

Abstract
Background and Purpose
The κ receptor antagonists have potential for treating neuropsychiatric disorders. We have investigated the in vivo pharmacology of a novel buprenorphine analogue, BU10119, for the first time.
Experimental Approach
To determine the opioid pharmacology of BU10119 (0.3–3 mg·kg−1, i.p.) in vivo, the warm‐water tail‐withdrawal assay was applied in adult male CD1 mice. A range of behavioural paradigms was used to investigate the locomotor effects, rewarding properties and antidepressant or anxiolytic potential of BU10119. Additional groups of mice were exposed to a single (1 × 2 h) or repeated restraint stress (3× daily 2 h) to determine the ability of BU10119 to block stress‐induced analgesia.
Key Results
BU10119 alone was without any antinociceptive activity. BU10119 (1 mg·kg−1) was able to block U50,488, buprenorphine and morphine‐induced antinociception. The κ antagonist effects of BU10119 in the tail‐withdrawal assay reversed between 24 and 48 h. BU10119 was without significant locomotor or rewarding effects. BU10119 (1 mg·kg−1) significantly reduced the latency to feed in the novelty‐induced hypophagia task and reduced immobility time in the forced swim test, compared to saline‐treated animals. There were no significant effects of BU10119 in either the elevated plus maze or the light–dark box. Both acute and repeated restraint stress‐induced analgesia were blocked by pretreatment with BU10119 (1 mg·kg−1). Parallel stress‐induced increases in plasma corticosterone were not affected.
Conclusions and Implications
BU10119 is a mixed κ/μ receptor antagonist with relatively short‐duration κ antagonist activity. Based on these preclinical data, BU10119 has therapeutic potential for the treatment of depression and other stress‐induced conditions.
Linked Articles
This article is part of a themed section on Emerging Areas of Opioid Pharmacology. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.14/issuetoc
Abbreviations
- %MPE
percentage maximum possible effect
- CCAM
clocinnamox
- CPP
conditioned place preference
- EPM
elevated plus maze
- LDB
light–dark box
- NOP receptor
nociception/orphanin FQ receptor
- norBNI
norbinaltorphimine
Introduction
There is growing interest in the possibility of targeting κ opioid receptors (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=318, also known as KOP; Toll et al., 2017) for the treatment of depression (Carroll and Carlezon, 2013; Lalanne et al., 2014). The endogenous opioid neuropeptide dynorphin is released in response to stress and activates κ receptors to produce pro‐depressive behaviours (Shirayama et al., 2004; Schwarzer, 2009). In humans, activation of κ receptors has been shown to be dysphoric (Pfeiffer et al., 1986). In contrast, blockade of κ receptors or κ receptor gene deletion have shown antidepressant‐like effects in mice (McLaughlin et al., 2003; 2006b).
A number of high affinity, selective κ antagonists exist (Carroll and Carlezon, 2013), which have an unusual pharmacokinetic profile that perhaps limits their translatability to the clinic. For example, following a single injection, the effects of the selective κ receptor antagonist http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1642 last for several weeks (Endoh et al., 1992). A number of strategies have arisen to develop alternative means of targeting the κ receptor by producing short‐acting κ antagonists (Aldrich et al., 2009; Peters et al., 2011; Verhoest et al., 2011; Ross et al., 2012; Casal‐Dominguez et al., 2014; Rorick‐Kehn et al., 2014).
We have recently shown that the combination of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1670 (a partial opioid http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=319 agonist/κ antagonist) with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1639 (a non‐selective μ/κ antagonist) produces a functional short‐acting κ antagonist in vivo that has antidepressant‐like activity in mice (Almatroudi et al., 2015). A similar approach of a combination of buprenorphine with a μ antagonist samidorphan has been shown to have antidepressant effects in treatment‐resistant depressed patients (Ehrich et al., 2015). While buprenorphine and naltrexone are both licensed for other indications, so may be attractive to translate to the clinic, the approach of using combination treatments may have limitations. Where buprenorphine/naltrexone combination has been trialled clinically for the treatment of opioid dependence (Rothman et al., 2000; Gerra et al., 2006), naltrexone was administered orally and buprenorphine sublingually, so achieving the correct dose combination to achieve an antidepressant effect may not be trivial (Cordery et al., 2014; Almatroudi et al., 2015). Furthermore, the risk of diversion of buprenorphine and its abuse liability as a partial μ receptor agonist must be considered. The non‐therapeutic misuse of buprenorphine has recently been reported to be rising among drug users, as it serves as a substitute for other drugs of abuse (Cicero et al., 2014).
An alternative strategy to targeting κ receptors, building on this combination approach, is to design novel chemical entities that combine the pharmacology of buprenorphine and naltrexone into one molecule. This would overcome both the abuse liability issue and the dosing issues. BU10119 is one of a novel series of orvinol analogues in which the C20‐methyl group has been moved to the C7‐b position (Cueva et al., 2015; Figure 1). In vitro pharmacology studies have established that BU10119 has high affinity for both κ and μ receptors with little efficacy at either of these receptors indicating an antagonist‐like profile (Cueva et al., 2015; Table 1). Here, we report the initial characterization of BU10119 in vivo and behavioural studies that demonstrate the therapeutic antidepressant‐like potential in mice.
Figure 1.
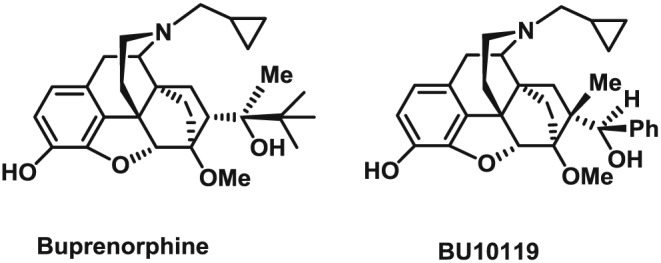
Chemical structures of buprenorphine and BU10119.
Table 1.
Maximal stimulation of [35S]‐GTPγS binding of buprenorphine and BU10119 to κ, μ and NOP receptors
| Compound | κ | μ | NOP |
|---|---|---|---|
| Buprenorphine (1a*) | 0 ± 6% (Ke = 0.14 ± 0.06 nM) | 20 ± 6% (EC50: 0.7 ± 0.3 nM) | 39 ± 12% (EC50: 1480 ± 980 nM) |
| BU10119 (15a*) | −2 ± 1% (Ke = 0.09 ± 0.04 nM) | 2 ± 4% (Ke = 0.28 ± 0.04 nM) | 56 ± 1% (EC50: 147 ± 33 nM) |
Results show % maximal stimulation at a single high concentration (10 μM) with respect to the standard agonists U69,593 (κ), DAMGO (μ) and nociception (NOP). Agonist EC50 (nM) or antagonist Ke (nM), the antagonist dissociation constant determined against the same agonists. Neither buprenorphine nor BU10119 has any appreciable efficacy at δreceptors. All data taken from Cueva et al., 2015. *1a and *15a are the identifiers used for these compounds in Cueva et al., 2015.
Methods
Animals
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Adult male CD‐1 mice weighing 25 to 43 g (8–10 weeks) were used throughout these experiments. CD‐1 mice were initially from Charles River (Crl:CD1(ICR)) and bred in‐house at the University of Bath for more than 10 years. At weaning, mice were housed as mixed litter groups of four to five in cages (30 × 16 × 14 cm) with wood shavings and nesting material and with ad libitum access to food (RM1 E, Special Diet Services) and water. Mice were maintained on a 12 h light–dark cycle (lights on 07:00 h, lights off 19:00 h) at 20 ± 2°C. All experiments were performed in accordance with the UK Home Office guidelines, including local ethical review, and the Animals (Scientific Procedures) Act 1986/ Directive 2010/63/EU. Mice were randomly assigned to treatment groups. All behavioural experiments were performed between 09:00 and 16:00 h by a male experimenter (Sorge et al., 2014), and mice were habituated to the behavioural room for 1 h before starting an experiment. For each behavioural task, separate groups of animals were used (n = 5–18 per treatment group).
Warm‐water tail‐withdrawal test
The warm‐water tail‐withdrawal assay was carried out as described previously (Almatroudi et al., 2015). Water temperature was maintained at 52°C, and the latency for tail withdrawal was recorded. A 15 s cut‐off was imposed to avoid tissue damage and antinociception calculated as percentage maximum possible effect (%MPE) = (test latency–control latency)/(15 s–control latency) × 100. To counteract any possible confounding effects of injection‐induced stress, in all experiments, animals received 0.9% w.v‐1 saline injections so that the total number of injections an individual mouse received, whether in control or in drug treated groups, was equivalent.
