

Abstract
Background and Purpose
The histaminergic system is a promising target for the development of new analgesics, as histamine H3 and H4 receptors are expressed in regions concerned with nociceptive transmission. Here we have determined the analgesic effects of new H3 and H4 receptor antagonists in naive and neuropathic mice.
Experimental Approach
We used chronic constriction injury (CCI) to the sciatic nerve in mice to model neuropathy. Effects of a new H3 receptor antagonist, E‐162(1‐(5‐(naphthalen‐1‐yloxy)pentyl)piperidine) and H4 receptor antagonist, TR‐7(4‐(4‐chlorophenyl)‐6‐(4‐methylpiperazin‐1‐yl)‐1,3,5‐triazin‐2‐amine) were assessed on mechanical (von Frey) and thermal (cold plate, tail flick) stimuli in mice with and without CCI (7 days after injury). Effects of these antagonists on morphine analgesia were also evaluated, along with the possible participation of H1 receptors in their effects. We analysed the compounds in binding and functional cAMP assays at the H3 and H4 receptors and determined metabolic stability.
Key Results
E‐162 and TR‐7 attenuated nociceptive responses and profound morphine analgesia in males with CCI. These antagonists showed analgesia in naive mice (tail flick test) and produced prolonged analgesia in neuropathic females. E‐162‐induced analgesia was reversed by pyrilamine, an H1 receptor antagonist. E‐162 bound potently to H3 receptors (K i = 55 nM) and inhibited cAMP accumulation (IC50 = 165 nM). TR‐7 showed lower affinity for H4 receptors (K i = 203 nM) and IC50 of 512 nM.
Conclusions and Implications
We describe a therapeutic use for new H3 (E‐162) and H4 receptor (TR‐7) antagonists in neuropathy. Targeting H3 and H4 receptors enhanced morphine analgesia, consistent with multimodal pain therapy.
Abbreviations
- CCI
chronic constriction injury
- E‐162
1‐(5‐(naphthalen‐1‐yloxy)pentyl)piperidine)
- SNI
spared nerve injury
- TR‐7
4‐(4‐chlorophenyl)‐6‐(4‐methylpiperazin‐1‐yl)‐1,3,5‐triazin‐2‐amine)
Introduction
Effective pain therapy is one of society's principal needs. Neuropathic pain represents a significant clinical problem and, as a chronic condition, it can cause distress and seriously affect a patient's quality of life. This condition is often refractory to conventional therapy. Analgesics that are used to treat this pathology produce considerable side effects and, moreover, the great majority of patients obtain only partial relief (Kingery, 1997; Finnerup et al., 2005). Therefore, there is a clear need for new analgesics.
A promising target for the development of new analgesics are the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=264 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=265. Histamine exerts its effects on many physiological and pathological processes, such as inflammation and gastric acid secretion, and as a neurotransmitter. It also appears to play a role in modulation of nociceptive transmission. The H3 and H4 receptors are GPCRs, which are associated with Gi/o proteins and consequently down‐regulate cAMP signalling and enhance calcium mobilization. H3 autoreceptors are involved in negative feedback of histamine levels (Torrent et al., 2005; Moreno‐Delgado et al., 2006), while heteroreceptors are involved in crosstalk with other neurotransmitters, such as ACh, dopamine, 5‐HT and noradrenaline (Schlicker et al., 1988; Schlicker et al., 1989; Clapham and Kilpatrick, 1992; Schlicker et al., 1993; Blandina et al., 1996; Hsieh et al., 2010a). H4 receptors are predominantly expressed within immune cells (O'Reilly et al., 2002; Hofstra et al., 2003; Zampeli and Tiligada, 2009). However, recently reported data showing the presence of H4 receptors on neurons highlight its participation in neuronal functions (Connelly et al., 2009; Sanna et al., 2015). As expression of both receptors has been reported in regions of the CNS related to nociceptive transmission, such as dorsal root ganglia and the spinal cord (Connelly et al., 2009; Kajihara et al., 2010; Sanna et al., 2015, 2017a), H3 and H4 receptors have emerged as promising targets for pharmacological intervention in the development of new analgesics. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1267 – which is a ligand that is extensively used in H3 receptor male and female ‐related pain studies – is both a H3 and H4 receptor inverse agonist (Gbahou et al., 2006). Therefore, there is a strong need for new, effective and selective pharmacological tools to study the role of the histaminergic system in pain.
Consequently, we investigated the analgesic effect of the i.p. administration of newly synthesized antagonists of the H3 receptor (E‐162) and of the H4 receptor (TR‐7) in naive animals and in a preclinical model of neuropathic pain [chronic constriction injury (CCI)] in mice. We also investigated the effects of co‐administration of the tested compounds in neuropathic males. Differences between males and females in pain perception have recently been intensively studied (Vacca et al., 2014). Therefore, in the present study, we analysed sex‐dependent differences in analgesic effects of the tested compounds.
Interestingly, some data have revealed that ligands of histamine receptors may modulate the action of the opioid system, which is unquestionably an essential control system in nociceptive transmission (Owen et al., 1994; Mobarakeh et al., 2009; Galeotti et al., 2013). Therefore, in our study, we carried out experiments to investigate the participation of H3 and H4 receptors in http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627 analgesia using the new receptor antagonists, E‐162 and TR‐7. Moreover, to evaluate the mechanism of action of the new compounds, we analysed the participation of spinal http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=262 in the effects of E‐162 and TR‐7.
To assess the in vivo metabolic stability of these histamine receptor ligands in mice, we used an in vitro model with mouse liver microsomes. The metabolic stability of drug candidates is an important parameter due to the key roles that pharmacokinetics and drug metabolism play as determinants of a drug's in vivo efficacy. Additionally – in order to determine the structure of the metabolites – a precise analysis of the fragment ions produced by substrates and metabolites under ion fragment analysis conditions was undertaken, supported by in silico data.
Methods
Animals
All animal care and experimental procedures complied with the recommendations of the International Association for the Study of Pain (Zimmermann, 1983) and the NIH Guide for the Care and Use of Laboratory Animals and were approved by the II Local Ethics Committee Branch of the National Ethics Committee for Experiments on Animals based at the Institute of Pharmacology, Polish Academy of Sciences (approval number: 37/2016, 123/2017, Cracow, Poland). Care was taken to minimize animal suffering and minimize the number of animals used (3R policy). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Adult male and female albino Swiss CD‐1 mice initially weighing between 20 and 25 g were purchased from Charles River Laboratories (Hamburg, Germany). The animals were housed in groups of six under controlled conditions (temperature 21 ± 2°C; 12 h light/dark cycle – lights on at 06:00 h) with ad libitum food and water.
Sciatic nerve surgery
The model of neuropathy was generated by CCI to the sciatic nerve, performed under isoflurane anaesthesia (2% isoflurane in 100% oxygen with a flow of 1.5 L·min−1) according to the procedure described by Bennett and Xie (1988) and modified for mice by Mika et al. (2007). Briefly, an incision was made below the right hipbone, parallel to the sciatic nerve. The sciatic nerve was exposed, and three ligatures (3/0 silk) were tied loosely around the nerve, distal to the sciatic notch, with 1 mm spacing until a brief twitch in the respective hind limb was observed. No procedure was conducted on the control animals. After CCI, all mice developed allodynia and hyperalgesia.
Drug administration
E‐162 and TR‐7 were dissolved in 25% DMSO/water and administered i.p. 1–20 mg·kg−1 (injection volume 10 mL·kg−1 of body weight) at day 7 after CCI (Scheme 1) or to naive animals. The control group received the vehicle (25% DMSO/water) according to the same schedule. Morphine and pyrilamine were dissolved in water for injection, and the control group received this solvent. Pyrilamine (10 μg; (Wei et al., 2016) was injected i.t. (volume 5 μL) through a lumbar puncture between L5 and L6 to non‐anaesthetized mice, as described by Fairbanks (2003). The i.t. injection was performed with disposable 30‐gauge 0.5 in. needles (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) matched to a 25 μL syringe (Hamilton, OH, USA).
Scheme 1.
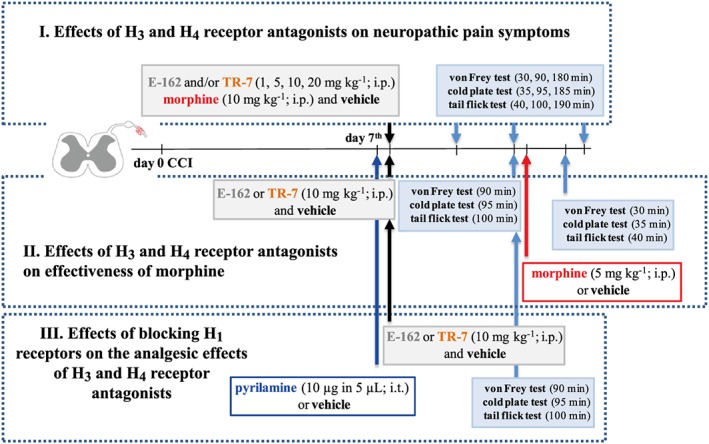
Behavioural tests schedule.
Behavioural tests
Behavioural experiments were performed between 08:00 and 12:00 h. Behavioural tests were conducted on naive (Supporting Information Figures S1–S3) or neuropathic animals (at day 7 after CCI) according to the schedule presented in Scheme 1. Behavioural evaluations were performed by a blinded observer: i.e., without knowledge of drug administration. Across the experiments, we compared vehicle‐treated animals (referred to as control animals) with E‐162‐treated and TR‐7‐treated animals at different time points after treatment. For the behavioural study, mice were assessed before (pretest) and after intervention and served as their own controls. Experiments were conducted 30, 90 and 180 min (von Frey test), 35, 95 and 185 min (cold plate test) and 40, 100 and 190 min (tail flick test) after the i.p. administration of each drug (Figures 1A–F, 2A–F and 3A, C, E).
