAMP‐dependent FRET (Figure 8B, C, F), which was even more pronounced when we focused on the relative PDE contribution (Figure 8G). Notably, neither fenoterol alone nor PDE4 inhibition with rolipram revealed any difference between cAMP FRET responses in PCLS in control media or after exposure to CS extract(Figure 8D–G).
Figure 8.
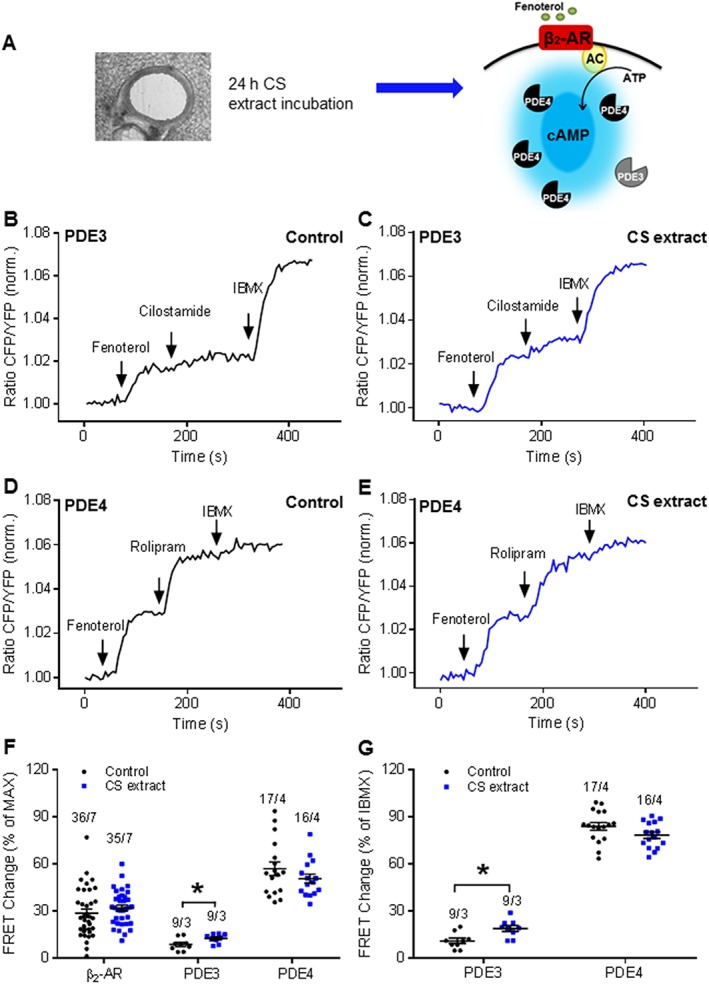
CS extract induces local PDE changes in the presence of fenoterol in the ex vivo model. (A) Experimental design. Lung slices were treated with CS extract ex vivo exposure for 24 h, and PDE3 and PDE4 FRET activities were monitored after β2‐adrenoceptor stimulation with fenoterol. β2‐AR, β2‐adrenoceptor; AC, adenylyl cyclase. (B, D) Representative FRET traces from slices after 24 h incubation with control medium or (C, E) medium containing CS extract (2.5% v/v; final concentration) . After treatment with fenoterol, PDE3 and PDE4 were inhibited with cilostamide (B, C) and rolipram (D, E) respectively. Finally, the non‐selective PDE inhibitor IBMX was used to inhibit multiple PDEs. (F, G) Quantification of FRET experiments is shown in B–E. Maximal FRET response was considered as the response of fenoterol, cilostamide/rolipram and IBMX. IBMX FRET response was calculated as the response of cilostamide/rolipram and IBMX. It indicated a significant increase of PDE3 contribution to cAMP hydrolysis after β2‐adrenoceptor stimulation. No differences could be obtained on the PDE4 level in either maximal FRET response or IBMX‐induced FRET change. Number of slices/mice is indicated above the bars. On average n = 9–36 independent slices from three to seven mice have been used. Data are expressed as mean ± SEM, *P < 0.05; significantly different from control; One‐way ANOVA was used for simple two groups comparison. Fenoterol, 1 μM; cilostamide, 10 μM; rolipram, 10 μM; IBMX, 100 μM.
Furthermore, we observed that, in PCLS exposed to CS extract, the abundance of both mRNA (PDE4A, PDE4D, β2‐adrenoceptor) (Figure 9A) and protein (Figure 9D, E) (PDE4D) were increased, as we observed no effects of exposure to CS extract on mRNA or protein levels of PDE3A (Figure 9A–C).
Figure 9.
