

Abstract
Trichloroethylene (TCE) is a widespread environmental pollutant associated with immunotoxicity and autoimmune disease. Previous studies showed that mice exposed from gestation through early life demonstrated CD4+ T cell alterations and autoimmune hepatitis. Determining the role of one environmental risk factor for any disease is complicated by the presence of other stressors. Based on its known effects, we hypothesized that developmental overnutrition in the form of a moderately high-fat diet (HFD) consisting of 40% kcal fat would exacerbate the immunotoxicity and autoimmune-promoting effects of low-level (<10 μg/kg/day) TCE in autoimmune-prone MRL+/+ mice over either stressor alone. When female offspring were evaluated at 27 weeks of age we found that a continuous exposure beginning at 4 weeks preconception in the dams until 10 weeks of age in offspring that TCE and HFD promoted unique effects that were often antagonistic. For a number of adiposity endpoints, TCE significantly reversed the expected effects of HFD on expression of genes involved in fatty acid synthesis/insulin resistance, as well as mean pathology scores of steatosis. Although none of the animals developed pathological signs of autoimmune hepatitis, the mice generated unique patterns of antiliver antibodies detected by western blotting attributable to TCE exposure. A majority of cytokines in liver, gut, and splenic CD4+ T cells were significantly altered by TCE, but not HFD. Levels of bacterial populations in the intestinal ileum were also altered by TCE exposure rather than HFD. Thus, in contrast to our expectations this coexposure did not promote synergistic effects.
Keywords: trichloroethylene, autoimmune, high-fat diet, microbiome, adiposity
Recent decades have seen a steady rise in immune-mediated inflammatory diseases. These are clinically diverse disorders that share a chronic and debilitating phenotype with an estimated 5%–7% prevalence in the United States encompassing over 100 different diseases including multiple sclerosis, rheumatoid arthritis, and inflammatory bowel disease (El-Gabalawy et al., 2010). These diseases can manifest during childhood or adolescence (Kappelman et al., 2011), and it is possible that those diagnosed in adults may have been initiated during childhood or even earlier in development.
Various aspects of a 21st century lifestyle have been proposed as immune-mediated disease risk factors including the unwitting exposure to environmental chemicals. There is increasing evidence that developmental and early life environmental exposures to toxicants at doses not immunotoxic in adults can have even more profound and/or sustained effects even after removal of the stressor. In rodents, persistent immune suppression could be generated by developmental exposure to chemicals such as dioxin (TCDD), or pesticide (Smialowicz, 2002). Prenatal exposure to TCDD dampened the immune response to immunizations in children, and decreased lymphocyte numbers that persisted into adolescence (Leijs et al., 2009; Stølevik et al., 2013).
Although toxicant-induced immunosuppression can impair host resistance to infection and cancer, toxicant-induced immune activation can inappropriately increase risk for hypersensitivity disorders and autoimmune diseases. Human exposure to trichloroethylene (TCE), whether occupational or environmental, has been linked to autoimmune and hypersensitivity diseases (Cooper et al., 2009; Huang et al., 2015; Parks and De Roos, 2014; Zhao et al., 2016). Chronic adult exposure to TCE at occupationally relevant concentrations altered CD4+ T cell function and induced T cell-mediated liver inflammation similar to autoimmune hepatitis (AIH) in humans (Gilbert et al., 2009; Griffin et al., 2000). Recently, developmental exposure to TCE, at concentrations 10 000-fold lower than adult exposure promoted autoimmunity even after the TCE was removed from the drinking water (Gilbert et al., 2017a). Thus, developmental exposure to TCE increased the risk for developing immune-mediated disease in later life, and the exposure does not need to be continuous.
Perinatal overnutrition/obesity has become an increasing concern as a risk factor for adverse health effects for both mother and child. Fifty percent of American women of childbearing age are overweight or obese (Ogden et al., 2015). Adverse outcomes of maternal and postnatal obesity were observed in animal models of autoimmunity and allergy (Dinger et al., 2016; Xue et al., 2014). In humans, maternal obesity negatively influenced programming of the neonatal immune system (Wilson et al., 2015) potentially enhancing risk to inflammatory disease such as asthma (Forno et al., 2014).
Although the biological mechanisms for the effects of obesity on the immune system are not completely understood, it is known that adipose tissue secretes a wide variety of adipokines that interface with the immune system (Cohen et al., 2017). Altered leptin has been linked with rheumatoid arthritis disease activity (Batun-Garrido et al., 2017), and several other autoimmune disorders (Procaccini et al., 2015). Patients with idiopathic AIH (the type of autoimmune disease induced by TCE in our mouse model) present with significantly higher body fat than controls (Pereira et al., 2011).
Little is known about the impact of obesity in pregnancy or early life on autoimmunity in the offspring. Higher childhood BMI was shown to be a risk factor for the onset of pediatric multiple sclerosis (Bove et al., 2016). Patients with childhood-onset lupus show a positive correlation between serum TNF-α and percent body fat (Sinicato et al., 2014). Pregnant obese mothers have increased circulating levels of inflammatory cytokines and adipokines (Schmatz et al., 2010). These mediators in the uterine microenvironment have been invoked to explain the link between maternal obesity/overnutrition and metabolic disorders in the offspring. It is likely that an increase in these proinflammatory factors may similarly predispose the offspring in later life for certain types of autoimmune diseases. Moreover, intestinal microbiome and inflammation in gut mucosa are known to be extra-articular triggers for autoimmune disorders such as rheumatoid arthritis (Belkaid and Hand, 2014; Brusca et al., 2014).
Confirming the contribution of a particular environmental risk factor for autoimmune disease is complicated by the fact that it is almost always encountered in the presence of other effect-modifying environmental stressors. Controlled animal studies represent a logical way to study the contribution of multiple factors to an alteration in phenotype. We hypothesized that developmental overnutrition would exacerbate the autoimmune-promoting effects of low-level TCE exposure in female offspring. Due to known adverse effects of inflammation on intestinal homeostasis, we predicted that coexposure, over either stressor alone, would augment gut cytokines, and bacterial populations in the intestinal ileum. It was expected that biomarkers associated with inflammation and adiposity would be further increased by coexposure.
MATERIALS AND METHODS
Mice
The experiment used MRL+/+ mice that spontaneously develop a relatively mild lupus-like disease late in life (50% mortality at 17 months), but can also develop other autoimmune disorders such as Sjogren’s syndrome and T cell-infiltrating pancreatitis (Qu et al., 2002; Saito et al., 2013). The cause of the predisposition for autoimmunity in MRL+/+ mice is not known. Before they reach 1 year of age most female MRL+/+ mice do not exhibit autoimmune tissue pathology and indications of autoimmunity are minor. The main goal of this study was to understand how TCE in combination with high-fat diet (HFD) promoted or accelerated autoimmunity. Since autoimmune diseases are found 4 times as often in women than men, we confined this investigation to female mice. The male offspring were not discarded but utilized in a separate study with different endpoints (manuscript in preparation). Thus, young adult female MRL+/+ mice, with their propensity for autoimmunity but absence of overt disease, are a good model to test whether environmental stressors can boost autoimmunity.
Exposure strategy
The exposure strategy is shown in Figure. 1. Starting at 4 weeks of age, female MRL+/+ mice (Jackson Laboratories; Bar Harbor, Maine) to be used as breeding dams (10 mice/treatment group) were randomly assigned to 1 of 4 exposure groups. The 4 experimental groups consisted of: (1) mice receiving vehicle control drinking water and 10% kcal fat diet or “control” mice (TCE−/HFD−); (2) mice receiving vehicle control drinking water 40% kcal fat diet (TCE−/HFD+); (3) mice receiving TCE in the drinking water and 10% kcal fat diet (TCE+/HFD−); (4) mice receiving TCE in the drinking water and 40% kcal fat diet (TCE+/HFD+). Mice were exposed for approximately 10 weeks (4 weeks prior to mating, 3 weeks during pregnancy, and an additional 3 weeks during lactation). The female dams were mated with previously untreated male MRL+/+ mice. The TCE was administered (0 or 0.05 μg/ml) in drinking water as previously described (Blossom and Doss, 2007). Starting at weaning (postnatal day [PND] 21) the resulting pups were exposed to TCE (0 or 0.05 μg/ml) in drinking water and/or HFD directly for an additional 7 weeks. At that point, TCE and/or HFD was removed, and the mice received untreated drinking water and standard rodent chow (Harlan 7027) ad libitum for an additional 17 weeks at which time the experiment was terminated.
Figure 1.

Experimental Design. Starting at 4 weeks of age female MRL+/+ mice were randomly assigned to 1 of 4 treatment groups (10 mice/treatment group). Each group consisted of (1) Vehicle and 10% kcal fat diet (TCE−/HFD−); (2) Vehicle and 40% kcal fat diet (TCE−/HFD+); (3) TCE and 10% kcal fat diet (TCE+/HFD−), and (4) TCE and 40% kcal fat diet (TCE+/HFD+). All exposures started 4 weeks prior to breeding and continued during gestation and lactation. Female offspring were weaned at 3 weeks and exposed to the same treatment directly for an additional 7 weeks after which TCE was removed from the purified drinking water and a standard diet was implemented. Mice were euthanized at 27 weeks of age and tissues/cells assessed for parameters as described in detail in “Materials and Methods” section.
TCE and HFD exposure
Like our previous studies, TCE was administered in the drinking water. The TCE-containing drinking water was changed 3 times/week to offset degradation of TCE. Non TCE groups were given water containing only vehicle or 1% Alkamuls EL-620, the reagent used to maintain TCE in a solution. All drinking water was Ultrapure and unchlorinated (Milli-Q) to ensure that chlorination or its by-products did not confound the results. To calculate the dose of TCE in μg/kg/day, both female breeders and resulting female offspring were weighed weekly and water consumption was monitored. TCE exposure (μg/kg/day) was based on average amount of TCE-containing water consumed per cage (2–4 mice/cage) divided by the average mouse weight per cage and a previously determined 20% degradation of TCE in the water bottles. During the same time period as the TCE exposure, mice were fed a moderately high fat “western” diet consisting of 40% kcal fat diet. Those mice not receiving a HFD were given a protein, cholesterol, and sucrose-matched control diet consisting of 10% kcal fat. All diets were purchased from Research Diets, New Brunswick NJ.