Figure 1.
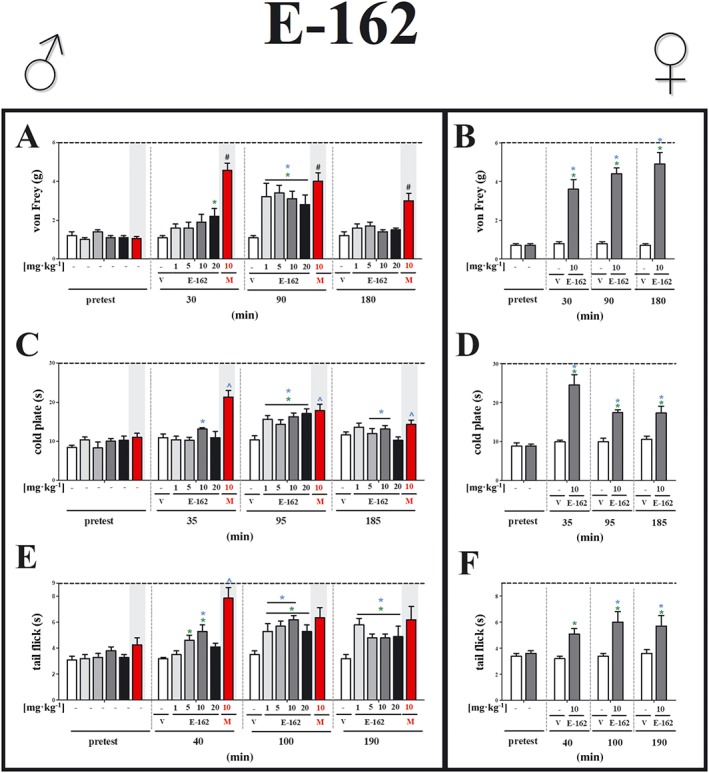
Effects of the H3 receptor antagonist, E‐162, on neuropathic pain symptoms in male and female mice with CCI. The effects of single i.p. administration of E‐162 (males: 1, 5, 10 and 20 mg·kg−1; females: 10 mg·kg−1) on mechanical (A, von Frey) and thermal (B, cold plate; C, tail flick) stimulus on day 7 following CCI to the sciatic nerve, were evaluated. The data are presented as means ± SEM. The group receiving morphine (M; 10 mg·kg−1, i.p.), was used as a positive control in this study. The analgesic effects of morphine were compared with vehicle (V; water)‐treated mice, which received water for injection as a vehicle. The results of the experiments were statistically evaluated using unpaired sample t‐test (B, D, F), a repeated measures ANOVA (A, C, E; blue symbols) Changes in tested doses at each time point were performed using one‐way ANOVA (A, C, E; green symbols) and were further analysed with Bonferroni's post hoc test. * P < 0.05, significantly different from vehicle (25% DMSO/water)‐treated mice; # P < 0.05, significantly different from vehicle (water)‐treated mice. The number of animals in each group was as follows: von Frey test [males: E‐162 1, 5, 10 mg·kg−1 (n = 7) and 20 mg·kg−1 (n = 6), V (n = 8) and M 10 mg·kg−1 (n = 7); females: E‐162 10 mg·kg−1 (n = 7) and V (n = 7)]; cold plate test [males: E‐162 1 mg·kg−1 (n = 7) 5, 10 and 20 mg·kg−1 (n = 6), V (n = 8) and M 10 mg·kg−1 (n = 7); females: E‐162 10 mg·kg−1 (n = 6) and V (n = 6)]; and tail flick test [males: E‐162 1, 5, 10 and 20 mg·kg−1 (n = 6), V (n = 8) and M 10 mg·kg−1 (n = 6); females: E‐162 10 mg·kg−1 (n = 7) and V (n = 7)].
Figure 2.
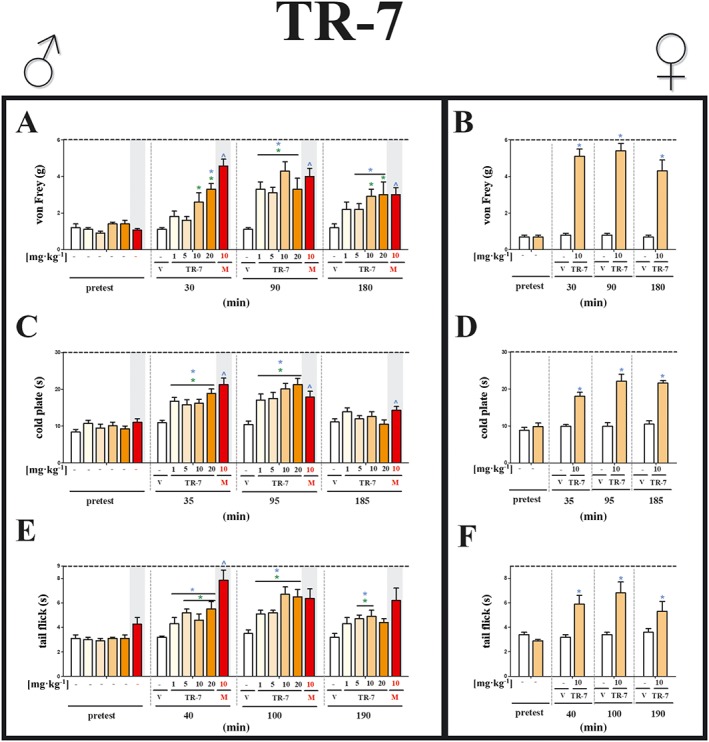
Effects of TR‐7, the H4 receptor antagonist, on neuropathic pain symptoms in male and female mice with CCI. The effects of single i.p. administration of TR‐7 (males: 1, 5, 10 and 20 mg·kg−1; females: 10 mg·kg−1) on mechanical (A, von Frey) and thermal (B, cold plate; C, tail flick) stimulus on day 7 following CCI to the sciatic nerve, were evaluated. The data are presented as means ± SEM. The group, which received morphine (M; 10 mg·kg−1, i.p.), was used as a positive control in this study. The analgesic effects of morphine were compared with vehicle (V; water)‐treated mice, which received water for injection as a vehicle. The results of the experiments were statistically evaluated using unpaired sample t‐test (B, D, F), a repeated measures ANOVA (A, C, E; blue symbols) Changes in tested doses at each time point were performed using one‐way ANOVA (A, C, E; green symbols) and were further analysed with Bonferroni's post hoc test. ^ P < 0.05, significantly different from vehicle (25% DMSO/water)‐treated mice; # P < 0.05, significantly different from vehicle (water)‐treated mice. The number of animals each group was as follows: von Frey test [males: TR‐7 1 (n = 6), 5 (n = 7), 10 (n = 8) and 20 (n = 6) mg·kg−1, V (n = 8) and M 10 mg·kg−1 (n = 7); females: TR‐7 10 mg·kg−1 (n = 7) and V (n = 7)]; cold plate test [males: TR‐7 1 (n = 8) 5, 10 (n = 7) and 20 (n = 8) mg·kg−1, V (n = 8) and M 10 mg·kg−1 (n = 7); females: TR‐7 10 mg·kg−1 (n = 6) and V (n = 6)]; and tail flick test [males: TR‐7 1, 5, 10 and 20 mg·kg−1 (n = 7), V (n = 8) and M 10 mg·kg−1 (n = 6); females: TR‐7 10 mg·kg−1 (n = 7) and V (n = 7)].
Figure 3.
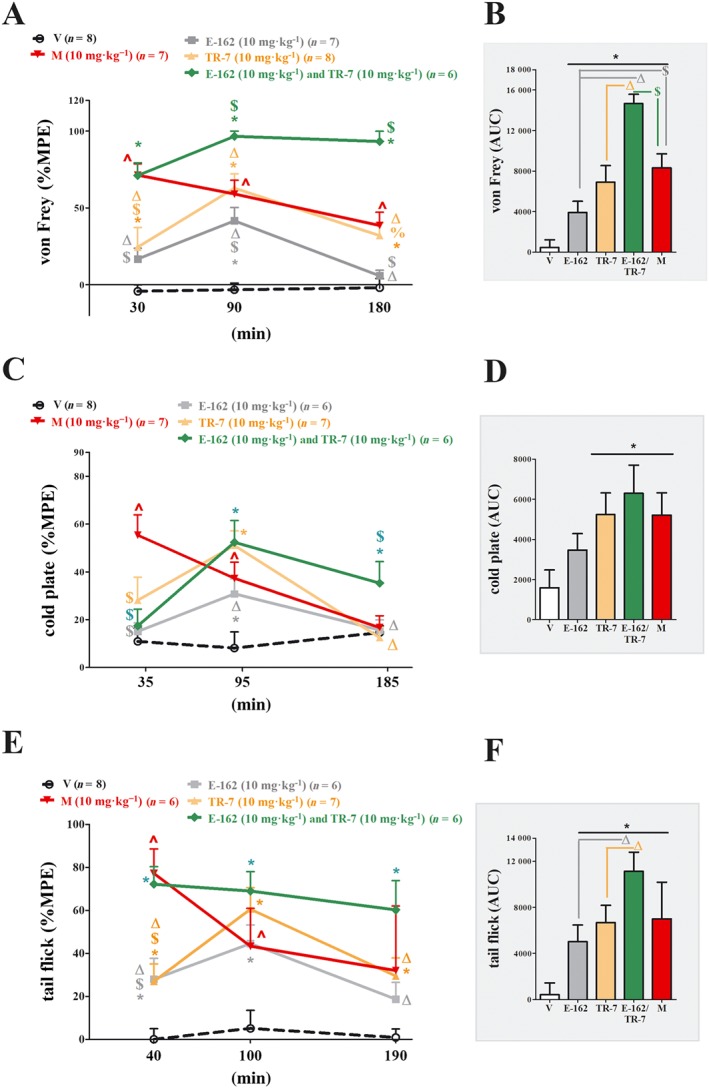
Comparison of the analgesic effects of administration of E‐162, TR‐7 or co‐administration of E‐162+TR‐7, with morphine in mice with CCI. The effects of single i.p. injection of E‐162 and TR‐7 or co‐administration of E‐162+TR‐7 (10 mg·kg−1 each) on mechanical (A, von Frey) and thermal (B, cold plate; C, tail flick) stimulus on day 7 following CCI to the sciatic nerve, were evaluated. The data are presented as means ± SEM . The results are presented as a percentage of the maximal possible effect [%MPE = 100% × (measured response − basal)∕(cut‐off value − basal)] of drug action (A, C, E), and the AUC for all tests was calculated (B, D, F). The group receiving morphine (M; 10 mg·kg−1, i.p.), was used as a positive control in this study. The analgesic effects of morphine were compared with vehicle (V; water)‐treated mice, which received water for injection as a vehicle . The results of the experiments were statistically evaluated using one‐way ANOVA and were further analysed with Bonferroni's post hoc test. * P < 0.05, significantly different from vehicle (25% DMSO/water)‐treated mice; # P < 0.05, significantly different from vehicle (water)‐treated mice; % P < 0.05, significant differences between E‐162‐treated and TR‐7‐treated mice; $ P < 0.05, significant differences between antagonist‐treated and morphine‐treated mice; Δ P < 0.05, significant differences between E‐162‐treated or TR‐7‐treated and E‐162+TR‐7‐treated mice. The number of animals used in the study is highlighted on the graph.