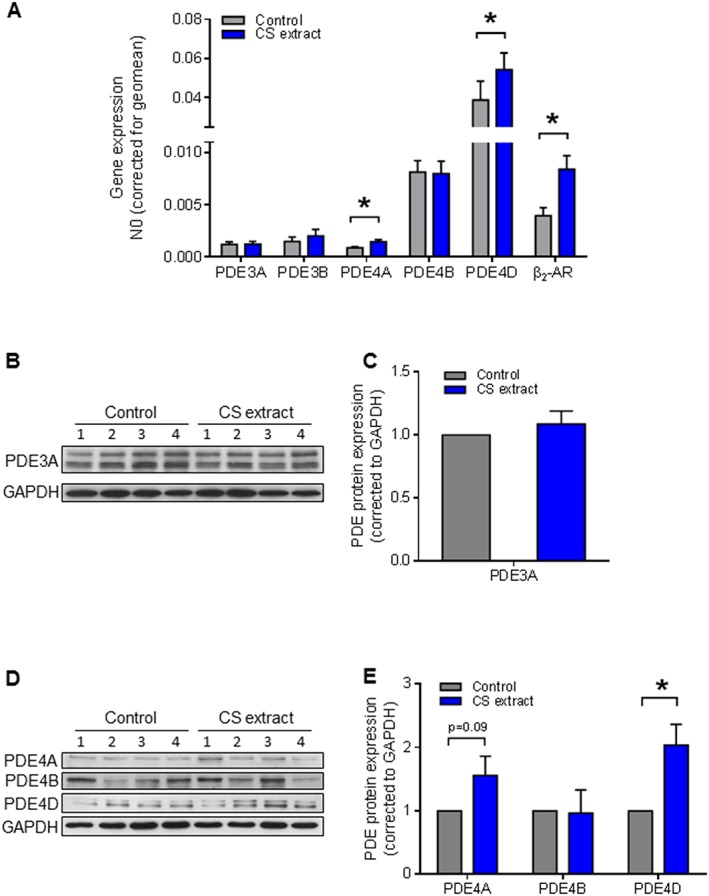
CS extract increases, primarily, RNA and protein expression of PDE4 in the ex vivo model. (A) Gene expression of PDE3 isoforms (PDE3A and PDE3B) PDE4 isoforms (PDE4A, PDE4B and PDE4D) and β2‐AR were analysed by real‐time quantitative PCR in lysates of lung slice treatment with or without CS extract for 24 h. PDE4A, PDE4D and β2‐AR were up‐regulated by CS. Data shown are from six animals. (B–E) Protein expression of PDE3A and PDE4 isoforms (PDE4A, PDE4B and PDE4D) were determined by Western blot analysis in lung homogenates. Quantification and representative blots are shown. PDE3A: air n = 4, smoke n = 4; PDE4A, PDE4B and PDE4D: air n = 6, smoke n = 6. Data are expressed as mean ± SEM, *P < 0.05; significantly different from control; paired t‐test.
Physiological responses of epithelial and ASM layers in ex vivo PCLS
To correlate cAMP changes with physiological responses, we measured CBF, as an indication of epithelial cell function (Figure 10A, B) and relaxation of methacholine pre‐contracted airways, as an indication of ASM function (Figure 10E, F) in ex vivo PCLS exposed to CS extract. As reported earlier (Milara et al., 2012), CBF was reduced significantly by the CS extract (Figure 10B) and the PDE4 inhibitor rolipram (10 μM) fully reversed the reduction of CBF (Figure 10B). As illustrated in Figure 10C, methacholine did not significantly alter cAMP levels in both control and CS extract‐exposed airways. In addition, methacholine did not affect cAMP FRET responses induced by fenoterol, cilostamie or rolipram (Figure 10D). As shown in Figure 10E, methacholine pre‐contracted airways were relaxed (although to a different degree) with fenoterol, cilostamide or rolipram in ex vivo PCLS, exposed to both control and CS extract. However, only the airway relaxation induced by cilostamide was significantly increased after exposure to CS extract, compared with the control group, and no differences were observed for either fenoterol or rolipram treated airways (Figure 10F).
Figure 10.