Experimental endpoints
At 27 weeks of age female mice were euthanized and randomly selected mice in each litter (n = 6–10) were examined for histopathological signs of inflammation, liver steatosis, and autoimmune disease. Biomarkers of adiposity, fat biosynthesis, and energy balance in adipose and liver tissue were evaluated. In addition, several immune parameters including autoantibody production (serum), CD4+ T cell cytokine production (spleen), and inflammation/repair gene expression (liver) were assessed. In addition, gut-mucosa-associated cytokines and bacterial population (ileal mucosa-associated) were also evaluated. All the samples that were not utilized immediately were flash frozen in liquid nitrogen and stored at −80°C for later use. This study was approved by the Animal Care and Use Committee at the University of Arkansas for Medical Sciences.
Histopathology
Parametrial fat (PMF) and liver tissue were harvested and fixed in 10% neutral buffered formalin, and paraffin-embedded. PMF sections were stained with hematoxylin and eosin (H&E) and imaged using a light microscope. The adipose tissue size was evaluated using FIJI Adiposoft software. Liver tissue sections were also stained with H&E and imaged using a 100× oil immersion. Steatosis was identified based on presence of lipid accumulation, and classified as macrovesicular if the nucleus was displaced by the vacuole or microvasicular if the nucleus remained in the center of the hepatocyte. Steatosis was scored using a 4-point scoring system (0= very little to no steatosis; 1 = observable steatosis (25%–50%); 2 = observable steatosis (50%–75%); 3 = >75% observable steatosis with lipid fully surrounding nucleus.
Liver, skin, and kidney tissue harvested from mice at 27 weeks of age was stained with H&E and examined for pathology related to autoimmune tissue damage. Liver sections were examined microscopically and scored in a blinded manner by a veterinary pathologist for the severity of inflammation and fibrosis based on a 4-point scale (0–3), ranging from no change to severe, respectively) as described (Gilbert et al., 2009). Kidney sections were examined for 4 criteria, each with individual scores ranging from 1 to 4, for a total possible score of 13 as described (Gilbert et al., 2017a). Based on previously observed results in which chronic exposure to a TCE metabolite in the drinking induced alopecia, skin was also evaluated as described (Blossom et al., 2006).
Immunohistochemistry
Immunohistochemistry was used to detect adipophilin (ADFP), also known as Perilipin-2, in the liver. Paraffin-embedded liver tissue were serially sectioned, dewaxed, and rehydrated through a graded alcohol series as described previously in Al-Dwairi et al. (2014). Antigen unmasking was conducted by boiling the sections in a microwave using Citra Plus (Biogenex, San Ramon, California) for a duration of 2 min at power 10 and then for 10 min at power 1. Samples were cooled for 20 min at room temperature before treating the sections with 3% hydrogen peroxide to quench endogenous peroxidase activity and then incubating in blocking solution (Vectastain Elite ABC kit, Vector Laboratories, Inc.; Burlingame, California) for 30 min. Sections were first incubated overnight with AntiADFP antibody (antiPerilipin2/ADFP antibody; Novus Biologicals) followed by incubation with secondary antibody (Vectastain Elite ABC kit; Vector Laboratories) for 30 min. Sections were stained with 3, 3’-diaminobenzidine tetra-hydrochloride (Dako Inc.; Carpinteria, California) and counterstained with hematoxylin. Slides were dehydrated and cleared by passing them through alcohol series and xylene, respectively.Quantification of staining was performed using Aperio Positive Pixel Count Algorithm (Leica Biosystems).
Quantitative reverse transcriptase polymerase chain reaction
Fluorescence-based quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was conducted as described in Gilbert et al. (2012) using the iScript cDNA synthesis kit (Bio-Rad, Hercules, Califorina). Total RNA isolated from naïve or effector/memory CD4+ T cells, PMF tissue, and liver was reverse-transcribed into cDNA. Gene expression was evaluated by qRT-PCR using Bio-Rad iTaq SYBR Green Supermix Primers procured from Integrated DNA Technologies. For CD4+ T cells, fold change in expression was determined using expression levels of resting (unactivated) CD4+ T cells of the appropriate subset of control mice as the control (1×) expression level. For PMF and liver tissue, samples were run in duplicate or triplicate and averaged to obtain a mean fold change expression level.
Isolation and activation of CD4+ T cells from spleen
CD4+ T cells were isolated from spleen cell suspensions using Dynabeads FlowComp Mouse CD4 kit (Invitrogen). CD4+ T cells were seeded into 24-well plates and stimulated with immobilized antiCD3 antibody and soluble antiCD28 antibody for 18 h. Cells were harvested from the wells, centrifuged and reconstituted in a solution of RLT buffer (RNeasy, Quiagen). Pellets were frozen at −80°C until qRT-PCR.
Antiliver antibody production
Using previously described methodology (Gilbert et al., 2017a), microsomal liver protein (30 µg) obtained from an untreated MRL+/+ mouse was separated on 12% SDS-PAGE, electrotransferred onto nitrocellulose, and subsequently probed with sera (1:500) obtained from each of 4 groups of mice followed by HRP-conjugated polyclonal goat antimouse IgG (1:4000). Densitometric analysis of mouse myeloma IgG run in adjoining lanes and detected by the HRP-conjugated polyclonal goat antimouse IgG was used to normalize exposure times for the individual Western blots.
Detection of gut cytokines
Ileal tissues were removed from −80°C, weighed, lysed and extracted using a gentleMACS dissociator. After centrifugation (4°C for 10 min; at 750 rpm), clear supernatant was transferred to Eppendorf tubes and centrifuged at 4°C for 15 min at 10 000 × g. Supernatants were stored at −80°C until cytokine profiling. The protein concentration was measured in the spectrophotometer using BioRad Protein Assay. All samples were diluted to a final concentration of 900 mg/ml for cytokine measurement and stored at −80°C until use for cytokine analysis as described earlier in Gokulan et al. (2016). Cytokines relevant to mucosal immunity were measured (IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 [p40], IL-12 [p70], IL-13, IL-17, eotaxin, G-CSF, GM-CSF, IFN-γ, KC, monocyte chemoattractant protein [MCP-1], MIP-1α, MIP-1β, RANTES, TNF-α IL-17, IL-21, IL-22, IL-23p19, IL-31, IL-33, and MIP-3α) in duplicate using Bio-Plex Mouse Cytokine multiplex kits (Bio-Rad) per the manufacturer’s instructions.
Identification of bacterial groups from ileal tissue
Ileal tissue lysate was prepared by bead-beating in a Fast Prep Machine. Proteins and RNA were removed by incubating with proteinase K, followed by a second incubation at 37°C for 15 min with RNAse-A. An equal volume of phenol-chloroform-isopropanol was added to the tube containing lysate and mixed. After centrifuging at 12 000 × g for 30 min, the aqueous layer was transferred to a new tube, and DNA was precipitated using standard techniques. The cell pellet was washed with 70% ethanol and air dried. DNA was suspended and stored in nuclease-free water. Real-time PCR was utilized to amplify DNA fragments of bacteria present in intestinal mucosa using an ABI 7500 machine. Primer Express software (Applied Biosystems, Foster City, California) was used to design primers for the identification of predominant phyla and representative genera and species of bacteria present in intestinal mucosa as previously reported in Williams et al. (2015).
Statistical analysis
Assays were conducted using samples from 6 to 10 individual mice in each treatment group. Summary statistics such as mean and SD are presented for each treatment group. Data were initially evaluated with a 2-way analysis of variance (ANOVA) with interactions to determine the overall effects of TCE and diet (HFD), and the interaction between the 2 main factors. All analyses were followed by Tukey’s post hoc tests to protect the overall significance level of 0.05. The 6 prespecified pairwise comparisons included (1) (TCE−/HFD−) versus (TCE− HFD+), (2) (TCE−/HFD−) versus (TCE+/HFD−), (3) (TCE− HFD−) versus (TCE+/HFD+), (4) (TCE−/HFD+) versus (TCE+/HFD−), (5) TCE−/HFD+ versus TCE+/HFD+, and (6) (TCE+/HFD−) versus (TCE+/HFD+). Statistical significance resulting from this comparison is reported in the graphs and the tables. Pregnancy data were compared using Fisher’s exact method. Steatosis grading, which is an ordinal categorical variable, was analyzed using Kruskal-Wallis H test. Subsequent pairwise comparisons between pair specified a priori were tested using Wilcoxon rank-sum test. All analyses were completed in Stata v15.1 (StataCorp, College Station, Texas).
RESULTS
Dams: Pregnancy and Birth Parameters
Neither terminal body weights nor a longitudinal evaluation of body weight during the treatment regimen revealed any statistically significant group-specific changes in the dams when evaluated approximately 3 weeks after their 10-week exposure to TCE and/or HFD. Similarly, when maternal fat depots were examined and normalized to body weight (ie, maternal retroperitoneal fat [RPF] and PMF) there were no differences among the groups (data not shown). Thus, the relatively short duration of the exposure to HFD did not alter body weight or distribution of fat depots in the dams. In addition, none of the treatments had a significant effect on CD4+ T cell function or serum markers of autoimmunity (data not shown). Thus, the dams were not evaluated further for autoimmune pathology or other mediators related to fat metabolism, lipogenesis, or inflammation in other tissues.
Interestingly, the combination of the 2 environmental stressors appeared to significantly decrease breeding effectiveness. When compared with 100% successful pregnancies for dams in the control group (10/10 dams), only 60% (6/10) of the dams in the coexposure group had successful pregnancies. When compared with a total of 68 pups generated by the dams in the (TCE−/HFD−) group, dams in the (TCE+/HFD+) group only generated 20 pups, and this effect was statistically significant (Table 1). However, other pregnancy endpoints such as average litter size (No. of pups per born from each litter) were not statistically different among the groups. Thus, although not conclusive, some aspect of pregnancy or birth appeared to be altered in the coexposure group despite no evidence of obesity, CD4+ T cell cytokine effects, or autoimmunity in the dams.
Table 1.
Summary of Pregnancy Data
| TCE− HFD− | TCE− HFD+ | TCE+ HFD− | TCE+ HFD+ | |
|---|---|---|---|---|
| Number of litters | 10/10 (100%) | 9/10 (90%) | 9/10 (90%) | 6/10 (60%) |
| Litter size (no. of pups born) | 7.3 (1.3) | 5.7 (2.3) | 6.2 (2.8) | 5.7 (2.5) |
| Live pups out of total pups | 68/86 (79%) | 42/56 (75%) | 51/71 (72%) | 20/39 (51%) * |
Summary statistics of pregnancy data of dams by group. Two sample proportion tests comparing the percentages indicated statistical significance *(p = .0016) when compared with controls.
