

Abstract
The ATP binding site located on the subunit B of DNA gyrase is an attractive target for the development of new antibacterial agents. In recent decades, several small-molecule inhibitor classes have been discovered but none has so far reached the market. We present here the discovery of a promising new series of N-phenylpyrrolamides with low nanomolar IC50 values against DNA gyrase, and submicromolar IC50 values against topoisomerase IV from Escherichia coli and Staphylococcus aureus. The most potent compound in the series has an IC50 value of 13 nM against E. coli gyrase. Minimum inhibitory concentrations (MICs) against Gram-positive bacteria are in the low micromolar range. The oxadiazolone derivative 11a, with an IC50 value of 85 nM against E. coli DNA gyrase displays the most potent antibacterial activity, with MIC values of 1.56 μM against Enterococcus faecalis, and 3.13 μM against wild type S. aureus, methicillin-resistant S. aureus (MRSA) and vancomycin-resistant Enterococcus (VRE). The activity against wild type E. coli in the presence of efflux pump inhibitor phenylalanine-arginine β-naphthylamide (PAβN) is 4.6 μM.
Keywords: Antibacterial, DNA gyrase, GyrB, Inhibitor, N-phenylpyrrolamide, ParE, Topoisomerase IV
1. Introduction
The discovery of antibacterials is considered to be one of the greatest medical achievements of all time. However, since the 1960s only a small number of new-class antibacterial agents have reached clinical practice while, on the other hand, the number of multi-drug resistant (MDR) bacteria is rising [1,2]. Nowadays, we are increasingly faced with life-threatening infections due to resistant Gram-positive and Gram-negative pathogens that belong to the “ESKAPE” group (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species). Thus, the ESKAPE pathogens were included by the World Health Organisation in the »WHO priority pathogens list for R&D of new antibiotics« [3,4].
DNA gyrase (gyrase) is a member of bacterial type IIA topoisomerase enzymes [5] that control the topology of DNA during processes of transcription, replication and recombination by introducing transient breaks to both DNA strands [6,7]. DNA gyrase helps relieve torsional tension by introducing negative supercoils to the DNA molecule during replication. It is a heterotetrameric protein composed of two GyrA subunits where the DNA cleavage site is located, and two GyrB subunits that provide the energy necessary for the catalytic function of the enzyme through ATP hydrolysis [6,8,9]. The four subunits form a functional tetramer A2B2. The other member of bacterial type IIA topoisomerases, topoisomerase IV (topo IV), is composed of two ParC and two ParE subunits that possess homologous structures to GyA and GyrB subunits of DNA gyrase. The main function of topo IV is decatenation of two daughter chromosomal DNA molecules after replication [8]. The structural and functional similarities of the two enzymes indicate that there is a possibility of designing inhibitors that target active sites of both gyrase and topo IV. This could reduce the capacity of bacteria to develop target-based drug resistance, since the probability of concurrent mutations on both targets is low [10]. Drugs targeting bacterial type IIA topoisomerases act by two main mechanisms, either by stabilizing the complex between a DNA molecule and the ParC/GyrA active site of the enzyme (e.g. quinolones), or by inhibiting the ATPase activity of the ParE/GyrB subunit (e.g. aminocoumarin class of inhibitors) [5,6]. Novobiocin, a representative of the natural aminocoumarins, is the only ATP-competitive inhibitor to have been used in the clinic but it has been withdrawn due to its toxicity and low effectiveness [11]. Studies of many co-crystal structures of ParE and GyrB subunits with small ligands and fragment-based design campaigns have led to several new classes of GyrB/ParE inhibitors being discovered (Fig. 1) [8,10]. However, none of them have advanced beyond phase I clinical trials, and most are only active against Gram-positive bacteria [10–12].
Fig. 1.

Representative DNA gyrase B inhibitors: benzothiazole A [13], pyrrolopyrimidine B [14], indazole C [15], pyrrolamide D [16], pyrimidoindole E [17], azaindole urea F [18], and their IC50 or Ki, and MIC values.
2. Results and discussion
2.1. Design
The design of the presented set of compounds was based on our previous N-phenylpyrrolamide inhibitors which had low nanomolar inhibitory activities against E. coli gyrase (IC50 < 100 nM). An example is compound D (Fig. 2a) [16]. The aim was to improve the GyrB/ParE binding affinity of compounds and their antibacterial activity by structural modifications, resulting in type I (Schemes 1–3) and type II (Scheme 4) compounds (Fig. 2b). For easier discussion, we have divided the structures into three parts: Part A, Part B and Part C (Fig. 2a).
Fig. 2.

a) A representative N-phenylpyrrolamide D and its inhibitory activities on DNA gyrase and topoisomerase IV [16]; b) Structures of type I and type II N-phenylpyrrolamide inhibitors; c) Docking binding mode of inhibitor D coloured according to the atom chemical type (C, yellow; N, blue; O, red; Cl, green) in the ATP binding site of E. coli GyrB (in cyan, PDB code: 4DUH [22]). The water molecule is presented as a red sphere. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Scheme 1.

Reagents and conditions: (a) thionyl chloride, MeOH, 0 °C → rt, 15 h; (b) isopropanol, triphenylphosphine, diisopropyl azodicarboxylate, THF, rt, 15 h (for the synthesis of 3a), tert-butyl (2-chloroethyl)carbamate, K2CO3, KI, DMF, rt → 60 °C, 15 h (for the synthesis of 3b); (c) 1 M NaOH, MeOH, rt, 15 h; (d) corresponding amino acid methyl ester hydrochloride, TBTU, NMM, CH2Cl2, rt, 15 h; (e) H2, Pd-C, MeOH, rt, 2–4 h; (f) corresponding pyrrole carboxylic acid, oxalyl chloride, CH2Cl2, rt, 15 h, then ii) 6a-g, pyridine, CH2Cl2, rt, 15 h; (g) 1 M NaOH, MeOH/THF, rt, 15 h (for the synthesis of 8a-e and 8g-i) or 1 M LiOH, MeOH/THF, rt, 4 h (for the synthesis of 8f); (h) 4 M HCl in 1,4-dioxane, THF, rt, 2 h.
Scheme 3.

Reagents and conditions: (a) pyridin-2-ylmethanamine (for the synthesis of 13a) or 2-(pyridin-2-yl)ethan-1-amine (for the synthesis of 13b), TBTU, NMM, CH2Cl2, rt, 15 h; (b) H2, Pd-C, MeOH, rt, 5 h; (c) 4,5-dibromopyrrole-2-carboxylic acid, oxalyl chloride, CH2Cl2, rt, 15 h, then ii) 14a-b, pyridine, CH2Cl2, rt, 15 h.
Scheme 4.