To examine the duration of the κ antagonist properties of BU10119 (0.3, 1 and 3 mg·kg−1), tail‐withdrawal latencies were determined at intervals 1–48 h post‐injection, with antinociceptive agonist challenge (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1652) administered 30 min prior to each time point (Figure 2B). The μ antagonist properties of BU10119 (0.3, 1 and 3 mg·kg−1) were established by blockade of the μ agonist‐induced antinociceptive effects of buprenorphine (1 mg·kg−1) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1627 (10 mg· kg−1) and compared to the irreversible, selective μ antagonist CCAM (3 mg·kg−1) (Broadbear et al., 2000) (Figure 2C). Baseline latencies were measured immediately before injecting BU10119 or saline at time zero. Buprenorphine or morphine was injected 30 min later, and 1 h elapsed before ‘test’, tail‐withdrawal latencies were measured. CCAM was injected 24 h before time zero (Broadbear et al., 2000).
Figure 2.
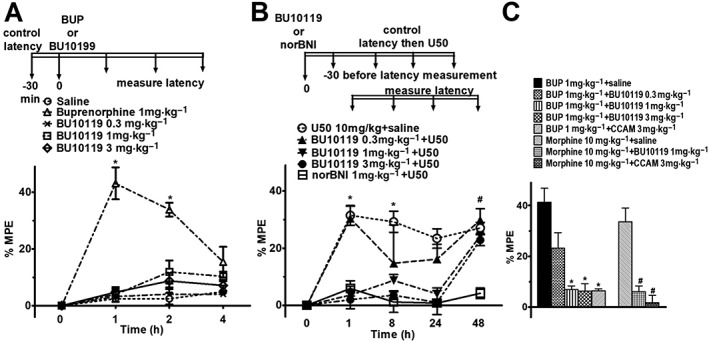
Effects of BU10119 in adult male CD1 mice in the warm‐water tail‐withdrawal assay. The time course of the experiments is indicated, and tail‐withdrawal latencies are expressed as %MPE. (A) Antinociceptive effects of buprenorphine (1 mg·kg−1) and BU10119 (0.3, 1 and 3 mg·kg−1). *P < 0.05 as compared between buprenorphine and all other groups. (B) Antinociceptive effects of the κ‐agonist U50,488 (10 mg·kg−1) were blocked by BU10119 (1 and 3 mg·kg−1) and by norBNI (1 mg·kg−1). *P < 0.05 as compared to BU10119 (1 and 3 mg·kg−1) and norBNI (1 mg·kg−1), #P < 0.05 as compared between all groups and norBNI (1 mg·kg−1). (C) Antinociceptive effects of buprenorphine and morphine at 60 min post‐administration were blocked by BU10119 (1 mg·kg−1) and by the irreversible μ‐antagonist CCAM (3 mg·kg−1). *P < 0.05 compared to buprenorphine; #P < 0.05 compared to morphine. All values are mean ± SEM, n = 5 per group, separate experimental groups in each figure.
To assess stress‐induced analgesia, baseline latencies were measured before drug injection on the first and third day of restraint. These baseline latencies did not change across the duration of the experiment (Supporting Information Figure S1). BU10119 (1 mg·kg−1) or buprenorphine/naltrexone (1 mg·kg−1) combination was given daily 1 h before restraint [naltrexone (1 mg·kg−1) was injected 10 min prior to buprenorphine]. Tail‐withdrawal latency was measured 5 min after the end of the restraint session on the first and third day. For mice treated with norBNI (10 mg·kg−1), the drug was given once only 24 h before the first restraint session.
Locomotor activity
To investigate any potential sedative effect of BU10119 (0.3, 1 mg and 3 mg·kg−1), locomotor activity was assessed 1 h post‐administration in a 10 min open‐field test (72 × 72 cm) under low light conditions (30 lux) via infrared photobeams (Almatroudi et al., 2015).
Forced swim test
Antidepressant‐like effects of BU10119 (1 mg·kg−1), buprenorphine/naltrexone combination (1 mg·kg−1) and the selective 5‐HT reuptake inhibitor http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=203 were investigated in a 6 min forced swim test (Almatroudi et al., 2015). All drugs were administered 1 h prior to testing, and the behaviour during the last 4 min of the test was reported.
Novelty‐induced hypophagia
The novelty‐induced hypophagia paradigm used was as previously described (Almatroudi et al., 2015). Mice were trained on three consecutive days to consume condensed milk. The latency to drink milk was recorded on day 4 in the home cage and on day 5 in a novel cage environment. Mice received fluoxetine (20 mg·kg−1), BU10119 (1 mg·kg−1) and buprenorphine/naltrexone combination (1 mg·kg−1) (naltrexone was injected 10 min prior to buprenorphine) 1 h prior to testing. For mice receiving the κ receptor antagonist norBNI (10 mg·kg−1), the drug was injected immediately after training on day 3, 24–48 h prior to testing.
Elevated plus maze (EPM)
The time spent in, and entries into, the open and closed arms and total ambulation during a 5 min EPM test were recorded via infrared photobeams (Almatroudi et al., 2015). Illumination was 150 lux in the open arms and <1 lux in the closed arms. Mice were treated with saline, BU10119 (1 mg·kg−1), buprenorphine/naltrexone (1 mg·kg−1) combination and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3364 (2 mg·kg−1) 60 min prior to testing.
Light–dark box (LDB)
The number of entries into and time spent in the lit compartment (400 lux) and the distance travelled during a 10 min LDB test were recorded via beam‐breaks (Almatroudi et al., 2015). Mice were treated with saline, BU10119 (1 mg·kg−1), buprenorphine/naltrexone (1 mg·kg−1) combination and diazepam (2 mg·kg−1) 60 min prior to testing.
Conditioned place preference (CPP)
Place preference conditioning was conducted in a CPP chamber with an auto‐monitoring tracking system (Ethovision XT version 8.0, Tracksys, Nottingham, UK) as described previously (Almatroudi et al., 2015). Experiments were performed between 09:00 and 16:00 h under dim light (approximately 15 lux). During all test sessions, the time each mouse spent in each compartment was recorded. Mice were randomly assigned to treatment groups, and the pairing was counterbalanced (i.e. within each treatment group, equal numbers of mice were drug‐paired to each compartment type). On days 1 and 2, mice were habituated to the entire chamber for 15 min (one session a day), during which baseline preference scores were taken. On days 3–8, mice were conditioned (40 min) to one of the two compartments, and daily sessions alternated between drug treatment and saline (In all treatment groups, mice received both drug and saline). Mice were given buprenorphine (1 mg·kg−1), BU10119 (1 mg·kg−1), morphine (10 mg·kg−1) or saline (0.9% w.v‐1). Where CCAM (3 mg·kg−1) was injected, this was 24 h before conditioning and mice were immediately returned to the home cage. After buprenorphine injection, the mice were transferred directly to the place preference box, and at the end, mice were returned to their home cage. Chamber floors and trays were removed and cleaned with ethanol 70% and left for 5 min for ethanol to evaporate before the next trial. On day 9, mice were not injected with saline or drugs. In a free‐to‐explore test, lasting 15 min, mice had free access to both compartments and their preference was determined by recording the time spent in the drug‐paired chamber. Data are presented as preference for drug‐paired side of CPP chamber, determined as the time spent in drug‐paired side minus time spent in drug‐paired side pre‐conditioning (baseline).
Restraint stress
Mice in restraint‐stressed groups were restrained in a well‐ventilated modified 50 mL syringe for 2 h on three consecutive days from 09:00–11:00 h (Sadler and Bailey, 2016). Stressed mice were weighed daily, monitored and scored for signs of stress. Non‐stressed control mice were weighed daily and returned to their home cage.
Measurement of corticosterone level
All blood samples were collected from the lateral tail vein using the tail nick method and the minimal blood volume (~40 μL) collected in a heparin‐treated capillary tube (Sadler and Bailey, 2013). Samples were taken at baseline, 24 h before the first restraint session, and immediately following the end of restraint stress (11:00 to 13:00 h). Blood was collected in centrifuge tubes containing EDTA (final concentration in sample 3 μg·μL−1) and kept on ice until being centrifuged for 20 min at 4°C at 3500× g. Plasma was taken and stored at −20°C until analysed using an elisa kit (IBL International, Hamburg, Germany) to determine the level of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2869.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data were analysed using two‐way repeated measures mixed model analysis or single measures one‐way ANOVA followed by unadjusted least significant difference post hoc test (InVivoStat 2.3). Only planned pairwise tests were carried out and P values adjusted for multiple comparisons with Benjamin–Hochberg correction. In the CCAM, buprenorphine and morphine CPP study Student's unpaired t‐test was used. P < 0.05 was taken to indicate a significant difference. Values are reported as mean ± SEM for each treatment group.