TCE Opposed the Effect of HFD on Body Weight and Adiposity Biomarkers in Female Offspring
The offspring from the 4 groups of dams were maintained on the same treatment regimen as dams from weaning age (3 weeks of age) until 10 weeks of age. At that point, all treatments were removed and offspring were given standard chow and unchlorinated drinking water for the duration of the experiment (Figure 1). At 27 weeks of age all of the female pups were euthanized and examined for multiple parameters. Body weight and fat depots are presented in Table 2. The mice fed HFD (either alone or in combination with TCE) weighed significantly more than mice in the (TCE−/HFD−) group. Mice coexposed to TCE and HFD weighed significantly more compared with mice in the (TCE+/HFD−) group. At 27 weeks of age, and after a period of 17 weeks of exposure cessation, (TCE−/HFD+) offspring weighed significantly more than mice in the (TCE−/HFD−) and (TCE+/HFD−) groups. Although the effect of coexposure on body weight at 27 weeks was less dramatic relative to the 10-week time point, there was a significant interaction effect between TCE and HFD (p = .02), which was not observed at the 10-week point (p = .18). However, there were significant main effects for HFD consumption at 10 weeks as well as 27 weeks that were significant (p < .001) and not observed for TCE exposure. Together these results suggested that HFD was associated with increased body weight and TCE appeared to mitigate this effect.
Table 2.
HFD Consumption Increased Body Weight and Fat Depot Weights in Offspring
| TCE− | TCE− | TCE+ | TCE+ | (1) | (1) | (1) | (2) | (2) | (3) | |
|---|---|---|---|---|---|---|---|---|---|---|
| HFD− | HFD+ | HFD− | HFD+ | vs | vs | vs | vs | vs | vs | |
| (1) | (2) | (3) | (4) | (2) | (3) | (4) | (3) | (4) | (4) | |
| 10 weeks of age | ||||||||||
| BW | 33.2 (2.7) | 39.8 (3.9) | 33.7 (3.8) | 38.1 (2.9) | <0.001 | 0.9 | 0.001 | <0.001 | 0.6 | 0.005 |
| 27 weeks of age | ||||||||||
| BW | 44.0 (5.0) | 50.7 (6.1) | 44.9 (4.3) | 46.5 (2.4) | <0.001 | 0.7 | 0.06 | 0.001 | 0.1 | 0.2 |
| PMF | 3.7 (1.2) | 4.9 (1.6) | 3.6 (1.3) | 3.8 (0.7) | 0.008 | 0.9 | 0.9 | 0.01 | 0.1 | 0.9 |
| PMF/BW | 0.08 (0.02) | 0.09 (0.02) | 0.08 (0.02) | 0.081 (0.01) | 0.09 | 0.9 | 0.9 | 0.06 | 0.3 | 0.9 |
| RPF | 0.05 (0.3) | 0.07 (0.4) | 0.5 (0.2) | 0.5 (0.10) | 0.003 | 0.9 | 0.8 | 0.04 | 0.2 | 0.9 |
| RPF/BW | 0.010 (0.004) | 0.014 (0.005) | 0.011 (0.003) | 0.012 (0.002) | 0.01 | 0.8 | 0.8 | 0.2 | 0.5 | 0.9 |
Female offspring were weighed once weekly starting at weaning. Body weights (BWs) of the female offspring are represented in grams and are shown at 10 weeks of age when their exposures stopped and at study terminus (27 weeks of age). Mice were euthanized and adipose tissue (PMF and RPF) was collected and represented independently or in body weight ratios. Numbers represent mean and SD of the 4 exposure groups. Data were analyzed by 2-way ANOVA to test for significant interaction and main effects of the 2 variables (TCE × HFD) and these results are reported in the text. Shown in the table are p values for the 6 pairwise Tukey’s post hoc comparisons numbered by group and conducted as described in the “Materials and Methods” section. Results are statistically significant (p < .05) indicated in bold.
Also shown in Table 2, in addition to body weight, fat depots which reflect a more accurate assessment of obesity were examined at 27 weeks. Similar to body weights, animals from the (TCE−/HFD+) group had significantly more PMF and RPF compared with (TCE−/HFD−) and (TCE+/HFD−) groups. When represented as a ratio to body weight, the mean RPF of (TCE−/HFD+) offspring was significantly increased relative to no treatment controls. PMF/body weight ratios were not significantly different. Although there were no significant interaction effects on these endpoints, for PMF, RPF, and RPF/body weight ratio there were statistically significant main effects of HFD (p = .03, .02, and .04, respectively) but not TCE. Thus, similar to body weight, HFD was associated with increased fat depots, and TCE appeared to lessen this effect.
TCE Decreased HFD-Mediated Expression of Genes Involved in Fat Synthesis and Insulin Resistance in Adipose Tissue and Liver
Several genes related to obesity, overnutrition, and insulin resistance were examined in adipose tissue and liver. Adipose tissue was evaluated for mRNA expression of adipokines, leptin, and adiponectin (adipoq). In both adipose tissue and liver, genes involved in fatty acid synthesis including fasn which encodes fatty acid synthase and cytosolic malic enzyme (me1) which contributes to de novo fatty acid synthesis and HFD-induced adiposity (Al-Dwairi et al., 2012) were evaluated at study terminus. Results of qRT-PCR are depicted in Figures 2A and 2B. For adipose tissue, Adipoq is negatively correlated with body fat and was decreased in all groups relative to no treatment controls, an effect that was not specific to HFD-fed mice. In contrast, TCE had an antagonistic effect on HFD-induced leptin levels. Leptin expression levels were decreased as much as 5-fold in both TCE exposure groups (± HFD) compared with (TCE−/HFD+) mice. This suppressive effect of TCE carried over to fasn expression in adipose tissue. Me1 expression pattern was similar but also significantly decreased in (TCE+/HFD+) coexposed mice relative to the (TCE−/HFD−) control group. Treatment with HFD alone inhibited fasn and me1 expression relative to controls albeit not significantly (p = .06). Although there were no statistically significant interaction effects on expression of these genes, there were statistically significant main effects of TCE on adipoq (p = .04), leptin (p = .0003), fasn (p = .0004), and me1 (p < .0001). The only significant main effect for HFD was observed for adipoq expression (p = .03). The average area of adipocytes in offspring as a function of adipose tissue growth (Jo et al., 2009) was measured and was not significantly different when compared among the 4 groups (data not shown).
Figure 2.

TCE decreased gene expression related to fatty acid synthesis and insulin resistance. A, Adipose (PMF) and B, liver tissues were collected from female offspring at study terminus. Expression of genes in PMF and liver were measured by qRT-PCR as described in the “Materials and Methods” section. Results are represented in the graphs as mean (SD) fold change. Data were analyzed by 2-way ANOVA to test for significant interaction and main effects between the 2 variables (TCE × HFD). Shown in the graphs are p values for the 6 pairwise Tukey’s post hoc comparisons. Results are statistically significant (*p < .05 and **p < .001).
In addition to adipose tissue, q-RT-PCR was conducted in liver to assess expression of me1 and fasn, which are important biomarkers of metabolic changes associated with obesity in liver. As shown in Figure 2B, unlike that observed in adipose tissue, mean levels of fasn increased significantly by nearly 50% in (TCE−/HFD+) offspring compared with the (TCE−/HFD−) group. Mean expression of fasn decreased in (TCE+/HFD−) groups compared with both (TCE−/HFD+) and (TCE+/HFD+) groups. In contrast, me1 was not only significantly increased by approximately 63% in the HFD-only group; it was also significantly increased in the TCE-alone group (by approximately 50%) and with coexposure (by approximately 52%) compared with (TCE−/HFD−) controls. Although TCE appeared to enhance the expression of this enzyme, mice in the (TCE−/HFD+) group expressed significantly more me1 compared with all TCE-exposed groups, and there was a significant interaction of HFD and TCE (p = .0001). In contrast, for fasn there was not a significant interaction, but both TCE and HFD imparted significant main effects (p = .02 and .0004, respectively). Thus, overall TCE appeared to suppress HFD-induced lipogenic gene expression in adipose tissue and liver.
TCE Decreased HFD-Induced Effects on Hepatic Steatosis and ADFP
Despite changes in liver gene expression, liver weight, when normalized to body weight, was not significantly different among the 4 groups indicating absence of overt liver toxicity (Figure 3A). However, histopathology assessment revealed liver steatosis-like pathology associated with HFD consumption (Figure 3B). When individual pathology scores were compared, 90% of the liver samples from mice fed HFD received a score of 3 compared with only 22% of controls and this difference was statistically significant. TCE exposure alone was associated with little to no steatosis (median pathology score of 0) compared with a median score of 1 in (TCE−/HFD−) controls, an effect that was also significant. Animals coexposed to both TCE and HFD exhibited a significant reduction in steatosis compared with the (TCE−/HFD+) group (median score of 1 vs 3, respectively). As reported by others, small cytoplasmic vacuoles were present in no-treatment control mice, resulting in steatosis scores of >0.
Figure 3.

TCE-inhibited HFD-mediated steatosis-like liver pathology. Liver tissues were collected from female offspring at study terminus. A, Liver weights (g) are presented in graphs (mean [SD]). B, Liver was processed as described in the “Materials and Methods” section and scored to evaluate steatosis and represented as mean (SD). Each plot in the graph represents a liver sample from a single mouse within each group. C, Representative H&E staining of liver samples (100× oil immersion). The red arrow points to fat deposits which are largely absent in TCE groups. Results are statistically significant (*p < .05 and **p < .001).
Representative images of H&E staining from each of the 4 treatment groups revealed significant lipid accumulation in a slide from a mouse in the (TCE−/HFD+) group (Figure 3C). The pattern of the hepatic steatosis appeared to be microvesicular where the cytoplasm is replaced by bubbles of fat that do not displace the nucleus and thus are not characteristic of the more severe form of steatosis characterized by lobular inflammation, hepatocellular ballooning, and fibrosis found in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis (Kleiner, 2017). In contrast, TCE exposure ±HFD indicated little or no fat content.
