

Abstract
Opioid receptors are 7‐transmembrane domain receptors that couple to heterotrimeric G proteins. The endogenous ligands for opioid receptors are peptides which bind to the orthosteric site on the receptors. The μ‐opioid receptor is the target for opioid analgesics, while the δ‐opioid receptor has been suggested as a target for pain management, migraine and depression. Similarly, κ‐opioid receptors are involved in pain and depression and nociceptin receptors in pain and mood behaviours. However, exogenous orthosteric ligands for opioid receptors cause a myriad of on‐target side effects. Recently, selective allosteric ligands for μ‐ and δ‐opioid receptors have been described. These compounds bind to a site on the receptor distinct from the orthosteric site. Occupation of this allosteric site leads to modulation of orthosteric ligand binding affinity and/or efficacy. Allosteric modulators may be positive, negative or silent (neutral) (PAMs, NAMs or SAMs respectively). PAMs may have in vivo activity by enhancing the activity of exogenous drugs or endogenous opioid peptides. Enhancing endogenous opioid peptide activity maintains the temporal and spatial distribution of these molecules but improves, and potentially qualitatively changes, activity at their cognate receptors which could limit side effects compared with traditional opioid drugs. In this review, we describe the rationale and promise for the development of allosteric modulators for opioid receptors, the discovery of selective allosteric modulators, the identification of potential allosteric sites on opioid receptors and the mode of action of the modulators.
Linked Articles
This article is part of a themed section on Emerging Areas of Opioid Pharmacology. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.14/issuetoc
Abbreviations
- GPCR
G proteins coupled receptor
- MD
Molecular dynamics
- NAM
Negative allosteric modulator
- PAM
Positive allosteric modulator
- Pdb
Protein data bank
- SAM
Silent allosteric modulator
- TM
Transmembrane
Introduction
The role of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 (Alexander et al., 2015a), in the modulation of pain makes this receptor one of the most pharmacologically targeted GPCRs in modern medicine and μ‐receptor agonists such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7093 are invaluable in the clinic. As a result, the number of prescriptions written for opioid analgesics is rising rapidly. However, while activation of μ‐receptors provides pain relief, it also results in a wide range of unwanted effects including constipation and life‐threatening respiratory depression, as well as rewarding effects that lead to addiction liability (Matthes et al., 1996). This in turn has led to the current opioid abuse epidemic in the UK, USA and other countries. In the USA specifically, there has been a fourfold increase in the number of deaths from licit and illicit opioids since 1999 (Volkow, 2014; Heron, 2016). In addition, the effectiveness of traditional opioid drugs such as morphine in the management of neuropathic pain is controversial (Smith et al., 2012; McNicol et al., 2013). Consequently, there remains an unmet need for safer efficacious analgesics that circumvent the issues associated with activation of μ‐receptors by opioid drugs.
Alternative approaches have included the development of compounds acting at other members of the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=50 (OR) family (Alexander et al., 2015a), particularly http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=317 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=318&familyId=50&familyType=GPCR agonists, as well as compounds with activity at more than one opioid receptor. While δ‐receptor agonists suffer from a lack of efficacious analgesia and proconvulsive effects, they may be effective as antidepressants (Jutkiewicz, 2006) and in migraine treatment (Charles and Pradhan, 2016). Additionally, κ‐receptor agonists possess analgesic properties (Chavkin, 2011) but are linked with dysphoria (Pfeiffer et al., 1986) and are pro‐depressant to the extent that κ‐receptor antagonists may find use in the management of depression (Shippenberg, 2009; Chavkin, 2011; Lalanne et al., 2014). Finally, agonists for the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=320 (Alexander et al., 2015a) may be analgesic or pro‐nociceptive, depending on the circumstances (Lambert, 2008). Selective δ‐, κ‐ and NOP receptor compounds have not successfully found their way into the clinic, although mixed μ‐/δ‐ receptor (Harland et al., 2015) and mixed μ‐ /NOP receptor compounds (Toll, 2013) show promise.
Despite best efforts, the μ‐receptor system remains the most efficacious target for the treatment of pain. In this review, we discuss allosteric modulators as a novel way to harness the analgesic efficacy of these receptors and the potentially beneficial therapeutic actions of other opioid receptors. This article focuses on small molecule (low molecular weight) exogenous ligands as allosteric modulators of the μ‐receptors. In addition, allosteric modulation of the μ‐receptor (and other GPCRs) via receptor heteromers has also been proposed. For information on this aspect, the reader is referred to reviews by Fujita et al. (2015) and Ferré et al. (2014).
Allosteric ligands of opioid receptors as potential therapeutic agents
Morphine and other traditional opioid drugs act at the orthosteric site on the opioid receptors, defined as the site for the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627, including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1613‐ and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1614, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1643 and the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1620. Opioid receptors, like all 7‐transmembrane domain (7‐TM) GPCRs, are allosteric proteins. The simplest idea of 7‐TM domain receptor action can be explained by the Monod–Wyman–Changeux two‐state allostery model (Monod et al., 1965) where receptors are distributed into inactive (R) and active (R*) conformations that exist in equilibrium. These do not represent individual conformations but rather ensembles of R and R* states (Kenakin, 2013). R* states are distinguished from R states by an ability to bind agonists with high affinity and activate heterotrimeric G proteins, triggering downstream intracellular signalling pathways. The conformational state of a GPCR, including the opioid receptors, is controlled not only by agonist occupying the orthosteric site but also by endogenous substances acting at other sites on the receptor. These include sodium ions (Pert and Snyder, 1974; Simon and Groth, 1975; Yabaluri and Medzihradsky, 1997; Fenalti et al., 2014; Shang et al., 2014) and interacting proteins, especially heterotrimeric G proteins that stabilize the R* state (DeVree et al., 2016). Furthermore, there is evidence that the lipid environment regulates GPCR function. In particular, cholesterol modulates the function of both μ‐ and δ‐receptors (Xu et al., 2006; Levitt et al., 2009; Zheng et al., 2012) as well as other GPCRs, probably by a combination of direct actions at a conserved motif on the receptors and membrane effects (see Oates and Watts, 2011).
A burgeoning field in drug discovery at GPCRs is the development of small molecule allosteric modulators that bind to druggable pockets on receptors separate from the orthosteric sites. These spatially distinct allosteric sites are defined by the ability of molecules binding at these sites to regulate the activity of molecules binding at the orthosteric site (Figure 1). Allosteric modulators can alter affinity, potency and efficacy of orthosteric ligands. Positive allosteric modulators (PAMs) improve the activity of orthosteric ligands. Negative allosteric modulators (NAMs) do the opposite, and SAMs or silent allosteric modulators occupy the site without activity and as such act as antagonists to PAMs and NAMs. Ideally, PAMs and NAMs would enhance or inhibit respectively the affinity and/or efficacy of an orthosteric ligand while failing to directly activate or inhibit the receptor on its own. However, some compounds may have direct agonist activity; such compounds are known as ‘ago‐PAMs’ (Figure 1). Allosteric activity is dependent on the binding affinity (KB) of the modulator and the allosteric cooperativity (αβ) which describes the ability of the modulator to change the affinity and/or efficacy of an orthosteric ligand (Figure 1; Christopoulos and Kenakin, 2002; Melancon et al., 2012; Christopoulos, 2014). Allosteric modulators also have differing effects depending on the orthosteric ligand, a phenomenon called ‘probe dependence’. It is thought that allosteric modulators provide better selectivity and could provide better therapeutic indexes with fewer side effects. For more on this topic, see (Christopoulos and Kenakin, 2002; Christopoulos, 2014; Christopoulos et al., 2014; Burford et al., 2015a).