Von Frey test
The von Frey test was used for the assessment of tactile allodynia (Mika et al., 2007). Mice were placed in plastic cages with a wire net floor 5 min before the experiment. Mechanical sensitivity to non‐noxious stimuli was measured by applying a set of calibrated nylon monofilaments (0.6–6 g; Stoelting, Wood Dale, IL, USA) in serial increments on the ipsilateral and contralateral hind paw (Supporting Information Figure S2) midplantar surface until a behavioural response was observed. These responses included paw withdrawal, shaking and licking. The contralateral paw was not affected by compound treatment in male and female mice with CCI (Supporting Information Figure S2). In the von Frey test, results are expressed as pressure (g) applied with the filament; the cut‐off filament was 6 g.
Cold plate test
Sensitivity to noxious thermal stimuli was assessed with the cold plate test (Cold/Hot Plate Analgesia Meter, Columbus Instruments) as previously described (Mika et al., 2007). The temperature of the plate was kept at 2°C, and the cut‐off latency was 30 s. The mice were placed on the cold plate, and the time until lifting of the injured paw was recorded.
Tail flick test
The tail flick test was used to measure spinal nociceptive responses to thermally induced pain. The responsiveness to a thermal stimulus was determined using a tail flick analgesic meter (Analgesia Meter; Ugo Basile, Monvalle, Italy). Tail flick latency was measured on the tail at two‐thirds of its length by applying a focused beam of light (thermal stimulus). The time interval between onset of the thermal stimulus and withdrawal of the tail from the beam was recorded; the cut‐off latency was 9 s to prevent tissue damage.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The behavioural data are presented as means ± SEM. The unpaired sample t‐test (Figures 1B, D, F and 2B, D, F), repeated measures ANOVA (Figures 1 and 2) and one‐way ANOVA (Figures 1, 2, 3, 4, 5) were performed. Bartlett's test for homogeneity of variances was used to check that the assumption of equal variances is true before running further statistical tests. The behavioural data in Figure 3A, C, E are presented as a percentage of the maximal possible effect [%MPE = 100% × (measured response − basal)∕(cut‐off value − basal)] of drug action and additionally as AUC in Figure 3B, D, F. The differences between the groups were further analysed with Bonferroni's post hoc test. Significant differences between group means are indicated when P < 0.05. The analysis and charts were prepared using GraphPad Prism v.5.04 (GraphPad Software, La Jolla, CA, USA).
Figure 4.
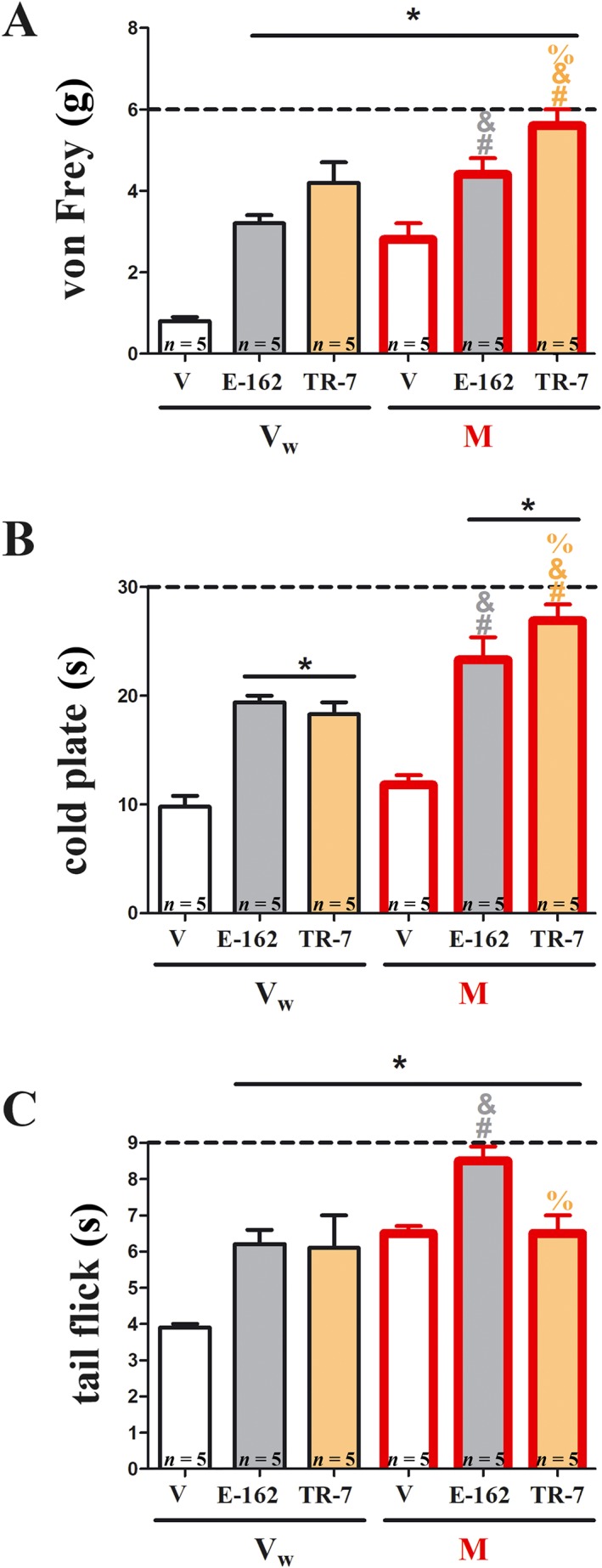
Effects of E‐162 or TR‐7 on morphine analgesia in mice with CCI. The effects of single i.p. administration of E‐162 or TR‐7 (10 mg·kg−1) on morphine (M; 5 mg·kg−1, i.p.) analgesia on day 7 following CCI to the sciatic nerve, were evaluated. Ninety minutes after antagonist administration, mice received a single injection of morphine or vehicle (water; Vw); behavioural tests were performed 30 min after injection. Mechanical allodynia was assessed by (A) von Frey test and thermal hyperalgesia by (B) cold plate test, and spinal nociceptive responses to heat‐induced pain were measured by (C) tail flick test. The data are presented as means ± SEM. The results were statistically evaluated using one‐way ANOVA and were further analysed with Bonferroni's post hoc test. * P < 0.05, significantly different from vehicle (25% DMSO/water)‐treated mice; # P < 0.05, significant differences between vehicle (25% DMSO/water)‐treated and antagonist‐treated mice that had received a single dose of morphine; & P < 0.05, significant differences between antagonist‐treated mice that received a single dose of vehicle (water)‐treated and antagonist‐treated mice that received a single dose of morphine; % P < 0.05, significant differences between E‐162‐treated and TR‐7‐treated mice. The number of animals used in the study is highlighted on the bar graph.
Figure 5.