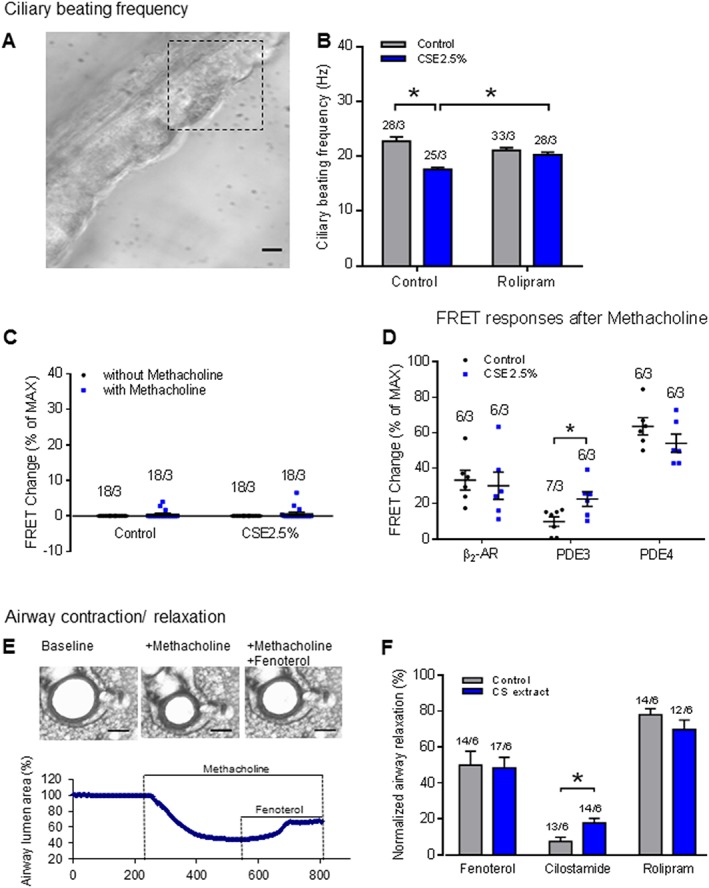
CS extract induces airway physiological responses: effects on ciliary beating frequency (CBF) and airway dilution. (A, B) Measurement of the effect of CS extract on CBF in airways of mice. (A) Epithelial layers of lung slices were imaged with digital high‐speed imaging technique. Region of interest used to measure CBF is indicated. Scale bar: 20 μm. (B) exposure of PCLS to CS extract for 24 h significantly decreased CBF. The PDE4 inhibitor rolipram (10 μM) reversed the CBF reduction. On average n = 25–33 independent slices from three mice have been used. Data are expressed as mean ± SEM, *P < 0.05; significantly different from control. One‐way ANOVA was used for simple two groups comparison. (C) Methacholine pre‐contraction did not affect the cAMP levels in the airways. (D) FRET responses to fenoterol, cilostamide and rolipram after pre‐contraction induced by methacholine indicated a trend, similar to the responses from not‐contracted slices. Quantification of FRET data demonstrated a significant increase of PDE3 contributions to basal cAMP hydrolysis after treatment with medium containing CS extract (2.5% v/v; final concentration). (E, F) Measurement of the effect of ex vivo CS extract exposure on airway contraction/relaxation in mice. (E) Illustration of the methodology used in airway contraction/relaxation study. Methacholine (1 μM) was used to induce about 50% airway narrowing followed by addition of fenoterol (1 μM), cilostamide (10 μM) or rolipram (10 μM) to dilate the airways. The percentage relaxation induced by each compound is normalized to its own control contraction (measured after methacholine treatment). Scale bar: 200 μm. (F) Methacholine pre‐contracted airways were relaxed (although to a different degree) with fenoterol, cilostamide or rolipram in ex vivo PCLS exposed to both control and CS extract. However, only airway relaxation induced by cilostamide was dramatically (by about 235%) increased after exposure to CS extract. Number of slices and mice is indicated above the bars. On average n = 12–17 independent slices from six mice have been used. Data are expressed as mean ± SEM, *P < 0.05; significantly different from control. One‐way ANOVA was used for simple two groups comparison.
Discussion
To evaluate the effect of the most important risk factor for COPD, CS, on regulation of intracellular cAMP in airway cells, we monitored real‐time changes in cAMP levels in mouse PLCS and cultured human ASM and epithelial cells. Our work showed that CS caused an increased capacity of PDE3 and PDE4 activity that was likely to explain the alterations of intracellular cAMP levels resulting from CS exposure. Indeed, we demonstrated that CS altered the PDE expression profile, increasing PDE4 mRNA, protein and activity whereas, although the activity of PDE3 was increased, expression of this isoform was not changed by exposure to CS. Furthermore, we showed that exposure to CS extract altered the PDE activity profile in a manner dependent on cell type, with cilostamide‐sensitive PDE3 activity primarily increased in human ASM cells, whereas rolipram‐sensitive PDE4 activity was increased in both human ASM and 16HBE 14o− cells. Consistent with these findings, inhibition of PDE4 reversed the reduction of CBF induced by exposure to CS extract. In addition, fenoterol, rolipram and cilostamide relaxed airways pre‐contracted by methacholine, but only airway relaxation induced by cilostamide was increased in mouse PCLS exposed to CS extract.
It is difficult to monitor intracellular cAMP levels and dynamics using standard biochemical techniques. Therefore, biophysical methods, including FRET‐based biosensors, have been developed to facilitate real‐time measurement. FRET allows the visualization of cAMP fluctuations in living cells with high temporal and spatial resolution (Adams et al., 1991; DiPilato et al., 2004; Nikolaev et al., 2004; Violin et al., 2008; Sprenger et al., 2015). Although FRET has previously been used to estimate cAMP levels in human airway epithelial cells (Schmid et al., 2006, 2015), airway smooth muscle cells (Billington and Hall, 2011) and endothelial cells (Yañez‐Mó et al., 2008), real‐time monitoring cAMP levels in the lung tissue has not been reported so far. Due to the fact that PCLS retain the complex micro‐composition and environment of the airways, they are recognized as a reliable model to study airway responsiveness and drug toxicity (Schlepütz et al., 2012; Oenema et al., 2013; Watson et al., 2015). In the present study, we report that PCLS from Epac1‐camps FRET reporter mice represent a useful model to monitor airway cAMP levels in real‐time. Importantly, using PCLS ex vivo, we have revealed that CS extract exposure largely mimics results using lung preparations from live mice pre‐exposed to CS (Figures 2, 3, 4, Figures 7, 8, 9). Our data indicate that the combination between FRET measurements and PCLS offers a unique opportunity to study the airway as a whole structural unit. Intriguingly, we were able to monitor cellular cAMP dynamics with distinct functional responses.