Drugs
BU10119 was synthesized and supplied at the University of Bath (Cueva et al., 2015). Buprenorphine hydrochloride and morphine sulphate were purchased from MacFarlan Smith (Edinburgh, UK). U50,488 was obtained from Sigma (Dorset, UK). Clocinnamox (CCAM) mesylate and norBNI dihydrochloride were supplied by Tocris Bioscience (Bristol, UK). Fluoxetine hydrochloride and naltrexone hydrochloride were purchased from Abcam Biochemicals (Cambridge, UK). All drugs were injected via the i.p. route at a volume of 10 mL·kg−1, except CCAM, which was administered at 20 mL·kg−1, and were dissolved in 0.9% w.v‐1 saline (Hameln Pharmaceuticals, Gloucester, UK).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Establishing the opioid receptor pharmacology of BU10119 in vivo
Pilot studies in mice revealed no overt signs of toxic effects of BU10119 at doses up to 10 mg·kg−1 using a minimal numbers approach (n = 3 per dose) (Hillhouse et al., 2016). The in vivo pharmacology of BU10119 was established using the warm‐water tail‐withdrawal test in adult male CD1 mice. BU10119 at 0.3, 1 and 3 mg·kg−1 produced no significant antinociceptive action up to 4 h post‐injection (Figure 2A; n = 5 per group). Two‐way repeated measures mixed model analysis revealed a significant interaction of Treatment*Time [F(12,60) = 19.35, P < 0.05]. Post hoc testing showed that only buprenorphine (1 mg·kg−1) produced a significant antinociceptive effect compared to saline treated controls that peaked at 60 min post‐administration (P < 0.05) and returned to baseline after 240 min.
To determine the κ antagonist properties of BU10119 (0.3, 1 and 3 mg·kg−1), its ability to block U50,488‐induced antinociception was determined at 1, 8, 24 and 48 h post‐administration (Figure 2B; n = 5 per group). Two‐way repeated measures mixed model analysis revealed that there was a significant interaction of Treatment*Time [F(28140) = 5.46, P < 0.05]. U50,488 produced a pronounced antinociceptive effect that was significantly reduced by BU10119 (1 and 3 mg·kg−1) at 1, 8 and 24 h post‐administration (P < 0.05) but not at 48 h. The high affinity, selective κ antagonist norBNI (1 mg·kg−1) was able to block U50,488‐induced antinociception at all‐time intervals tested (all P values < 0.05, compared to U50,488).
In addition to κ antagonist properties, BU10119 demonstrated μ antagonist properties in the tail‐withdrawal assay (Figure 2C). Two‐way repeated measures mixed model analysis revealed that there was a significant interaction of Treatment*Time [F(7,28) = 18.68, P < 0.05]. Post hoc testing showed that BU10119 (≥1 mg·kg−1) significantly blocked the antinociceptive effect of buprenorphine (1 mg·kg−1) (P < 0.05) and morphine (10 mg·kg−1) (P < 0.05) 60 min post‐administration. The irreversible μ antagonist CCAM (3 mg·kg−1), administered 24 h before buprenorphine or morphine, was also able to block antinociception significantly (P values < 0.05). Taken together, these data show that, in vivo, BU10119 is a mixed κ/μ antagonist with relatively short acting κ antagonist activity (24–48 h).
Effects of BU10119 on locomotor activity
The open‐field arena was used to assess the locomotor effects of BU10119 (0.3, 1 and 3 mg·kg−1) in CD‐1 male mice. There were no significant effects of BU10119 on locomotion [F(3,16) = 1.65, P = 0.218] (Figure 3). However, there was an apparent trend for increased locomotion at 3 mg·kg−1. For this reason, in subsequent behavioural tasks, BU10119 was investigated at 1 mg·kg−1 alone.
Figure 3.
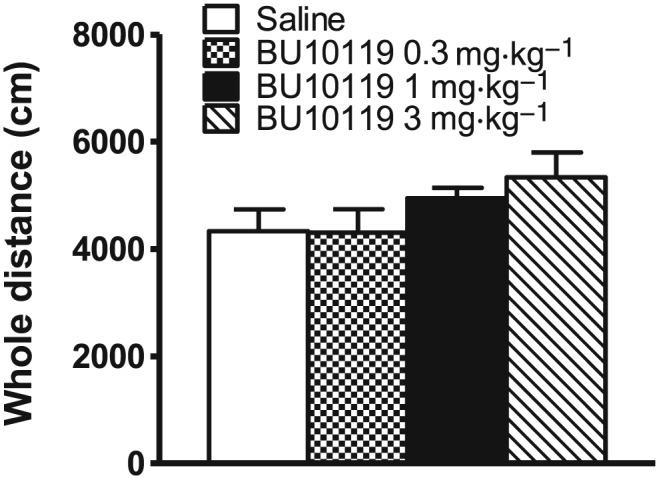
Locomotor activity in the open field in adult male CD1 mice treated with BU10199 (0.3, 1 and 3 mg·kg−1). All values are mean ± SEM, n = 5 per group. No statistically significant effects on locomotion were observed.
Effects of BU10119 in the conditioned place preference (CPP) task
One‐way ANOVA revealed a significant effect of Treatment on the preference for the drug‐paired compartment of the CPP chamber [F(5,48) = 6.78, P < 0.05]. Mice receiving 1 mg·kg−1 of buprenorphine exhibited significant CPP compared to saline‐treated mice (P < 0.05, n = 9 per treatment group; Figure 4A). BU10119 at the same dose appeared to increase preference for the drug‐paired side, although this effect was not significant (P = 0.08). The irreversible μ‐antagonist CCAM (3 mg·kg−1) was administered 24 h before saline, buprenorphine or BU10119. Interestingly, CCAM appeared to reduce the effects of BU10119 in the CPP chamber (P = 0.058). However, CCAM failed to block or to reduce the effects of buprenorphine in the CPP chamber (P = 0.914). CCAM alone was neither rewarding nor aversive. There were no significant differences in baseline times, determined pre‐conditioning, between any of the treatment groups.
Figure 4.
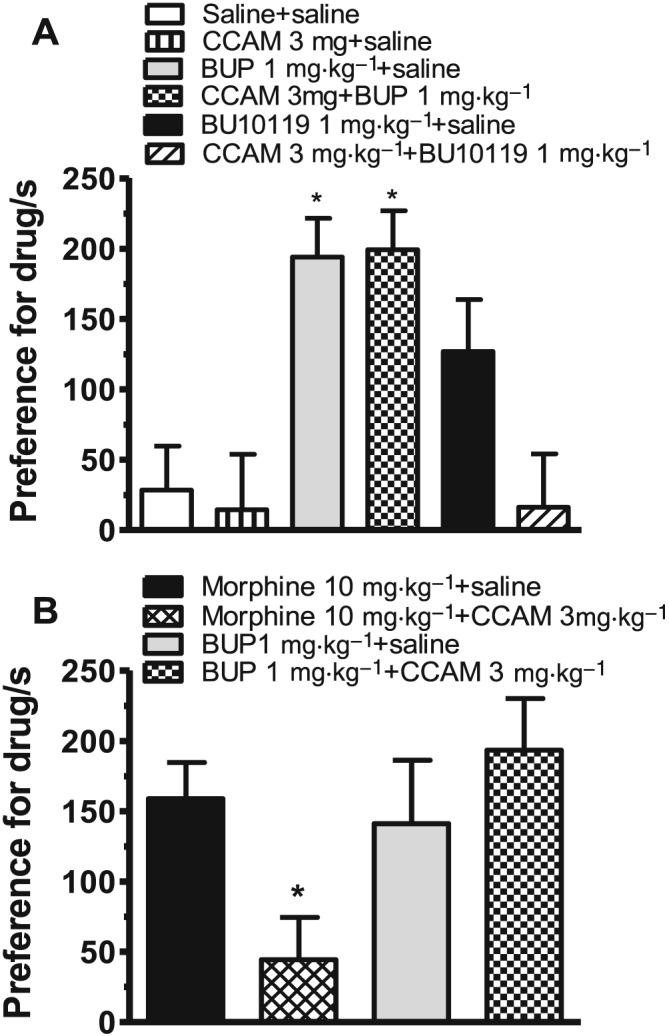
Conditioned place preference (CPP) to buprenorphine (BUP, 1 mg·kg−1), BU10119 (1 mg·kg−1) and morphine (10 mg·kg−1). Two separate experiments were conducted. (A) BU10119 did not produce significant CPP. CPP to buprenorphine was not blocked in the presence of the irreversible μ antagonist CCAM (3 mg·kg−1). (B) The activity of CCAM as a μ antagonist was confirmed by its ability to block morphine‐induced CPP. Data are presented as preference for drug‐paired side of CPP chamber, determined as the time spent in drug‐paired side (post‐conditioning) minus time spent in drug‐paired side pre‐conditioning (baseline). All data points are mean ± SEM. (A) *P < 0.05 compared to saline, n = 9 per treatment group. (B) *P < 0.05 compared to morphine, n = 8 per treatment group.