Figure 1.
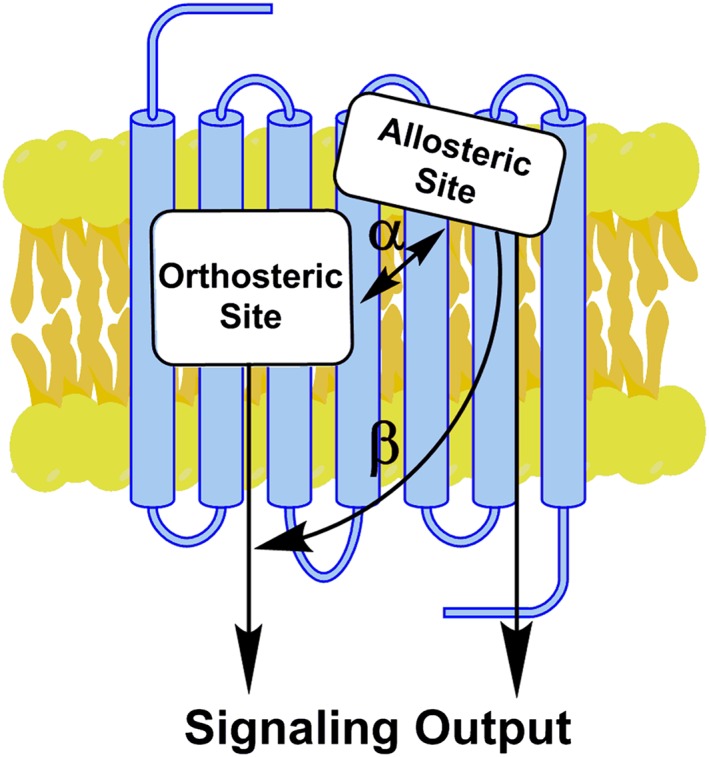
Small molecule allosteric modulation at GPCRs. Allosteric ligands bind to a site distinct from the orthosteric site to modulate orthosteric agonist affinity and/or efficacy. α is the co‐operativity factor between the two sites and represents the degree of an enhancement by a PAM (if a value > 1) or reduction by a NAM (if a value < 1) of the affinity of the orthosteric ligand. β is the modulation factor and describes allosteric modulation of orthosteric ligand efficacy. β will have a value > 1 for a PAM or < 1 for a NAM. Allosteric modulators may activate intracellular messengers directly as “ago‐PAMs” (modified from Conn et al., 2009).
A prime example of the potential power of allosteric modulators is PAM activity at the μ‐receptor (μ‐PAM). Such a compound could serve to increase the potency and/or efficacy of opioid drugs like morphine and lower the dose requirement. Perhaps more importantly, a μ‐PAM can be predicted to enhance the activity of endogenous opioid peptides which are elevated during stress and in pain states (Hughes, 1983). This activity would be confined to μ‐receptors that have access to released endogenous opioids at specific times and so maintain their spatial as well as temporal selectivity pattern. This is in sharp contrast to traditional opioid agonists which activate μ‐receptors across many tissues with limitations set only by pharmacokinetic parameters. There is evidence that such an approach would be feasible since non‐selectively increasing opioid peptide levels by blocking enzymes responsible for their degradation with inhibitors of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1611 (neutral endopeptidase;NEP) provides preclinical analgesia (Roques et al., 2012), but not constipation (Noble et al., 2008), respiratory depression (Boudinot et al., 2001), antinociceptive tolerance (Noble et al., 1992b) or abuse liability (Noble et al., 1992a; Valverde et al., 1996).
An additional potential advantage of using small molecule allosteric modulators for the opioid receptors is to introduce a signalling bias downstream of the receptors. Biased agonism is the preferential activation of one signalling pathway over another and has been demonstrated at μ‐receptors between β‐arrestin recruitment and G protein activation (McPherson et al., 2010; Thompson et al., 2015). The goal of biased signalling is to activate pathways downstream of opioid receptors responsible for the beneficial effects (e.g. pain relieving in the case of the μ‐receptors) without activating pathways producing undesirable effects. For example, the β‐arrestin pathway has been implicated in the constipatory and respiratory depressive actions of opioids (Raehal and Bohn, 2011), and newly developed biased ligands including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7334; Dewire et al., 2013) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9286 (Manglik et al., 2016) avoid activation of this pathway. It is tempting to speculate that the μ‐receptor occupied by a PAM might behave as a novel receptor, compared with an unoccupied μ‐receptor and so be envisaged to signal differently. Similarly, introducing bias at δ‐receptors could promote antidepressant actions over proconvulsant actions and at κ‐receptors could enhance analgesia at the expense of dysphoria. For a more comprehensive discussion of the potential benefits of opioid PAMs as therapeutic agents, see Burford et al., (2015a).
Discovery of small molecule allosteric modulators of opioid receptors
The BMS series of compounds
The first selective positive allosteric modulators of μ‐receptors, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9156 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9157, were identified in 2013 (Table 1; Figure 2; Burford et al., 2013) using a high‐throughput screen (HTS) monitoring for ability to enhance a low concentration of the putative endogenous μ‐receptor agonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1623, to recruit β‐arrestin to μ‐receptors. The HTS methodology has been described in detail (Burford et al., 2014; Bertekap et al., 2015). Further studies with BMS‐986122 showed that it can enhance the affinity and/or efficacy of various opioid agonists, including opioid peptides Leu‐ and Met‐enkephalin, β‐endorphin as well as endomorphin‐1. Along with BMS‐986122, a number of structurally similar μ‐PAMs were identified, plus SAMs such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9160 (Figure 2). BMS‐986122 exhibits dramatic probe dependence in that its effects are reliant on the ligand occupying the orthosteric site. For agonists such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5458, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1647 and the endogenous opioid peptides, BMS‐986122 enhances the potency and affinity, while for morphine and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1663, it enhances agonist efficacy with no alteration in the affinity. There is no effect on the binding of antagonists (Livingston and Traynor, 2014). This is discussed in more detail later under ‘Mechanism of allostery at opioid receptors’ in the subsection ‘Role of orthosteric ligand and Na+ ions’ as it points to a potential explanation for the action of the modulators. BMS‐986122 does not have PAM activity at δ‐ receptors, a fact which has been taken into account in structure–activity studies.
Table 1.