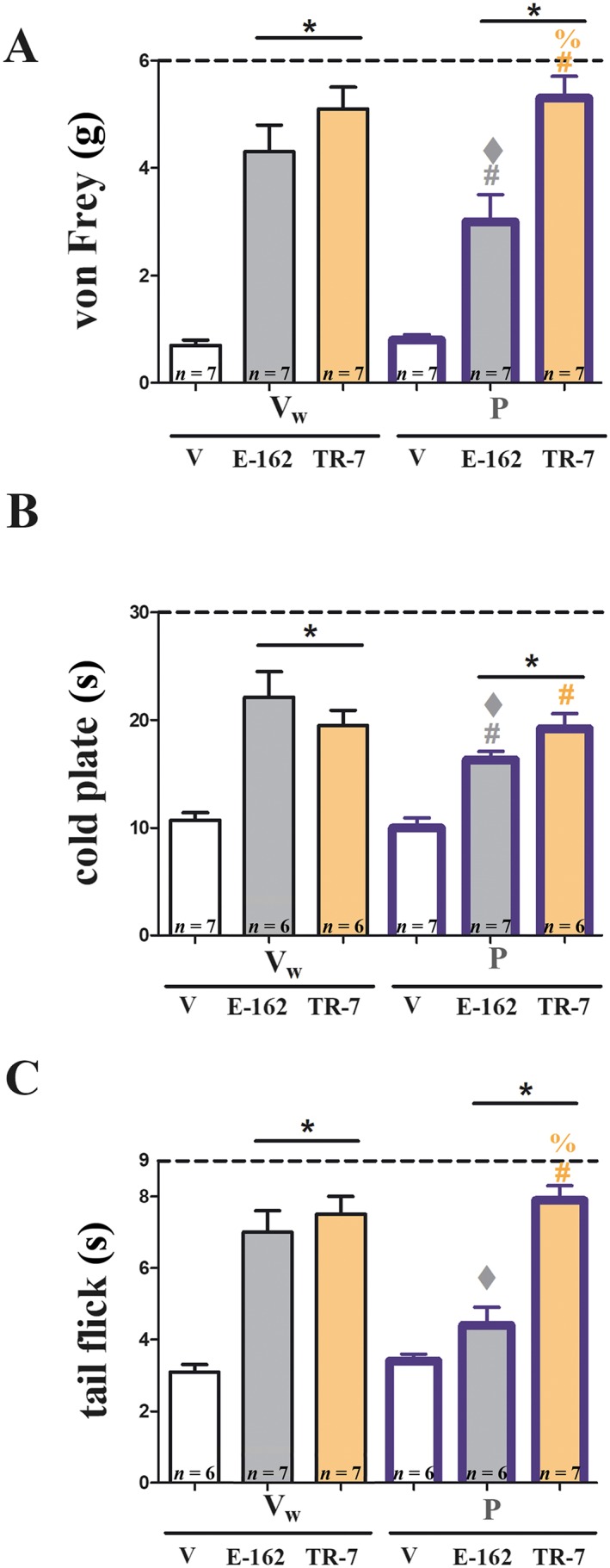
The influence of blockade of spinal H1 receptors on analgesic effects of E‐162 and TR‐7 in mice with CCI. The participation of spinal H1 receptors in analgesic effects of E‐162 (10 mg·kg−1, i.p.) and TR‐7 (10 mg·kg−1, i.p.) on day 7 following sciatic nerve injury (CCI) was analysed. The H1 receptor antagonist, pyrilamine (P; 10 μg 5 μL−1, i.t.), or solvent (Vw; water) were injected i.t. 30 min before drug treatment. Behavioural tests were performed 90 min after drug exposure. Mechanical allodynia was assessed by (A) von Frey test and thermal hyperalgesia by (B) cold plate test, and spinal nociceptive responses to heat‐induced pain were measured by (C) tail flick test. The data are presented as means ± SEM. The results of the experiments were statistically evaluated using one‐way ANOVA and were further analysed with Bonferroni's post hoc test. * P < 0.05, significantly different from vehicle (25% DMSO/water)‐treated mice; # P < 0.05, significant differences between vehicle (25% DMSO/water)‐treated and drug‐treated mice that had received a single dose of pyrilamine; ♦ P < 0.05, significant differences between antagonist‐treated and pyrilamine‐treated mice; and % P < 0.05, significant differences between E‐162‐treated and TR‐7‐treated mice. The number of animals used in the study is highlighted on the bar graph.
Materials
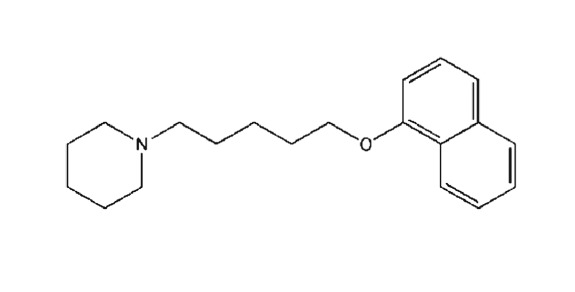
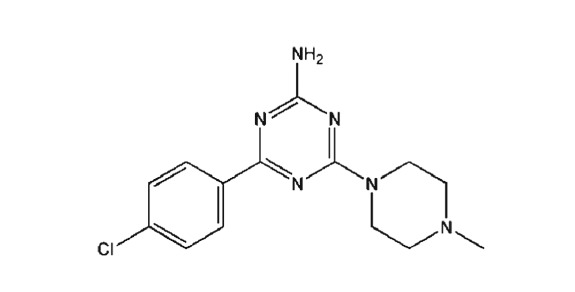
Compounds used in these experiments were supplied as follows: morphine hydrochloride (Polfa Kutno, Poland) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1227 maleate (Sigma‐Aldrich, Poznań, Poland). E‐162 and TR‐7 were synthesized in the Department of Technology and Biotechnology of Drugs (Jagiellonian University Medical College, Krakow, Poland), as described in the Supporting Information. Chemical structures and in vitro histamine H3, H4 and H1 receptor data of tested compounds are presented in Table 1.
Table 1.
Structures and in vitro pharmacological profiles of E‐162 and TR‐7 in binding and functional assays (cAMP) at human H3 receptors, H4 receptors, and H1 receptors
| In vitro assays | Compounds | ||
|---|---|---|---|
| E‐162 | TR‐7 | ||
|
|
|
||
| H3R | Binding assay | K i = 55 ± 15 nMa | K i = 13 200 ± 800 nMa |
| Functional cAMP assay | IC50 = 165 nMb | nt | |
| H4R | Binding assay | K i = 58 500 ± 6600 nMc | K i = 203 ± 65 nMe |
| Functional cAMP assay | nt | IC50 = 512 nMf | |
| H1R | Binding assay | K i = 1824 ± 203 nMd | nt |
| Functional assay in guinea pig ileum | nt | pA2 = 6.3g | |
nt, not tested.
[3H]N α‐Methylhistamine binding assay performed with cell membrane preparation of HEK293 cells stably expressing the human histamine H3 receptor. Data are means ± SD of three independent experiments.
cAMP accumulation assay in HEK293 cells expressing human histamine H3 receptors, co‐treated with forskolin, (R)‐(−)‐α‐methylhistamine and the tested compound.
[3H]Histamine binding assay in membrane preparations of CHO cells stably expressing the human histamine H4 receptor. Data are means ± SD of three independent experiments.
[3H]Pyrilamine binding assay performed with CHO‐K1 cells stably transfected with the human H1 receptor. Data are means ± SD of three independent experiments.
[3H]Histamine binding assay performed with membrane preparation of Sf9 cells expressing the human H4 receptor, co‐expressed with G‐protein Gαi2 and Gβ1γ2 subunits (data from Łażewska et al., 2014).
cAMP accumulation assay in CHO cells expressing the human H4 receptor, co‐treated with forskolin, histamine and the tested compound (Łażewska et al., 2014).
Data from Mogilski et al. (2017).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
In vitro profile of E‐162 and TR‐7 in binding and functional assays (cAMP) at the H3, H4 and H1 receptors
The molecular and cellular action of the new compounds, E‐162 and TR‐7, on H3 and H4 receptors was evaluated in binding and functional (cAMP) assays. E162 demonstrated good affinity for human H3 receptors in HEK293 cells (Table 1). This compound also blocked the decrease in http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2352 induced by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1236, in HEK293‐hH3R‐cAMPzen cells co‐treated with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5190, and was therefore classified as an antagonist at H3 receptors. In terms of this cAMP assay, the IC50 for E‐162 (Table 1) was about 100‐fold higher than that for the known H3 receptor antagonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8924 (IC50 of 1.3 ± 0.6 nM; Supporting Information Figures S1). Moreover, E‐162 demonstrated selectivity for H3 over H4 receptors, as shown in the [3H]histamine binding assay in membrane preparations of CHO cells stably expressing the human H4 receptor and in the [3H]pyrilamine binding assays, performed with CHO‐K1 cells stably transfected with the human H1 receptor, E‐162 showed only weak affinity for these receptors (Table 1).
TR‐7 was classified as an antagonist at H4 receptors (Table 1) and it blocked the histamine‐induced cAMP reduction in CHO‐hH4R‐cAMPzen cells, co‐treated with forskolin (10 μM; Łażewska et al., 2014), determined by performing a dose–response curve in presence of a known H4 receptor agonist (histamine, 140 nM, corresponding to its EC80) and forskolin (Table 1). TR‐7 showed low affinity for human H3 receptors and therefore good (65 fold) selectivity towards H4 receptors over H3 receptors. The affinity of TR‐7 for the H1 receptors has been measured in the guinea pig ileum (Mogilski et al., 2017) and the pA2 value they obtained (Table 1) indicated a weak competitive interaction of this compound with the H1 receptors present in this tissue.
Effects of E‐162 and TR‐7 on neuropathic pain symptoms in male and female mice with CCI
Behavioural assessment of drugs was performed 7 days after CCI. Males were randomly assigned to 11 treatment groups: vehicle (25% DMSO/water) treated (i.p.), E‐162‐treated (1, 5, 10 and 20 mg·kg−1, i.p.), TR‐7‐treated (1, 5, 10 and 20 mg·kg−1, i.p.), vehicle (water)‐treated (i.p.) and morphine‐treated (10 mg·kg−1, i.p.) and females into three groups: vehicle (25% DMSO/water)‐treated (i.p.), E‐162‐treated (10 mg·kg−1, i.p.) and TR‐7‐treated (10 mg·kg−1, i.p.). Baseline measurements (pretest) for all tests were obtained before compound administration. There were no significant baseline differences between the groups. Application of the vehicle (25% DMSO/water) had no significant effect on the pain response (Figures 1A–F and 2A–F). The group receiving morphine was used as a positive control in this study (Figures 1A, C, E and 2A, C, E). The analgesic effects of morphine were compared with vehicle (water)‐treated mice, which received water for injection as a vehicle. There were no significant differences between vehicle (25% DMSO/water)‐treated and vehicle (water)‐treated mice (Supporting Information Table S1).
In male mice, the H3 receptor antagonist E‐162 exhibited significant analgesic effects at 90 min after injection for all tested doses, compared with pre‐stimulation data (pretest) (Figure 1A). Analysis of the data, at each time point separately, showed that a single injection of E‐162 increased the withdrawal thresholds in the von Frey test at all four doses (1 , 5 , 10 and 20 mg·kg−1) at 90 min after drug administration (Figure 1A). Only the highest dose of E‐162 was analgesic at 30 min after administration. In the cold plate test (Figure 1C), significant analgesic effects were observed at 35 min (only for 10 mg·kg−1), at 95 min for all tested doses and at 185 min for 5 and 10 mg·kg−1 after injection, compared with pretest data
The repeated measurements analysis of the tail flick test results (Figure 1E) showed significant analgesic effect of E‐162 at 40 min (only for dose 10 mg·kg−1), 100 min (for doses 1, 5 and 10 mg·kg−1) and 190 min (for all doses) after injection as compared with pre‐stimulation values.