Active cigarette smoking is the most common risk factor in COPD, and it is associated with accelerated decline in FEV1 (forced expiratory volume in 1 s) and a higher mortality rate in COPD patients (Tamimi et al., 2012). In animal studies, CS exposure leads to a reduced lung function, emphysema, mucus hypersecretion and induction of proinflammatory processes (Rangasamy et al., 2009; Rinaldi et al., 2012; Oldenburger et al., 2014; Page, 2014; Heulens et al., 2015). PDE4 inhibitors are currently used for the treatment of COPD, and additional compounds are under development (Page, 2014). The PDE4 inhibitor roflumilast N‐oxide partly reverses CS‐induced epithelial dysfunction (Milara et al., 2014, 2012; Schmid et al., 2015; Tyrrell et al., 2015), and the PDE4 inhibitors, GPD‐1116 and piclamilast can prevent the development of CS‐induced emphysema and pulmonary hypertension in mice (Mori et al., 2008; Seimetz et al., 2015, p. 4). Zl‐n‐91, a selective PDE4 inhibitor can also suppress CS‐induced lung inflammatory in rats (Wang et al., 2010; Bucher et al., 2016). The aim of the present study was to investigate whether CS may directly induce changes in PDE expression or activity, thus contributing to lung pathophysiology.
Using an acute CS exposure in vivo and ex vivo exposure models, we demonstrate that CS increased the expression and activity of PDE4. This up‐regulation was chiefly associated with increased PDE4D mRNA and protein. Our findings are in line with previous reports that PDE4 is increased in mice that were prenatally exposed to cigarette smoke (Singh et al., 2009, p. 5, 2003). Moreover, in a genome‐wide association study, Yoon and co‐workers identified a novel single nucleotide polymorphism in the PDE4D gene, rs16878037, which is significantly associated with a risk of COPD (Yoon et al., 2014). As part of this effort, our new findings point to a key role of PDE4D as a contributor to CS‐induced lung diseases. We observed an increase in PDE4A and PDE4B in the CS exposure models, and we thus propose that PDE4 subtypes may be effective drug targets, for example, to improve cilia motility of ciliated cells (Milara et al., 2012), to inhibit the secretion of proinflammatory cytokines (Jin and Conti, 2002; Ariga et al., 2004; Ma et al., 2014) and to diminish cell proliferation (Selige et al., 2011). To correlate cAMP changes with physiological responses, we measured CBF, as a marker for epithelial function. We found that inhibition of PDE4 fully reversed CBF down‐regulation induced by ex vivo exposure to CS extract (Figure 10B), which was in agreement with previous studies (Milara et al., 2012).
Next to PDE4, PDE3 is the primary PDE in airway smooth muscle cells (Rabe et al., 1993; Page and Spina, 2012) and PDE3 inhibitors induce ASM relaxation in vitro (Rabe et al., 1993; Schmidt et al., 2000) and in vivo (Hirota et al., 2001). Even though RPL554 is considered as one of the most selective PDE3 inhibitors based on an about 3000× higher IC50 value for purified human platelet PDE3 compared to neutrophil PDE4 (PDE3: 0.4 nM; PDE4:1479 nM) (Boswell‐Smith et al., 2006), a growing body of evidence suggest that dual inhibition of PDE3 and PDE4, using higher levels of inhibitor (10 μM or 0.018 mg·kg−1), can more effectively induce bronchodilation with potential additive effects of suppressing release of inflammatory mediators (Calzetta et al., 2013; Franciosi et al., 2013). Moreover, dual inhibition of PDE3 and PDE4 sensitized cells to long acting β2‐adrenoceptor agonists (LABAs) and corticosteroid (ICS)/LABA combinations, in cell‐based models (BinMahfouz et al., 2015), indicating that dual inhibition of PDE3 and PDE4 acted as an add‐on effect to LABAs and ICS/LABA combinations, to further enhance their therapeutic benefits (Giembycz and Maurice, 2014). Here, we demonstrate that PDE3 activity, together with PDE4, is up‐regulated by exposure to CS. However, the effect on PDE3 was not associated with accumulation of PDE3 protein. Intriguingly, we also observed that only airway relaxation to cilostamide was increased in ex vivo PCLS exposed to CS extract. Thus, our findings indicate that whereas both PDE3 and PDE4 are involved in CS induced cAMP dynamics, the mechanism(s) for the effects of CS on PDE3 and PDE4 is not completely overlapping. Considering that PDE3 also plays a central role in balancing http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2347/cAMP crosstalk (Murray et al., 2002), even though our current experimental design do not allow any conclusive statement on the effects of CS on cGMP levels, our data may indicate that CS simultaneously affected the modulatory mechanisms for both cAMP and cGMP.