Confirmed or putative small molecule allosteric modulators of opioid receptorsa
| μ‐OR | δ‐OR | κ‐OR | References | |
|---|---|---|---|---|
| Salvinorin A | x | – | – | Rothman et al., 2007 |
| Cannabidiol | x | x | – | Vaysse et al., 1987; Kathmann et al., 2006 |
| THC | x | x | – | Vaysse et al., 1987; Kathmann et al., 2006 |
| BMS‐986122 | x | – | – | Burford et al., 2013; Livingston and Traynor, 2014 |
| BMS‐986121 | x | – | – | Burford et al., 2013 |
| BMS‐986124 | – | – | – | Burford et al., 2013 |
| BMS‐986187 | x | x | – | Burford et al., 2015b |
| MS1 | x | – | – | Bisignano et al., 2015 |
| Ignavine | x | – | – | Ohbuchi et al., 2016 |
| SCH‐202676 | x | x | x | Fawzi et al., 2001 (but see Göblyös et al., 2005; Lewandowicz et al., 2006) |
To date, no modulators have been identified for NOP receptors.
Figure 2.
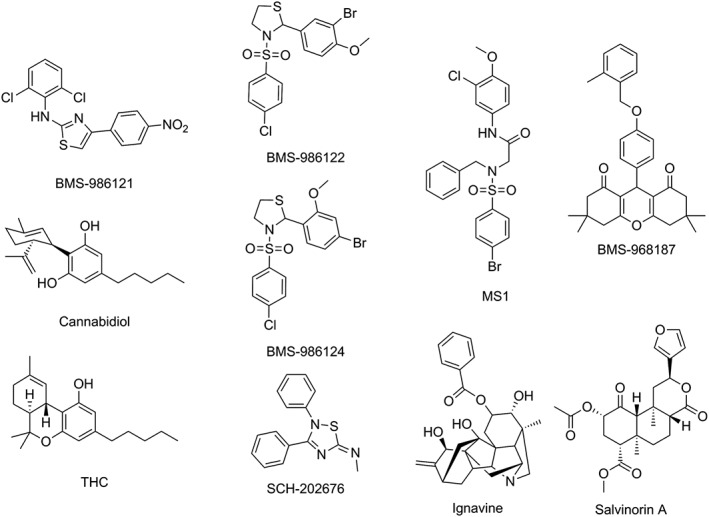
Structures of known or putative allosteric modulators of opioid receptors discussed in the text. The Figure shows compounds discovered by high‐throughput screening that exhibit PAM (BMS‐986122 and BMS‐986121) or SAM (BMS‐986124) activity at μ‐receptors plus the similarly structured MS1 which was identified by chemoinformatic analysis. BMS‐986187 is a δ‐PAM discovered by high‐throughput screening. Other compounds that have been suggested as modulators include the natural products cannabidiol, THC, ignavine and salvinorin A as well as the low MW compound SCH‐202676.
The structure–activity relationships of the BMS series of μ‐receptor allosteric modulators published so far is unclear. Subtle changes have profound effects on defining a compound as a PAM or a SAM (compare BMS‐986122 and BMS‐986124 in Figure 2). No NAMs have been described in this series. With this in mind, Bisignano et al. (2015) searched the eMolecules (www.emolecules.com) and ZINC (Irwin et al., 2012) databases for structural analogues. Of the compounds identified, 28 were evaluated in the β‐arrestin recruitment assay, 14 were found to be PAMs and 12 were identified as SAMs. None of the compounds had higher affinity than the original molecules, although one compound, MS1 (Table 1; Figure 2), was chosen for a more extensive study. MS1 did not bind to the μ‐receptor orthosteric site but improved the affinity of methadone and the potency of methadone to activate heterotrimeric G proteins. Surprisingly, neither the affinity nor potency to activate G proteins was enhanced for http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1623 or DAMGO, in spite of the fact that MS1 was discovered using endomorphin‐1 as the orthosteric probe. This anomaly could be due to the fact that endomorphins may be β‐arrestin‐biased molecules (McPherson et al., 2010). On the other hand, the conflicting probe dependence may be explained by the relatively weak allosteric cooperativity of MS1, even against methadone which thus far is the most sensitive orthosteric ligand to allosteric modulation (Livingston and Traynor, 2014).
Using the β‐arrestin recruitment HTS assay (Burford et al., 2014; Bertekap et al., 2015) allosteric modulators of the closely related δ‐receptor have been discovered (Burford et al., 2015b). These compounds are structurally dissimilar to BMS‐986122 being tetramethyl substituted hexahydro‐xanthine‐1,8‐diones. The lead compound BMS‐986187 (Table 1; Figure 2) is effective as a δ‐PAM in the <100 nM range, while showing 100‐fold weaker PAM activity at the μ‐receptor. The high potency of BMS‐986187 is somewhat surprising given its affinity for the unoccupied receptor (KB) of approximately 1 μM. On the other hand, this demonstrates that allostery is bidirectional and so the orthosteric agonist enhances PAM affinity and also highlights the importance of efficacy interactions (β) as well as affinity interactions (α). Indeed, BMS‐986187 is able to stimulate signalling downstream of δ‐ receptors even in the absence of orthosteric agonist. However, the compound does not bind to the orthosteric site, as determined by its inability to displace http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1612, and so, it is activating the receptor through its allosteric site. Consequently, it is designated as an ‘ago‐PAM’ (Figure 1). Like the μ‐PAMs, BMS‐986187 also exhibited probe dependence when tested on a limited number of compounds with a greatest effect on the affinity of the peptide Leu‐enkephalin (32‐fold shift) and smaller shifts for http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1611 (14‐fold shift) and Tan‐67 (threefold shift). BMS‐986187 acted as a δ‐PAM for several downstream measures including the [35S]GTPγS binding assay, inhibition of adenylate cyclase, recruitment of β‐arrestin and phosphorylation of ERK1/2 for all three of these ligands (Burford et al., 2015b). Moreover, BMS‐986187 has been demonstrated to potentiate endogenous opioid signalling at δ‐ receptors in intercalated cells that modulate output from the amygdala (Winters et al., 2017).
Other putative allosteric modulators
In addition to the small molecules described above, other putative, structurally unrelated modulators of ORs have been described (Table 1; Figure 2).