In female mice, we found an analgesic effect of E‐162 (10 mg·kg−1) at all time points, compared with pre‐stimulation values, in the von Frey and cold plate tests (Figure 1B, D) and at 100 and 190 min after injection in the tail flick test (Figure 1F). No analgesic effects could be detected 24 h after drug injection (data not shown).
Assays, in male mice, of the H4 receptor antagonist TR‐7, using the von Frey test (Figure 2A), showed analgesic effects at 30 min (for 10 and 20 mg·kg−1), 90 min (for all doses) and 180 min (10 and 20 mg·kg−1) after a single injection, compared with pretest data. In the cold plate test (Figure 2C), TR‐7 showed analgesic effects for all doses, at 35 and 95 min, compared with pretest data. TR‐7 also exhibited analgesic effects in the tail flick test (Figure 2E), at 40 and 100 min (for all doses) and 190 min (for 5 and 10 mg·kg−1), compared with pretest data.
TR‐7 in female mice showed analgesic effects at all time points after a single injection of 10 mg·kg−1, in the von Frey, cold plate and tail flick tests (Figure 2B, D, F), compared with pretest data. No analgesic effects were observed 24 h after drug injection (data not shown).
Comparison of the analgesic effects of E‐162 and TR‐7 and co‐administration of both compounds, with morphine in mice with CCI
As revealed by the above experiments, we did not observe changes in analgesic action between tested doses. However, both antagonist compounds at a dose of 10 mg·kg−1 had significant analgesic effects in all tests. We then investigated the effects of the compounds injected simultaneously. In this set of experiments we chose a single effective dose (10 mg kg‐1) of both antagonists and for the three tests already used we have calculated the analgesic effects as %MPE and AUC (Figure 3A, C, E). We gave each antagonist alone and in combination, i.e. in the same injection, using morphine (10 mg kg‐1) as a positive control.
As assessed by the %MPE from the von Frey test (Figure 3A), both compounds given alone showed analgesic action at 90 min after injection, but only TR‐7 was effective at 30 min and 180 min. The combination of antagonists showed analgesic action at all time points (Figure 3A).
Compared with morphine, both drugs given alone, were less effective at 30 min and in case of E‐162 also at 180 min. However, there were no significant differences between morphine and the antagonists, at 90 min after injection. By contrast, the combination of E‐162 and TR‐7 was more effective than morphine at 90 and 180 min (Figure 3A) and consequently, the combination was also more effective than the antagonists given alone at different times (for E‐162 at 30 and 180 min; for TR‐7 at 30, 90 and 180 min) (Figure 3A).The antiallodynic effect of TR‐7 was greater than that of E‐162 at 180 min (Figure 3A). Calculating the AUC for the von Frey tests confirmed that both drugs were effective analgesics but, by this measure, TR‐7 was as effective as morphine analgesia (Figure 3B). Moreover, the combination was more effective than single administration and more effective than morphine (Figure 3B).
Using the %MPE calculated from the cold plate assay (Figure 3C), both antagonists given alone were strongly antihyperalgesic only at 95 min, compared with the vehicle. The combination was effective at 95 min and at the last time point, 185 min. Also, ,compared with single administration, the combination was more effective than E‐162 at 95 and 185 min and more effective than TR‐7 at 185min. Relative to the effects of morphine, both antagonists given alone and in combination were less effective analgesics than morphine at the first time point (35min) but at both later times the combination was more effective. Analysis of AUC data from the cold plate assay (Figure 3D) showed that TR‐7 alone, the combination of TR‐7 and E‐162, and morphine were equally effective.
In the tail flick test (Figure 3E), the calculated %MPE showed that the analgesic effects of the antagonists given alone varied with time (from 40 to 190 min) with a peak at 100 min. However, the combination was more effective and was maintained essentially unchanged over the experimental period. The analgesic effects of morphine were most marked at 40 min and then decreased at the two later time points. These different temporal patterns of activity meant that, at 40 min, E‐162 and TR‐7 alone were less potent than morphine but at 100 and 190min they were as effective as morphine. The combination treatment was as effective as morphine at 40 min but remained highly analgesic at the later times (Figure 3E). AUC analysis from tail flick results (Figure 3F) also showed the analgesic action of E‐162 and TR‐7 and that both drugs were less effective when injected alone, compared with the combination.
Effects of E‐162 and TR‐7 on morphine analgesia in mice with CCI
We next studied the effects of pre‐treatment with the H3 and H4 receptor antagonists on the analgesia induced by morphine to assess possible interactions between their analgesic actions. Mice, on day 7 after CCI, were randomly assigned into three groups: vehicle (25% DMSO/water)‐treated, E‐162 (10 mg·kg−1, i.p.)‐treated or TR‐7 (10 mg·kg−1, i.p.)‐treated. Ninety minutes after this pre‐treatment, all animals received a single injection of morphine (5 mg·kg−1, i.p.) or vehicle (water), and behavioural tests were performed 30 min later. The results of the von Frey test (Figure 4A) showed that the analgesic effects of morphine were greater after treatment with E‐162 or TR‐7 than with morphine alone. Similar potentiation of the morphine response was shown in the cold plate test (Figure 4B). However, in the tail flick test (Figure 4C), only E‐162 improved the response to morphine. A comparison of the analgesia produced by morphine after pretreatment with the antagonists with the sum of their separate analgesic results suggested an additive effect, for both antagonists.
We also observed significant differences in the potentiation of morphine analgesia between E‐162 and TR‐7 pretreatment. This potentiation was greater after TR‐7 in the von Frey and cold plate tests (Figure 4A, B) but E‐162 was more effective in the tail flick test (Figure 4C).
The influence of blockade of spinal H1 receptors on analgesic effects of E‐162 and TR‐7 in mice with CCI
The contribution of spinal H1 receptors to the analgesic effects of E‐162 (10 mg·kg−1, i.p.) or TR‐7 (10 mg·kg−1, i.p.) was investigated using the H1 receptor antagonist, pyrilamine, given by i.t. injection (10 μg in 5 μL), 30 min before the H3 and H4 receptor antagonists. Behavioural tests were performed 90 min later. As shown in Figure 5, the analgesic effects of E‐162 were reduced by pyrilamine pretreatment in the von Frey, cold plate and tail flick tests, although the analgesic effects of TR‐7 were not affected by pyrilamine. Pyrilamine injected alone did not show any analgesic action (Figure 5).
Discussion
Our results have demonstrated the analgesic effects of the new H3 receptor (E‐162) and H4 receptor (TR‐7) antagonists in naive animals and in a preclinical model of neuropathic pain, in male and female mice. Moreover, both compounds significantly enhanced morphine analgesia. The analgesic effects of E‐162, but not TR‐7, were reduced after pretreatment with pyrilamine, a H1 receptor antagonist. Our results are consistent with earlier data showing that H3 and H4 receptors are present in regions related to nociceptive transmission (Connelly et al., 2009; Kajihara et al., 2010; Sanna et al., 2015, 2017a).
Histamine exerts its effects on many physiological and pathological processes, for example, nociceptive transmission, and its diverse biological actions are mediated via four receptors, H1, H2, H3 and H4. Many studies have shown that histamine injected directly into the brain decreases nociceptive transmission in naive (Chung et al., 1984; Bhattacharya and Parmar, 1985; Braga et al., 1992; Malmberg‐Aiello et al., 1994; Thoburn et al., 1994) as well as neuropathic (Sanna et al., 2015; Wei et al., 2016) animals. It is believed that the analgesic effects of histamine are mediated by H1 and H2 receptors (Thoburn et al., 1994; Braga et al., 1996; Lamberti et al., 1996; Wei et al., 2016). Some authors have also shown roles for H3 and H4 receptor in pain processing, although these results are somewhat conflicting. These discrepancies are mainly caused by differences in the pain models used in the given study, in the routes of drug administration and in the nature of the nociceptive stimulus (heat, pressure and chemicals) (Wei et al., 2016). The pharmacological variables in these studies are the selectivity of the compounds used, such as thioperamide (an extensively used ligand), which is both a H3 and H4 receptor inverse agonist (Gbahou et al., 2006). Therefore, effective and selective pharmacological tools to study the role of the histaminergic system are needed.
Our results revealed that systemic administration of E‐162, an H3 receptor antagonist, reduced symptoms of neuropathic pain. H3 autoreceptors are involved in negative feedback control of histamine levels, and an increase in histamine concentration in the CNS may account for the analgesic effects of E‐162. Moreover, blockade of the H3 receptors increased the release of other neurotransmitters (ACh, dopamine, 5‐HTand noradrenaline) in the CNS (Schlicker et al., 1988; Schlicker et al., 1989; Clapham and Kilpatrick, 1992; Schlicker et al., 1993; Blandina et al., 1996; Hsieh et al., 2010a). Depolarization activates histamine synthesis in nerve endings and this process is controlled by H3 autoreceptors (Torrent et al., 2005; Moreno‐Delgado et al., 2006). Here, we have shown that blockade of spinal H1 receptors by the antagonist pyrilamine decreased the analgesia induced by E‐162. Our new data support the hypothesis that the analgesic effects of E‐162 are partly due to regulation of the histamine level in the CNS. This possibility is consistent with other studies, where selective H3 receptor antagonists, GSK189254 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9103 (in repeated, orally delivered doses), decreased paw withdrawal thresholds. However, in rats with CCI (Medhurst et al., 2008), as far we know, there have been no studies of the effects of these compounds on thermal stimuli. There are also no data showing how these H3 receptor antagonists modulate pain transmission after a single dose. We have shown, for the first time, that i.p. injection of E‐162 attenuated nociceptive responses to heat (tail flick) and cold (cold plate), in a mouse model of neuropathic pain. The analgesic effect of E‐162 was also observed in naive animals in the tail flick test (Supporting Information Figure S1). Our in vitro metabolic stability study showed that E‐162 is a stable compound, as only trace amounts of hydroxylated metabolites were found after incubation with liver microsomes (Supporting Information S7). In summary, E‐162 seems to be a promising drug candidate in view of its analgesic effects and metabolic stability. Moreover, E‐162 is a structural homologue of pitolisant (Wakix™), an antagonist/inverse agonist of H3 receptors, which has been approved in the European Union for the treatment of narcolepsy (European Medicines Agency, 2015).