Reagents and conditions: (a) thionyl chloride, MeOH, 0 °C / rt, 15 h; (b) K2CO3, benzyl bromide, CH3CN, 60 °C, 15 h (for the synthesis of 18a), β-dimethyl-amino-ethylchloride hydrochloride, K2CO3, THF, 60 °C, 72 h (for the synthesis of 18b), isopropanol, triphenylphospine, DIAD, THF, rt, 15 h (for the synthesis of 18c); (c) 1 M NaOH, MeOH, rt, 15 h; (d) glycine methyl ester hydrochloride, TBTU, NMM, CH2Cl2, rt, 15 h; (e) SnCl2, EtOAc/MeOH, 55 °C, 15 h (for the synthesis of 21a), H2, Pd-C, MeOH, rt, 3 h, (for the synthesis of 21b-c); (f) the corresponding pyrrole carboxylic acid, oxalyl chloride, CH2Cl2, rt, 15 h, then ii) 21a-c, pyridine, CH2Cl2, rt, 15 h; (g) 1 M NaOH, MeOH/THF, rt, 15 h.
To Part A, we introduced either a 4,5-dibromo-1H-pyrrole or a 3,4-dichloro-5-methyl-1H-pyrrole group. The pyrrole NH and pyrrolamide C=O groups are important hydrogen bond donor and acceptor groups that interact with Asp73 (E. coli numbering) and with a conserved water molecule (Fig. 2c) in the GyrB binding site. Halogen atoms on the pyrrole moiety lower the pKa of the NH group and thus strengthen the formed H-bond. Chlorine and bromine atoms also increase the lipophilicity of the pyrrole moiety and thus increase hydrophobic interactions with the amino acid residues in the hydrophobic pocket of the enzyme (Val43, Ala47, Val71, Val167).
On the Part B benzene ring, compound D contains a lipophilic isopropoxy substituent that can form hydrophobic interactions with residues Ile78 and Ile94 in the lipophilic floor of the enzyme. Since these interactions were found to be favourable for the binding affinity, in some type I compounds (7a-f, 8a-f, 10a, 11a and 15a-b) the isopropoxy group on the 3-position of the benzene ring was retained. To other type I compounds (9a-e and 12) we introduced a 2-aminoethoxy substituent at this position, with the aim of improving the solubility of the compounds and/or broadening their antibacterial spectrum. Recently, the presence of an amino group, especially a primary amino group, was found to contribute to the ability of compounds to accumulate in Gram-negative E. coli [19]. With the type II compounds, we further explored the lipophilic floor of the enzyme by changing the position of substituents on the benzene ring from position 3- to 2-position. Similarly as in some type I compounds, in some type II compounds (22c-d and 23c-d) we introduced the isopropoxy substituent to the 2-position of the benzene ring. In other type II compounds we introduced a sterically larger benzyloxy substituent (compounds 22a and 23a) or a basic 2-(dimethylamino)ethoxy substituent (compounds 22b and 23b), with the aim of enabling additional interactions with the enzyme and improving the water solubility of the compounds.
To Part C of type I compounds, groups that are able to form either ionic interactions with Arg136 side chain or π-stacking interactions with the Glu50-Arg76 salt bridge were attached. The influence of different α-amino acids attached to Part C was studied, aiming to determine how the stereochemistry and the size of the amino acid side chain affect the binding affinity. Thus, glycine, l- and d-alanine, l-valine, and l-phenylalanine derivatives were prepared. The activities of these derivatives, in the form of methyl esters, free carboxylic acids, and hydrazides, were compared. Additionally, three compounds (11a, 11b and 12), each with a 1,3,4-oxadiazol-2-one ring as bioisosteric replacement for the carboxylic acid functionality, were prepared to reduce the acidity and polarity and thus improve bacterial cell membrane penetration of the compounds [20,21]. Furthermore, two compounds, each with a pyridine containing substituent in Part C, pyridin-2-ylmethanamine (n = 1, compound 15a) and 2-(pyridin-2-yl) ethan-1-amine (n = 2, compound 15b) were synthesized, and the optimal length for the activity determined. The pyridine moiety was intended to enable π-stacking interactions with the Glu50-Arg76 salt bridge.
2.2. Chemistry
The synthesis of type I compounds (8a-i, 9a-e, 11a-b, 12, 15a-b) is presented in Schemes 1–3. 3-Hydroxy-4-nitrobenzoic acid (1) was reacted with thionyl chloride in methanol to give methyl ester 2, which was converted to 3a in a Mitsunobu reaction using isopropanol, and to 3b in the presence of potassium carbonate using tert-butyl (2-chloroethyl)carbamate as an alkylating agent. Compounds 3a-b were hydrolysed with 1 M sodium hydroxide to give carboxylic acids 4a-b, which were coupled with different amino acid methyl esters using TBTU (N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate) in order to prepare amides 5a-g. The nitro groups of 5a-g were reduced by catalytic hydrogenation to give amines 6a-g. Compounds 6a-g were then coupled with 4,5-dibromopyrrole-2-carboxylic acid (to give 7a-c and 7g-h) or with 3,4-dichloro-5-methylpyrrole-2-carboxylic acid to give 7d-f and 7i in a two-step reaction. Oxalyl chloride was used to form pyrrole-2-carboxylic acid chloride in the first step followed by its aminolysis in pyridine in the second step. Products 8a-i were prepared by alkaline hydrolysis of 7a-i. Removal, by acidolysis, of the Boc protecting group from compounds 7h-i and 8g-i led to the final compounds 9a-e. Compounds 7e and 7i were additionally reacted with hydrazine monohydrate under reflux to prepare hydrazides 10a-b. These were converted to 11a-b using 1,1′-carbonyldiimidazole (CDI) at 100 °C. The Boc protecting group was removed from 11b to yield the target compound 12.
To prepare products 15a-b, 4a was first coupled with pyridin-2-ylmethanamine (to prepare 13a) or 2-(pyridin-2-yl)ethan-1-amine (to prepare 13b) using TBTU. Reducing the nitro groups of 13a-b by catalytic hydrogenation led to compounds 14a-b. The final products 15a-b were prepared by coupling amines 14a-b with 4,5-dibromopyrrole-2-carboxylic acid.