In a subsequent experiment (Figure 4B), CCAM (3 mg·kg−1) was able to significantly block the rewarding effects of morphine in the CPP task. One‐way ANOVA revealed that there was a significant effect of treatment on the preference for the drug‐paired side (P < 0.05). However, CCAM (3 mg·kg−1) was again not able to significantly reduce the time spent in the drug‐paired side for buprenorphine (1 mg·kg−1) (P = 0.3838, n = 8 per treatment group, unpaired t‐test). Taken together, these data show that in contrast to the tail‐withdrawal assay, BU10119 may have some weak μ agonist activity in the CPP task. The apparent increase in preference for the drug‐paired side shown by BU10119, although not significant, may suggest weak μ agonist properties that are blocked by the irreversible μ antagonist CCAM. Interestingly, buprenorphine's rewarding effects in the CPP task are not apparently mediated via its partial μ agonist activity.
Effects of BU10119 on depression‐ and anxiety‐related behaviours
The antidepressant‐like effects of BU10119 were assessed using the forced swim test and the novelty‐induced hypophagia task. One‐way ANOVA revealed a significant effect of Treatment on the time spent swimming [F(3, 36) = 6.58, P < 0.05] and immobile [F(3, 36) = 7.02, P < 0.05] during the last 4 min of a forced swim test session (Figure 5A). Post hoc analysis revealed that BU10119 (1 mg·kg−1), combination buprenorphine/naltrexone (1 mg·kg−1) and fluoxetine (20 mg·kg−1) increased the time spent swimming and decreased the time spent immobile compared to saline‐treated controls (n = 10 per treatment group, all P values < 0.05).
Figure 5.
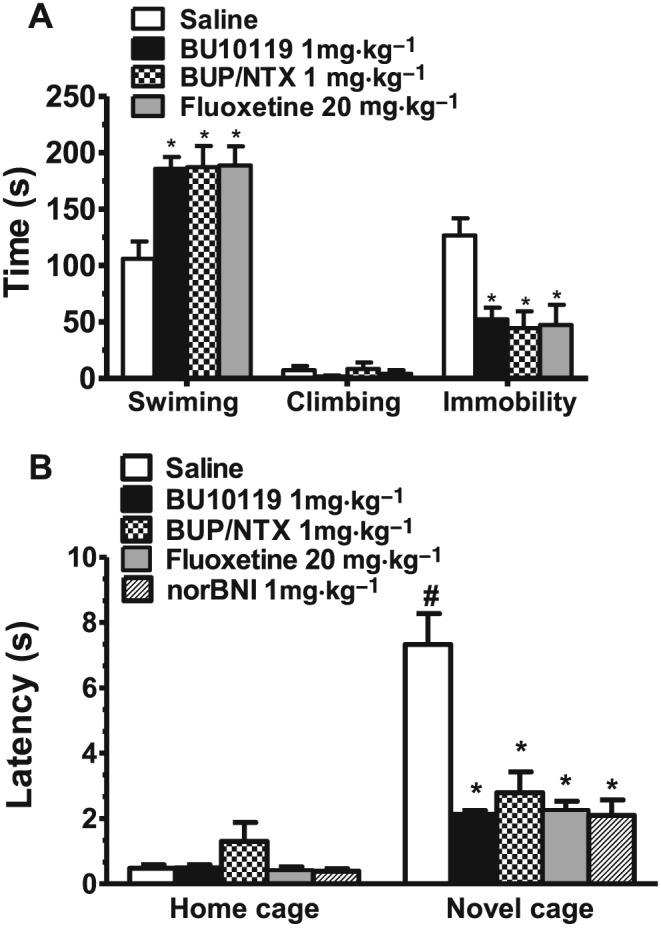
Effects of BU10119 (1 mg·kg−1) in adult male CD1 mice in the forced swim test (A) and in the novelty‐induced hypophagia task (B). The SSRI fluoxetine (20 mg·kg−1) was administered as a positive control. The effects of combination of buprenorphine with naltrexone (both at 1 mg·kg−1, BUP/NTX) and the κ antagonist norBNI are also shown. (A) The total time spent swimming, climbing or immobile during the last 4 min of the forced swim session are shown. (B) The latency to drink milk in both the home and novel cage environments is shown. All values are the mean ± SEM (n = 10 per group, separate experimental groups in each figure). *P < 0.05 as compared to saline; #P < 0.05 for comparison between groups.
In the novelty‐induced hypophagia task (Figure 5B), two‐way repeated measures mixed model analysis of the latency to drink times revealed significant main effects of Treatment [F(4,75) = 6.13, P < 0.05] and a significant Treatment*Environment interaction [F(4,75) = 5.92, P < 0.05]. The novel cage was shown to be aversive as saline control‐treated mice showed a significant increased latency to drink milk in the novel cage (latency = 7.32 ± 0.94 min) versus the home cage (latency = 0.48 ± 0.10 min, P < 0.05). Within treatment, post hoc comparisons to saline‐treated controls revealed that all drug‐treated groups decreased the latency to drink milk in the novel cage (n = 10 per treatment group, all P values < 0.05).
In the EPM, one‐way ANOVA showed significant effects of Treatment on the time spent in [F(3, 36) = 3.29, P < 0.05] and number of entries into [F(3, 36) = 3.89, P < 0.05] the open arms (Figure 6A–C, n = 10 per group). Post hoc comparisons to saline‐treated controls revealed that only the benzodiazepine diazepam (2 mg·kg−1) significantly increased these parameters (P < 0.05). Interestingly, both buprenorphine/naltrexone (1 mg·kg−1) combination and BU10119 (1 mg·kg−1) did not show any significant changes in behaviours in the EPM. Total ambulation in the EPM was not affected by drug treatment [F(3,36) = 1.15 P = 0.342], showing an absence of any sedative effects. Furthermore, in the LDB, there were no significant effects of either BU10119 or combination buprenorphine/naltrexone (Figure 6D–F, n = 18 per group). One‐way ANOVA revealed a significant main effect of Treatment on the time spent in the light [F(3, 60) = 3.59, P < 0.05] and dark [F(3,60) = 3.59, P < 0.01] compartment of the LDB. However, within‐treatment analysis to saline controls showed that only diazepam (2 mg·kg−1) significantly increased the total time spent in the lit compartment (P < 0.05). As with the EPM, total ambulation in the LDB was not significantly affected by drug treatment [F(3,60) = 1.26, P = 0.29].
Figure 6.
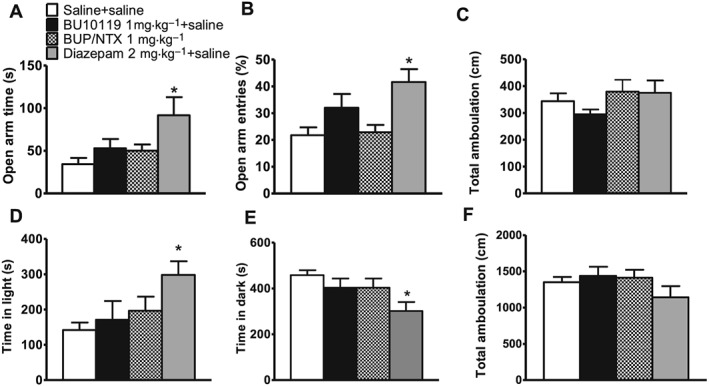
Effects of BU10119 (1 mg·kg−1) in adult CD1 male mice in the elevated plus maze (EPM; A–C) and in the light‐dark box (LDB; D–F). The effects of combination of buprenorphine with naltrexone (both at 1 mg·kg−1, BUP/NTX) are also shown. The benzodiazepine diazepam (2 mg·kg−1) was used as a positive control. The time spent in the open arms (A), number of entries into the open arms (as a percentage of the total entries into open and closed arms) (B) and total ambulation (C) in the EPM are shown (n = 10 per group). The time spent in the light box (D), in the dark box (E) and total ambulation (F) in the LDB are shown (n = 18 per treatment group, n = 10 for BU10119). All values are the mean ± SEM. *P < 0.05 compared to saline.