A recently discovered member of the histamine receptor family is the H4 receptor (Nakamura et al., 2000), which is mainly expressed within the cells of the immune system, such as mast cells, leukocytes, dendritic cells and T‐cells. There is a growing body of evidence for a role of H4 receptors in inflammation andalso, surprisingly, in neuronal action (Sanna et al., 2015). Data from knockout mice have illustrated an important role of these receptors in neuropathic pain, as H4 receptor‐KO mice with spared nerve injury (SNI) showed more severe thermal and mechanical hypersensitivity than control mice (Sanna et al., 2017b). The new H4 receptor antagonist, TR‐7, used in our study, had strong antinociceptive effects as, at a dose of 10 mg·kg−1, it was as effective as morphine, a gold standard in chronic pain treatment. This antagonist also exhibits anti‐inflammatory properties, as i.p. injection decreased carrageenan‐induced oedema in mice (Łażewska et al., 2014). Other anti‐inflammatory effects of TR‐7 were also found in a model of zymosan‐induced peritonitis, in which this compound decreased vascular permeability and the influx of inflammatory cells into the peritoneum. Further, TR‐7 reduced ROS, TNF‐α and IL‐1β production in RAW 264.7 macrophages (Mogilski et al., 2017). The strong relationship of H4 receptors with the immune system suggests that the antinociceptive action of TR‐7 may be due to a reduction in ongoing inflammatory processes at the site of injury during neuropathy and diminished activation of immune cells (especially mast cells), which release histamine in the periphery. Such persistent inflammatory conditions may contribute to maintenance of pain (Clatworthy et al., 1995). Sensitization of nociceptors may be caused by inflammatory mediators released by leukocytes, which are known to accumulate around the site of an injured peripheral nerve (Clatworthy et al., 1995; Perkins and Tracey, 2000). Mast cells are another cell type crucial to the development of neuropathic pain development, because degranulation or depletion of these cells reduces hyperalgesia (Perkins and Tracey, 2000; Thacker et al., 2007). Hsieh et al. (2010a) studied the effects of the H4 receptor antagonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=1279, and found that it diminished mechanical allodynia, given i.p., to rats with CCI (Hsieh et al., 2010b). These data are consistent with results obtained by our group. However, the antagonist (TR‐7) used by us also decreased thermally‐induced nociception and, used as a pre‐treatment, added to the analgesic effect of morphine. Interestingly, Sanna et al. (2015) showed that i.c.v. injection of two H4 receptor agonists (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=8983 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1274) reversed mechanical allodynia in mice with SNI. In the same study, an orally administered H4 receptor antagonist, JNJ10151984, did not affect the mechanical pain threshold, although it reversed the analgesic effect of histamine (i.c.v.) (Sanna et al., 2015). Most recently, i.t. injection of VUF8430 was shown to prevent mechanical allodynia in mice with SNI (Sanna et al., 2017a). Subcutaneous injection of thioperamide directly into the injured paw also increased mechanical hyperalgesia in rats with sciatic nerve ligation (Smith et al., 2007). All of these data indicate that the effects of H4 receptor ligands are dependent on the site of administration.
In contrast to E‐162, TR‐7 was metabolically unstable (Supporting Information S7) and the most probable structures of its metabolites and main metabolic pathways were identified. Therefore, taking into account that active metabolites may be more potent than the parent compound, we plan to synthesize and examine the activity of the main metabolites of TR‐7, in further studies.
Sex‐dependent differences in pain perception have recently been intensively studied. In our present study, we observed strong and long‐lasting analgesic action of both antagonists in female mice. In contrast to male mice, female mice with CCI exhibit long‐lasting allodynia and other sex‐related differences in nerve regeneration and glial cell activation (Vacca et al. 2014). Li et al. (2013) demonstrated sex differences in the chemosensitivity of vagal ganglion neurons to histamine as, in contrast to male rats, in female rats, histamine affected not only unmyelinated C‐type, but also myelinated A‐nerve fibres. These results may explain the stronger analgesic effects of E‐162 and TR‐7 tested in our study, while females seem to be more sensitive to histamine. However, there is a paucity of information concerning changes in the histaminergic system under neuropathy between males and females. We have shown sex‐related differences in pain sensation after administration of H3 and H4 receptor antagonists. However, this interesting issue needs further investigation.
In neuropathic pain therapy, opioids are still the drugs of choice, but this type of pain is relatively less responsive to opioid treatment and causes different side effects (Arnér and Meyerson, 1988; Eisenberg et al., 2005; Obara et al., 2009; Hirsch and Dickenson, 2014). Therefore, the treatment paradigm points to minimizing the therapeutic doses of opioids, which diminishes their side effects, while improving their analgesic action. In our present study, we performed a set of experiments to evaluate the effects of two new H3 and H4 receptor antagonists on morphine analgesia. Blockade of both receptors significantly enhanced morphine antinociceptive action after mechanical and thermal (cold) stimulus. Our results suggest dditive effects of these antagonists on morphine‐induced analgesia. Moreover, the H3 receptor antagonist E‐162 intensified morphine analgesia after heat‐induced nociception. An interaction between histaminergic and opioidergic systems within the CNS has already been suggested (Barke and Hough, 1992; Suh et al., 1999; Mobarakeh et al., 2009; Tamaddonfard et al., 2011). Using H3 receptor knockout mice, Mobarakeh et al. (2009) showed profound morphine‐induced analgesia, which suggests that these receptors may have a crucial role in the response to opioids. The antinociceptive effects of i.t. administered morphine were greater in H3 receptor‐KO mice than in the wild type in the inflammatory pain model. Moreover, spinal blockade of H3 receptors in C57BL/6J naive mice significantly increased morphine analgesia. These authors suggested that histamine exerts an inhibitory action on morphine‐induced analgesia through H3 receptors at the spinal level. These data are consistent with our results, which showed that blockade of the H3 receptors by E‐162 potentiated the analgesic effects of morphine. Moreover, Tamaddonfard et al. (2011) showed that morphine‐induced and histamine‐induced analgesia were prevented by pretreatment with H1 and H2 receptor antagonists. These data are in line with our newly obtained results, where we have shown that pretreatment with a H1 receptor antagonist, injected i.t., prevented the analgesic effects of E‐162.
To our knowledge, there are no data on the effects of H4 receptor antagonism on opioid analgesia. We and others have shown that decreased opioid analgesia during neuropathic pain is partly due to disrupted neuroimmune interaction (see Mika et al., 2013). Inflammatory mediators may affect peripheral terminals of primary sensory neurons, which leads to peripheral sensitization. Histamine release from mast cells in the periphery is also able to sensitize nociceptors, resulting in potentiated pain sensation (Zuo et al., 2003). Taking all the factors together, we suggest that enhancement of morphine analgesia by the antagonist TR‐7 might be due to reduced inflammation and activation of immune cells (especially mast cells), which release histamine in the periphery. However, this issue needs further research.
In conclusion, the results from the present research, using potent and selective H3 and H4 receptor antagonists – E‐162 and TR‐7, respectively – demonstrated that these histamine receptors are intimately involved in nociceptive transmission during neuropathy. Our work provides the first evidence for the analgesic potency of both antagonists and their potentiation of the effects of morphine in a model of neuropathic pain. E‐162 is especially promising because it is metabolically stable and its structural homologue, pitolisant (Wakix™), is already used in therapy. However, further studies are needed, especially on the important implications for opioid analgesia mediated by H3 and H4 receptor antagonists. Our present data provide strong evidence for the role of H1 receptors and spinal histamine in the analgesic effects of H3 receptor antagonists in neuropathy. We have also evaluated a new and important issue: sex‐dependent differences in antinociceptive effects of H3 and H4 receptor antagonists. Additionally, our study has shown that both antagonists can be considered as effective and selective pharmacological tools to study the role of the histaminergic system in other CNS pathologies.
Author contributions
K.P.‐B. conceived, designed and performed the study, analysed the data and contributed to writing the manuscript. D.Ł. conceived and designed the tested antagonists and contributed to writing the manuscript. G.L. and A.O. performed the study, analysed the data and contributed to writing the manuscript. W.M. performed the study. H.S. contributed to H3 and H4 receptor binding studies. K.K.‐K. and J.M. conceived and designed the study and contributed to writing the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effects of E‐162 (H3R antagonist) and TR‐7 (H4R antagonist) on pain symptoms in naive male and female mice. The effects of single intraperitoneal (i.p.) administration of E‐162 (10 mg kg‐1) and TR‐7 (10 mg kg‐1) on mechanical (A,B) and thermal (C‐F) stimulus in naive males and females mice were evaluated. Mechanical allodynia was assessed 30, 90 and 180 min after drug injection by von Frey test (A,B). Sensitivity to noxious cold stimuli (thermal hyperalgesia) was assessed with cold plate test 35, 95, 185 min after drug injection (C,D). Spinal nociceptive responses to heat‐induced pain was measure by tail flick test 40, 100, 190 min after drug administration (E,F). The data are presented as the mean ± S.E.M. The results of the experiments were statistically evaluated using a repeated measures ANOVA (blue symbols) and one‐way ANOVA (green symbols) and were further analyzed with Bonferroni's post‐hoc test. Significant differences in comparisons with vehicle25%DMSO/water‐treated mice are indicated by *P<0.05 The number of animals used in the study is highlighted on the bar graph.