The synthesis of type II compounds (23a-d) is outlined in Scheme 4. 2-Hydroxy-4-nitrobenzoic acid (16) was converted to its methyl ester 17 using thionyl chloride in methanol. Compounds 18a-b were prepared by reacting 17 with benzyl bromide or β-dimethyl-aminoethylchloride hydrochloride using potassium carbonate, while 18c was synthesized under Mitsunobu conditions, using isopropyl alcohol. Alkaline hydrolysis of methyl esters 18a-c led to carboxylic acids 19a-c, which were coupled with glycine methyl ester hydrochloride, using TBTU, to give 20a-c. The amines 21a-c obtained after reduction of nitro groups of 20a-c were coupled with either 4,5-dibromopyrrole-2-carboxylic acid to give 22a-c, or 3,4-dichloro-5-methylpyrrole-2-carboxylic acid to give 22d. Finally, methyl esters of 22a-d were hydrolysed with 1 M sodium hydroxide to give target compounds 23a-d.
2.3. Inhibitory activities against DNA gyrase and topoisomerase IV
All final compounds were evaluated for their inhibitory activity against E. coli DNA gyrase in a supercoiling assay. Results are presented as residual activities (RAs) of the enzyme at 1 μM of compounds or as IC50 values for compounds with RA < 50% (Tables 1 and 2). Compounds with submicromolar IC50 values were evaluated against S. aureus DNA gyrase, and E. coli and S. aureus topoisomerase IV (Table 3). To determine the possible binding modes of compounds, molecular docking of all tested compounds to the E. coli GyrB binding site was performed, using GOLD software [23].
Table 1.
Inhibitory activity of type I compounds 7a-i, 8a-i, 9a-e, 10a-b, 11a-b, 12 and 15a-b against DNA gyrase from E. coli.
 | ||||||
|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | R3 | R4 | * | IC50 (nM)a or RA (%)b |
| E. coli gyrase | ||||||
| 7a | 4,5-diBr | iPr | iPr | COOMe | l | 94% |
| 7b | 4,5-diBr | iPr | Bn | COOMe | l | 100% |
| 7c | 4,5-diBr | iPr | CH3 | COOMe | l | 93% |
| 7d | 3,4-diCl-5-Me | iPr | Bn | COOMe | l | 100% |
| 7e | 3,4-diCl-5-Me | iPr | CH3 | COOMe | l | 66% |
| 7f | 3,4-diCl-5-Me | iPr | CH3 | COOMe | d | 87% |
| 7g | 4,5-diBr | CH2CH2NHBoc | H | COOMe | / | 53% |
| 7h | 4,5-diBr | CH2CH2NHBoc | Bn | COOMe | l | 100% |
| 7i | 3,4-diCl-5-Me | CH2CH2NHBoc | CH3 | COOMe | l | 100% |
| 8a | 4,5-diBr | iPr | iPr | COOH | l | 70% |
| 8b | 4,5-diBr | iPr | Bn | COOH | l | 100% |
| 8c | 4,5-diBr | iPr | CH3 | COOH | l | 370 ± 160 nM |
| 8d | 3,4-diCl-5-Me | iPr | Bn | COOH | l | 91% |
| 8e | 3,4-diCl-5-Me | iPr | CH3 | COOH | l | 38 ± 9 nM |
| 8f | 3,4-diCl-5-Me | iPr | CH3 | COOH | d | 41 ± 16 nM |
| 8g | 4,5-diBr | CH2CH2NHBoc | H | COOH | / | 96% |
| 8h | 4,5-diBr | CH2CH2NHBoc | Bn | COOH | l | 100% |
| 8i | 3,4-diCl-5-Me | CH2CH2NHBoc | CH3 | COOH | l | 590 ± 20 nM |
| 9a | 4,5-diBr | Bn | COOMe | l | 85% | |
| 9b | 3,4-diCl-5-Me | CH3 | COOMe | l | 34 ± 1 nM | |
| 9c | 4,5-diBr | H | COOH | l | 50% | |
| 9d | 4,5-diBr | Bn | COOH | l | 370 ± 10 nM | |
| 9e | 3,4-diCl-5-Me | CH3 | COOH | l | 28 ± 7 nM | |
| 10a | 3,4-diCl-5-Me | iPr | CH3 | CONHNH2 | l | 280 ± 80 nM |
| 10b | 3,4-diCl-5-Me | CH2CH2NHBoc | CH3 | CONHNH2 | l | 68% |
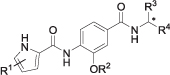
| 11a | 3,4-diCl-5-Me | iPr | CH3 | 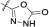 |
l | 85 ± 7 nM |
| 11b | 3,4-diCl-5-Me | CH2CH2NHBoc | CH3 | 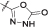 |
l | 1600 ± 200 nM |
| 12 | 3,4-diCl-5-Me | CH3 | 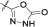 |
l | 13 ± 4 nM | |
| 15a | 4,5-diBr | iPr | H | 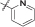 |
/ | 100% |
| 15b | 4,5-diBr | iPr | H | 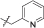 |
/ | 87% |
| novobiocin | 170 ± 20 nM | |||||
Concentration of compound that inhibits the enzyme activity by 50%.
Residual activity of the enzyme at 1 μM of the compound.
Table 2.
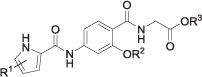
Inhibitory activity of type II compounds 22a-d and 23a-d against DNA gyrase from E. coli.
 | ||||
|---|---|---|---|---|
| Compd. | R1 | R2 | R3 | IC50 (nM)a or RA (%)b |
| E. coli gyrase | ||||
| 22a | 4,5-diBr | Bn | Me | 100% |
| 22b | 4,5-diBr | CH2CH2N(Me)2 | Me | 88% |
| 22c | 4,5-diBr | iPr | Me | 91% |
| 22d | 3,4-diCl-5-Me | iPr | Me | 59% |
| 23a | 4,5-diBr | Bn | H | 88 ± 0 nM |
| 23b | 4,5-diBr | H | 82% | |
| 23c | 4,5-diBr | iPr | H | 400 ± 90 nM |
| 23d | 3,4-diCl-5-Me | iPr | H | 91 ± 29 nM |
| novobiocin | 170 ± 20 nM | |||
Concentration of compound that inhibits the enzyme activity by 50%.
Residual activity of the enzyme at 1 μM of the compound.
Table 3.