Stress‐induced analgesia and elevation in corticosterone level
To assess whether BU10119 or combination buprenorphine/naltrexone could block the effects of stress in adult male mice, stress‐induced analgesia was assessed in the warm‐water tail‐withdrawal assay (Figure 7A). Two‐way repeated measures mixed model analysis revealed a significant interaction of Treatment*Time [F(12,75) = 23.3, P < 0.05]. Exposure to acute restraint (1 × 2 h restraint) or 3 days repeated restraint stress (3 × 2 h restraint) produced a significant stress‐induced analgesia evident as an antinociceptive effect in restraint‐stressed mice compared to non‐stressed controls (P < 0.05, n = 6 per treatment group). Pretreatment with BU10119 (1 mg·kg−1), combination buprenorphine/naltrexone (1 mg·kg−1) or the selective κ antagonist norBNI (10 mg·kg−1) blocked stress‐induced analgesia (P < 0.05), compared to respective acute or repeated stressed saline control groups. Furthermore, there was no significant difference between the baseline tail‐withdrawal latency in non‐stressed control groups in the first and third day of experiment (P = 0.3344).
Figure 7.
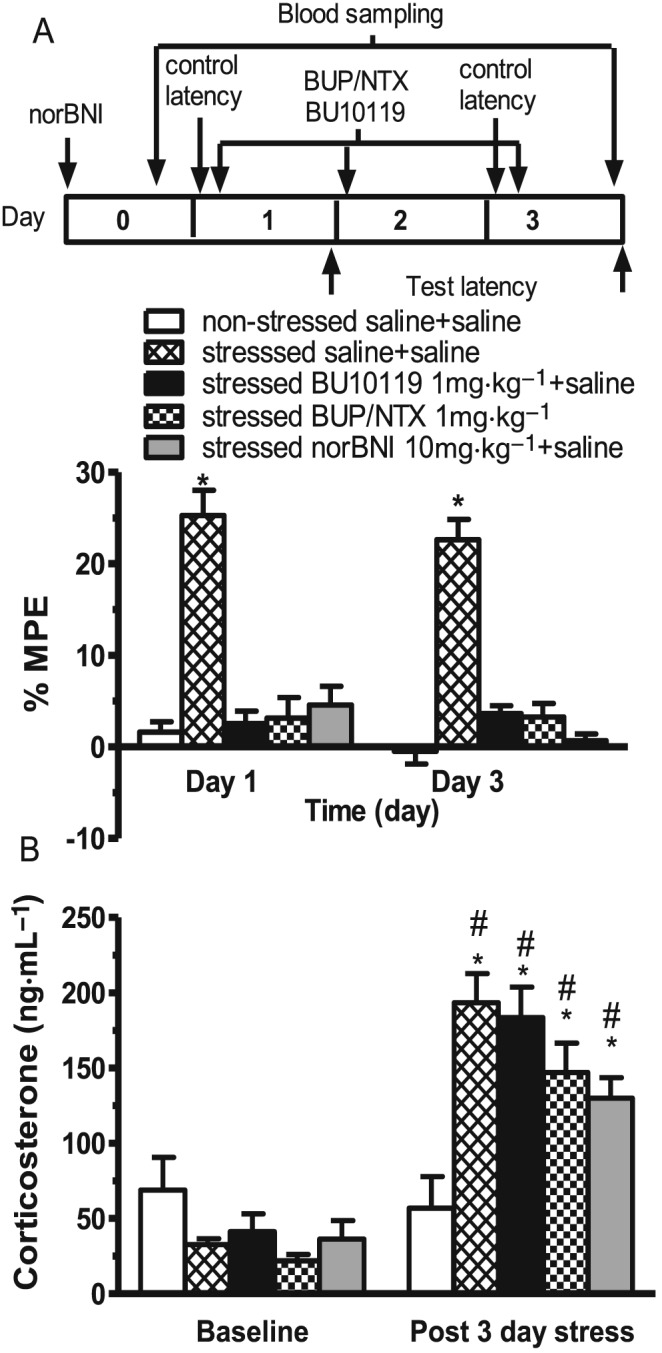
Ability of BU10119 (1 mg·kg−1), the combination of buprenorphine/naltrexone (both at 1 mg·kg−1, BUP/NTX) and the κ antagonist norBNI to block stress‐induced effects. Drug treatments were administered 1 h prior to acute restraint stress (1 × 2 h, Day 1) and daily repeated restraint stress (3 × 2 h, Day 3) in adult male CD1 mice. (A) Stress‐induced analgesia was evident, in the warm‐water tail‐withdrawal assay, as increased %MPE. Test latencies were assessed immediately following the restraint session. *P < 0.05 compared to all groups within the same day. (B) Blood samples were taken at baseline and immediately following the last session of restraint to assess plasma corticosterone. All blood samples were taken during the light phase 11:00–13:00 h. *P < 0.05, compared to non‐stress saline post day 3; #P < 0.05, compared to baseline for the same treated group. All values are the mean ± SEM (n = 6 per group, separate experimental groups in A and B).
Blood samples were collected at baseline (24 h pre‐stress) and after 3 days repeated restraint stress, from the same animals, for analysis of corticosterone levels (Figure 7B). Two‐way repeated measures mixed model analysis revealed that there was a significant effect of stress [F(1,21) = 62.62, P < 0.05], no significant effects of treatment [F(4,25) = 1.2, P < 0.3299] but a significant interaction Stress * Treatment [F(4,21) = 13.14, P < 0.05] on plasma corticosterone levels. Post hoc comparisons revealed no significant difference in baseline plasma corticosterone levels in all mice prior to treatment. However, following 3 days of repeated restraint stress, there was a significant effect of stress in all drug‐treated groups compared to non‐stressed saline‐injected controls (all P < 0.05), but none of the drug treatments were able to block stress‐induced increases in plasma corticosterone (all P values >0.05).
Discussion
We have previously shown that the combined administration of buprenorphine and naltrexone produces antidepressant‐like effects in mice (Almatroudi et al., 2015). BU10119 is a recently reported novel compound with an in vitro pharmacology that resembles the combination of buprenorphine/naltrexone: high affinity/zero efficacy at κ receptors, high affinity/little efficacy at μ receptors and a weak partial agonist profile at NOP receptors (Cueva et al., 2015). Here, we report for the first time the in vivo pharmacology of BU10119 in adult male CD1 mice. The data show that BU10119 has a mixed κ/μ antagonist profile and the κ antagonist effects are relatively short‐acting with no activity evident at 24–48 h post‐administration. We also show for the first time the antidepressant‐like properties of BU10119 in the forced swim test and the novelty‐induced hypophagia task. Importantly, BU10119 is without significant locomotor effects at the doses used and has no significant rewarding effects in the conditioned placed preference task. Finally, we have shown that BU10119 is able to block stress‐induced analgesia, although it did not reduce stress‐induced increases in plasma corticosterone.
In the tail‐withdrawal assay, BU10119 (0.3 to 3 mg·kg−1) was without significant analgesic effects suggesting that it has no agonist efficacy at any of the opioid receptors at the doses studied. Furthermore, BU10119 (1 and 3 mg·kg−1) was able to significantly reduce the analgesic effects of the κ agonist U50,488 and of the μ agonists buprenorphine and morphine. These data clearly indicate that BU10119 has a mixed κ/μ antagonist profile in vivo. These results are consistent with in vitro studies using the rodent vas deferens, which found that BU10119 acts as a κ receptor antagonist, with an average pA2 value of 9.831 (9.084–10.58) and a competitive reversible antagonist at μ receptors with a pA2 value of 10.08 (9.847–10.310) (Ridzwan, 2012). We have previously shown that naltrindole‐derived compounds with a similar mixed κ/μ antagonist profile may have antidepressant and anxiolytic potential (Casal‐Dominguez et al., 2013). However, those compounds had long lasting κ antagonist effects; 21–35 days following a single injection (Casal‐Dominguez et al., 2013). Experiments in the tail‐withdrawal assay show the time course of BU10119's κ antagonist effects. Interestingly, BU10119's effects in the tail‐withdrawal assay are not evident 24–48 h post‐administration making it a relatively short‐acting κ antagonist. This is slightly longer than the duration of action of combination buprenorphine/naltrexone or naltrexone alone, which have a duration of effect <24 h (Almatroudi et al., 2015) but shorter than the prototypical κ antagonist norBNI (Casal‐Dominguez et al., 2013).