The cannabinoids (CBs) http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2424 (THC) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4150 (CBD) (Figure 2) were suggested many years ago (Vaysse et al., 1987) to be NAMs of both μ‐ and δ‐ receptors. This assertion was based on the ability of the CBs to fully inhibit 3H‐orthosteric agonist binding to the μ‐receptors and δ‐ receptors in rat brain membranes, in a non‐competitive manner, by reducing the Bmax but not altering ligand affinity. To avoid the caveat that the compounds might be working via http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=13 in the rat brain membranes and so acting indirectly through receptor–receptor interactions, the authors showed THC to be just as effective at solubilized, partially purified μ‐receptors This also suggests the effect was on the receptor itself or a closely associated lipid, but not due to a non‐specific effect on the lipid bilayer. In support of this CB receptor‐independent effect on μ‐receptors, several other CBs displayed a wide variety of activities that were not correlated with their activity at CB receptors. For instance, levonantradol and dextronatradol were equiactive at inhibiting binding of the μ‐receptor orthosteric agonist 3H‐http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1616 (DHM) to rat brain membranes but showed 100‐fold difference in behavioural potencies as CBs (Johnson et al., 1981). Also, 11‐hydroxy‐THC displayed comparable CB potency to THC but had less than 20% of the activity of THC at displacing 3H‐DHM. Later kinetic experiments comparing the effect of THC and CBD at μ‐ and δ‐ receptors in rat cortical membranes (Kathmann et al., 2006) were claimed to support the idea of the CBs as allosteric modulators by demonstrating that CBD and THC at high concentrations (30‐100 μM) increased the dissociation rate for the μ‐receptor agonist 3H‐DAMGO in the presence of a high concentration of naloxone. Similar but much smaller shifts in the dissociation rate of the antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3829 (NTI) from δ‐ receptors were seen. On the other hand, both THC and CBD alone enhanced 3H‐DAMGO and 3H‐NTI dissociation and also displaced DAMGO binding in competition assays, giving affinities in the 10 μM range, albeit with a reported Hill slope ~ 1.5, though only CBD inhibited 3H‐NTI binding. Moreover, no functional studies of allosterism have been reported. Thus, it cannot be ruled out that the CBs at these very high concentrations are acting non‐specifically or even binding to the orthosteric site rather than acting as true allosteric modulators. The http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=56 antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=743 shows a similar profile (Kathmann et al., 2006), and this compound has been reported to have an affinity at μ‐receptors of 650 nM and to be an antagonist at these receptors in vivo and in vitro (Seely et al., 2012). The concentrations of the CBs acting at opioid receptors are much higher than their affinity for the CB1 receptor, suggesting that activity at opioid receptors is not likely to play a role in the in vivo activities of the CBs.
The neoclerodanediterpene http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1666 (Sal A; Figure 2) is a selective κ‐receptor agonist which lacks a positively charged nitrogen atom for interaction with the conserved Asp in TM‐III of the κ‐receptor (Roth et al., 2002). Based on the observation that Sal A has a weak ability to compete with orthosteric ligands at μ‐receptors, Rothman and colleagues examined the compound as a possible allosteric modulator of this receptor (Rothman et al., 2007). Their data suggested that Sal A might be a negative allosteric modulator of μ‐receptors, based on its ability to only partly inhibit binding of the agonists 3H‐DAMGO or 125I‐[IOXY] or the antagonist 3H‐diprenorphine to the orthosteric site of the receptor in both μ‐receptor expressing CHO cells and rat brain membranes. Binding experiments showed that Sal A decreased the affinity of the orthosteric ligands by twofold to threefold, reduced Bmax values and had complex effects on ligand dissociation. In the [35S]GTPγS assay, which measures μ‐receptor activation of heterotrimeric G proteins, Sal A decreased both the potency (EC50) and Bmax for DAMGO. The concentrations of Sal A used in these experiments were in the high μM range, much higher than the affinity of Sal A for the κ‐receptor (~4 nM; Roth et al., 2002). This will make in vivo studies challenging, although a study in κ‐ receptor knockout animals might be informative.
Ignavine (Figure 2) is a diterpene alkaloid isolated from the plant Aconitum japonica (Saito et al., 1982; Ohbuchi et al., 2016). There is evidence that the ‘processed aconite tuber’ has analgesic activity mediated by κ‐receptors, although the specific κ‐receptor agonist has not been isolated (Ohbuchi et al., 2016). Ignavine itself gives a biphasic antinociceptive dose–response curve in the mouse tail‐flick and tail pressure tests. The title of a recent publication (Ohbuchi et al., 2016) states ignavine is a ‘novel allosteric modulator of the μ‐receptor’. This claim is based on the finding that the compound both enhances and inhibits the activity of the μ‐receptor orthosteric agonist DAMGO to inhibit cAMP accumulation and to cause internalization of μ‐receptors in HEK 293 cells depending on the ignavine concentration (1 or 10 μM respectively). However, binding studies reported in the same publication indicate that the compound fully displaces 3H‐diprenorphine from the orthosteric site of the μ‐receptor in an apparently competitive manner and docking studies suggest that the compound binds at the orthosteric site. Thus, this compound would seem to be incorrectly identified as an allosteric modulator but may have other actions at μ‐receptors, for example, as a μ‐receptor partial agonist.
Finally, a thiazolidine compound, SCH‐202676 (Figure 2), has been claimed to be a non‐specific allosteric modulator of many GPCRs including the μ‐, δ‐ and κ‐receptors (Fawzi et al., 2001). However, this compound covalently binds to GPCRs by sulfhydryl bond formation and so is not a true allosteric modulator (Göblyös et al., 2005; Lewandowicz et al., 2006).
The above evidence suggests certain CBs and Sal A are NAMs of μ‐receptors. A negative modulator, unless it can be specially targeted at reducing the side effects of orthosteric μ‐receptor agonists, for example, by introducing a bias into downstream signalling as discussed above, may not make a useful clinical compound. Nonetheless, it will be important to re‐evaluate these putative modulators (as well as ignavine) of opioid receptors, using more rigorous analysis methods for the determination of allostery (Melancon et al., 2012; Christopoulos, 2014), as these natural products could provide scaffolds for the future design of modulators.
Mechanism of allostery at opioid receptors
Allosteric binding site(s) on opioid receptors
There is no definitive structural work that accurately defines the nature and location of allosteric sites on the opioid receptors. However, there have been several attempts to identify site(s) on μ‐ and δ‐receptors by computational methods using docking and molecular dynamics (MD) simulations.
Using molecular docking, Bartuzi et al. (2016) obtained several poses for BMS‐986122 within the μ‐receptor although two had very similar orientations and interaction energies. These data indicated an allosteric site involving amino acids above the orthosteric binding pocket and towards the extracellular surface in TM domains II and VII (Figure 3). At the δ‐receptor, Shang et al. (2016) using metadynamic calculations (Schneider et al., 2015) of the δ‐ receptor bound to the orthosteric ligand SNC80 and in a water‐lipid environment found two metastable binding poses for BMS‐986187 occupying the same site that was in close proximity to the orthosteric site but, as in the μ‐receptor, towards the extracellular surface (Figure 3). Both metastable states formed direct polar, water‐mediated polar, hydrophobic and/or aromatic interactions with amino acids residues in TM domains I, II and VII, with several residues specific to a particular pose. Mutational studies of several amino acid residues in the putative site affected either the binding of the modulator and/or the degree of cooperativity between the modulator and the othosteric ligand, therefore giving some credence to this as an allosteric site (Shang et al., 2016), although with the caveat that mutagenesis can alter orthosteric ligand affinity and basal activity of the receptor, thus providing confounds.
Figure 3.