Figure S2 Effects of intraperitoneal (i.p.) E‐162 and TR‐7 administration on ipsilateral and contralateral hindlimb in naive and CCI‐treated male and female mice. The effects of single intraperitoneal (i.p.) administration of E‐162 (10 mg kg‐1) and TR‐7 (10 mg kg‐1) on mechanical stimuli in naive (A,B) and neuropathic (C,D) males and females mice. In animals with CCI behavioral evaluation was performed on day 7 following sciatic nerve injury. Mechanical allodynia was assessed 90 min after drug injection by von Frey test. The results of the experiments were statistically evaluated using The results of the experiments were statistically evaluated using one‐way ANOVA (green symbols) and were further analyzed with Bonferroni's post‐hoc test. Significant differences between ipsilateral and contrlateral hindlimb are indicated by *P<0.05.
Figure S3 Effects of intraperitoneal (i.p.) E‐162 and TR‐7 administration on motor coordination in naive and CCI‐treated male and female mice. The effects of single intraperitoneal (i.p.) administration of E‐162 (10 mg kg‐1) and TR‐7 (10 mg kg‐1) on motor coordination in naive (A,B) and neuropathic (C,D) males and females mice. In animals with CCI, behavioral evaluation was performed on day 7 following sciatic nerve injury. Motor coordination was assessed 200 min after drug injection by RotaRod test. The number of animals used in the study is highlighted on the bar graph.
Figure S4 The UPLC spectra of E‐162 (H3R antagonist) and TR‐7 (H4R antagonist) after incubation with MLMs. The metabolic stability of E‐162 (A) and TR‐7 (B) was evaluated in vitro by incubation with mouse liver microsomes (MLMs) for 90 min. The identification of structures of metabolites and determination of the most probably E‐162 and TR‐7 metabolism pathways were performed next using MS fragmentation analysis supporting by MetaSite software.
Figure S5 The plot of MetaSite predictions for sites of metabolism and ion fragments analysis of compound E‐162 (A) and its metabolites M1 (B) and M2 (C).
Figure S6 The plot of MetaSite predictions for sites of metabolism and ion fragments analysis of compound TR‐7 (A) and its main metabolites M1 (B) and M2 (C).
Figure S7 Ion fragments analysis of compound's TR‐7 metabolites M3 (A) and M4 (B).
Figure S8 MS analysis of contamination found in the compound's TR‐7 reaction mixture after incubation with MLMs (retention time = 3.14 min).
Acknowledgements
The authors are grateful to Agata Siwek and Anna Gryboś (Krakow, Poland) for their evaluation of the binding affinity for the human H1R. This work was partially financed by a grant from the National Science Center, Poland (MAESTRO DEC‐2011/02/A/NZ4/00031) and statutory funds from the Institute of Pharmacology Polish Academy of Sciences, Department of Pain Pharmacology.
Popiolek‐Barczyk K., Łażewska D., Latacz G., Olejarz A., Makuch W., Stark H., Kieć‐Kononowicz K., and Mika J. (2018) Antinociceptive effects of novel histamine H3 and H4 receptor antagonists and their influence on morphine analgesia of neuropathic pain in the mouse, British Journal of Pharmacology, 175, 2897–2910, https://doi.org/10.1111/bph.14185.
Contributor Information
Katarzyna Kieć‐Kononowicz, Email: mfkonono@cyf-kr.edu.pl.
Joanna Mika, Email: joamika@if-pan.krakow.pl.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnér S, Meyerson B (1988). Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain 33: 11–23. [DOI] [PubMed] [Google Scholar]
- Barke KE, Hough LB (1992). Morphine‐induced increases of extracellular histamine levels in the periaqueductal grey in vivo: a microdialysis study. Brain Res 572: 146–153. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK (1988). A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33: 87–107. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Parmar S (1985). Antinociceptive effect of intracerebroventricularly administered histamine in rats. Res Commun Chem Pathol Pharmacol 49: 125–136. [PubMed] [Google Scholar]
- Blandina P, Giorgetti M, Bartolini L, Cecchi M, Timmerman H, Leurs R et al (1996). Inhibition of cortical acetylcholine release and cognitive performance by histamine H3 receptor activation in rats. Br J Pharmacol 119: 1656–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga P, Sibilia V, Guidobono E, Pecile A, Netti C (1992). Electrophysiological correlates for antinociceptive effects of histamine after intracerebral administration to the rat. Neuropharmacology 31: 937–941. [DOI] [PubMed] [Google Scholar]
- Braga P, Soldavini E, Pecile A, Sibilia V, Netti C (1996). Involvement of H1 receptors in the central antinociceptive effect of histamine: pharmacological dissection by electrophysiological analysis. Experientia 52: 60–65. [DOI] [PubMed] [Google Scholar]
- Chung Y, Miyake H, Kamei C, Tasaka K (1984). Analgesic effect of histamine induced by intracerebral injection into mice. Agents Actions 15: 137–142. [DOI] [PubMed] [Google Scholar]
- Clapham J, Kilpatrick G (1992). Histamine H3 receptors modulate the release of [3H]‐acetylcholine from slices of rat entorhinal cortex: evidence for the possible existence of H3 receptor subtypes. Br J Pharmacol 107: 919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clatworthy A, Illich P, Castro G, Walters E (1995). Role of peri‐axonal inflammation in the development of thermal hyperalgesia and guarding behavior in a rat model of neuropathic pain. Neurosci Lett 184: 5–8. [DOI] [PubMed] [Google Scholar]
- Connelly W, Shenton F, Lethbridge N, Leurs R, Lees G (2009). The histamine H4 receptor is functionally expressed on neurons in the mammalian CNS. Br J Pharmacol 157: 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, McNicol ED, Carr DB (2005). Efficacy and safety of opioid agonists in the treatment of neuropathic pain of nonmalignant origin: systematic review and meta‐analysis of randomized controlled trials. JAMA 293: 3043–3052. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency (2015) Narcolepsy treatment recommended for approval Wakix to target excessive daytime sleepiness and cataplexy. 44(November).
- Fairbanks CA (2003). Spinal delivery of analgesics in experimental models of pain and analgesia. Adv Drug Deliv Rev 55: 1007–1041. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Otto M, McQuay HJ, Jensen TS, Sindrup SH (2005). Algorithm for neuropathic pain treatment: an evidence based proposal. Pain 118: 289–305. [DOI] [PubMed] [Google Scholar]
- Galeotti N, Sanna MD, Ghelardini C (2013). Pleiotropic effect of histamine H4 receptor modulation in the central nervous system. Neuropharmacology 71: 141–147. [DOI] [PubMed] [Google Scholar]
- Gbahou F, Vincent L, Humbert‐Claude M, Tardivel‐Lacombe J, Chabret C, Arrang J (2006). Compared pharmacology of human histamine H3 and H4 receptors: structure–activity relationships of histamine derivatives. Br J Pharmacol 147: 744–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch SJ, Dickenson AH (2014). Morphine sensitivity of spinal neurons in the chronic constriction injury neuropathic rat pain model. Neurosci Lett 562: 97–101. [DOI] [PubMed] [Google Scholar]
- Hofstra CL, Desai PJ, Thurmond RL (2003). Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. J Pharmacol Exp Ther 305: 1212–1221. [DOI] [PubMed] [Google Scholar]
- Hsieh GC, Chandran P, Salyers AK, Pai M, Zhu CZ, Wensink EJ et al (2010a). H4 receptor antagonism exhibits anti‐nociceptive effects in in fl ammatory and neuropathic pain models in rats. Pharmacol Biochem Behav 95: 41–50. [DOI] [PubMed] [Google Scholar]
- Hsieh GC, Honore P, Pai M, Wensink EJ, Chandran P, Salyers AK et al (2010b). Antinociceptive effects of histamine H3 receptor antagonist in the preclinical models of pain in rats and the involvement of central noradrenergic systems. Brain Res 1354: 74–84. [DOI] [PubMed] [Google Scholar]
- Kajihara Y, Murakami M, Imagawa T, Otsuguro K, Ito S, Ohta T (2010). Histamine potentiates acid‐induced responses mediating transient receptor potential V1 in mouse primary sensory neurons. Neuroscience 166: 292–304. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingery WS (1997). A critical review of controlled clinical trials for peripheral neuropathic pain and complex regional pain syndromes. Pain 73: 123–139. [DOI] [PubMed] [Google Scholar]
- Lamberti C, Bartolini A, Ghelardini C, Malmberg‐Aiello P (1996). Investigation into the role of histamine receptors in rodent antinociception. Pharmacol Biochem Behav 53: 567–574. [DOI] [PubMed] [Google Scholar]
- Li J, Qian Z, Xu W, Xu B, Lu X, Yan Z et al (2013). Gender differences in histamine‐induced depolarization and inward currents in vagal ganglion neurons in rats. Int J Biol Sci 9: 1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łażewska D, Więcek M, Ner J, Kamińska K, Kottke T, Schwed J et al (2014). Aryl‐1,3,5‐triazine derivatives as histamine H4 receptor ligands. Eur J Med Chem 83: 534–546. [DOI] [PubMed] [Google Scholar]
- Malmberg‐Aiello P, Lamberti C, Ghelardini C, Giotti A, Bartolini A (1994). Role of histamine in rodent antinociception. Br J Pharmacol 111: 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol. 172, 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medhurst SJ, Collins SD, Billinton A, Bingham S, Dalziel RG, Brass A et al (2008). Novel histamine H3 receptor antagonists GSK189254 and GSK334429 are efficacious in surgically‐induced and virally‐induced rat models of neuropathic pain. Pain 138: 61–69. [DOI] [PubMed] [Google Scholar]