Inhibitory activity of selected compounds against DNA gyrase from S. aureus and topoisomerase IV from E. coli and S. aureus.
| Compd. | IC50 (μM)a or RA (%)b |
||
|---|---|---|---|
| S. aureus gyrase | E. coli topo IV | S. aureus topo IV | |
| 8c | 5.9 ± 0.7 μM | 78% | 98% |
| 8e | 0.093 ± 0.065 μM | 0.45 ± 0.12 μM | 0.28 ± 0.20 μM |
| 8f | 1.6 ± 0.6 μM | 7.4 ± 1.5 μM | 1.3 ± 0.1 μM |
| 8i | 1.3 ± 0.0 μM | 100% | 99% |
| 9b | 0.39 ± 0.09 μM | 89% | 3.7 ± 0.3 μM |
| 9d | 89% | 96% | 98% |
| 9e | 0.11 ± 0.04 μM | 0.53 ± 0.06 μM | 3.2 ± 0.4 μM |
| 10a | 22 ± 10 μM | 100% | 92% |
| 11a | 0.75 ± 0.15 μM | 11 ± 0 μM | 12 ± 0 μM |
| 12 | 1.0 ± 0.3 μM | 0.57 ± 0.16 μM | 0.96 ± 0.09 μM |
| 23a | 79% | 42 ± 11 μM | 39 ± 0 μM |
| 23c | 85% | 100% | 96% |
| 23d | 0.14 ± 0.02 μM | 100% | 12 ± 0 μM |
| novobiocin | 0.041 ± 0.07 μM | 11 ± 2 μM | 27 ± 7 μM |
Concentration of compound that inhibits the enzyme activity by 50%.
Residual activity of the enzyme at 1 μM of the compound.
Compounds 8e, 8f, 9b, 9e, 11a, 12, 23a and 23d showed inhibitory activities stronger than that of novobiocin (IC50 = 170 nM) against E. coli DNA gyrase, with low nanomolar inhibitory values (IC50 ≤ 91 nM). Seven of the eight most active compounds contained the 3,4-dichloro-5-methyl-1H-pyrrole moiety in Part A of the molecules, which thus proved to be more suitable than the 4,5-dibromo-1H-pyrrole moiety. The reason for this is probably the slightly smaller size of the chloro and methyl substituents than of the bromo substituents which bind to the hydrophobic pocket of the enzyme more strongly [16]. Compounds with free carboxylic acid groups in the eastern part of the molecules showed more potent activities than did their methyl ester analogues. For example, methyl ester 7f was almost inactive at 1 μM concentration (RA = 87%), while its carboxylic acid derivative 8f had an IC50 value of 41 nM and was among the most potent compounds of the series. Free carboxylic acids are able to form ionic interactions and/or hydrogen bonds with the Arg136 side chain in the binding site of the enzyme, while esters can only form hydrogen bonds, thus resulting in their weaker activity. A similar trend can be observed for other carboxylic acid – methyl ester pairs presented in Tables 1 and 2 The only methyl ester derivative with activity in the low nanomolar range was compound 9b, with an IC50 value of 34 nM. Further, in some type I compounds (compounds 7e and 7i), methyl ester groups were converted to hydrazides (compounds 10a and 10b), which were then further converted to oxadiazolone rings (compounds 11a-b and 12). The activities of hydrazides (10a, IC50 = 280 nM) were weaker than those of the corresponding carboxylic acids (8e, IC50 = 38 nM) or oxadiazolones (11a, IC50 = 85 nM), probably because they cannot form ionic interactions with Arg136. On the other hand, both compound 8e with a carboxylic acid group (IC50 = 38 nM) and its oxadiazolone containing analogue 11a (IC50 = 85 nM) inhibited the enzyme in the low nanomolar range, although compound 8e was approximately two-fold more potent. A different result was observed when oxadiazolone 12 (IC50 = 13 nM) was compared with its carboxylic acid analogue 9e (IC50 = 28 nM), where the former was more potent. The only compound containing an oxadiazolone ring that did not show low nanomolar potency was 11b, with an IC50 value of 1.6 μM, probably because it contains a side chain with the sterically larger tert-butyl carbamate group.
As reported recently [16], lipophilic substituents at the 3-position of the Part B benzene ring (e.g. methoxy, isopropoxy and benzyloxy substituents) form favourable interactions with the hydrophobic floor of the enzyme. In type I compounds, three different substituents have thus been introduced at this position: isopropoxy, 2-aminoethoxy and N-Boc-2-aminoethoxy substituents. Analogues with either an isopropoxy (8e, IC50 = 38 nM) or a 2-aminoethoxy substituent (9e, IC50 = 28 nM) had greater activity than analogues with an N-Boc-2-aminoethoxy substituent (8i, IC50 = 590 nM), probably because the tert-butyl carbamate group is too large to fit into the binding site of the enzyme. When compounds with isopropoxy and 2-aminoethoxy substituents were compared, the latter were generally more potent, as seen by comparing the isopropoxy derivatives 8d (RA = 91% at 1 μM) and 7e (RA = 66% at 1 μM) with their 2-aminoethoxy counterparts 9d (IC50 = 370 nM) and 9b (IC50 = 34 nM). Based on the docking experiments, it was predicted that the amino group of the 2-aminoethoxy substituent could form an H-bond with Ala100 that is part of the flexible loop formed by Gly97-Ser108 (compound 9e, Fig. 3), thus increasing the inhibitory potency. Similarly, in the crystal structure of a bithiazole inhibitor in complex with E. coli GyrB reported by Brvar et al. (PDB code: 4DUH [22]), a hydrogen bond was seen with Gly101 that is also a part of this loop. These interactions could stabilize the loop, reduce its flexibility and lead to stronger binding of the inhibitor.
Fig. 3.

The GOLD-predicted binding pose of inhibitor 9e (in orange sticks) in the E. coli GyrB ATP-binding site (PDB entry: 4DUH [22], in grey). Hydrogen bonds are presented as green dashed lines. The water molecule is shown as a red sphere. The figure was prepared with PyMOL [24]. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To Part C of the type I compounds, we attached various amino acids: glycine, l- and d-alanine, l-valine and l-phenylalanine. Additionally, two compounds (15a and 15b) were prepared with side chains containing a pyridine ring bound through either a methylene or an ethylene linker to the central benzamide core. Compounds with alanine side chains displayed the strongest inhibitory activity of the series while l-phenylalanine containing compounds were the weakest. For example, l-alanine analogue 8c exhibited an IC50 value of 370 nM, while its l-phenylalanine containing counterpart 8b was inactive at 1 μM concentration (RA = 100%). From these results it can be concluded that the benzyl group of the l-Phe side chain does not form favourable hydrophobic or π-π interactions with the enzyme. Furthermore, the size of the L-Phe group may force the molecule to take up an unfavourable conformation. Because of its size and flexibility, the benzyl group might be oriented out of the binding site towards the solvent, as was predicted with molecular docking of compound 8b into the E. coli GyrB ATP-binding site (Fig. 1Sa, Supporting information). There were no marked differences in the activities of l-alanine (compound 8e, IC50 = 38 nM) or d-alanine containing compounds (compound 8f, IC50 = 41 nM). Compounds 15a and 15b, with pyridine containing groups, were not active against E. coli gyrase (RA = 100% or 87%). Even though the interaction between the pyridine nitrogen of compound 15a and Arg76 was predicted by molecular docking, this side chain is probably too flexible to form a stable contact with Arg76 (Fig. 1Sb, Supporting information).