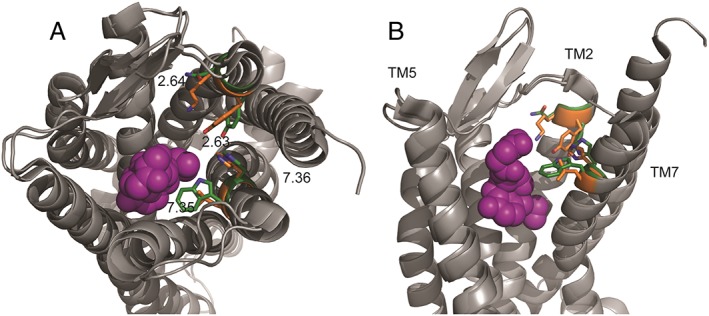
Theoretical binding site for BMS‐986122 on both μ‐ and δ‐receptors. Inactive state μ‐receptors (pdb 4DKL; Huang et al., 2015) and inactive state δ‐ receptors (pdb 4N6H; Fenalti et al., 2014) were aligned. The residues proposed (Bartuzi et al., 2016; Shang et al., 2016) to be involved in allosteric ligand binding are highlighted in green for the μ‐receptor and orange for the δ‐ receptor. (A) view of aligned receptors from the extracellular side, (B) side view. The orthosteric site is shown occupied by the irreversible μ‐receptor antagonist β‐funaltrexamine (purple). Extracellular loop 2 and TM6 have been removed from image B for clarity.
MD simulations of an active μ‐receptor homology model in complex with Gαs protein in a raft‐like membrane suggested a common binding pocket for lipophilic modulators CBD and THC at the top of TM domains I, II and VI (Bartuzi et al., 2015). Cannabinoids occupying this site appear to oppose the action of agonists by moving the TM domains closer together towards an inactive receptor state. In addition, a second site for Sal A at μ‐receptors that overlapped with the binding site for DAMGO was suggested, possibly explaining its NAM activity.
Overall, computational evidence suggests the μ‐ and δ‐ receptors are predicted to have similarly positioned allosteric sites (Figure 3). It is worth noting that this putative site is correspondingly situated to the allosteric site on the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=2 including M1(Abdul‐Ridha et al., 2014), M2 (Jäger et al., 2007; Haga et al., 2012; Dror et al., 2013) and M4 receptors (Thal et al., 2016) and the site for the modulator http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=806 on the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=62 chemokine receptor (Tan et al., 2013), suggesting that this region of class A GPCRs could be a common site for allosterism.
Role of orthosteric ligand and Na+ ions
Na+ ions play a major role in stabilizing inactive R states of 7‐TM domain receptors. This was first shown by the ability of NaCl to inhibit the binding of orthosteric agonists to μ‐receptors while having no effect on orthosteric antagonist binding (Pert et al., 1973), due to a shift in equilibrium to inactive R conformational states. Thus, Na+ can be considered an endogenous NAM of 7‐TM receptors. Current understanding of the mechanisms by which Na+ stabilizes R is better appreciated due to recent high‐resolution crystallographic work performed first with the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=19 (Liu et al., 2012) and later with other receptors including the δ‐receptor (Fenalti et al., 2014). The binding site of Na+ is conserved across many GPCRs, including all the opioid receptors. This site is located in the middle of the 7‐TM bundle and involves coordination with an aspartate residue in TM II (Asp 2.50) and a Ser residue in TM II (Ser 3.39) plus a number of highly organized water molecules across TM domains II, III, VI and VII (Katritch et al., 2014). Importantly, the Na+ ion is absent in the structures of active GPCRs including μ‐receptors (Huang et al., 2015) because movement of the TM domains upon receptor activation provides insufficient space for the Na+ ion and its associated water molecules.
The Monod–Wyman–Changeux two‐state model has been applied to describe the action of small molecule allosteric modulators of muscarinic receptors (Canals et al., 2012) and of the μ‐receptor (Livingston and Traynor, 2014) where they act to promote formation of R*. There is evidence that the allosteric activity of BMS‐986122 at the μ‐receptor is related to the negative modulatory activity of Na+ ions (Livingston and Traynor, 2014). The degree of allosteric activity of BMS‐986122 is dependent on the orthosteric probe, such that antagonists are insensitive and agonists are generally highly sensitive in line with their efficacy. There is a strong inverse correlation between the sensitivity of a μ‐receptor agonist to Na+ ions and the sensitivity of the same ligand to positive allosteric modulation by BMS‐986122, with methadone being the orthosteric ligand most sensitive to μ‐PAM activity (Figure 4). Moreover, the action of BMS‐986122 antagonizes the ability of Na+ ions to inhibit agonist binding such that BMS‐986122 and Na+ ions oppose each other's action. As BMS‐986122 is selective for μ‐ over δ‐receptors while the Na+ binding site is conserved, we have proposed a model in which the μ‐PAM binds at a distinct site from Na+ to allosterically disrupt the binding of Na+ (Figure 5). In support of this, ligands that target the Na+ binding site on GPCRs, such as amiloride, are not selective amongst GPCRs that are sensitive to Na+ ions (Gao and Ijzerman, 2000; Hoare et al., 2000; Schetz and Sibley, 2001; Heitman et al., 2008). It is notable that ‘superagonists’ at μ‐receptors such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9363 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1625 do not fit this pattern. These compounds are insensitive to the actions of the modulators (Livingston and Traynor, 2014) and much less affected by Na+ ions (Lee et al., 1999).
Figure 4.
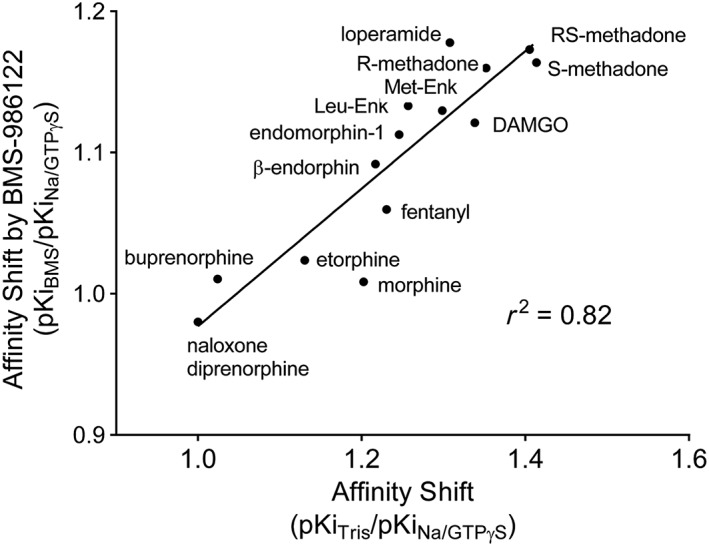
Relationship between the effect of the μ‐PAM, BMS‐986122 and the action of Na+ ions plus guanine nucleotide on the binding affinity of opioid ligands to the orthosteric site on μ‐receptors. The abscissa represents a reduction in affinity values (as a shift ratio) for each opioid ligand in the presence of Na+ ions and guanine nucleotide. The ordinate represents the increase in affinity (as a shift ratio) in the presence of Na+ ions and guanine nucleotide in the presence of BMS‐986122 (adapted from Livingston and Traynor, 2014).
Figure 5.