ADFP, also known as Perilipin-2, is a biomarker of lipid droplet formation, fatty liver, adipocyte differentiation, and steatosis (Carr et al., 2014). There was a modest, yet statistically insignificant increase in ADFP staining of fat droplets from (TCE−/HFD+) group versus (TCE−/HFD−) controls (Figure 4A). Although TCE exposure (either in the presence or absence of HFD) appeared to decrease ADFP staining, this effect was not statistically significant, as there was some variability among samples. There were no significant interactions or main effects of either TCE or HFD. However, considering that 70% of the (TCE+/HFD−) and 67% of the (TCE+/HFD+) liver samples had an ADFP staining value of 0.05 or less compared with (TCE−/HFD−) and (TCE−/HFD+) samples (25% for both groups) suggested that TCE suppressed ADFP staining. Shown are representative images of ADFP staining depicted in the (TCE/HFD+) exposure group (Figure 4B). Thus, similar to effects of HFD-induced lipogenic gene expression, TCE exposure appeared to oppose the effect of HFD on lipid droplet staining and pathology indicative of steatosis.
Figure 4.
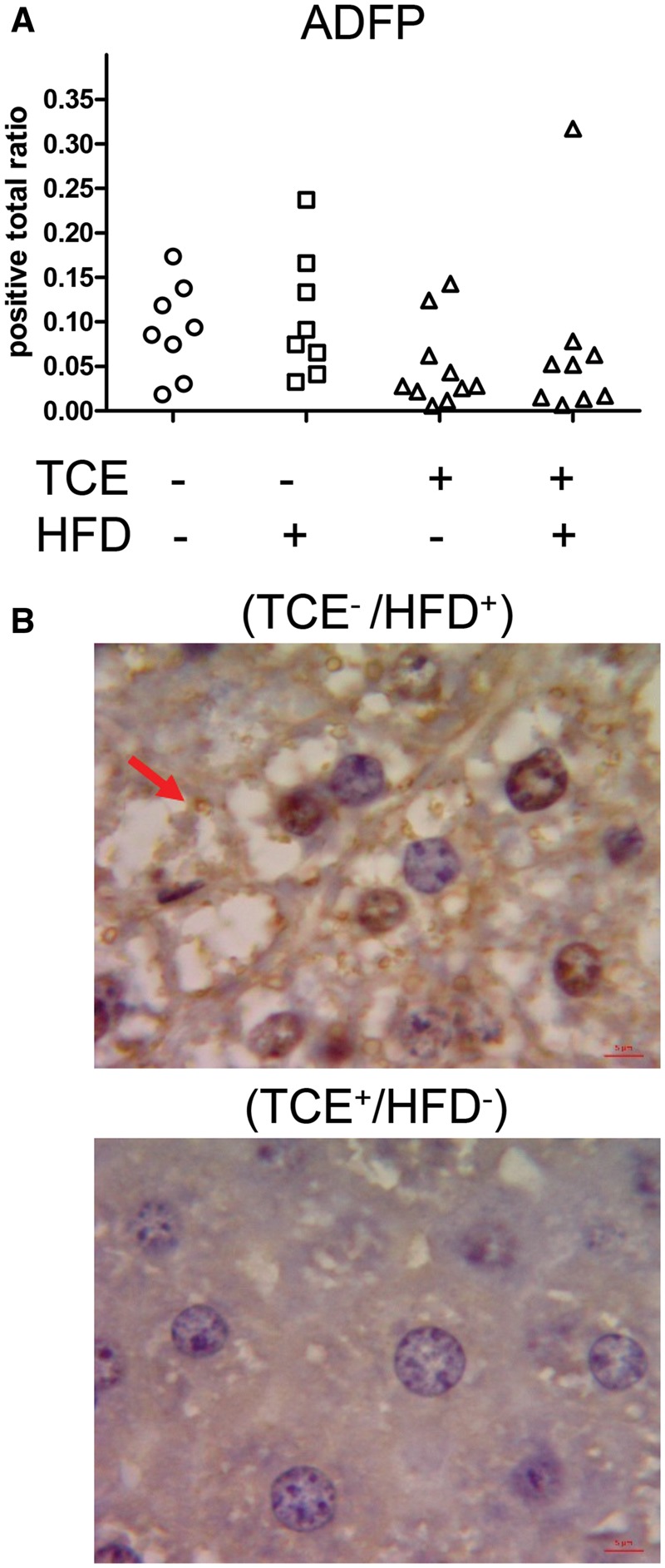
TCE decreased ADFP staining. Liver tissues were collected from female offspring at study terminus and subjected to Immunohistochemical staining of ADFP or Perilipin-2 as described in the “Materials and Methods” section, quantified using Aperio Positive Pixel Count Algorithm (Leica Biosystems) and represented as positive total ratio. A, Each plot in the graph represents a liver sample from a single mouse within each group. B, Representative staining (100× oil immersion). The arrow points to the ADFP-stained fat droplets in the HFD-exposure group which are significantly decreased in the TCE group.
TCE Increased Cytokine and IL-6 Signaling Genes Involved in Inflammation and Liver Repair/Regeneration in Liver
Despite antagonistic effects of TCE and HFD on liver steatosis, H&E histopathological assessment did not reveal significant changes related to AIH-like pathology in the form of lymphoplasmacytic portal infiltrate and lobular inflammation in any of the groups examined (data not shown). Earlier studies have demonstrated that liver AIH-like histopathology does not appear until 26 weeks of exposure in adult animals (Gilbert et al., 2009), and in approximately 37-week-old mice that were exposed to TCE during development (Gilbert et al., 2017a). We have shown later-occurring liver pathology caused by TCE was associated with earlier-occurring hepatic events related to inflammation and repair (Gilbert et al., 2014). As shown in Table 3, TCE exposure (either alone or in combination with HFD) was associated with a significant increase in mRNA of proinflammatory chemokines (C-C motif ligand 2 (ccl2) relative to (TCE−/HFD−) controls. This expression was also significantly increased in coexposed mice compared with the (TCE−/HFD+) and (TCE−/HFD−) groups. As for ccl5 expression, TCE had a similar effect when administered alone, but not in combination with HFD compared with controls. TCE increased ccl5 relative to HFD exposure and, interestingly, a decrease in levels from the coexposed group relative to (TCE+/HFD−) demonstrating HFD’s apparent ability to decrease this chemokine with coexposure. A significant interaction effect was detected for ccl5 (p = .02) but likely not as important as the main effect of TCE exposure (p = .001).
Table 3.
TCE Increases Liver Cytokines/Chemokines
| TCE− | TCE− | TCE+ | TCE+ | (1) | (1) | (1) | (2) | (2) | (3) | |
|---|---|---|---|---|---|---|---|---|---|---|
| HFD− | HFD+ | HFD− | HFD+ | vs | vs | vs | vs | vs | vs | |
| (1) | (2) | (3) | (4) | (2) | (3) | (4) | (3) | (4) | (4) | |
| ccl2 | 0.96 (0.5) | 1.3 (0.7) | 2.5 (1.2) | 3.4 (1.3) | 0.9 | 0.01 | <0.001 | 0.06 | 0.001 | 0.3 |
| ccl5 | 1.1 (0.4) | 1.1 (0.7) | 4.1 (1.8) | 2.1 (0.8) | 1.0 | <0.001 | 0.3 | <0.001 | 0.3 | 0.008 |
| tnfa | 0.88 (0.3) | 0.45 (0.2) | 1.5 (0.4) | 0.94 (0.3) | 0.04 | 0.003 | 0.9 | <0.001 | 0.03 | 0.01 |
| Il1b | 0.95 (0.5) | 1.2 (0.8) | 2.1 (0.8) | 1.5 (0.4) | 0.9 | 0.008 | 0.3 | 0.07 | 0.8 | 0.3 |
| tgfb | 1.0 (0.4) | 1.7 (0.9) | 2.5 (0.7) | 2.3 (1.1) | 0.1 | 0.001 | 0.03 | 0.5 | 0.3 | 0.7 |
Liver tissue from female offspring was harvested and flash frozen at study terminus. RNA was isolated and converted to cDNA for qRT-PCR as described in the “Materials and Methods” section. Fold change values for each gene relative to EEF2 housekeeping gene are represented in the table. Numbers represent mean and SD of the 4 exposure groups. Data were analyzed by 2-way ANOVA to test for significant interaction and main effects between the 2 variables (TCE × HFD). Shown in the table are p values for the 6 pairwise Tukey’s post hoc comparisons numbered by group and conducted as described in the “Materials and Methods” section. Results are statistically significant (p < .05) indicated in bold.
Cytokines, tnfa, il1b, and tgfb were also similarly increased by TCE exposure, but not in the context of coexposure relative to controls except for tgfb. HFD exposure alone significantly decreased tnfa relative to controls directly opposing TCE’s effects. There were no statistically significant interactions among these cytokines. However, significant main effects of TCE were observed for ccl2 (p < .001), tnfa (p < .001), il1b (p = .004), and tgfb (p = .001) whereas significant main effects for HFD were only observed for tnfa (p = .0002).
IL-6 signaling has been shown to be important in liver regeneration following damage (Tao et al., 2017). The IL-6 signaling complex consists of IL-6 receptor, glycoprotein (gp) 130, a transmembrane signaling protein, and Egr-1. Based on previous studies of altered IL-6 signaling with TCE exposure (Gilbert et al., 2014) mRNA expression of il6r, gp130, and egr1 were evaluated by q-RT-PCR. As shown in Figure 5, TCE exposure increased expression of IL-6 signaling complex genes while the HFD groups had little effect even in the context of coexposure. There was a statistically significant interaction effect with egr1 and il6ra expression (p = .03 and .04) but with a more significant main effect with TCE exposure (p < .001 for both genes). There was also a significant main effect of TCE, but not HFD, with gp130 (p < .001) implying that TCE impacts most IL-6 signaling complex components dependent upon the level of HFD.
Figure 5.

TCE enhances IL-6 signaling in liver. Expression of liver IL-6 signaling components was measured by qRT-PCR as described in the “Materials and Methods” section. Results are represented in the graphs as mean (SD) fold change. Data were analyzed by 2-way ANOVA to test for significant interaction and main effects between the 2 variables (TCE × HFD). Shown in the graphs are p values for the 6 pairwise Tukey’s post hoc comparisons. Results are statistically significant (*p < .05 and **p < .001).