In summary, our study demonstrated that CS decreased the airway cAMP levels by distinct up‐regulation of PDE3 and PDE4, in whole tissue and in airway smooth muscle and epithelial cells. CS‐induced changes in cAMP levels by PDE3 and PDE4 may thus contribute to the increased beneficial effect of cAMP. In addition, inhibition of PDE3 and PDE4 exerted benefit on distinct physiological responses of epithelial (CBF) and smooth muscle layers (airway relaxation) in PCLS from mice. Thus, PDE3 and PDE4 represent promising targets for further development of precise and effective therapeutics.
Author contributions
M.S. and V.O.N. designed the project. H.Z. conceived study design, performed live tissue and cell FRET imaging, immunoblot, real‐time PCR experiments and manuscript writing. B.H. and W.J.P. conceived experiments, study design and technique support. L.R. performed live tissue FRET imaging and real time PCR experiments. L.E.M.K. and R.G. conceived study design. A.J.H. provided HASM cell line. All authors reviewed the manuscript and conceived manuscript polishing.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 (A‐C) Fenoterol concentration‐responses in mouse PCLS and human airway structural cells. Mouse PCLS (A), human airway structural cells HASM (B) and 16 HBE 14o‐ cells (C) were stimulated with the indicated fenoterol concentrations. EC50 values: mouse PCLS, 0.3±0.09 μM (n=8 independent slices from 3 mice); HASM, 19.0±3.3 nM (n=8 independent cells); 16 HBE 14o‐, 3,7±0,76 nM (n=7 independent cells). (D) Cilostamide concentration‐response in mouse PCLS model (EC50 value: 0.51±0.2 μM, n=9 independent slices from 3 mice). (E) Rolipram concentration‐response in mouse PCLS model (EC50 value: 0.11±0.03 μM, n=9 independent slices from 3 mice). (F‐I) Specificity of antibodies for PDE3 and PDE4. The lung tissues from specific PDE isoform knockout mice were lysed followed by western blotting (F‐I) to determine the antibodies specificity. (J) Representative FRET trace in PCLS. After treatment with fenoterol (1 μM), rolipram (10 μM) was added followed by addition of the non‐selective PDE inhibitor IBMX (100 μM) and forskolin (10 μM) to define the maximal FRET response. (K) Forskolin stimulation did not give any further increase after fenoterol and IBMX treatment in both control and CS extract exposed PCLS, indicating that fenoterol and IBMX induce a maximal FRET response. Fenoterol+IBMX+forskolin was considered as 100%; n=16 independent slices from 4 mice. Data are expressed as mean ± SEM.
Acknowledgements
We thank Sophie Sprenger for support with Western blotting, Sophie T. Bos for the CS in vivo exposure experiment and Marco Conti for anti‐PDE4A, anti‐PDE4B and anti‐PDE4D antibodies. The immunoblot data showing specificity of PDE4A antibody were kindly provided by Marco Conti. Also, we would like to thank Alex KleinJan for PDE3 KO mouse tissue to test the antibody specificity and Michaela Schweizer for sharing the vibratome used in PCLS preparation. This work was supported by the Ubbo Emmius Programme (grant to H.Z.), the Deutsche Forschungsgemeinschaft (grant to M.S.) and the Gertraud und Heinz‐Rose Stiftung (grant to V.O.N.).
Zuo H., Han B., Poppinga W. J., Ringnalda L., Kistemaker L. E. M., Halayko A. J., Gosens R., Nikolaev V. O., and Schmidt M. (2018) Cigarette smoke up‐regulates PDE3 and PDE4 to decrease cAMP in airway cells, British Journal of Pharmacology, 14, 2988–3006, https://doi.org/10.1111/bph.14347.