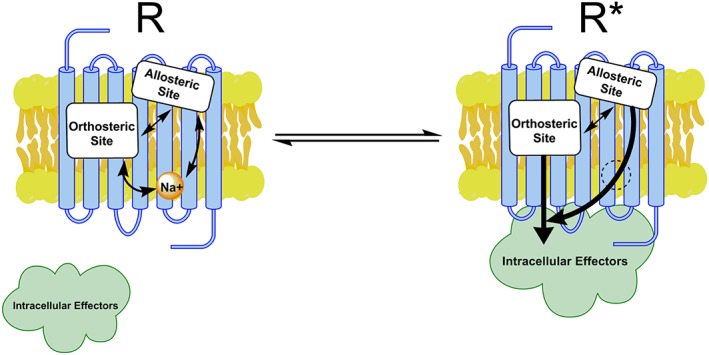
Allosteric interactions within μ‐receptors. Inactive receptor (R) contains a Na+ ion. Orthosteric agonist captures an active state (R*) that does not contain Na+ ion (dotted circle) and allows for receptor interaction with intracellular signalling proteins (e.g. heterotrimeric G protein or β‐arrestin). It is proposed that the μ‐PAM improves the affinity and potency of the orthosteric agonist by its incompatibility with Na+ binding, thereby promoting a state that more readily binds and responds to agonist.
Following publication of the experimental data discussed above, Bartuzi and colleagues (Bartuzi et al., 2016) performed principal component analysis of μ‐receptors in a native membrane environment. Their calculations showed that BMS‐986122 bound to a putative allosteric site (see above) and interacted with a Trp at the top of TM VII (Trp 7.35) to alter the conformation of this TM domain resulting in stabilization of the binding of the orthosteric ligand methadone as determined by its interaction with Asp 3.32 in the orthosteric pocket and destabilization of Na+ ion binding as measured by the distance of this ion from Asp 2.50. Similarly, recent MD simulations of the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=27 identified a potential allosteric site involving TM domains 2 and 3 and extracellular loops 1 and 2 that the authors propose could disrupt Na+ binding (Hui et al., 2016).
It should be understood that the putative binding sites used for these calculations are defined by docking procedures and may not represent the true allosteric sites. Nonetheless, there are multiple binding sites on μ‐receptors that allosterically communicate, including the orthosteric site, allosteric site for BMS‐986122, the Na+ binding site and the G protein‐binding site (Figure 5). The interplay between the sites differs depending on the orthosteric ligand. As Na+ regulates a number of GPCRs, and the Na+ site is highly conserved and MD simulations suggest that allosteric sites are similarly situated close to the orthosteric binding pocket, this may be a common mechanism of action for small molecule allosteric modulation across GPCRs.
Conclusions
Selective allosteric modulators of the μ‐ and δ‐ receptors have been described, but there are no specific modulators published to date for the κ‐ or the NOP receptors. Knowledge of allosteric modulation of opioid receptors is still in its infancy. However, we know that the μ‐ and δ‐ receptor PAMs show a marked probe dependence that appears to relate to the efficacy of the probe (the ligand occupying the orthosteric site) and to the sensitivity of the probe to Na+ ions that stabilize inactive R states of the receptors. At least for the μ‐ and δ‐ receptors, proof‐of‐principle for in vivo efficacy of allosteric modulators is needed. This will require the development of more potent and drug‐like molecules. Although some ideas about structural requirements and identity of the allosteric site on opioid receptors have been developed using computational methods, the field will benefit immensely from confirmation of the location and nature of allosteric binding site(s) and the conduit by which occupation of this site leads to dissociation of the bound Na+ ion and formation of R*. This will come from biophysical methods such as hydrogen–deuterium exchange mass spectrometry, NMR and X‐ray crystallography. Given the recent success in crystallizing GPCRs, including crystal structures of muscarinic receptors bound to allosteric modulators, this information should soon be available, allowing for the rational design of a new generation of allosteric modulators acting at opioid receptors.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by a grant from the National Institutes of Health, USA (DA 033397 to J.R.T.). We thank Veronica Taylor, Nicholas Griggs and Matthew Stanzyk for critical reading of the manuscript and Veronica Taylor and Evan Schramm for help with the figures.
Livingston K. E., and Traynor J. R. (2018) Allostery at opioid receptors: modulation with small molecule ligands, British Journal of Pharmacology, 175 2846–2856, https://doi.org/10.1111/bph.13823.
References
- Abdul‐Ridha A, Lane JR, Mistry SN, López L, Sexton PM, Scammells PJ et al. (2014). Mechanistic insights into allosteric structure‐function relationships at the M1 muscarinic acetylcholine receptor. J Biol Chem 289: 33701–33711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartuzi D, Kaczor AA, Matosiuk D (2015). Activation and allosteric modulation of human μ opioid receptor in molecular dynamics. J Chem Inf Model 55: 2421–2434. [DOI] [PubMed] [Google Scholar]
- Bartuzi D, Kaczor AA, Matosiuk D (2016). Interplay between two allosteric sites and their influence on agonist binding in human μ opioid receptor. J Chem Inf Model 56: 563–570. [DOI] [PubMed] [Google Scholar]
- Bertekap RL, Burford NT, Li Z, Alt A (2015). High‐throughput screening for allosteric modulators of GPCRs. Methods Mol Biol 1335: 223–240. [DOI] [PubMed] [Google Scholar]
- Bisignano P, Burford NT, Shang Y, Marlow B, Livingston KE, Fenton AM et al. (2015). Ligand‐based discovery of a new scaffold for allosteric modulation of the μ‐opioid receptor. J Chem Inf Model 55: 1836–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudinot E, Morin‐Surun M, Foutz AS, Fournié‐Zaluski M, Roques BP, Denavit‐Saubié M (2001). Effects of the potent analgesic enkephalin‐catabolizing enzyme inhibitors RB101 and kelatorphan on respiration. Pain 90: 7–13. [DOI] [PubMed] [Google Scholar]
- Burford NT, Clark MJ, Wehrman TS, Gerritz SW, Banks M, O'Connell J et al. (2013). Discovery of positive allosteric modulators and silent allosteric modulators of the μ‐opioid receptor. Proc Natl Acad Sci U S A 110: 10830–10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford NT, Livingston KE, Canals M, Ryan MR, Budenholzer LM, Han Y et al. (2015b). Discovery, synthesis, and molecular pharmacology of selective positive allosteric modulators of the δ‐opioid receptor. J Med Chem 58: 4220–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford NT, Traynor JR, Alt A (2015a). Positive allosteric modulators of the μ‐opioid receptor: a novel approach for future pain medications. Br J Pharmacol 172: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford NT, Wehrman T, Bassoni D, O'Connell J, Banks M, Zhang L et al. (2014). Identification of selective agonists and positive allosteric modulators for μ‐ and δ‐opioid receptors from a single high‐throughput screen. J Biomol Screen 19: 1255–1265. [DOI] [PubMed] [Google Scholar]
- Canals M, Lane JR, Wen A, Scammells PJ, Sexton PM, Christopoulos A (2012). A Monod–Wyman–Changeux mechanism can explain G protein‐coupled receptor (GPCR) allosteric modulation. J Biol Chem 287: 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles A, Pradhan AA (2016). Delta‐opioid receptors as targets for migraine therapy. Curr Opin Neurol 29: 314–319. [DOI] [PubMed] [Google Scholar]