To further explore the binding site, type II compounds with substituents at the 2-position of the Part B benzene ring were prepared. Compounds with isopropoxy, benzyloxy and 2-dimethylaminoethoxy groups at this position were evaluated. As in type I compounds, only compounds with free carboxylic acid groups in the eastern part of the molecules showed nanomolar enzymatic inhibition. Compound 23b, with a 2-dimethylaminoethoxy substituent (RA = 82% at 1 μM), showed the weakest activity of the series. The benzyloxy analogue 23a (IC50 = 88 nM) was the most active type II compound, being 4-fold more active than its isopropoxy analogue 23c (IC50 = 400 nM), indicating that it can form stronger hydrophobic interactions with the enzyme. A second very potent compound in the type II series was compound 23d (IC50 = 91 nM) with an isopropoxy substituent on the benzene ring and a 3,4-dichloro-5-methylpyrrole group attached as Part A.
In general, the activities on DNA gyrase from S. aureus and on topo IV from E. coli and S. aureus were weaker than those on E. coli gyrase, but compounds 8e, 9e and 12 that were among the most potent inhibitors of E. coli gyrase also displayed promising results on these three enzymes (Table 3). Compounds 8e, 9e and 12 had IC50 values in the submicromolar range (<1 μM) against E. coli topo IV, which is much lower than that for novobiocin (IC50 = 11 μM). Compound 8e, with an isopropoxy substituent, and compound 9e, with aminoethoxy substituent on the 3-position of the benzene ring, additionally showed nanomolar IC50 values (93 nM and 110 nM, respectively) against S. aureus gyrase. Compound 8e displayed the most balanced activities against all four tested enzymes, with a low IC50 of 280 nM also against S. aureus topo IV (IC50 of novobiocin is 27 μM) and thus appears to be a promising dualtarget inhibitor. Interestingly, compound 8f, a d-alanine analogue of 8e, that showed an activity comparable to that of 8e on DNA gyrase from E. coli, did not display the same potency on the other three enzymes. Since there are certain structural differences between the enzymes, the stereochemistry could be more important in the latter. A difference between 9e and its methyl ester analogue 9b was also observed, with weaker activities of the latter on all enzymes. These results correlate well with those for E. coli gyrase, leading to the assumption that an acidic functional group is also important for the binding to S. aureus gyrase and to topo IV from E. coli and S. aureus. Similarly, the carboxylic acid analogue 8e displayed stronger activity than either the corresponding hydrazide 10a or the oxadiazolone 11a on these enzymes. Furthermore, the activity of the 3,4-dichloro-5-methyl-pyrrolamides (8e, 8f, 8i, 9b, 9e, 10a, 11a, 12 and 23d) was stronger than that of the 4,5-dibromopyrrolamides (8c, 9d, 23a and 23c). The reason probably lies in the smaller size of the binding pockets of S. aureus DNA gyrase and of topo IV that results from differences in certain amino acid residues in the ATP binding site [25,26]. 3,4-Dichloro-5-methylpyrrolamides with smaller groups on the pyrrole ring bind more effectively to the enzyme.
2.4. Antibacterial activity
Compounds were tested for their antibacterial activity against two Gram-positive (S. aureus ATCC 25923 and E. faecalis ATCC 29212) and two Gram-negative (E. coli ATCC 25922 and P. aeruginosa ATCC 27853) wild type bacterial strains and, additionally, against two E. coli mutant strains (JW5503 ΔtolC and JD17464 ΔlpxC) at inhibitor concentrations of 50 μM. The tolC deletion mutant represents Gram-negative bacteria with defective efflux mechanisms and the lpxC deletion mutant represents a Gram-negative strain with disrupted bacterial cell wall. Results are presented in Table 1S (Supporting information) as percentages of growth inhibition. For compounds that inhibited bacterial strain growth by ≥ 80%, MIC values were also determined (Table 4).
Table 4.
Minimum inhibitory concentrations (MICs) of compounds 7c, 8a, 8d, 8e, 8f, 9a, 9b, 10a, 11a, 11b and 12 against S. aureus (ATCC 25923), E. faecalis (ATCC 29212) and E. coli (JW5503, a tolC deletion mutant).
| Compd. | MIC (μM)a | ||
|---|---|---|---|
| S. aureus (ATCC 25923) | E. faecalis (ATCC 29212) | E. coli (JW5503) ΔtolCb | |
| 7c | n.d.c | n.d. | 25 μM |
| 8a | n.d. | >75 μM | n.d. |
| 8d | n.d. | 3.13 μM | n.d. |
| 8e | n.d. | 3.13 μM | 50 μM |
| 8f | n.d. | 6.25 μM | n.d. |
| 9a | 50 μM | 50 μM | 25 μM |
| 9b | 50 μM | 25 μM | 25 μM |
| 10a | n.d. | 6.25 μM | n.d. |
| 11a | 3.13 μM | 1.56 μM | n.d. |
| 11b | n.d. | 100 μM | n.d. |
| 12 | n.d. | n.d. | 50 μM |
| ciprofloxacin | 1.5 μM | 3.0μM | 0.015 μM |
MIC (minimum inhibitory concentration that inhibits the growth of bacteria by ≥ 90%) values against E. faecalis, S. aureus and E. coli JW5503 (ΔtolC). Ciprofloxacin was used as a positive control.
E. coli strain with mutated efflux pump.
Not determined.
Overall, the compounds displayed stronger inhibitory activity against Gram-positive than against Gram-negative bacteria. Nine compounds (8a, 8d, 8e, 8f, 9a, 9b, 10a, 11a and 11b) showed more than 80% growth inhibition of E. faecalis, and three (9a, 9b and 11a) inhibited the growth of S. aureus by more than 80%. Compound 9b, with an aminoethoxy substituent on the 3-position of the central benzene ring, inhibited the growth of wild type E. coli by 67% (Table 1S) and completely suppressed the E. coli ΔtolC deletion mutant at 50 μM. For compounds 7c, 8e, 8f, 9a, 11a and 12, significant differences in growth inhibition between wild type E. coli and the E. coli mutant strain having a defective efflux pump were also observed, while growth inhibition of the E. coli strain with impaired outer membrane was similar to that of the wild type for all compounds (Table 1S). These results suggest that some compounds are subject to bacterial efflux, which is probably the main reason for their weaker activity against Gram-negative bacteria.