References
- Abbott‐Banner KH, Page CP (2014). Dual PDE3/4 and PDE4 inhibitors: novel treatments for COPD and other inflammatory airway diseases. Basic Clin Pharmacol Toxicol 114: 365–376. [DOI] [PubMed] [Google Scholar]
- Adams SR, Harootunian AT, Buechler YJ, Taylor SS, Tsien RY (1991). Fluorescence ratio imaging of cAMP in single cells. Nature 349: 694–697. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga M, Neitzert B, Nakae S, Mottin G, Bertrand C, Pruniaux MP et al (2004). Nonredundant function of phosphodiesterases 4D and 4B in neutrophil recruitment to the site of inflammation. J Immunol Baltim Md 1950 173: 7531–7538. [DOI] [PubMed] [Google Scholar]
- Barnes PJ (2000). Chronic obstructive pulmonary disease. N Engl J Med 343: 269–280. [DOI] [PubMed] [Google Scholar]
- Billington CK, Hall IP (2011). Real time analysis of β(2)‐adrenoceptor‐mediated signaling kinetics in human primary airway smooth muscle cells reveals both ligand and dose dependent differences. Respir Res 12: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BinMahfouz H, Borthakur B, Yan D, George T, Giembycz MA, Newton R (2015). Superiority of combined phosphodiesterase PDE3/PDE4 inhibition over PDE4 inhibition alone on glucocorticoid‐ and long‐acting β2‐adrenoceptor agonist‐induced gene expression in human airway epithelial cells. Mol Pharmacol 87: 64–76. [DOI] [PubMed] [Google Scholar]
- Börner S, Schwede F, Schlipp A, Berisha F, Calebiro D, Lohse MJ et al (2011). FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat Protoc 6: 427–438. [DOI] [PubMed] [Google Scholar]
- Boswell‐Smith V, Spina D, Oxford AW, Comer MB, Seeds EA, Page CP (2006). The pharmacology of two novel long‐acting phosphodiesterase 3/4 inhibitors, RPL554 [9,10‐dimethoxy‐2(2,4,6‐trimethylphenylimino)‐3‐(n‐carbamoyl‐2‐aminoethyl)‐3,4,6,7‐tetrahydro‐2H‐pyrimido[6,1‐a]isoquinolin‐4‐one] and RPL565 [6,7‐dihydro‐2‐(2,6‐diisopropylphenoxy)‐9,10‐dimethoxy‐4H‐pyrimido[6,1‐a]isoquinolin‐4‐one]. J Pharmacol Exp Ther 318: 840–848. [DOI] [PubMed] [Google Scholar]
- Bucher H, Duechs MJ, Tilp C, Jung B, Erb KJ (2016). Tiotropium attenuates virus‐induced pulmonary inflammation in cigarette smoke‐exposed mice. J Pharmacol Exp Ther 357: 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C et al (2009). Persistent cAMP‐signals triggered by internalized G‐protein‐coupled receptors. PLoS Biol 7: e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzetta L, Page CP, Spina D, Cazzola M, Rogliani P, Facciolo F et al (2013). Effect of the mixed phosphodiesterase 3/4 inhibitor RPL554 on human isolated bronchial smooth muscle tone. J Pharmacol Exp Ther 346: 414–423. [DOI] [PubMed] [Google Scholar]
- Chen J, Sanderson MJ (2017). Store‐operated calcium entry is required for sustained contraction and Ca2+oscillations of airway smooth muscle. J Physiol 595: 3203–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmotte P, Sanderson MJ (2006). Ciliary beat frequency is maintained at a maximal rate in the small airways of mouse lung slices. Am J Respir Cell Mol Biol 35: 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent G, White SR, Tenor H, Bodtke K, Schudt C, Leff AR et al (1998). Cyclic nucleotide phosphodiesterase in human bronchial epithelial cells: characterization of isoenzymes and functional effects of PDE inhibitors. Pulm Pharmacol Ther 11: 47–56. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J (2004). Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A 101: 16,513–16,518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Chung K (2006). Phosphodiesterase inhibitors in airways disease. Eur J Pharmacol 533: 110–117. [DOI] [PubMed] [Google Scholar]
- Franciosi LG, Diamant Z, Banner KH, Zuiker R, Morelli N, Kamerling IMC et al (2013). Efficacy and safety of RPL554, a dual PDE3 and PDE4 inhibitor, in healthy volunteers and in patients with asthma or chronic obstructive pulmonary disease: findings from four clinical trials. Lancet Respir Med 1: 714–727. [DOI] [PubMed] [Google Scholar]
- Francis R, Lo C (2013). Ex vivo method for high resolution imaging of cilia motility in rodent airway epithelia. J Vis Exp JoVE . https://doi.org/10.3791/50343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giembycz MA, Maurice DH (2014). Cyclic nucleotide‐based therapeutics for chronic obstructive pulmonary disease. Curr Opin Pharmacol, Respiratory Musculoskeletal 16: 89–107. [DOI] [PubMed] [Google Scholar]
- Gosens R, Stelmack GL, Dueck G, McNeill KD, Yamasaki A, Gerthoffer WT et al (2006). Role of caveolin‐1 in p42/p44 MAP kinase activation and proliferation of human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 291: L523–L534. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heulens N, Korf H, Cielen N, De Smidt E, Maes K, Gysemans C et al (2015). Vitamin D deficiency exacerbates COPD‐like characteristics in the lungs of cigarette smoke‐exposed mice. Respir Res 16: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Yoshioka H, Kabara S, Kudo T, Ishihara H, Matsuki A (2001). A comparison of the relaxant effects of olprinone and aminophylline on methacholine‐induced bronchoconstriction in dogs. Anesth Analg 93: 230–233. [DOI] [PubMed] [Google Scholar]