- Chavkin C (2011). The therapeutic potential of κ‐opioids for treatment of pain and addiction. Neuropsychopharmacology 36: 369–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A (2014). Advances in G protein‐coupled receptor allostery: from function to structure. Mol Pharmacol 86: 463–478. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Changeux J‐P, Catterall WA, Fabbro D, Burris TP, Cidlowski JA et al. (2014). International Union of Basic and Clinical Pharmacology. XC. Multisite Pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev 66: 918–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T (2002). G protein‐coupled receptor allosterism and complexing. Pharmacol Rev 54: 323–374. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW (2009). Allosteric modulation of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8: 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVree BT, Mahoney JP, Vélez‐Ruiz GA, Rasmussen SGF, Kuszak AJ, Edwald E et al. (2016). Allosteric coupling from G protein to the agonist‐binding pocket in GPCRs. Nature 535: 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM et al. (2013). A G protein‐biased ligand at the μ‐opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 344: 708–717. [DOI] [PubMed] [Google Scholar]
- Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC et al. (2013). Structural basis for modulation of a G‐protein‐coupled receptor by allosteric drugs. Nature 503: 295–299. [DOI] [PubMed] [Google Scholar]
- Fawzi AB, Macdonald D, Benbow LL, Smith‐Torhan A, Zhang H, Weig BC et al. (2001). SCH‐202676: an allosteric modulator of both agonist and antagonist binding to G protein‐coupled receptors. Mol Pharmacol 59: 30–37. [DOI] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang X‐P, Thompson AA, Cherezov V et al. (2014). Molecular control of δ‐opioid receptor signalling. Nature 506: 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ et al. (2014). G protein–coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev 66: 413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita W, Gomes I, Devi LA (2015). Heteromers of μ‐δ opioid receptors: new pharmacology and novel therapeutic possibilities. Br J Pharmacol 172: 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Ijzerman A (2000). Allosteric modulation of A(2A) adenosine receptors by amiloride analogues and sodium ions. Biochem Pharmacol 60: 669–676. [DOI] [PubMed] [Google Scholar]
- Göblyös A, de Vries H, Brussee J, Ijzerman AP (2005). Synthesis and biological evaluation of a new series of 2,3,5‐substituted [1,2,4]‐thiadiazoles as modulators of adenosine A1 receptors and their molecular mechanism of action. J Med Chem 48: 1145–1151. [DOI] [PubMed] [Google Scholar]
- Haga K, Kruse AC, Asada H, Yurugi‐Kobayashi T, Shiroishi M, Zhang C et al. (2012). Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482: 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harland AA, Yeomans L, Griggs NW, Anand JP, Pogozheva ID, Jutkiewicz EM et al. (2015). Further optimization and evaluation of bioavailable, mixed‐efficacy μ‐opioid receptor (MOR) agonists/δ‐opioid receptor (DOR) antagonists: balancing MOR and DOR affinities. J Med Chem 58: 8952–8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman LH, Ye K, Oosterom J, Ijzerman AP (2008). Amiloride derivatives and a nonpeptidic antagonist bind at two distinct allosteric sites in the human gonadotropin‐releasing hormone receptor. Mol Pharmacol 73: 1808–1815. [DOI] [PubMed] [Google Scholar]
- Heron M (2016). Deaths: leading causes for 2013. Natl Vital Stat Rep 65: 1–95. [PubMed] [Google Scholar]
- Hoare SR, Coldwell MC, Armstrong D, Strange PG (2000). Regulation of human D1, D2(long), D2(short), D3 and D4 dopamine receptors by amiloride and amiloride analogues. Br J Pharmacol 130: 1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL et al. (2015). Structural insights into μ‐opioid receptor activation. Nature 524: 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J (1983). Biogenesis, release and inactivation of enkephlains and dynorphins. Br Med Bull 39: 17–24. [DOI] [PubMed] [Google Scholar]
- Hui W‐Q, Cheng Q, Liu T‐Y, Ouyang Q (2016). Homology modeling, docking, and molecular dynamics simulation of the receptor GALR2 and its interactions with galanin and a positive allosteric modulator. J Mol Model 22: 90. [DOI] [PubMed] [Google Scholar]
- Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG (2012). ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52: 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger D, Schmalenbach C, Prilla S, Schrobang J, Kebig A, Sennwitz M et al. (2007). Allosteric small molecules unveil a role of an extracellular E2/transmembrane helix 7 junction for G protein‐coupled receptor activation. J Biol Chem 282: 34968–34976. [DOI] [PubMed] [Google Scholar]
- Johnson KM, Melvin LS, Althuis TH, Bindra JS, Harbart CA, Milne GM et al. (1981). Selective and potent analgetics derived from cannabinoids. J Clin Pharmacol 21: 2715–2825. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz EM (2006). The antidepressant‐like effects of delta‐opioid receptor agonists. Mol Interv 6: 162–169. [DOI] [PubMed] [Google Scholar]
- Kathmann M, Flau K, Redmer A, Tränkle C, Schlicker E (2006). Cannabidiol is an allosteric modulator at μ‐ and δ‐opioid receptors. Naunyn Schmiedebergs Arch Pharmacol 372: 354–361. [DOI] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC (2014). Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39: 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T (2013). New concepts in pharmacological efficacy at 7TM receptors: IUPHAR review 2. Br J Pharmacol 168: 554–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalanne L, Ayranci G, Kieffer BL, Lutz P‐E (2014). The κ‐opioid receptor: from addiction to depression, and back. Front Psychiatry 5: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DG (2008). The nociceptin/orphanin FQ receptor: a target with broad therapeutic potential. Nat Rev Drug Discov 7: 694–710. [DOI] [PubMed] [Google Scholar]
- Lee KO, Akil H, Woods JH, Traynor JR (1999). Differential binding properties of oripavines at cloned μ‐ and δ‐opioid receptors. Eur J Pharmacol 378: 323–330. [DOI] [PubMed] [Google Scholar]
- Levitt ES, Clark MJ, Jenkins PM, Martens JR, Traynor JR (2009). Differential effect of membrane cholesterol removal on μ‐ and δ‐opioid receptors: a parallel comparison of acute and chronic signaling to adenylyl cyclase. J Biol Chem 284: 22108–22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowicz AM, Vepsäläinen J, Laitinen JT (2006). The ‘allosteric modulator’ SCH‐202676 disrupts G protein‐coupled receptor function via sulphydryl‐sensitive mechanisms. Br J Pharmacol 147: 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V et al. (2012). Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston KE, Traynor JR (2014). Disruption of the Na+ ion binding site as a mechanism for positive allosteric modulation of the μ‐opioid receptor. Proc Natl Acad Sci 111: 18369–18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G et al., Da Duan (2016). Structure‐based discovery of opioid analgesics with reduced side effects. Nature 537: 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I et al. (1996). Loss of morphine‐induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ‐opioid‐receptor gene. Nature 383: 819–823. [DOI] [PubMed] [Google Scholar]