While BU10119 (1 mg·kg−1) was without any agonist efficacy in the tail‐withdrawal assay, or in in vitro GTPγS assays (Table 1), the CPP task revealed that BU10119 may be a weak partial μ agonist. Measures of partial agonist activity in GTPγS assays are dependent on specific experimental design such as expression levels. Indeed, naltrexone has been reported to have partial μ agonist activity under conditions of high receptor expression (Kelly et al., 2015). BU10119 appeared to increase the time spent in the drug‐paired compartment of the CPP apparatus, although not to the same extent as buprenorphine, and this effect was not statistically significant. Interestingly, the irreversible μ antagonist CCAM (3 mg·kg−1) was able to reduce the apparent rewarding effects of BU10119 suggesting that these effects are mediated by BU10119 activating μ receptors. We confirmed that CCAM was acting to block μ receptors as it reduced the ability of morphine to produce CPP in a separate experiment. Interestingly, in both CPP experiments performed here, the rewarding properties of buprenorphine were not reversed by CCAM. This is consistent with findings in μ receptor knockout mice that buprenorphine maintains its rewarding properties (Ide et al., 2004). However, in another study using different CPP protocols and different μ‐opioid receptor knockout mice, CPP to buprenorphine was not demonstrated (Marquez et al., 2007). Ide et al. (2004) also showed that the non‐selective opioid antagonist naloxone, and to a lesser extent the δ antagonist naltrindole, and the κ antagonist norBNI were able to reduce buprenorphine conditioning suggesting that multiple opioid receptors are involved in mediating the rewarding effects of buprenorphine. It is interesting that the antidepressant drug fluoxetine has also been shown to produce CPP (Collu et al., 1997), suggesting that perhaps any drug with a positive, stress‐relieving action may somewhat increase preference for the drug‐paired compartment.
BU10119 shows antidepressant‐like activity in both the forced swim test and the novelty‐induced hypophagia test. The acute administration of the SSRI fluoxetine has been shown previously to reduce the time spent immobile in the mouse forced swim test; a widely used behavioural screen for antidepressant efficacy (Lucki, 1997; Cryan et al., 2002). Our data show that BU10119 produces effects equivalent to fluoxetine in the forced swim test. We have previously reported the effects of norBNI (10 mg·kg−1) in adult male CD‐1 mice in the forced swim test 6, 13 and 20 days post‐administration (Casal‐Dominguez et al., 2013) and 24–48 h post administration (Almatroudi et al., 2015). In both these experiments, norBNI produced responses equivalent to fluoxetine. The novelty‐induced hypophagia task is a procedure developed to assess anxiety‐related behaviours using a conflict‐based approach‐avoidance task that is sensitive to a range of anxiety‐relieving drugs including chronic, but not subchronic, administration of fluoxetine (Dulawa and Hen, 2005). Here, we have shown that acute fluoxetine (20 mg·kg−1) delivered by intraperitoneal injection, 1 h prior to testing in both the home and novel cage, did reduce the latency to feed in the novel cage in CD‐1 mice, as we have previously reported (Almatroudi et al., 2015). Several procedural variations could account for these observations including mouse strain and route of administration: Dulawa and Hen (2005) examined a range of doses of fluoxetine (0–25 mg·kg−1) delivered via drinking water to BALB/cJ mice for 4–5 days (subchronic) or for 28/29 days (chronic). Furthermore, Dulawa and Hen administered fluoxetine throughout the training period whereas fluoxetine was only administered here during the testing period. The behavioural effects of BU10119 in both the forced swim test and novelty‐induced hypophagia tasks resembled that of the SSRI fluoxetine and the combination of buprenorphine/naltrexone suggesting that it has antidepressant‐like activity.
Interestingly, BU10119 was without any appreciable effects in the EPM and LDB tasks. This is similar to our previous studies with combination buprenorphine/naltrexone and naltrexone alone (Almatroudi et al., 2015). This is perhaps surprising given that dynorphin has been shown to induce significant anxiogenic‐like effects in mice in the EPM (Narita et al., 2005), while high affinity selective κ antagonists, norBNI and JDTic, have been shown to produce anxiolytic like effects (Knoll et al., 2007). A lack of anxiolytic‐like effect in the EPM may be related to the duration of κ antagonist effects. We have previously shown that long‐acting mixed μ/κ antagonists, and the selective κ antagonist nor‐BNI, have anxiolytic‐like activity in these tasks when tested 7 and 14 days post‐administration (Casal‐Dominguez et al., 2013). BU10119, naltrexone and combination buprenorphine/naltrexone are relatively short‐acting κ antagonists and do not display this anxiolytic‐like effect in the EPM and LDB (Almatroudi et al., 2015). Similar findings have been reported with the short‐acting κ‐antagonists zyklophin and LY2444296, which have effects in a novelty‐induced hypophagia paradigm but no anxiolytic‐like activity in the EPM (Huang et al., 2016). This was in contrast to norBNI, which they showed to be effective in both behavioural paradigms, as we have shown previously, also in CD1 mice (Casal‐Dominguez et al., 2013; Almatroudi et al., 2015). The duration of activity of κ‐receptor antagonists has been demonstrated to correlate with c‐Jun N‐terminal kinase‐1 activation (Melief et al., 2011). It is not clear whether long duration of κ antagonist action is required to produce behavioural effects in the EPM as other short‐acting κ antagonists, AZ‐MTAB and LY‐DMPF, have been shown in prenatally stressed rats to exhibit anxiolytic type responses (Peters et al., 2011). The absence of a robust anxiolytic‐like effect of BU10119 in naïve mice may be due to the fact that the EPM and LDB are not sufficiently stressful paradigms to activate dynorphin release and alter anxiety behaviours (Shirayama et al., 2004; McLaughlin et al., 2006a; Wittmann et al., 2009).
The phenomenon of stress‐induced analgesia is an endogenous protective mechanism that occurs in response to stressful stimuli and involves activation of the descending inhibitory pain pathways (Butler and Finn, 2009). In this study, we have used the warm‐water tail‐withdrawal assay to assess stress‐induced analgesia. This model may be complicated by stress‐induced changes in skin temperature arising from vasoconstriction, but this does not preclude an interpretation of stress‐induced analgesia (Butler and Finn, 2009). Acute stress has been shown to produce antinociception whereas prolonged or repeated exposure to the stress results in tolerance or even hyperalgesic responses (Gamaro et al., 1998; Seo et al., 2006; Seo et al., 2011). Different types of stressors including restraint, cold‐swim or presence of a predator can elicit opioid‐mediated stress‐induced analgesia, which activates distinct neuroanatomical structures (Keay and Bandler, 2001). In this study, we show that both acute and repeated restraint stress resulted in stress‐induced analgesia that was blocked by pretreatment with BU10119 and the combination buprenorphine/naltrexone. Restraint stress‐induced analgesia is blocked by pretreatment with the κ antagonist norBNI in female rats (Botelho et al., 2010) and in mice exposed to forced swim stress (McLaughlin et al., 2003). Interestingly, forced swim stress‐induced analgesia has been shown to be absent in prodynorphin −/− mice (McLaughlin et al., 2003) but is not altered in κ receptor knockout mice (Contet et al., 2005). As these authors discuss, κ receptor and prodynorphin knockouts are not equivalent animal models since the prodynorphin gene encodes a number of opioid peptides that activate opioid receptors (Contet et al., 2005). There are also differences in these studies in the application of the forced swim stress and in the methodology to assess stress‐induced analgesia. Here, we have demonstrated that restraint stress‐induced analgesia is blocked by κ antagonists suggesting the possibility that these compounds may have a role as prophylactic stress treatments (Van't Veer and Carlezon, 2013).
Interestingly, BU10119 was not able to block stress‐induced increases in plasma corticosterone. There are contradictory reports in the literature about the impact of κ‐receptors on plasma corticosterone levels. McLaughlin et al. (2006a) used repeated forced swim stress in C57BL/6 mice, which produced a threefold increase in plasma corticosterone. They went on to show that following pretreatment with the κ antagonist norBNI, and in prodynorphin knockout mice, swim stress still produced an increase in plasma corticosterone. However, Wittmann et al. (2009) reported that basal corticosterone serum levels were reduced in prodynorphin knockout animals and in wild‐type mice pretreated with 10 mg·kg−1 norBNI. In food‐restricted rats, elevated plasma corticosterone levels were reduced by treatment with norBNI (Allen et al., 2013). However, swim stress in rats induced elevated plasma corticosterone that was not blocked by norBNI treatment (Polter et al., 2014). Contet et al. (2005) used a swim stress in warm water in κ‐receptor knockout mice and suggested the dissociation of stress‐induced analgesia and stress‐induced increases in plasma corticosterone levels. These apparently contradictory findings could be explained in part by the different stressors used, which would likely implicate different circuitry in the stress coping behaviours (Pacák and Palkovits, 2001). It is noteworthy that in all of these studies, stress‐induced behaviours were blocked by treatment with norBNI, or in dynorphin −/− mice, even though stress‐induced corticosterone levels may or may not have been affected. Thus, κ antagonists may have therapeutic potential in treating stress‐induced behaviours independent of effects on corticosterone levels (Carroll and Carlezon, 2013; Van't Veer and Carlezon, 2013).