Compound 11a exhibited the strongest antibacterial activity of the series, with an MIC value of 1.56 μM against E. faecalis, which is almost two-fold lower than that of ciprofloxacin (3 μM), and of 3.13 μM against S. aureus. Compound 11a, that contains an oxadiazolone ring in the eastern part of the molecule, was twice as active as its carboxylic acid analogue 8e (E. faecalis MIC = 3.13 μM) and four times more active than the corresponding hydrazide 10a (E. faecalis MIC = 6.25 μM). This result is in agreement with our design strategy that compounds with a less acidic and less polar bioisostere for the carboxylic acid group could permeate bacterial membrane more easily and display stronger antibacterial activity. Compounds with an aminoethoxy substituent at the 3-position of the benzene ring (9a, 9b and 12) exhibited no or weaker activity against E. faecalis (MIC values > 25 μM for 9a and 9b and no growth inhibition for compound 12) than isopropoxy derivatives, presumably because the polarity of the amino group reduces the ability of compounds to enter the Gram-positive bacteria. Compounds containing both a free carboxylic acid group at the eastern part of the molecule and an aminoethoxy substituent at the 3-position of the benzene ring (9c, 9d and 9e) were inactive against bacteria (less than 36% of growth inhibition, Table 1S) although compound 9e was among the most active in enzyme assays (S. aureus gyrase IC50 = 110 nM). On the other hand, compound 9b, a methyl ester analogue of 9e with a higher IC50 value (S. aureus gyrase IC50 = 390 nM), inhibited the growth of Gram-positive bacteria more strongly than 9e, with MIC values of 50 μM against S. aureus and 25 μM against E. faecalis (Table 4). Additionally, compound 9b showed a MIC value of 25 μM against an E. coli strain with defective efflux pump. Similar activities against an E. coli strain with a defective efflux pump were obtained for compound 9a (MIC = 25 μM) and for compound 12 (MIC = 50 μM) that also contain an aminoethoxy substituent. These results suggest that the aminoethoxy substituent improves activity against Gram-negative bacteria, but the higher activity is not observed because the compounds are possible substrates for efflux pumps.
2.5. Advanced antibacterial evaluation of compound 11a
Compound 11a was selected as the most promising of the series, since it had the low nanomolar IC50 values in enzyme tests and the lowest MIC values against Gram-positive strains of all the compounds. For this reason, compound 11a was further evaluated against a diverse panel of Gram-positive and Gram-negative bacterial strains (Table 5). In line with our previous observations, compound 11a displayed an MIC value of 4.17 μM against the Gram-positive human pathogen S. aureus ATCC 29213. Furthermore, it was active against methicillin-resistant Staphylococcus aureus (MRSA, ATCC 43300) and vancomycin-resistant Enterococcus (VRE, ATCC 70022), with MICs of 3.13 μM (Table 5). On the other hand, compound 11a displayed no bioactivity against wild type E. coli strains (ATCC 25922, MG1655 and BW 25113). We therefore sought to explore systematically the limiting factors for lack of anti-Gram-negative activity of compound 11a. To investigate possible difficulties with its penetration into the bacterial cell, we tested compound 11a on a diverse panel of mutant E. coli strains carrying loss-of-function mutations in either dapF, mrcB or surA genes that disrupt the bacterial cell wall integrity [27]. As a parallel approach, to test the susceptibility of compounds to efflux, we treated the parental strains and their corresponding mutants with an efflux pump inhibitor phenylalanine-arginine β-naphthylamide (PAβN), which is in fact not a typical inhibitor but a substrate for efflux pumps that is preferentially effluxed. Furthermore, we tested compound 11a against E. coli BW 25113 ΔacrB and ΔtolC deletion mutants with defective efflux pumps. Mutations in the genes involved in the formation of the cell wall (dapF, mrcB and surA) did not increase the antibacterial activity (MIC > 50 μM) but, in the case of ΔsurA strain, stronger inhibition was observed in the presence of PAβN (MIC = 13.8 μM). The presence of PAβN also increased the activity against wild type E. coli strains ATCC 25922 (MIC = 4.6 μM) and MG1655 (MIC = 10.2 μM). Compound 11a did not inhibit the efflux pump deficient E. coli BW 25113 strains ΔacrB and ΔtolC (MIC > 50 μM, Table 5), but it displayed weak activity against the E. coli JW5503 ΔtolC strain (51% inhibition at 50 μM, Table 1S). These results indicate that the most probable reason for the inactivity of compound 11a against Gram-negative strains lies in bacterial efflux.
Table 5.
Minimum inhibitory concentrations (MICs) of compound 11a against a broad panel of Gram-positive and Gram-negative bacterial strains.
| Compound 11a | MIC (μM)a |
|---|---|
| Gram-positive bacteria | |
| S. aureus (ATCC 29213) | 4.17 μM |
| S. aureus (MRSA, ATCC 43300) | 3.13 μM |
| E. faecium (VRE, ATCC 70022) | 3.13 μM |
| Gram-negative bacteria | |
| E. coli (ATCC 25922) | No inhibition up to 50 μM |
| E. coli (MG1655) | No inhibition up to 50 μM |
| E. coli (BW 25113) | No inhibition up to 50 μM |
| E. coli (BW 25113) ΔtolCb | No inhibition up to 50 μM |
| E. coli (BW 25113) ΔacrBb | No inhibition up to 50 μM |
| E. coli (BW 25113) ΔdapFc | No inhibition up to 50 μM |
| E. coli (BW 25113) ΔmrcBc | No inhibition up to 50 μM |
| E. coli (BW 25113) ΔsurAc | No inhibition up to 50 μM |
| E. coli (ATCC 25922) + 50 μg/mL PAβN | 4.6 μM |
| E. coli (MG1655) + 50 μg/mL PAβN | 10.2 μM |
| E. coli (BW 25113) + 50 μg/mL PAβN | No inhibition up to 20 μM |
| E. coli (BW 25113) ΔdapFc + 50 μg/mL PAβN | No inhibition up to 20 μM |
| E. coli (BW 25113) ΔmrcBc + 50 μg/mL PAβN | No inhibition up to 20 μM |
| E. coli (BW 25113) ΔsurAc + 50 μg/mL PAβN | 13.8 μM |
| E. coli (MG1655) R136-to-C mutant + 50 μg/mL PAβN | No inhibition up to 50 μM |
MIC (minimum inhibitory concentration that inhibits the growth of bacteria by ≥ 90%) measurements were performed according to the EUCAST guidelines in 3 independent measurements.
E. coli strain with mutated efflux pump.
E. coli strain with loss-of-function mutation in the gene involved in the cell wall formation.
Additionally, in order to validate the interaction between the oxadiazolone ring of 11a and Arg136 in the DNA gyrase binding site, compound 11a was tested, in the presence of PAβN, on E. coli MG1655 strain with the Arg136 to Cys mutation. The compound was not active on the mutated bacteria (MIC > 50 μM), but displayed activity against wild type E. coli in the presence of PAβN (MIC = 10.2 μM), indicating that the compound interacts with Arg136.