- McNicol ED, Midbari A, Eisenberg E (2013). Opioids for neuropathic pain In: McNicol ED. (ed). Cochrane Database of Systematic Reviews. John Wiley & Sons, Ltd: Chichester, UK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson J, Rivero G, Baptist M, Llorente J, Al‐Sabah S, Krasel C et al. (2010). μ‐opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol 78: 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A et al. (2012). Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem 55: 1445–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP (1965). On the nature of allosteric transitions: a plausible model. J Mol Biol 12: 88–118. [DOI] [PubMed] [Google Scholar]
- Noble F, Benturquia N, Bilkei‐Gorzo A, Zimmer A, Roques BP (2008). Use of preproenkephalin knockout mice and selective inhibitors of enkephalinases to investigate the role of enkephalins in various behaviours. Psychopharmacology (Berl) 196: 327–335. [DOI] [PubMed] [Google Scholar]
- Noble F, Coric P, Fournié‐Zaluski MC, Roques BP (1992a). Lack of physical dependence in mice after repeated systemic administration of the mixed inhibitor prodrug of enkephalin‐degrading enzymes, RB101. Eur J Pharmacol 223: 91–96. [DOI] [PubMed] [Google Scholar]
- Noble F, Turcaud S, Fournié‐Zaluski MC, Roques BP (1992b). Repeated systemic administration of the mixed inhibitor of enkephalin‐degrading enzymes, RB101, does not induce either antinociceptive tolerance or cross‐tolerance with morphine. Eur J Pharmacol 223: 83–89. [DOI] [PubMed] [Google Scholar]
- Oates J, Watts A (2011). Uncovering the intimate relationship between lipids, cholesterol and GPCR activation. Curr Opin Struct Biol 21: 802–807. [DOI] [PubMed] [Google Scholar]
- Ohbuchi K, Miyagi C, Suzuki Y, Mizuhara Y, Mizuno K, Omiya Y et al. (2016). Ignavine: a novel allosteric modulator of the μ opioid receptor. Sci Rep 6: 31748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pert C, Snyder S (1974). Opiate receptor binding of agonists and antagonists affected differentially by sodium. Mol Pharmacol 1: 868–879. [Google Scholar]
- Pert CB, Pasternak G, Snyder SH (1973). Opiate agonists and antagonists discriminated by receptor binding in brain. Science 182: 1359–1361. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM (1986). Psychotomimesis mediated by κ‐opiate receptors. Science 233: 774–776. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM (2011). The role of beta‐arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 60: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roques BP, Fournié‐Zaluski M‐C, Wurm M (2012). Inhibiting the breakdown of endogenous opioids and cannabinoids to alleviate pain. Nat Rev Drug Discov 11: 292–310. [DOI] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S et al. (2002). Salvinorin A: a potent naturally occurring nonnitrogenous κ‐opioid selective agonist. Proc Natl Acad Sci U S A 99: 11934–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Murphy DL, Xu H, Godin JA, Dersch CM, Partilla JS et al. (2007). Salvinorin A: allosteric interactions at the μ‐opioid receptor. J Pharmacol Exp Ther 320: 801–810. [DOI] [PubMed] [Google Scholar]
- Saito H, Ueyama T, Naka N, Yagi J, Okamoto T (1982). Pharmacological studies of ignavine, an aconitum alkaloid. Chem Pharm Bull(Tokyo) 30: 1844–1850. [DOI] [PubMed] [Google Scholar]
- Schetz JA, Sibley DR (2001). The binding‐site crevice of the D4 dopamine receptor is coupled to three distinct sites of allosteric modulation. J Pharmacol Exp Ther 296: 359–563. [PubMed] [Google Scholar]
- Schneider S, Provasi D, Filizola M (2015). The dynamic process of drug‐GPCR binding at either orthosteric or allosteric sites evaluated by metadynamics. Methods Mol Biol 1335: 277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seely KA, Brents LK, Franks LN, Rajasekaran M, Zimmerman SM, Fantegrossi WE et al. (2012). AM‐251 and rimonabant act as direct antagonists at μ‐opioid receptors: implications for opioid/cannabinoid interaction studies. Neuropharmacology 63: 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, LeRouzic V, Schneider S, Bisignano P, Pasternak GW, Filizola M (2014). Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions. Biochemistry 53: 5140–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Yeatman HR, Provasi D, Alt A, Christopoulos A, Canals M et al. (2016). Proposed mode of binding and action of positive allosteric modulators at opioid receptors. ACS Chem Biol 11: 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippenberg TS (2009). The dynorphin/kappa opioid receptor system: a new target for the treatment of addiction and affective disorders? Neuropsychopharmacology 34: 247. [DOI] [PubMed] [Google Scholar]
- Simon EJ, Groth J (1975). Kinetics of opiate receptor inactivation by sulfhydryl reagents: evidence for conformational change in presence of sodium ions. Proc Natl Acad Sci U S A 72: 2404–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BH, Torrance N, Johnson M (2012). Assessment and management of neuropathic pain in primary care. Pain Manag 2: 553–559. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I et al. (2013). Structure of the CCR5 chemokine receptor‐HIV entry inhibitor maraviroc complex. Science 341: 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DM, Sun B, Feng D, Nawaratne V, Leach K, Felder CC et al. (2016). Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 531: 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson GL, Lane JR, Coudrat T, Sexton PM, Christopoulos A, Canals M (2015). Biased agonism of endogenous opioid peptides at the μ‐opioid receptor. Mol Pharmacol 88: 335–346. [DOI] [PubMed] [Google Scholar]
- Toll L (2013). The use of bifunctional NOP/mu and NOP receptor selective compounds for the treatment of pain, drug abuse, and psychiatric disorders. Curr Pharm Des 19: 7451–7460. [PubMed] [Google Scholar]
- Valverde O, Fournie‐Zaluski MC, Roques BP, Maldonado R (1996). Similar involvement of several brain areas in the antinociception of endogenous and exogenous opioids. Eur J Pharmacol 312: 15–25. [DOI] [PubMed] [Google Scholar]
- Vaysse PJ, Gardner EL, Zukin RS (1987). Modulation of rat brain opioid receptors by cannabinoids. J Pharmacol Exp Ther 241: 534–539. [PubMed] [Google Scholar]
- Volkow ND (2014). America's addiction to opioids: heroin and prescription drug abuse. Available at: https://www.drugabuse.gov/about-nida/legislative-activities/testimony-to-congress/2016/americas-addiction-to-opioids-heroin-prescription-drug-abuse.
- Winters BL, Gregoriou GC, Kissiwaa SA, Wells OA, Medagoda DI, Hermes SM et al. (2017). Endogenous opioids regulate moment‐to‐moment neuronal communication and excitability. Nat Commun 8: 14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yoon SI, Huang P, Wang Y, Chen C, Chong PL et al. (2006). Localization of the kappa opioid receptor in lipid rafts. J Pharmacol Exp Ther 317: 1295–1306. [DOI] [PubMed] [Google Scholar]
- Yabaluri N, Medzihradsky F (1997). Regulation of mu‐opioid receptor in neural cells by extracellular sodium. J Neurochem 68: 1053–1061. [DOI] [PubMed] [Google Scholar]
- Zheng H, Pearsall EA, Hurst DP, Zhang Y, Chu J, Zhou Y et al. (2012). Palmitoylation and membrane cholesterol stabilize μ‐opioid receptor homodimerization and G protein coupling. BMC Cell Biol 13: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]