- Jin S‐LC, Conti M (2002). Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS‐activated TNF‐alpha responses. Proc Natl Acad Sci U S A 99: 7628–7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistemaker LEM, Bos IST, Hylkema MN, Nawijn MC, Hiemstra PS, Wess J et al (2013). Muscarinic receptor subtype‐specific effects on cigarette smoke‐induced inflammation in mice. Eur Respir J 42: 1677–1688. [DOI] [PubMed] [Google Scholar]
- Ma H, Shi J, Wang C, Guo L, Yulei G, Li J et al (2014). Blockade of PDE4B limits lung vascular permeability and lung inflammation in LPS‐induced acute lung injury. Biochem Biophys Res Commun 450: 1560–1567. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milara J, Armengot M, Bañuls P, Tenor H, Beume R, Artigues E et al (2012). Roflumilast N‐oxide, a PDE4 inhibitor, improves cilia motility and ciliated human bronchial epithelial cells compromised by cigarette smoke in vitro. Br J Pharmacol 166: 2243–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milara J, Peiró T, Serrano A, Guijarro R, Zaragozá C, Tenor H et al (2014). Roflumilast fN‐oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm Pharmacol Ther 28: 138–148. [DOI] [PubMed] [Google Scholar]
- Mori H, Nose T, Ishitani K, Kasagi S, Souma S, Akiyoshi T et al (2008). Phosphodiesterase 4 inhibitor GPD‐1116 markedly attenuates the development of cigarette smoke‐induced emphysema in senescence‐accelerated mice P1 strain. Am J Physiol Lung Cell Mol Physiol 294: L196–L204. [DOI] [PubMed] [Google Scholar]
- Murray F, MacLean MR, Pyne NJ (2002). Increased expression of the cGMP‐inhibited cAMP‐specific (PDE3) and cGMP binding cGMP‐specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br J Pharmacol 137: 1187–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Bünemann M, Hein L, Hannawacker A, Lohse MJ (2004). Novel single chain cAMP sensors for receptor‐induced signal propagation. J Biol Chem 279: 37215–37218. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Lohse MJ (2006). Monitoring of cAMP synthesis and degradation in living cells. Phys Ther 21: 86–92. [DOI] [PubMed] [Google Scholar]
- Oenema TA, Maarsingh H, Smit M, Groothuis GMM, Meurs H, Gosens R (2013). Bronchoconstriction induces TGF‐β release and airway remodelling in guinea pig lung slices. PLoS One 8: e65580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburger A, Timens W, Bos S, Smit M, Smrcka AV, Laurent A‐C et al (2014). Epac1 and Epac2 are differentially involved in inflammatory and remodeling processes induced by cigarette smoke. FASEB J Off Publ Fed Am Soc Exp Biol 28: 4617–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page CP (2014). Phosphodiesterase inhibitors for the treatment of asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol 165: 152–164. [DOI] [PubMed] [Google Scholar]
- Page CP, Spina D (2012). Selective PDE inhibitors as novel treatments for respiratory diseases. Curr Opin Pharmacol 12: 275–286. [DOI] [PubMed] [Google Scholar]
- Poppinga WJ, Heijink IH, Holtzer LJ, Skroblin P, Klussmann E, Halayko AJ et al (2015). A‐kinase‐anchoring proteins coordinate inflammatory responses to cigarette smoke in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 308: L766–L775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qaseem A, Wilt TJ, Weinberger SE, Hanania NA, Criner G, van der Molen T et al (2011). Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med 155: 179–191. [DOI] [PubMed] [Google Scholar]
- Rabe KF, Tenor H, Dent G, Schudt C, Liebig S, Magnussen H (1993). Phosphodiesterase isozymes modulating inherent tone in human airways: identification and characterization. Am J Phys 264: L458–L464. [DOI] [PubMed] [Google Scholar]
- Rangasamy T, Misra V, Zhen L, Tankersley CG, Tuder RM, Biswal S (2009). Cigarette smoke‐induced emphysema in A/J mice is associated with pulmonary oxidative stress, apoptosis of lung cells, and global alterations in gene expression. Am J Physiol Lung Cell Mol Physiol 296: L888–L900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi M, Maes K, Vleeschauwer SD, Thomas D, Verbeken EK, Decramer M et al (2012). Long‐term nose‐only cigarette smoke exposure induces emphysema and mild skeletal muscle dysfunction in mice. Dis Model Mech 5: 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlepütz M, Rieg AD, Seehase S, Spillner J, Perez‐Bouza A, Braunschweig T et al (2012). Neurally mediated airway constriction in human and other species: a comparative study using precision‐cut lung slices (PCLS). PLoS One 7: e47344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A, Bai G, Schmid N, Zaccolo M, Ostrowski LE, Conner GE et al (2006). Real‐time analysis of cAMP‐mediated regulation of ciliary motility in single primary human airway epithelial cells. J Cell Sci 119: 4176–4186. [DOI] [PubMed] [Google Scholar]