2.6. Cytotoxic activity of compound 11a
The cytotoxicity of compound 11a was determined by MTS assay against a human hepatocellular carcinoma cancer cell line (HepG2) and against human endothelial cells (HUVEC). The decrease in cell proliferation after the treatment with 11a was compared with that after treatment with 50 μM of etoposide, as positive control. Compound 11a showed no cytotoxicity against HepG2 cells at 50 μM (85% cell proliferation), while it showed an IC50 value of 40.7 μM against HUVEC cells (Table 2S, Supporting information). However, the concentration at which the compound was active against HUVEC cells was much higher than that at which it is active against Gram-positive bacteria.
3. Conclusions
Overall, thirty-eight compounds were designed, synthesized and evaluated against DNA gyrase and topoisomerase IV, and against several Gram-positive and Gram-negative bacterial strains. The most active compound in enzymatic assays was 3,4-dichloro-5-methyl-pyrrolamide 8e, with an isopropoxy substituent at the 3-position of the benzene ring and a free carboxylic acid functionality in the eastern part of the molecule. Compound 8e had an IC50 value of 38 nM against E. coli gyrase and 93 nM against S. aureus gyrase, and was also active against E. coli topo IV (IC50 = 0.45 μM) and S. aureus topo IV (IC50 = 0.28 μM). Compound 11a, an oxadiazolone analogue of 8e, possessed low micromolar antibacterial activity against Gram-positive bacteria, with MIC values of 1.56 μM and 3.13 μM against E. faecalis and S. aureus, respectively. Further, compound 11a inhibited the growth of MRSA and VRE, key targets for antibiotic research, with MIC values of 3.13 μM [28]. Compound 11a was not active against wild type E. coli strains, but was active in the presence of PAβN (MIC = 4.6 μM). These results suggest that, in Gram-negative bacteria, the compounds are sensitive to the efflux mechanism. In recent years, however, new compounds that can be used as adjuvants to potentiate the activity of antibiotics against Gram-negative strains are becoming available [29]. In short, low nanomolar enzymatic activity against DNA gyrase and topo IV, low micromolar MIC values against drug-resistant Gram-positive bacteria, and low cytotoxicity make compound 11a a promising starting point for further optimization.
4. Experimental section
4.1. Determination of inhibitory activities on E. coli and S. aureus DNA gyrase
The assay for the determination of IC50 values was performed according to previously reported procedures [16].
4.2. Determination of inhibitory activities on E. coli and S. aureus topoisomerase IV
The assay for the determination of IC50 values was performed according to previously reported procedures [16].
4.3. Bacterial strains used in the study
Staphylococcus aureus ATCC 29213, Staphylococcus aureus ATCC 25923, S. aureus ATCC 43300, Enterococcus faecalis ATCC 29212, Enterococcus faecium ATCC 70022, Escherichia coli ATCC 25922, and Pseudomonas aeruginosa ATCC 27853 have been obtained from the American Type Culture Collection (ATCC) via Microbiologics Inc. (St. cloud, MN), E. coli MG1655 originated from the laboratory collection of Dr. Csaba Pál. E. coli BW 25113, and E. coli BW 25113 ΔacrB, ΔdapF, ΔmrcB and ΔsurA mutant strains originated from the KEIO collection [30]. Single-gene knock-out strains of E. coli, JW5503 ΔtolC [30] and JD17464 ΔlpxC were obtained from the NBRP-E.coli collection at the National Institute of Genetics (NIG, Japan).
4.4. Determination of antibacterial activity
The antibacterial activities against S. aureus ATCC 25923, E. faecalis ATCC 29212, E. coli ATCC 25922, P. aeruginosa ATCC 27853, E. coli JW5503, E. coli JD17464, S. aureus ATCC 43300, and E. faecium ATCC 70022 were determined following the CLSI guidelines and performed according to previously reported procedures [16].
MIC values against S. aureus ATCC 29213, E. coli ATCC 25922, E. coli MG1655, E. coli BW 25113, and E. coli BW 25113 ΔacrB, ΔdapF, ΔmrcB and ΔsurA were determined using a standard broth micro-dilution technique according to the EUCAST guidelines and ISO 20776-1:2006 [31]. In brief, 12-step serial dilutions of the test compound were prepared in 100 μL of cation adjusted Mueller-Hinton II Broth (Catalog n.o. 90922 from Merck KGaA, Darmstadt, Sigma) in 96-well microtiter plates. Approximately 5 × 104 bacteria were inoculated onto each well. Plates were incubated at 37 °C and shaken at 300 r.p.m. Measurements were performed in 3 replicates. After 18 h of incubation, optical density values were measured in a Biotek Synergy microplate reader at 600 nm wavelength for each well. MIC values were defined as the lowest concentration of the test substance where no visible growth can be observed, i.e. the background-normalized optical density of the culture at 600 nm was below 0.05.
4.5. In vitro cytotoxicity measurements
Cytotoxicity of compound 11a was determined in MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay [32] with a few modifications. HepG2 (ATCC) and HUVEC (ATCC) cells were cultured in Eagle's MEM medium supplemented with l-glutamine (2 mM), penicillin/streptomycin (100 UI/mL/100 μg/mL), and 10% FBS. The cells were incubated in a humidified atmosphere with 5% CO2 at 37 °C.
The cells were seeded in 96-well plates at densities 2000 cells per well in 100 μL of growth medium and incubated for 24 h to attach onto the wells. 50 μL of compounds at 50 μM in DMSO (0.5% final concentration) were added and incubated for 72 h. After 72 h 10 μL of CellTiter96® Aqueous One Solution Reagent, (Promega) [33] was added to determine the number of viable cells. The plates were incubated for another 3 h and absorbance (490 nm) was read with a BioTek's Synergy H4 microplate reader. Etoposide [IC50 = 20.1 μM (Ref. [34]: 30.2 μM) for HepG2 and IC50 = 5.21 μM (Ref. [35]: 1.25 μM) for HUVEC] at 50 μM was used as a positive control and 0.5% DMSO as a vehicle control. To determine cell viability, results from the wells that contained test compound-treated cells were compared to those with cells incubated in 0.5% DMSO. Independent experiments were run in triplicate and repeated two times. Statistical significance (p < 0.05) was calculated with two-tailed Welch's t-test between treated groups and DMSO. On HUVEC cell line, where compound showed higher activity, IC50 value (concentration of compound that inhibits cell proliferation by 50%) was determined using six concentrations of the tested compound. GraphPad Prism 5.0 software [36] was used to calculate the IC50 value.