TCE and HFD Imparted Distinct Antiliver Antibody Profiles in the Offspring
Although we reported that higher concentrations of TCE can increase levels of antinuclear antibodies (antissDNA) early in the exposure, later effects of TCE on ANA levels can be difficult to detect as baseline levels of these autoantibodies increase spontaneously in female MRL+/+ mouse as they age (Griffin et al., 2000). In this study, early signs of autoimmunity in the form of antiliver antibodies were found in the coexposed offspring to a higher degree than other groups (Figure 6) despite the finding that the relatively low levels of TCE were not sufficient to increase antissDNA levels beyond their already high baseline levels in the MRL+/+ mice (data not shown). Thus, although the relatively short-term and low-level coexposure to TCE and HFD did not generate overt AIH-like autoimmune tissue pathology or generalized serological signs of autoimmunity, the combined treatment did trigger early indications of autoimmunity directed towards liver.
Figure 6.

HFD/TCE coexposure increased antiliver antibody production. Sera from mice in each treatment group collected at study terminus were pooled (sera from 8 mice/group) and reacted with liver proteins separated by SDS-PAGE. A, representative blot showing the staining pattern. B, The blots from the pooled samples were repeated 3 times. Shown are densitometric analysis (average of 3 runs) of total antiliver antibodies normalized to mouse myeloma IgG run in adjoining lanes (not shown).
TCE Promoted CD4+ T Cell Proinflammatory Cytokine Profile in Splenic CD4+ T Cells
Previous evaluations of TCE exposure detected numerous alterations in splenic CD4+ T cells that preceded and/or accompanied liver pathology. The CD4+ T cells isolated from the 4 groups of offspring were activated in vitro, and examined for alterations in the upregulation of cytokine mRNA as a marker of peripheral inflammation. When samples from each of the 4 separate groups were compared, the coexposure significantly increased the expression of ifng approximately 2-fold and il17 approximately 5-fold relative to (TCE−/HFD−) controls (Table 4). Mean Ifng and il17 expression from the (TCE+/HFD−) group increased approximately 2-fold relative to controls, albeit not significantly. In contrast, HFD significantly decreased mean il17 levels relative to the coexposure group by approximately 6 fold. Likewise, HFD consumption significantly decreased il4 expression relative to controls by approximately 3-fold, and this effect carried over to the coexposure group with a approximately 6-fold decrease relative to controls. In contrast, unlike what was observed in liver tissue, expression of the gene that encodes TNF-α (tnfa) was not significantly different among the groups even though levels of tnfa increased in all groups relative to (TCE−/HFD−) group, but not significantly. Although our analysis did not reveal significant interaction effects, main effects of TCE treatment of ifng (p = .003) and il17 (p = .005) were detected. In contrast, main effects of HFD were found with il4 expression (p = .005). The results suggested that HFD and TCE had opposing effects on CD4+ T cell cytokine mRNA expression. Despite these functional effects, splenic cellular composition of CD4+ T cells and other lymphocyte populations was not significantly different among the treatment groups (data not shown).
Table 4.
TCE Promoted Th1/Th17 Phenotype
| TCE− | TCE− | TCE+ | TCE+ | (1) | (1) | (1) | (2) | (2) | (3) | |
|---|---|---|---|---|---|---|---|---|---|---|
| HFD− | HFD+ | HFD− | HFD+ | vs | vs | vs | vs | vs | vs | |
| (1) | (2) | (3) | (4) | (2) | (3) | (4) | (3) | (4) | (4) | |
| ifng | 33.2 (13.0) | 43.4 (19.8) | 65.8 (34.5) | 78.6 (11.9) | 0.8 | 0.08 | 0.02 | 0.4 | 0.1 | 0.8 |
| Il17 | 21.7 (14.1) | 17.5 (15.4) | 41.8 (26.0) | 111 (77.9) | 0.9 | 0.8 | 0.008 | 0.8 | 0.01 | 0.06 |
| Il4 | 692.6(1441.3) | 329.3 (507.9) | 799.6 (543.2) | 227.2 (189.7) | 0.03 | 0.2 | 0.008 | 0.8 | 0.9 | 0.4 |
| tnfa | 4.1 (1.8) | 26.7 (28.2) | 12.1 (6.1) | 15.4 (7.3) | 0.07 | 0.8 | 0.7 | 0.4 | 0.7 | 0.9 |
Splenic CD4+ T cells were isolated and activated as described in the “Materials and Methods” section. Cells were harvested and processed for RNA and cytokine expression was measured by qRT-PCR. Numbers represent mean and SD fold change values relative to unstimulated CD4+ T cells and normalized by EEF2 housekeeping gene. Data were analyzed by 2-way ANOVA to test for interaction and main effects of the 2 variables (TCE and HFD). Numbers represent mean and SD of the 4 exposure groups (1–4). Data were analyzed by 2-way ANOVA to test for significant interaction of the 2 variables (TCE × HFD). Shown in the table are p values for the 6 pairwise Tukey’s post hoc comparisons numbered by group and conducted as described in the “Materials and Methods” section. Results are statistically significant (p < .05) indicated in bold.
TCE and HFD Mediate Opposing Effects on Gut Mucosal Cytokines and Microbial Abundance
In view of the increasingly well-documented connection between the gut and the immune system, the levels of cytokines and microbial composition of the ileal region of the small intestine were examined. Peyer’s patches reside in this region of the gut, and its intestinal bacterial population most closely associated with the immune system function including inflammation (Geuking et al., 2014; Hashiguchi et al., 2015; Teng et al., 2016). A panel of cytokines was evaluated, and some either with statistically significant changes or others that were altered in liver or CD4+ T cells related to treatment are presented in Figure 7. Most alterations in gut cytokines were found in offspring that had been exposed to TCE, either alone or in the context of HFD coexposure. This included a statistically significant decrease in IL-1β, IL-3, GM-CSF, eotaxin, IL-6, and IFN-γ compared with (TCE−/HFD−) controls. The decline in IL-1α was unique to TCE-only exposure, and the decrease in IL-6 was found with coexposure relative to (TCE−/HFD−) controls. Although TCE facilitated an overall suppression of cytokines, the HFD-only group promoted increased levels of IL-1α, IL-3, GM-CSF, and IFN-γ relative to (TCE+/HFD−) and/or (TCE+/HFD+). IL-17, TNF-α, CCl2, CCl5, and IL-13, a cytokine that is similar in function to IL-4, were not statistically different. There were a few significant interaction effects including IL-3 (p = .009), GM-CSF (p = .008), and IFN-γ (p = .04), but these effects did not appear to be as important as the significant main effects of TCE (p < .001 for all genes). Significant main effects of TCE were also revealed for IL-1α (p = .002), IL-1β (p = .0005), eotaxin (p = .004), IL-6 (p = .02), and TNF-α (p = .007). Significant main effects of HFD were observed for CCL2 (p = .008). Thus, this result demonstrated a seemingly predominant effect of TCE exposure on decreasing gut cytokines relative to HFD exposure.
Figure 7.

TCE exposure decreased levels of gut cytokines. Ileal tissue was harvested from offspring at study terminus, protein was extracted, and multiplex cytokine assay was conducted as described in the “Materials and Methods” section. Means (SD) are represented in the graphs (ng/ml). Data were analyzed by 2-way ANOVA to test for significant interaction and main effects between the 2 variables (TCE × HFD). Shown in the graphs are p values for the 6 pairwise Tukey’s post hoc comparisons. Results are statistically significant (*p < .05 and **p < .001).
The connection between gut cytokines and peripheral immune responses is at least partially related to changes in the gut microbiome. Thus, the mucosa-associated bacteria in the ileal region were examined. The results showed a large intermouse variation and our statistical analysis did not reveal significant differences among the bacterial species (Figure 8A) Based on the downregulation of the mean of universal bacteria by the 2 TCE groups and a significant main effect (p = .05) with TCE exposure alone, we conducted a different comparison that can be viewed as another post hoc test, where the combined mean of the 2 TCE groups (TCE+/HFD−) and (TCE+/HFD+) was compared with the 2 non-TCE groups (TCE−/HFD− and TCE−/HFD+). Likewise, the combined means were also compared between the 2 HFD groups (TCE−/HFD±) and (TCE+/HFD+) and non-TCE groups (TCE−/HFD−) and (TCE+/HFD−). This comparison of the relative number of total bacteria (as measured by expression of universal 16s rRNA) was considerably decreased when the combined expression of the TCE groups was compared with the combined expression of the nonTCE groups (Figure 8B). TCE exposure, regardless of diet, also increased the ratio of firmicutes to bacteroidetes compared with controls and mice that received high fat alone (Figure 8C). Taken together, the alterations observed in the gut microbiome were modest but appeared to be mediated by TCE exposure with little influence of the diet.
Figure 8.
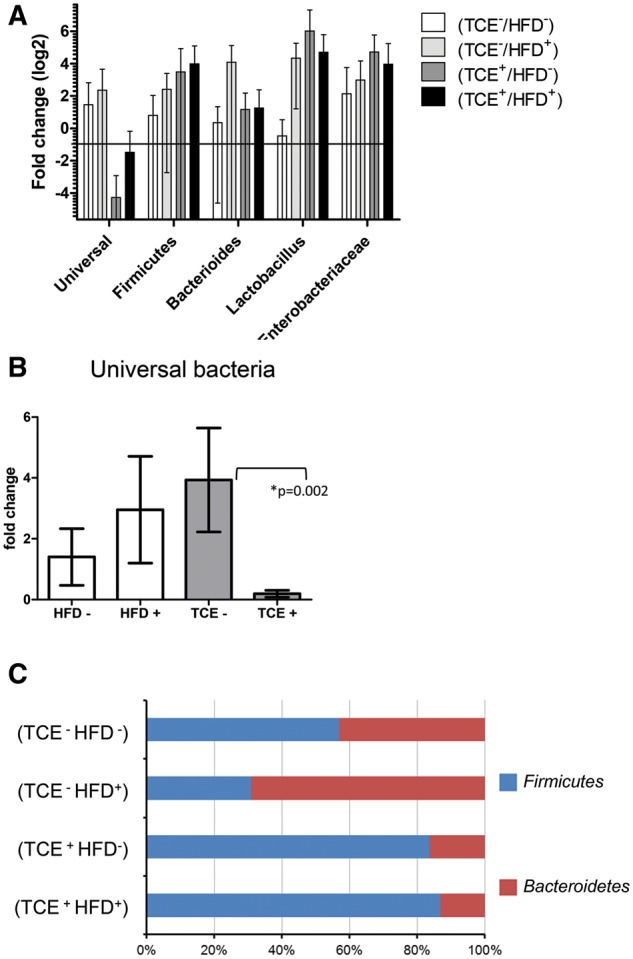
TCE altered the composition of the gut microbiome. Genomic DNA from the ileal tissue was extracted and real-time PCR was used to assess the cT value of each sample. β-actin was used as a housekeeping gene. A, Mean (SD) fold change (log2) values for bacterial species of ileal region of the gut. Data were normalized with β-actin. The results show the expression of genes representing entire bacterial population (Universal), 2 phylum (Bacteroidetes and Firmicutes), predominant genera (Bacteroides, Lactobacillus and Bifidobacterium) and Enterobacteria family. B, The data represent the mean (SD) fold change universal bacteria comparing all HFD-fed mice versus mice that received the control diet (HFD− vs. HFD +) and all TCE exposed mice versus mice that received vehicle in the drinking water (TCE − vs. TCE +). C, TCE exposure increased the ratio of firmicutes to bacteroidetes compared with controls and mice that received HFD. The results of the predominant phyla are represented as 100% stack bar diagram, in which each assessed phyla shows the contribution to the sum of the 2 predominant bacterial phyla.