- Schmid A, Baumlin N, Ivonnet P, Dennis JS, Campos M, Krick S et al (2015). Roflumilast partially reverses smoke‐induced mucociliary dysfunction. Respir Res 16: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt DT, Watson N, Dent G, Rühlmann E, Branscheid D, Magnussen H et al (2000). The effect of selective and non‐selective phosphodiesterase inhibitors on allergen‐ and leukotriene C(4)‐induced contractions in passively sensitized human airways. Br J Pharmacol 131: 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seimetz M, Parajuli N, Pichl A, Bednorz M, Ghofrani HA, Schermuly RT et al (2015). Cigarette smoke‐induced emphysema and pulmonary hypertension can be prevented by phosphodiesterase 4 and 5 inhibition in mice. PLoS One 10: e0129327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selige J, Hatzelmann A, Dunkern T (2011). The differential impact of PDE4 subtypes in human lung fibroblasts on cytokine‐induced proliferation and myofibroblast conversion. J Cell Physiol 226: 1970–1980. [DOI] [PubMed] [Google Scholar]
- Singh SP, Barrett EG, Kalra R, Razani‐Boroujerdi S, Langley RJ, Kurup V et al (2003). Prenatal cigarette smoke decreases lung cAMP and increases airway hyperresponsiveness. Am J Respir Crit Care MedAm J Respir Crit Care Med 168: 342–347. [DOI] [PubMed] [Google Scholar]
- Singh SP, Mishra NC, Rir‐Sima‐Ah J, Campen M, Kurup V, Razani‐Boroujerdi S et al (2009). Maternal exposure to secondhand cigarette smoke primes the lung for induction of phosphodiesterase‐4D5 isozyme and exacerbated Th2 responses: rolipram attenuates the airway hyperreactivity and muscarinic receptor expression but not lung inflammation and atopy. J Immunol Baltim Md 183: 2115–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger JU, Nikolaev VO (2013). Biophysical techniques for detection of cAMP and cGMP in living cells. Int J Mol Sci 14: 8025–8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger JU, Perera RK, Steinbrecher JH, Lehnart SE, Maier LS, Hasenfuss G et al (2015). In vivo model with targeted cAMP biosensor reveals changes in receptor‐microdomain communication in cardiac disease. Nat Commun 6: 6965. [DOI] [PubMed] [Google Scholar]
- Tamimi A, Serdarevic D, Hanania NA (2012). The effects of cigarette smoke on airway inflammation in asthma and COPD: therapeutic implications. Respir Med 106: 319–328. [DOI] [PubMed] [Google Scholar]
- Tyrrell J, Qian X, Freire J, Tarran R (2015). Roflumilast combined with adenosine increases mucosal hydration in human airway epithelial cultures after cigarette smoke exposure. Am J Physiol Lung Cell Mol Physiol 308: L1068–L1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, DiPilato LM, Yildirim N, Elston TC, Zhang J, Lefkowitz RJ (2008). β2‐adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J Biol Chem 283: 2949–2961. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jiang Y, Tang H, Zhao C, Chen J (2010). Zl‐n‐91, a selective phosphodiesterase 4 inhibitor, suppresses inflammatory response in a COPD‐like rat model. Int Immunopharmacol 10: 252–258. [DOI] [PubMed] [Google Scholar]
- Watson C, Damiani F, Ram‐Mohan S, Rodrigues S, de Moura Queiroz P, Donaghey TC et al (2015). Screening for chemical toxicity using cryopreserved precision cut lung slices. Toxicol Sci 150: 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yañez‐Mó M, Barreiro O, Gonzalo P, Batista A, Megías D, Genís L et al (2008). MT1‐MMP collagenolytic activity is regulated through association with tetraspanin CD151 in primary endothelial cells. Blood 112: 3217–3226. [DOI] [PubMed] [Google Scholar]
- Yoon H‐K, Hu H‐J, Rhee C‐K, Shin S‐H, Oh Y‐M, Lee S‐D et al (2014). Polymorphisms in PDE4D are associated with a risk of COPD in non‐emphysematous Koreans. COPD 11: 652–658. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Pozzan T (2002). Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295: 1711–1715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A‐C) Fenoterol concentration‐responses in mouse PCLS and human airway structural cells. Mouse PCLS (A), human airway structural cells HASM (B) and 16 HBE 14o‐ cells (C) were stimulated with the indicated fenoterol concentrations. EC50 values: mouse PCLS, 0.3±0.09 μM (n=8 independent slices from 3 mice); HASM, 19.0±3.3 nM (n=8 independent cells); 16 HBE 14o‐, 3,7±0,76 nM (n=7 independent cells). (D) Cilostamide concentration‐response in mouse PCLS model (EC50 value: 0.51±0.2 μM, n=9 independent slices from 3 mice). (E) Rolipram concentration‐response in mouse PCLS model (EC50 value: 0.11±0.03 μM, n=9 independent slices from 3 mice). (F‐I) Specificity of antibodies for PDE3 and PDE4. The lung tissues from specific PDE isoform knockout mice were lysed followed by western blotting (F‐I) to determine the antibodies specificity. (J) Representative FRET trace in PCLS. After treatment with fenoterol (1 μM), rolipram (10 μM) was added followed by addition of the non‐selective PDE inhibitor IBMX (100 μM) and forskolin (10 μM) to define the maximal FRET response. (K) Forskolin stimulation did not give any further increase after fenoterol and IBMX treatment in both control and CS extract exposed PCLS, indicating that fenoterol and IBMX induce a maximal FRET response. Fenoterol+IBMX+forskolin was considered as 100%; n=16 independent slices from 4 mice. Data are expressed as mean ± SEM.