Naltrexone is a relatively non‐selective opioid receptor antagonist with higher affinity for μ than κ receptors. In the UK, it is licensed as an abstinence promoter (Rosner et al., 2010). However, naltrexone is associated with aversive side effects. Combining naltrexone (50 mg daily) with buprenorphine (4 mg daily) has been shown to improve patient compliance in the treatment of opioid dependence, as this improves the dysphoria associated with drug withdrawal (Gerra et al., 2006). However, the tolerability of naltrexone has been shown to be significantly improved when given at lower doses and low dose naltrexone (<4 mg daily) is increasingly being used for the treatment of a number of chronic conditions (Segal et al., 2014; Younger et al., 2014). Low dose naltrexone has recently been trialled in a small cohort of major depressive disorder patients, maintained on antidepressant therapy but experiencing a relapse (Mischoulon et al., 2017). This study investigated whether augmentation of dopaminergic antidepressant regimens (mostly buproprion) with 1 mg naltrexone for 3 weeks would reduce Hamilton Depression Scale (HAM‐D17) scores by 50% compared to baseline. There was an apparent effect of low dose naltrexone although it was not significantly different from placebo in this small cohort study (Mischoulon et al., 2017). The potential of opioid antagonist mechanisms for the treatment of depression is also evident in the combination of buprenorphine with the μ antagonist samidorphan producing a functional mixed μ/κ antagonist (Ehrich et al., 2015). Following 7 days of once daily buprenorphine/samidorphan at a 1:1 ratio, patients with treatment‐resistant major depressive disorder showed a significant improvement in HAM‐D17 total score (Ehrich et al., 2015). These clinical findings, coupled with our preclinical data, suggest that molecules like BU10119 with a pharmacology resembling combination buprenorphine/naltrexone could have significant potential in the treatment of depression.
Author contributions
A.A., a PhD student, performed all of the experiments and data analysis, first draft of the manuscript. M.O., a postdoctoral researcher, synthesized BU10119 for these experiments. C.P.B. and S.M.H. contributed to the experimental design, research and drafting of the manuscript. S.J.B. contributed to the experimental design, research and was lead author on drafting the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 (A) Effect of 1 day and 3 day restraint stress (09:00–11:00) on stress‐induced analgesia in CD1 male mice. (B) Baseline latency on day 3. Results are expressed as mean ± SEM, n = 4. ***P < 0.001 as compared to non‐stressed controls. Analysis done was repeated measures mixed model analysis using InVivo Stat software.
Acknowledgements
This research is funded by the Government of Saudi Arabia through a PhD scholarship (A.A.), the Royal Society (S.J.B.) and NIDA DA07315 (S.M.H.).
Almatroudi A., Ostovar M., Bailey C. P., Husbands S. M., and Bailey S. J. (2018) Antidepressant‐like effects of BU10119, a novel buprenorphine analogue with mixed κ/μ receptor antagonist properties, in mice, British Journal of Pharmacology, 175 2869–2880, https://doi.org/10.1111/bph.14060.
References
- Aldrich J, Patkar K, McLaughlin J (2009). Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short duration of action. Proc Natl Acad Sci U S A 106: 18396–18401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CP, Zhou Y, Leri F (2013). Effect of food restriction on cocaine locomotor sensitization in Sprague–Dawley rats. Psychopharmacology (Berl) 226: 571–578. [DOI] [PubMed] [Google Scholar]
- Almatroudi A, Husbands SM, Bailey CP, Bailey SJ (2015). Combined administration of buprenorphine and naltrexone produces antidepressant‐like effects in mice. J Psychopharmacol 29: 812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho AP, Gameiro GH, Tuma CEDSN, Marcondes FK, Ferraz De Arruda Veiga MC (2010). The effects of acute restraint stress on nociceptive responses evoked by the injection of formalin into the temporomandibular joint of female rats. Stress 13: 269–275. [DOI] [PubMed] [Google Scholar]
- Broadbear JH, Sumpter TL, Burke TF, Husbands SM, Lewis JW, Woods JH et al (2000). Methocinnamox is a potent, long‐lasting, and selective antagonist of morphine‐mediated antinociception in the mouse: comparison with clocinnamox, beta‐funaltrexamine, and beta‐chlornaltrexamine. J Pharmacol Exp Ther 294: 933–940. [PubMed] [Google Scholar]
- Butler RK, Finn DP (2009). Stress‐induced analgesia. Prog Neurobiol 88: 184–202. [DOI] [PubMed] [Google Scholar]
- Carroll FI, Carlezon WA (2013). Development of κ opioid receptor antagonists. J Med Chem 56: 2178–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casal‐Dominguez JJ, Clark M, Traynor JR, Husbands SM, Bailey SJ (2013). In vivo and in vitro characterization of naltrindole‐derived ligands at the κ‐opioid receptor. J Psychopharmacol 27: 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casal‐Dominguez JJ, Furkert D, Ostovar M, Teintang L, Clark MJ, Traynor JR et al (2014). Characterization of BU09059: a novel potent selective κ‐receptor antagonist. ACS Chem Nerosci 5: 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicero TJ, Ellis MS, Surratt HL, Kurtz SP (2014). Factors contributing to the rise of buprenorphine misuse: 2008–2013. Drug Alcohol Depend 142: 98–104. [DOI] [PubMed] [Google Scholar]
- Collu M, Poggiu AS, Pani L, Serra G (1997). Fluoxetine‐induced conditioned place preference: a preliminary study. Synapse 25: 309–311. [DOI] [PubMed] [Google Scholar]
- Contet C, Gaveriaux‐Ruff C, Matifas A, Caradec C, Champy M‐F, Kieffer BL (2005). Dissociation of analgesic and hormonal responses to forced swim stress using opioid receptor knockout mice. Neuropsychopharmacology 31: 1733–1744. [DOI] [PubMed] [Google Scholar]
- Cordery SF, Taverner A, Ridzwan IE, Guy RH, Delgado‐Charro MB, Husbands SM et al (2014). A non‐rewarding, non‐aversive buprenorphine/naltrexone combination attenuates drug‐primed reinstatement to cocaine and morphine in rats in a conditioned place preference paradigm. Addict Biol 19: 575–586. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Markou A, Lucki I (2002). Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci 23: 238–245. [DOI] [PubMed] [Google Scholar]
- Cueva JP, Roche C, Ostovar M, Kumar V, Clark MJ, Hillhouse TM et al (2015). C7β‐methyl analogues of the orvinols: the discovery of kappa opioid antagonists with nociceptin/orphanin FQ peptide (NOP) receptor partial agonism and low, or zero, efficacy at Mu opioid receptors. J Med Chem 58: 4242–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulawa SC, Hen R (2005). Recent advances in animal models of chronic antidepressant effects: the novelty‐induced hypophagia test. Neurosci Biobehav Rev 29: 771–783. [DOI] [PubMed] [Google Scholar]
- Ehrich E, Turncliff R, Du Y, Leigh‐Pemberton R, Fernandez E, Jones R et al (2015). Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology 40: 1448–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh T, Matsuura H, Tanaka C, Nagase H (1992). Nor‐binaltorphimine: a potent and selective kappa‐opioid receptor antagonist with long‐lasting activity in vivo. Arch Int Pharmacodyn Ther 316: 30–42. [PubMed] [Google Scholar]
- Gamaro GD, Xavier MH, Denardin JD, Pilger JA, Ely DR, Ferreira MBC et al (1998). The effects of acute and repeated restraint stress on the nociceptive response in rats. Physiol Behav 63: 693–697. [DOI] [PubMed] [Google Scholar]
- Gerra G, Fantoma A, Zaimovic A (2006). Naltrexone and buprenorphine combination in the treatment of opioid dependence. J Psychopharmacol 20: 806–814. [DOI] [PubMed] [Google Scholar]
- Hillhouse TM, Hallahan JE, Jutkiewicz EM, Husbands SM, Traynor JR (2016). A buprenorphine analog attenuates drug‐primed and stress‐induced cocaine reinstatement. Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience, 2016. Online.