DISCUSSION
It was predicted that the combination of 2 proinflammatory stressors consisting of TCE and HFD administered during a critical time of development would have synergistic effects on a number of mediators of adiposity, immune function, autoimmunity, inflammation, and the microbiome over either treatment alone. In contrast to our expectations, many effects mediated by either exposure opposed one another, and appeared to antagonize the expected effects of either stressor. For instance, most of the obesity-related endpoints that were associated with known effects of HFD were seemingly reversed by TCE exposure. TCE decreased leptin, fasn, and me1 in adipose tissue relative to animals fed HFD. This effect carried over to me1 expression in liver tissue as well. Perhaps the most striking results of the study occurred when TCE reversed HFD-induced microvesicular hepatic steatosis-like pathology. Although this type of steatosis is considered to be more benign than the macrovesicular type, altered lipogenic gene expression and ADFP staining results by TCE exposure are suggestive of altered lipogenesis or lipid catabolism with TCE with the possibility of reduced partitioning/handling of the lipids by the liver which could result in a less apparent steatosis. Regardless, the results demonstrate an antagonism of HFD-mediated effects by TCE.
Although numerous studies of obesity and/or HFD on adiposity endpoints have been conducted, little is known how TCE impacts these effects. Studies examining some of these factors were conducted in adult rodents at very high doses after an acute exposure. For instance, ME1 levels were altered in kidney of rats given a high dose of TCE (1000 mg/kg/day) for 25 days (Khan et al., 2009). Ramdhan et al. (2010) examined the role of PPARα, a nuclear receptor important in fatty acid transport and β-oxidation, in TCE-induced liver steatosis. In this study, transgenic PPARα-humanized mice exposed to 2000 ppm TCE-exhibited increased steatosis relative to unexposed mice in both PPARα null and wildtype mice. The implications of these studies are not clear since the TCE dose in both studies were systemically toxic and not relevant to either human occupational or environmental TCE exposure.
The TCE concentration of 0.05 μg/ml (approximately 50 ppb) used in this study was considerably low, and should yield a total mouse exposure approximating human environmental exposure. The calculated TCE consumption in the pups averaged under 10 μg/kg/day. To put this dose into perspective, the 8-h permissible exposure limit for occupational exposure to TCE has been established by the Occupational Safety and Health Administration at 76 mg/kg/day. Although the dose used in the current study is higher than the maximum contaminant level (MCL) for TCE in municipal water supplies (5 ppb or 0.005 or 0.001 μg/ml for some states), it is not outside the realm of actual human environmental exposure to TCE. Cumulative exposure can be increased when environmental levels of TCE are above the MCL. For example, TCE has been detected in California drinking water sources with an average concentration up to 0.020 μg/ml in the contaminated samples (Williams et al., 2002). One could estimate that a cumulative exposure from ingestion, inhalation, and dermal contact could encompass around 120 μg/day or approximately 10.9 μg/kg/day for a toddler of average body weight and 1.7 μg/kg/day for an average body weight adult. Levels could be even higher if calculations were based on the maximum rather than average exposure levels.
Other significant findings included serological signs of autoimmunity in the form of antiliver antibodies. Tissue pathology that is associated with AIH in humans is often accompanied by the development of autoantibodies that recognize liver proteins. The generation of antiliver antibodies in the control mice, despite their well-known ability to generate ANA later in life, was minimal. Yet, similar to patients with AIH, a diverse autoantibody profile was detected in mice coexposed to TCE and HFD. The remaining groups of mice had different profiles. Most notably, a detectable band in the TCE only group at about 15 kda was absent in other groups. We recently reported that developmental exposure to a similar dose of TCE followed by a period of cessation promoted antiliver antibodies commensurate with AIH relative to control mice (Gilbert et al., 2017a). It is not clear why the result was not recapitulated here. However, the mice were several weeks younger in the current study. Thus, it is possible that with advancing age more profound autoimmune pathology would have manifested in the mice.
It is not obvious from the antiliver antibody profile that the effects are solely attributable to TCE. Little is known about the effects of HFD on AIH. One recent study in mice demonstrated that diet-induced obesity (DIO) increased the severity of AIH but with no effect on T cell responses (Gaur et al., 2017). HFD increased disease symptoms in a mouse model of experimental autoimmune encephalomyelitis (Hasan et al., 2017) and directly increased autoantibody production in C57BL/6 mice (Şelli et al., 2017). Although these studies underscore the importance of obesity/overnutrition in autoimmunity, no studies in the literature exist in HFD-fed autoimmune-prone mice after developmental exposure.
The alterations in biomarkers of liver inflammation and regeneration/repair genes appeared to be attributable to TCE, rather than HFD consumption. TCE significantly increased mRNA for ccl2 and ccl5, and HFD appeared to oppose these effects. Although the importance of this finding is not yet known, CCL2, also known as MCP-1 recruits immune cells to various organs including the liver, and induces proinflammatory cytokines at the site of tissue injury. CCL2 has been detected in sera in patients with AIH (Li et al., 2013). Importantly, CCL2 directly inhibited PPARα induction and impeded fatty acid oxidation in a model of alcoholic liver injury (Mandrekar et al., 2011) This finding may explain the mitigating effect of TCE on HFD-regulated gene expression and steatosis. Likewise, CCL5, or RANTES, was increased with TCE exposure. This chemokine is similar to CCL2 functionally (Lee et al., 2017), and may also be important in human AIH (Czaja, 2014). A corresponding increase in liver IL-1β expression with TCE exposure was observed with no effect of diet. Similarly, mRNA expression for TGF-β, which plays an important role in the progression of liver disease, was also elevated with TCE exposure (Dooley and ten Dijke, 2012). Interestingly, while TNF-α increased with TCE exposure, this proinflammatory cytokine decreased in HFD-consuming groups in liver and appeared to mitigate the effect of TCE. The effect of HFD on TNF-α carried over to CD4+ T cells where it increased rather than decreased the expression of this cytokine. Thus, HFD appeared to modulate the TNF-α response in our model. TNF-α is elevated in human obesity, and increased levels of leptin in obese individuals promotes the generation of proinflammatory cytokines such as TNF-α, IL-12, and IL-6 (Perez-Perez et al., 2017).
Liver IL-6 signaling components involved in tissue regeneration and repair increased with TCE exposure. HFD appeared to negate this effect. Like CCL2, the opposing effects of TCE on the adiposity endpoints (e.g., steatosis) could be explained by TCE’s effect in liver IL-6 signaling and inflammation. IL-6 related signaling can protect against the progression of hepatic steatosis, and blockade of IL-6 signaling ameliorated insulin resistance and adipokine levels in HFD-fed mice (Yamaguchi et al., 2015). This finding implies that, together, the increase in liver inflammation and repair markers may play a role in the TCE-mediated reversal of HFD-induced effects. In terms of AIH, it may be that a loss of this regenerative capacity may play a role in AIH progression, ultimately leads to actual disease in the mice with prolonged TCE exposure.
Based on the important role of the microbiome in DIO and immune function, we assessed the microbiome in all exposure groups. Despite some variability, clear patterns emerged, and TCE, unlike HFD-fed animals, appeared to significantly reduce the total number of bacteria and alter the ratio of some of the phyla. The reason for this effect is not clear since HFD exposure, obesity and dietary alterations can alter microbial composition. Distinct populations of intestinal microbiota can indirectly participate in the onset of obesity initiating low-grade inflammation, as well as fat deposition through metabolism of short chain fatty acids (Boulange et al., 2016). Future studies are needed to better understand the role of these exposures on the microbiome and its correlation with the short chain fatty acid metabolites. With regard to the liver, increased microbial abundance may alter steatosis and insulin resistance as shown in germ-free C57BL/6 mice administered HFD (Rabot et al., 2010). Germ-free mice were found to gain less weight than conventional mice, and colonization of these mice with microbiota from conventional mice resulted in replenished weight gain (Backhed et al., 2007) Thus, the finding that TCE basically reversed HFD-induced steatosis may also be linked with the microbiome. Especially relevant is that the consistent TCE-mediated decrease in gut cytokines is in line with what would be expected with decreased microbial abundance. Thus, several mechanisms may be involved in TCE’s ability to oppose HFD-mediated effects in our model.
Limitations include the baseline adiposity of the MRL+/+ mouse strain. The MRL mouse with its genetic propensity towards autoimmunity has been useful in understanding effects of environment on autoimmune disease. However, the MRL mouse itself already weighs approximately 10 g more than age-equivalent C57BL/6 mice, and the fat mass of the MRL mouse is almost 40% higher than C57BL/6 (Srivastava et al., 2006). Also noteworthy is the finding that MRL mice are resistant to metabolic changes and hyperglycemia related to HFD (60% kcal fat; Mull et al., 2014). It is possible that these baseline issues masked significant coexposure induced changes. Thus, while C57BL/6, the standard model for DIO (Wang and Liao, 2012) might be more sensitive model to evaluate HFD-related metabolic issues, it was necessary to conduct this assessment of autoimmune-promoting ability of TCE/HFD in MRL+/+ mice.