- Mika J, Osikowicz M, Makuch W, Przewlocka B (2007). Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur J Pharmacol 560: 142–149. [DOI] [PubMed] [Google Scholar]
- Mika J, Zychowska M, Popiolek‐Barczyk K, Rojewska E, Przewlocka B (2013). Importance of glial activation in neuropathic pain. Eur J Pharmacol 716: 106–119. [DOI] [PubMed] [Google Scholar]
- Mobarakeh J, Takahashi K, Yanai K (2009). Enhanced morphine‐induced antinociception in histamine H3 receptor gene knockout mice. Neuropharmacology 57: 409–414. [DOI] [PubMed] [Google Scholar]
- Mogilski S, Kubacka M, Łażewska D, Więcek M, Głuch‐Lutwin M, Tyszka‐Czochara M et al (2017). Aryl‐1,3,5‐triazine ligands of histamine H4 receptor attenuate inflammatory and nociceptive response to carrageen, zymosan and lipopolysaccharide. Inflamm Res 66: 79–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Delgado D, Torrent A, Gomez‐Ramirez J, de Esch I, Blanco I, Ortiz J (2006). Constitutive activity of H3 autoreceptors modulates histamine synthesis in rat brain through the cAMP/PKA pathway. Neuropharmacology 51: 517–523. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Itadani H, Hidaka Y, Ohta M, Tanaka K (2000). Molecular cloning and characterization of a new human histamine receptor, HH4R. Biochem Biophys Res Commun 279: 615–620. [DOI] [PubMed] [Google Scholar]
- O'Reilly M, Alpert R, Jenkinson S, Gladue RP, Foo S, Trim S et al (2002). Identification of a histamine H4 receptor on human eosinophils – role in eosinophil chemotaxis. J Recept Signal Transduct Res 22: 431–448. [DOI] [PubMed] [Google Scholar]
- Obara I, Parkitna JR, Korostynski M, Makuch W, Kaminska D, Przewlocka B et al (2009). Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain 141: 283–291. [DOI] [PubMed] [Google Scholar]
- Owen S, Sturman G, Freeman P (1994). Modulation of morphine‐induced antinociception in mice by histamine H3‐receptor ligands. Agents Actions 41Spec No: C62–C63. [DOI] [PubMed] [Google Scholar]
- Perkins NM, Tracey DJ (2000). Hyperalgesia due to nerve injury: role of neutrophils. Neuroscience 101: 745–757. [DOI] [PubMed] [Google Scholar]
- Sanna D, Lucarini L, Durante M, Ghelardini C (2017b). Histamine H4 receptor agonist‐induced relief from painful peripheral neuropathy is mediated by inhibition of spinal neuroinflammation and oxidative stress. Br J Pharmacol 174: 28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna M, Ghelardini C, Thurmond R, Masini E, Galeotti N (2017a). Behavioural phenotype of histamine H4 receptor knockout mice: focus on central neuronal functions. Neuropharmacology 114: 48–57. [DOI] [PubMed] [Google Scholar]
- Sanna M, Stark H, Lucarini L, Ghelardini C, Masini E, Galeotti N (2015). Histamine H4 receptor activation alleviates neuropathic pain through differential regulation of ERK, JNK, and P38 MAPK phosphorylation. Pain 156: 2492–2504. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Betz R, Göthert M (1988). Histamine H3 receptor‐mediated inhibition of serotonin release in the rat brain cortex. Naunyn Schmiedeberg's Arch Pharmacol 337: 588–590. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Fink K, Detzner M, Göthert M (1993). Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J Neural Transm 93: 1–10. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Fink K, Hinterthaner M, Göthert M (1989). Inhibition of noradrenaline release in the rat brain cortex via presynaptic H3 receptors. Naunyn Schmiedebergs Arch Pharmacol 340: 633–638. [DOI] [PubMed] [Google Scholar]
- Smith FM, Haskelberg H, Tracey DJ, Moalem‐Taylor G (2007). Role of histamine H3 and H4 receptors in mechanical hyperalgesia following peripheral nerve injury. Neuroimmunomodulation 14: 317–325. [DOI] [PubMed] [Google Scholar]
- Suh HW, Chung KM, Kim YH, Huh SO, Song DK (1999). Effects of histamine receptor antagonists injected intrathecally on antinociception induced by opioids administered intracerebroventricularly in the mouse. Neuropeptides 33: 121–129. [DOI] [PubMed] [Google Scholar]
- Tamaddonfard E, Erfanparast A, Farshid AA, Khalilzadeh E (2011). Interaction between histamine and morphine at the level of the hippocampus in the formalin‐induced orofacial pain in rats. Pharmacol Rep 63: 423–432. [DOI] [PubMed] [Google Scholar]
- Thacker M, Clark AK, Marchand F, McMahon SB (2007). Pathophysiology of peripheral neuropathic pain: immune cells and molecules. Anesth Analg 105: 838–847. [DOI] [PubMed] [Google Scholar]
- Thoburn KK, Hough LB, Nalwalk JW, Mischler SA (1994). Histamine‐induced modulation of nociceptive responses. Pain 58: 29–37. [DOI] [PubMed] [Google Scholar]
- Torrent A, Moreno‐delgado D, Jordi G, Rodrı D, Rodrı C, Francisca S et al (2005). H3 autoreceptors modulate histamine synthesis through calcium/calmodulin‐ and cAMP‐dependent protein kinase pathways. Mol Pharmacol 67: 195–203. [DOI] [PubMed] [Google Scholar]
- Vacca V, Marinelli S, Pieroni L, Urbani A, Luvisetto S, Pavone F (2014). Higher pain perception and lack of recovery from neuropathic pain in females: a behavioural, immunohistochemical, and proteomic investigation on sex‐related differences in mice. Pain 155: 388–402. [DOI] [PubMed] [Google Scholar]
- Wei H, Viisanen H, You H, Pertovaara A (2016). Spinal histamine in attenuation of mechanical hypersensitivity in the spinal nerve ligation‐induced model of experimental neuropathy. Eur J Pharmacol 772: 1–10. [DOI] [PubMed] [Google Scholar]
- Zampeli E, Tiligada E (2009). The role of histamine H4 receptor in immune and inflammatory disorders. Br J Pharmacol 157: 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M (1983). Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16: 109–110. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Perkins NM, Tracey DJ, Geczy CL (2003). Inflammation and hyperalgesia induced by nerve injury in the rat: a key role of mast cells. Pain 105: 467–479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of E‐162 (H3R antagonist) and TR‐7 (H4R antagonist) on pain symptoms in naive male and female mice. The effects of single intraperitoneal (i.p.) administration of E‐162 (10 mg kg‐1) and TR‐7 (10 mg kg‐1) on mechanical (A,B) and thermal (C‐F) stimulus in naive males and females mice were evaluated. Mechanical allodynia was assessed 30, 90 and 180 min after drug injection by von Frey test (A,B). Sensitivity to noxious cold stimuli (thermal hyperalgesia) was assessed with cold plate test 35, 95, 185 min after drug injection (C,D). Spinal nociceptive responses to heat‐induced pain was measure by tail flick test 40, 100, 190 min after drug administration (E,F). The data are presented as the mean ± S.E.M. The results of the experiments were statistically evaluated using a repeated measures ANOVA (blue symbols) and one‐way ANOVA (green symbols) and were further analyzed with Bonferroni's post‐hoc test. Significant differences in comparisons with vehicle25%DMSO/water‐treated mice are indicated by *P<0.05 The number of animals used in the study is highlighted on the bar graph.
Figure S2 Effects of intraperitoneal (i.p.) E‐162 and TR‐7 administration on ipsilateral and contralateral hindlimb in naive and CCI‐treated male and female mice. The effects of single intraperitoneal (i.p.) administration of E‐162 (10 mg kg‐1) and TR‐7 (10 mg kg‐1) on mechanical stimuli in naive (A,B) and neuropathic (C,D) males and females mice. In animals with CCI behavioral evaluation was performed on day 7 following sciatic nerve injury. Mechanical allodynia was assessed 90 min after drug injection by von Frey test. The results of the experiments were statistically evaluated using The results of the experiments were statistically evaluated using one‐way ANOVA (green symbols) and were further analyzed with Bonferroni's post‐hoc test. Significant differences between ipsilateral and contrlateral hindlimb are indicated by *P<0.05.
Figure S3 Effects of intraperitoneal (i.p.) E‐162 and TR‐7 administration on motor coordination in naive and CCI‐treated male and female mice. The effects of single intraperitoneal (i.p.) administration of E‐162 (10 mg kg‐1) and TR‐7 (10 mg kg‐1) on motor coordination in naive (A,B) and neuropathic (C,D) males and females mice. In animals with CCI, behavioral evaluation was performed on day 7 following sciatic nerve injury. Motor coordination was assessed 200 min after drug injection by RotaRod test. The number of animals used in the study is highlighted on the bar graph.
Figure S4 The UPLC spectra of E‐162 (H3R antagonist) and TR‐7 (H4R antagonist) after incubation with MLMs. The metabolic stability of E‐162 (A) and TR‐7 (B) was evaluated in vitro by incubation with mouse liver microsomes (MLMs) for 90 min. The identification of structures of metabolites and determination of the most probably E‐162 and TR‐7 metabolism pathways were performed next using MS fragmentation analysis supporting by MetaSite software.
Figure S5 The plot of MetaSite predictions for sites of metabolism and ion fragments analysis of compound E‐162 (A) and its metabolites M1 (B) and M2 (C).
Figure S6 The plot of MetaSite predictions for sites of metabolism and ion fragments analysis of compound TR‐7 (A) and its main metabolites M1 (B) and M2 (C).
Figure S7 Ion fragments analysis of compound's TR‐7 metabolites M3 (A) and M4 (B).
Figure S8 MS analysis of contamination found in the compound's TR‐7 reaction mixture after incubation with MLMs (retention time = 3.14 min).