- Huang P, Yakovleva T, Aldrich JV, Tunis J, Parry C, Liu‐Chen L‐Y (2016). Two short‐acting kappa opioid receptor antagonists (zyklophin and LY2444296) exhibited different behavioral effects from the long‐acting antagonist norbinaltorphimine in mouse anxiety tests. Neurosci Lett 615: 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide S, Minami M, Satoh M, Uhl GR, Sora I, Ikeda K (2004). Buprenorphine antinociception is abolished, but naloxone‐sensitive reward is retained, in [mu]‐opioid receptor knockout mice. Neuropsychopharmacology 29: 1656–1663. [DOI] [PubMed] [Google Scholar]
- Keay KA, Bandler R (2001). Parallel circuits mediating distinct emotional coping reactions to different types of stress. Neurosci Biobehav Rev 25: 669–678. [DOI] [PubMed] [Google Scholar]
- Kelly E, Mundell SJ, Sava A, Roth AL, Felici A, Maltby K et al (2015). The opioid receptor pharmacology of GSK1521498 compared to other ligands with differential effects on compulsive reward‐related behaviours. Psychopharmacology (Berl) 232: 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA, Jr. (2007). Anxiolytic‐like effects of kappa‐opioid receptor antagonists in models of unlearned and learned fear in rats. J Pharmacol Exp Ther 323: 838–845. [DOI] [PubMed] [Google Scholar]
- Lalanne L, Ayranci G, Kieffer BL, Lutz P‐E (2014). The kappa opioid receptor: from addiction to depression, and back. Front Psych 5: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucki I (1997). The forced swimming test as a model for core and component behavioral effects of antidepressant drugs. Behav Pharmacol 8: 523–532. [DOI] [PubMed] [Google Scholar]
- Marquez P, Baliram R, Kieffer BL, Lutfy K (2007). The mu opioid receptor is involved in buprenorphine‐induced locomotor stimulation and conditioned place preference. Neuropharmacology 52: 1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Marton‐Popovici M, Chavkin C (2003). Kappa opioid receptor antagonism and prodynorphin gene disruption block stress‐induced behavioral responses. J Neurosci 23: 5674–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C (2006a). Prior activation of kappa opioid receptors by U50,488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning. Neuropsychopharmacology 31: 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Li S, Valdez J, Chavkin TA, Chavkin C (2006b). Social defeat stress‐induced behavioral responses are mediated by the endogenous kappa opioid system. Neuropsychopharmacology 31: 1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Carroll FI, Beguin C, Carelzon WA, Cohen BM et al (2011). Duration of action of a broad range of selective kappa‐opioid receptor antagonists is positively correlated with c‐Jun N‐terminal kinase‐1 activation. Mol Pharmacol 80: 920–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischoulon D, Hylek L, Yeung AS, Clain AJ, Baer L, Cusin C et al (2017). Randomized, proof‐of‐concept trial of low dose naltrexone for patients with breakthrough symptoms of major depressive disorder on antidepressants. J Affect Disord 208: 6–14. [DOI] [PubMed] [Google Scholar]
- Narita M, Kaneko C, Miyoshi K, Nagumo Y, Kuzumaki N, Nakajima M et al (2005). Chronic pain induces anxiety with concomitant changes in opioidergic function in the amygdala. Neuropsychopharmacology 31: 739–750. [DOI] [PubMed] [Google Scholar]
- Pacák K, Palkovits M (2001). Stressor specificity of central neuroendocrine responses: implications for stress‐related disorders. Endocr Rev 22: 502–548. [DOI] [PubMed] [Google Scholar]
- Peters MF, Zacco A, Gordon J, Maciag CM, Litwin LC, Thompson C et al (2011). Identification of short‐acting κ‐opioid receptor antagonists with anxiolytic‐like activity. Eur J Pharmacol 661: 27–34. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM (1986). Psychotomimesis mediated by kappa opiate receptors. Science 233: 774–776. [DOI] [PubMed] [Google Scholar]
- Polter AM, Bishop RA, Briand LA, Graziane NM, Pierce RC, Kauer JA (2014). Poststress block of kappa opioid receptors rescues long‐term potentiation of inhibitory synapses and prevents reinstatement of cocaine seeking. Biol Psychiatry 76: 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridzwan IE (2012). A single compound alternative to a buprenorphine/naltrexone combination. PhD Thesis, University of Bath.
- Rorick‐Kehn LM, Witkin JM, Statnick MA, Eberle EL, Mckinzie JH, Kahl SD et al (2014). LY2456302 is a novel, potent, orally‐bioavailable small molecule kappa‐selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology 77: 131–144. [DOI] [PubMed] [Google Scholar]
- Rosner S, Hackl‐Herrwerth A, Leucht S, Vecchi S, Srisurapanont M, Soyka M (2010). Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev 12 CD001867.pub2. [DOI] [PubMed] [Google Scholar]
- Ross NC, Reilley KJ, Murray TF, Aldrich JV, McLaughlin JP (2012). Novel opioid cyclic tetrapeptides: Trp isomers of CJ‐15,208 exhibit distinct opioid receptor agonism and short‐acting κ opioid receptor antagonism. Br J Pharmacol 165: 1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman R, Gorelick D, Heishman S, Eichmiller PR, Hill BH, Norbeck J et al (2000). An open‐label study of a functional opioid kappa antagonist in the treatment of opioid dependence. J Subst Abuse Treat 18: 277–281. [DOI] [PubMed] [Google Scholar]
- Sadler AM, Bailey SJ (2013). Validation of a refined technique for taking repeated blood samples from juvenile and adult mice. Lab Anim 47: 316–319. [DOI] [PubMed] [Google Scholar]
- Sadler AM, Bailey SJ (2016). Repeated daily restraint stress induces adaptive behavioural changes in both adult and juvenile mice. Physiol Behav 167: 313–323. [DOI] [PubMed] [Google Scholar]
- Schwarzer C (2009). 30 years of dynorphins – new insights on their functions in neuropsychiatric diseases. Pharmacol Ther 123: 353–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal D, Macdonald JK, Chande N (2014). Low dose naltrexone for induction of remission in Crohn's disease. Cochrane Database Syst Rev CD010410. https://doi.org/10.1002/14651858.CD010410.pub2 [DOI] [PubMed] [Google Scholar]
- Seo Y‐J, Kwon M‐S, Shim E‐J, Park S‐H, Choi O‐S, Suh H‐W (2006). Changes in pain behavior induced by formalin, substance P, glutamate and pro‐inflammatory cytokines in immobilization‐induced stress mouse model. Brain Res Bull 71: 279–286. [DOI] [PubMed] [Google Scholar]
- Seo Y‐J, Kwon M‐S, Choi S‐M, Lee J‐K, Park S‐H, Jung J‐S et al (2011). Differential cross‐tolerance development between single and repeated immobilization stress on the antinociceptive effect induced by β‐endorphin, 5‐hydroxytryptamine, morphine, and WIN55,212‐2 in the inflammatory mouse pain mode. Arch Pharm Res 34: 269–280. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS (2004). Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant‐like effects. J Neurochem 90: 1258–1268. [DOI] [PubMed] [Google Scholar]
- Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH et al (2014). Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods 11: 629–632. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Calo G, Cox BM, Chavkin C Christie MJ, Civelli O et al (2017). Opioid receptors. IUPHAR/BPS guide to PHARMACOLOGY. Available at: http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=50 (accessed 28/08/2017).
- Van't Veer A, Carlezon W Jr (2013). Role of kappa‐opioid receptors in stress and anxiety‐related behavior. Psychopharmacology (Berl) 229: 435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoest PR, Sawant Basak A, Parikh V, Hayward M, Kauffman GW, Paradis V et al (2011). Design and discovery of a selective small molecule kappa opioid antagonist (2‐methyl‐N‐((2′‐(pyrrolidin‐1‐ylsulfonyl)biphenyl‐4‐yl)methyl)propan‐1‐am ine, PF‐4455242). J Med Chem 54: 5868–5877. [DOI] [PubMed] [Google Scholar]
- Wittmann W, Schunk E, Rosskothen I, Gaburro S, Singewald N, Herzog H et al (2009). Prodynorphin‐derived peptides are critical modulators of anxiety and regulate neurochemistry and corticosterone. Neuropsychopharmacology 34: 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger J, Parkitny L, McLain D (2014). The use of low‐dose naltrexone (LDN) as a novel anti‐inflammatory treatment for chronic pain. Clin Rheumatol 33: 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) Effect of 1 day and 3 day restraint stress (09:00–11:00) on stress‐induced analgesia in CD1 male mice. (B) Baseline latency on day 3. Results are expressed as mean ± SEM, n = 4. ***P < 0.001 as compared to non‐stressed controls. Analysis done was repeated measures mixed model analysis using InVivo Stat software.