This study has important consequences for human health. Both TCE and HFD imparted significant changes in adult offspring when exposures were applied during critical windows of development. Notably, these changes were observed approximately 17 weeks after the exposures were removed. Persistence of developmental programming effects have been reported for both TCE (Gilbert et al., 2017a; Blossom et al., 2017), and HFD (Lecoutre et al., 2017; Segovia et al., 2017) when administered alone. As for developmental coexposure, epigenetic mechanisms were found to be perturbed in a model of HFD and bisphenol A coexposure (Kochmanski et al., 2017) underscoring the potential importance of epigenetic effect as a mechanistic consideration. Programming effects can be observed in models of obesity/overnutrition for several endpoints including immune function, obesity, and growth in offspring (Johnson et al., 2017; Krasnow et al., 2011; Saad et al., 2016; Sferruzzi-Perri et al., 2013; Xie et al., 2017). TCE exposure has also been shown to modulate the epigenome in immune cell subpopulations (Gilbert et al., 2016a,b, 2017b). This study presents novel findings of several effects of developmental TCE and HFD coexposure in female MRL+/+ mice. Future research will explore mechanisms to understand how these long-lasting effects are maintained and how the individual stressors oppose one another.
FUNDING
This work was supported by grants from the Arkansas Bioscience Institute at the Arkansas Children’s Research Institute (ACRI) and the National Institutes of Health (NIEHS-1K02ES024387).
ACKNOWLEDGMENTS
We thank Keen Maher and Kanan Vyas at ACRI and Katherine Williams at the National Center for Toxicological Research for excellent technical assistance. The findings/opinions presented here represent the views of the authors. They do not reflect the views of the U.S. Food and Drug Administration.
REFERENCES
- Al-Dwairi A., Brown A. R., Pabona J. M., Van T. H., Hamdan H., Mercado C. P., Quick C. M., Wight P. A., Simmen R. C., Simmen F. A. (2014). Enhanced gastrointestinal expression of cytosolic malic enzyme (ME1) induces intestinal and liver lipogenic gene expression and intestinal cell proliferation in mice. PLoS One 9, e113058.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dwairi A., Pabona J. M., Simmen R. C., Simmen F. A. (2012). Cytosolic malic enzyme 1 (ME1) mediates high fat diet-induced adiposity, endocrine profile, and gastrointestinal tract proliferation-associated biomarkers in male mice. PLoS One 7, e46716.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F., Manchester J. K., Semenkovich C. F., Gordon J. I. (2007). Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. U.S.A. 104, 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batun-Garrido J. A. J., Salas-Magana M., Juarez-Rojop I. E., Hernandez-Nunez E., Olan F. (2017). Association between leptin and disease activity in patients with rheumatoid arthritis. Med. Clin. 26, 30819–30819. [DOI] [PubMed] [Google Scholar]
- Belkaid Y., Hand T. W. (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blossom S. J., Doss J. C. (2007). Trichloroethylene alters central and peripheral immune function in autoimmune-prone MRL(+/+) mice following continuous developmental and early life exposure. J. Immunotoxicol. 4, 129–141. [DOI] [PubMed] [Google Scholar]
- Blossom S. J., Doss J. C., Gilbert K. M. (2006). Chronic exposure to a trichloroethylene metabolite in autoimmune-prone MRL+/+ mice promotes immune modulation and alopecia. Toxicol. Sci. 95, 401–411. [DOI] [PubMed] [Google Scholar]
- Blossom S. J., Melnyk S. B., Li M., Wessinger W. D., Cooney C. A. (2017). Inflammatory and oxidative stress-related effects associated with neurotoxicity are maintained after exclusively prenatal trichloroethylene exposure. Neurotoxicology 59, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulange C. L., Neves A. L., Chilloux J., Nicholson J. K., Dumas M. E. (2016). Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 8, 016–0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove R., Musallam A., Xia Z., Baruch N., Messina S., Healy B. C., Chitnis T. (2016). Longitudinal BMI trajectories in multiple sclerosis: Sex differences in association with disease severity. Mult. Scler. Relat. Disord. 8, 136–140. [DOI] [PubMed] [Google Scholar]
- Brusca S. B., Abramson S. B., Scher J. U. (2014). Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr. Opin. Rheumatol. 26, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr R. M., Peralta G., Yin X., Ahima R. S. (2014). Absence of perilipin 2 prevents hepatic steatosis, glucose intolerance and ceramide accumulation in alcohol-fed mice. PLoS One 9, e97118.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S., Danzaki K., MacIver N. J. (2017). Nutritional effects on T-cell immunometabolism. Eur. J. Immunol. 47, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G. S., Makris S. L., Nietert P. J., Jinot J. (2009). Evidence of autoimmune-related effects of trichloroethylene exposure from studies in mice and humans. Environ. Health Perspect. 117, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaja A. J. (2014). Review article: chemokines as orchestrators of autoimmune hepatitis and potential therapeutic targets. Aliment Pharmacol. Ther. 40, 261–279. [DOI] [PubMed] [Google Scholar]
- Dinger K., Kasper P., Hucklenbruch-Rother E., Vohlen C., Jobst E., Janoschek R., Bae-Gartz I., van Koningsbruggen-Rietschel S., Plank C., Dotsch J. (2016). Early-onset obesity dysregulates pulmonary adipocytokine/insulin signaling and induces asthma-like disease in mice. Sci. Rep. 6, 24168.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley S., ten Dijke P. (2012). TGF-beta in progression of liver disease. Cell Tissue Res. 347, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gabalawy H., Guenther L. C., Bernstein C. N. (2010). Epidemiology of immune-mediated inflammatory diseases: Incidence, prevalence, natural history, and comorbidities. J. Rheumatol. Suppl. 85, 2–10. [DOI] [PubMed] [Google Scholar]
- Forno E., Young O. M., Kumar R., Simhan H., Celedon J. C. (2014). Maternal obesity in pregnancy, gestational weight gain, and risk of childhood asthma. Pediatrics 134, e535–e546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur P., Cebula M., Riehn M., Hillebrand U., Hauser H., Wirth D. (2017). Diet induced obesity has an influence on intrahepatic T cell responses. Metabolism 69, 171–176. [DOI] [PubMed] [Google Scholar]
- Geuking M. B., Koller Y., Rupp S., McCoy K. D. (2014). The interplay between the gut microbiota and the immune system. Gut Microbes 5, 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert K. M., Bai S., Barnette D., Blossom S. J. (2017a). Exposure cessation during adulthood did not prevent immunotoxicity caused by developmental exposure to low-level trichloroethylene in drinking water. Toxicol. Sci. 157, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert K. M., Blossom S. J., Erickson S. W., Broadfoot B., West K., Bai S., Li J., Cooney C. A. (2016a). Chronic exposure to trichloroethylene increases DNA methylation of the Ifng promoter in CD4+ T cells. Toxicol. Lett. 260, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert K. M., Blossom S. J., Erickson S. W., Reisfeld B., Zurlinden T. J., Broadfoot B., West K., Bai S., Cooney C. A. (2016b). Chronic exposure to water pollutant trichloroethylene increased epigenetic drift in CD4(+) T cells. Epigenomics 8, 633–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert K. M., Blossom S. J., Reisfeld B., Erickson S. W., Vyas K., Maher M., Broadfoot B., West K., Bai S., Cooney C. A. (2017b). Trichloroethylene-induced alterations in DNA methylation were enriched in polycomb protein binding sites in effector/memory CD4(+) T cells. Environ. Epigenet. 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert K. M., Nelson A. R., Cooney C. A., Reisfeld B., Blossom S. J. (2012). Epigenetic alterations may regulate temporary reversal of CD4(+) T cell activation caused by trichloroethylene exposure. Toxicol. Sci. 127, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert K. M., Przybyla B., Pumford N. R., Han T., Fuscoe J., Schnackenberg L. K., Holland R. D., Doss J. C., Macmillan-Crow L. A., Blossom S. J. (2009). Delineating liver events in trichloroethylene-induced autoimmune hepatitis. Chem. Res. Toxicol. 22, 626–632. [DOI] [PubMed] [Google Scholar]
- Gilbert K. M., Reisfeld B., Zurlinden T. J., Kreps M. N., Erickson S. W., Blossom S. J. (2014). Modeling toxicodynamic effects of trichloroethylene on liver in mouse model of autoimmune hepatitis. Toxicol. Appl. Pharmacol. 279, 284–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokulan K., Khare S., Williams K., Foley S. L. (2016). Transmissible plasmid containing salmonella enterica heidelberg isolates modulate cytokine production during early stage of interaction with intestinal epithelial cells. DNA Cell Biol. 35, 443–453. [DOI] [PubMed] [Google Scholar]
- Griffin J. M., Blossom S. J., Jackson S. K., Gilbert K. M., Pumford N. R. (2000). Trichloroethylene accelerates an autoimmune response by Th1 T cell activation in MRL +/+ mice. Immunopharmacology 46, 123–137. [DOI] [PubMed] [Google Scholar]
- Hasan M., Seo J. E., Rahaman K. A., Min H., Kim K. H., Park J. H., Sung C., Son J., Kang M. J., Jung B. H. (2017). Novel genes in brain tissues of EAE-induced normal and obese mice: Upregulation of metal ion-binding protein genes in obese-EAE mice. Neuroscience 343, 322–336. [DOI] [PubMed] [Google Scholar]
- Hashiguchi M., Kashiwakura Y., Kojima H., Kobayashi A., Kanno Y., Kobata T. (2015). Peyer’s patch innate lymphoid cells regulate commensal bacteria expansion. Immunol. Lett. 165, 1–9. [DOI] [PubMed] [Google Scholar]
- Huang Y., Xia L., Wu Q., Zeng Z., Huang Z., Zhou S., Jin J., Huang H. (2015). Trichloroethylene hypersensitivity syndrome is potentially mediated through its metabolite chloral hydrate. PLoS One 10, e0127101.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J., Gavrilova O., Pack S., Jou W., Mullen S., Sumner A. E., Cushman S. W., Periwal V. (2009). Hypertrophy and/or hyperplasia: Dynamics of adipose tissue growth. PLoS Comput. Biol. 5, e1000324.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. A., Javurek A. B., Painter M. S., Murphy C. R., Conard C. M., Gant K. L., Howald E. C., Ellersieck M. R., Wiedmeyer C. E., Vieira-Potter V. J. et al. , . (2017). Effects of a maternal high-fat diet on offspring behavioral and metabolic parameters in a rodent model. J. Dev. Orig. Health Dis. 8, 75–88. [DOI] [PubMed] [Google Scholar]
- Kappelman M. D., Galanko J. A., Porter C. Q., Sandler R. S. (2011). Association of paediatric inflammatory bowel disease with other immune-mediated diseases. Arch. Dis. Child 96, 1042–1046. [DOI] [PubMed] [Google Scholar]
- Khan S., Priyamvada S., Khan S. A., Khan W., Farooq N., Khan F., Yusufi A. N. (2009). Effect of trichloroethylene (TCE) toxicity on the enzymes of carbohydrate metabolism, brush border membrane and oxidative stress in kidney and other rat tissues. Food Chem. Toxicol. 47, 1562–1568. [DOI] [PubMed] [Google Scholar]
- Kleiner D. (2017). Histopathology, grading and staging of nonalcoholic fatty liver disease. Minerva Gastroenterol. Dietol. 25, 02445–0244X. [DOI] [PubMed] [Google Scholar]
- Kochmanski J., Marchlewicz E. H., Savidge M., Montrose L., Faulk C., Dolinoy D. C. (2017). Longitudinal effects of developmental bisphenol A and variable diet exposures on epigenetic drift in mice. Reprod. Toxicol. 68, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnow S. M., Nguyen M. L., Marks D. L. (2011). Increased maternal fat consumption during pregnancy alters body composition in neonatal mice. Am. J. Physiol. Endocrinol. Metab. 301, E1243.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoutre S., Oger F., Pourpe C., Butruille L., Marousez L., Dickes-Coopman A., Laborie C., Guinez C., Lesage J., Vieau D. et al. , . (2017). Maternal obesity programs increased leptin gene expression in rat male offspring via epigenetic modifications in a depot-specific manner. Mol. Metab. 6, 922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. M., Peng H. H., Yang P., Liou J. T., Liao C. C., Day Y. J. (2017). C-C Chemokine Ligand-5 is critical for facilitating macrophage infiltration in the early phase of liver ischemia/reperfusion injury. Sci. Rep. 7, 017–03956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leijs M. M., Koppe J. G., Olie K., van Aalderen W. M., de Voogt P., ten Tusscher G. W. (2009). Effects of dioxins, PCBs, and PBDEs on immunology and hematology in adolescents. Environ. Sci. Technol. 43, 7946–7951. [DOI] [PubMed] [Google Scholar]
- Li Y. L., Liu N., Zhao D. T., Li Z. M., Zhang H. P., Liu Y. M., Yan H. P., Zhao Y. (2013). Investigate circulating levels of chemokines and evaluate the correlation between these chemokines and liver function indicators in autoimmune hepatitis. Zhonghua Gan Zang Bing Za Zhi 21, 299–303. [DOI] [PubMed] [Google Scholar]
- Mandrekar P., Ambade A., Lim A., Szabo G., Catalano D. (2011). An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology 54, 2185–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mull A. J., Berhanu T. K., Roberts N. W., Heydemann A. (2014). The Murphy Roths Large (MRL) mouse strain is naturally resistant to high fat diet-induced hyperglycemia. Metabolism 63, 1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden C. L., Carroll M. D., Fryar C. D., Flegal K. M. (2015). Prevalence of obesity among adults and youth: United States, 2011-2014. NCHS Data Brief219, 1–8. [PubMed]
- Parks C. G., De Roos A. J. (2014). Pesticides, chemical and industrial exposures in relation to systemic lupus erythematosus. Lupus 23, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira T. C., Saron M. L., Carvalho W. A., Vilela M. M., Hoehr N. F., Hessel G. (2011). Research on zinc blood levels and nutritional status in adolescents with autoimmune hepatitis. Arq Gastroenterol. 48, 62–65. [DOI] [PubMed] [Google Scholar]
- Perez-Perez A., Vilarino-Garcia T., Fernandez-Riejos P., Martin-Gonzalez J., Segura-Egea J. J., Sanchez-Margalet V. (2017). Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev. 35, 71–84. [DOI] [PubMed] [Google Scholar]
- Procaccini C., Pucino V., Mantzoros C. S., Matarese G. (2015). Leptin in autoimmune diseases. Metabolism 64, 92–104. [DOI] [PubMed] [Google Scholar]
- Qu W. M., Miyazaki T., Terada M., Okada K., Mori S., Kanno H., Nose M. (2002). A novel autoimmune pancreatitis model in MRL mice treated with polyinosinic: polycytidylic acid. Clin. Exp. Immunol. 129, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabot S., Membrez M., Bruneau A., Gerard P., Harach T., Moser M., Raymond F., Mansourian R., Chou C. J. (2010). Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. Faseb J. 24, 4948–4959. [DOI] [PubMed] [Google Scholar]
- Ramdhan D. H., Kamijima M., Wang D., Ito Y., Naito H., Yanagiba Y., Hayashi Y., Tanaka N., Aoyama T., Gonzalez F. J. et al. , . (2010). Differential response to trichloroethylene-induced hepatosteatosis in wild-type and PPARalpha-humanized mice. Environ. Health Perspect. 118, 1557–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad A. F., Dickerson J., Kechichian T. B., Yin H., Gamble P., Salazar A., Patrikeev I., Motamedi M., Saade G. R., Costantine M. M. (2016). High-fructose diet in pregnancy leads to fetal programming of hypertension, insulin resistance, and obesity in adult offspring. Am. J. Obstet. Gynecol. 215, 378.e1.. [DOI] [PubMed] [Google Scholar]
- Saito K., Mori S., Date F., Ono M. (2013). Sjogren's syndrome-like autoimmune sialadenitis in MRL-Faslpr mice is associated with expression of glucocorticoid-induced TNF receptor-related protein (GITR) ligand and 4-1BB ligand. Autoimmunity 46, 231–237. [DOI] [PubMed] [Google Scholar]
- Schmatz M., Madan J., Marino T., Davis J. (2010). Maternal obesity: the interplay between inflammation, mother and fetus. J. Perinatol. 30, 441–446. [DOI] [PubMed] [Google Scholar]
- Segovia S. A., Vickers M. H., Gray C., Zhang X. D., Reynolds C. M. (2017). Conjugated linoleic acid supplementation improves maternal high fat diet-induced programming of metabolic dysfunction in adult male rat offspring. Sci. Rep. 7, 017–07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Şelli M. E., Wick G., Wraith D. C., Newby A. C. (2017). Autoimmunity to HSP60 during diet induced obesity in mice. Int. J. Obes. 41, 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sferruzzi-Perri A. N., Vaughan O. R., Haro M., Cooper W. N., Musial B., Charalambous M., Pestana D., Ayyar S., Ferguson-Smith A. C., Burton G. J. et al. , . (2013). An obesogenic diet during mouse pregnancy modifies maternal nutrient partitioning and the fetal growth trajectory. Faseb J. 27, 3928–3937. [DOI] [PubMed] [Google Scholar]
- Sinicato N. A., Postal M., Peres F. A., Peliçari K. d O., Marini R., dos Santos A. d O., Ramos C. D., Appenzeller S. (2014). Obesity and cytokines in childhood-onset systemic lupus erythematosus. J. Immunol. Res. 2014, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smialowicz R. J. (2002). The rat as a model in developmental immunotoxicology. Hum. Exp. Toxicol. 21, 513–519. [DOI] [PubMed] [Google Scholar]
- Srivastava A. K., Mohan S., Masinde G. L., Yu H., Baylink D. J. (2006). Identification of quantitative trait loci that regulate obesity and serum lipid levels in MRL/MpJ x SJL/J inbred mice. J. Lipid Res. 47, 123–133. [DOI] [PubMed] [Google Scholar]
- Stølevik S. B., Nygaard U. C., Namork E., Haugen M., Meltzer H. M., Alexander J., Knutsen H. K., Aaberge I., Vainio K., van Loveren H. et al. , . (2013). Prenatal exposure to polychlorinated biphenyls and dioxins from the maternal diet may be associated with immunosuppressive effects that persist into early childhood. Food Chem. Toxicol. 51, 165–172. [DOI] [PubMed] [Google Scholar]
- Tao Y., Wang M., Chen E., Tang H. (2017). Liver regeneration: Analysis of the main relevant signaling molecules. Mediators Inflamm. 2017, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng F., Klinger C. N., Felix K. M., Bradley C. P., Wu E., Tran N. L., Umesaki Y., Wu H. J. (2016). Gut microbiota drive autoimmune arthritis by promoting differentiation and migration of Peyer’s Patch T follicular helper cells. Immunity 44, 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Y., Liao J. K. (2012). A mouse model of diet-induced obesity and insulin resistance. Methods Mol. Biol. 821, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K., Milner J., Boudreau M. D., Gokulan K., Cerniglia C. E., Khare S. (2015). Effects of subchronic exposure of silver nanoparticles on intestinal microbiota and gut-associated immune responses in the ileum of Sprague-Dawley rats. Nanotoxicology 9, 279–289. [DOI] [PubMed] [Google Scholar]
- Williams P., Benton L., Warmerdam J., Sheehan P. (2002). Comparative risk analysis of six volatile organic compounds in California drinking water. Environ. Sci. Technol. 36, 4721–4728. [DOI] [PubMed] [Google Scholar]
- Wilson R. M., Marshall N. E., Jeske D. R., Purnell J. Q., Thornburg K., Messaoudi I. (2015). Maternal obesity alters immune cell frequencies and responses in umbilical cord blood samples. Pediatr. Allergy Immunol. 26, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L., Zhang K., Rasmussen D., Wang J., Wu D., Roemmich J. N., Bundy A., Johnson W. T., Claycombe K. (2017). Effects of prenatal low protein and postnatal high fat diets on visceral adipose tissue macrophage phenotypes and IL-6 expression in Sprague Dawley rat offspring. PLoS One 12, e0169581.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y., Wang H., Du M., Zhu M. J. (2014). Maternal obesity induces gut inflammation and impairs gut epithelial barrier function in nonobese diabetic mice. J. Nutr. Biochem. 25, 758–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K., Nishimura T., Ishiba H., Seko Y., Okajima A., Fujii H., Tochiki N., Umemura A., Moriguchi M., Sumida Y. et al. , . (2015). Blockade of interleukin 6 signalling ameliorates systemic insulin resistance through upregulation of glucose uptake in skeletal muscle and improves hepatic steatosis in high-fat diet fed mice. Liver Int. 35, 550–561. [DOI] [PubMed] [Google Scholar]
- Zhao J. H., Duan Y., Wang Y. J., Huang X. L., Yang G. J., Wang J. (2016). The influence of different solvents on systemic sclerosis: an updated meta-analysis of 14 case-control studies. J. Clin. Rheumatol. 22, 253–259. [DOI] [PubMed] [Google Scholar]