4.6. Molecular modeling
4.6.1. Protein and ligand preparation
3D compound models were built using ChemBio3D Ultra 16.0 [37]. MMFF94 force field [38] was used for the optimization of geometries and partial atomic charges were added. Energy was minimized to less than 0.001 kcal/(mol Å) gradient value. The structure was refined with GAMESS interface using PM3 method, QA optimization algorithm and Gasteiger Hückel charges for all atoms for 100 steps [37]. GOLD Suite v5.4 [23,39] was used for molecular docking calculations. GOLD graphical user interface was used for receptor preparation. To the protein hydrogen atoms were added and correct tautomers and protonation states were assigned. Except for HOH614, all water molecules and ligands were deleted from the crystal structure. Amino acid residues within 7 Å around the ligand (PDB entry: 4DUH [22]) were selected as the binding site.
4.6.2. Ligand docking
Compounds were docked to the defined binding site in 25 independent genetic algorithm (GA) runs by applying different GA parameters (population size = 100, selection pressure = 1.1, number of operations = 100,000, niche size = 2, number of islands = 5, mutation frequency = 95, crossover frequency = 95, migration frequency = 10) and scoring functions (GoldScore, ChemScore, CHEMPLP). The most representative results were obtained using GoldScore as a scoring function. Ligands with RMSD value less than 1.5 Å were joined in clusters and early termination was allowed if the top 3 solutions were within 1.0 Å of the RMSD value. Proposed binding modes of the top 5 highest scored docking poses were evaluated for each ligand and the highest scored pose was used for graphical representation with PyMOL [24].
4.7. Chemistry
Chemicals were obtained from Acros Organics (Geel, Belgium), Sigma-Aldrich (St. Louis, MO, USA) and Apollo Scientific (Stockport, UK) and used without further purification. Analytical TLC was performed on silica gel Merck 60 F254 plates (0.25 mm), using visualization with UV light and spray reagents. Column chromatography was carried out on silica gel 60 (particle size 240–400 mesh). HPLC analyses were performed on an Agilent Technologies 1100 instrument (Agilent Technologies, Santa Clara, CA, USA) with a G1365B UV–Vis detector, a G1316A thermostat, a G1313A autosampler, and a ChemStation data system or on a Thermo Scientific Dionex Ultimate 3000 Binary Rapid Separation LC System (Thermo Fisher Scientific, Waltham, MA, USA) with an autosampler, a binary pump system, a photodiode array detector, a thermostated column compartment, and a Chromeleon Chromatography Data System. The eluent for consisted of trifluoroacetic acid (0.1% in water) or 20 mM phosphate buffer (pH 6.8) as solvent A and acetonitrile as solvent B. Two methods were used, method A: Agilent Eclipse Plus C18 column (5 μm, 4.6 mm × 150 mm), mobile phase of 30–90% of acetonitrile in TFA (0.1%) in 16 min, 90% acetonitrile to 20 min, a flow rate of 1.0 mL/min and a sample injection volume of 10 μL, and method B: Phenomenex Luna C18 column (5 μm, 4.6 mm × 250 mm), mobile phase of 50–80% of acetonitrile in 20 mM phosphate buffer (pH 6.8) in 30 min, a flow rate of 1.0 mL/min and a sample injection volume of 10 μL. Melting points were determined on a Reichert hot stage microscope and are uncorrected. 1H and 13C NMR spectra were recorded at 400 and 100 MHz, respectively, on a Bruker AVANCE III 400 spectrometer (Bruker Corporation, Billerica, MA, USA) in DMSO-d6, CDCl3 or acetone–d6 solutions, with TMS as the internal standard. IR spectra were recorded on a Thermo Nicolet Nexus 470 ESP FT-IR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Mass spectra were obtained using a Q-TOF Premier mass spectrometer (Micromass, Waters, Manchester, UK) or ADVION expression CMSL mass spectrometer (Advion Inc., Ithaca, USA). Optical rotations were measured on a Perkin-Elmer 241 MC polarimeter. The reported values for specific rotation are average values of 10 successive measurements using an integration time of 5 s. The purity of the tested compounds was ≥95% as established by HPLC.
4.8. Synthetic procedures
4.8.1. General procedure A. Synthesis of Compounds 2 and 17 (with 2 as an Example)
To a suspension of 3-hydroxy-4-nitrobenzoic acid (1) (5.00 g, 27.3 mmol) in methanol (150 mL) cooled on ice bath thionyl chloride (5.90 mL, 81.9 mmol) was added dropwise. The mixture was stirred at rt for 15 h upon which a clear solution formed. The solvent was evaporated under reduced pressure. To the residue petroleum ether (50 mL) was added, the obtained suspension was sonicated, filtered and washed with petroleum ether (20 mL). The purification was repeated twice and the residue was dried to give compound 2 (4.92) as yellow crystals.
4.8.1.1. Methyl 3-hydroxy-4-nitrobenzoate (2) [40]
Yellow crystals; yield 92% (4.92 g); mp 87–88 °C (86–88 °C, lit [40]). IR (ATR) ν = 3309, 3053, 2961, 1720, 1623, 1586, 1434, 1321, 1220, 1146, 1097, 966, 842, 798, 743, 667 cm−1. 1H NMR (400 MHz, CDCl3) δ 3.99 (s, 3H, CH3), 7.64 (dd, 1H, 3J = 8.8 Hz, 4J = 1.6 Hz, Ar-H-6), 7.86 (d, 1H, 4J = 1.6 Hz, Ar-H-2), 8.20 (d, 1H, 3J = 8.8 Hz, Ar-H-5), 10.53 (s, 1H, OH).
4.8.2. General procedure B. Synthesis of Compounds 3a and 18c (with 3a as an Example)
To a stirred solution of compound 2 (3.00 g, 15.2 mmol) and triphenylphosphine (5.40 g, 20.6 mmol) in anhydrous tetrahydrofuran (30 mL) isopropanol (1.5 mL, 19.8 mmol) was added and the mixture was stirred at rt for 10 min. Diisopropyl azodicarboxylate (DIAD, 3.90 mL, 19.8 mmol) was added dropwise and the mixture was stirred at rt for 15 h under argon atmosphere. The solvent was evaporated under reduced pressure and the crude product was purified with flash column chromatography using ethyl acetate/petroleum ether (1/6) as an eluent to give 3a (3.50 g) as yellow solid.
4.8.2.1. Methyl 3-isopropoxy-4-nitrobenzoate (3a)
Yellow solid; yield 97% (3.50 g); mp 42–44°C (39–41 °C, lit [41]). IR (ATR) ν = 3117, 2989, 2950, 1724, 1610, 1525, 1422, 1284, 1103, 1004, 942, 835, 741 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.30 (d, 6H, 3J = 6.0 Hz, CH(CH3)2), 3.91 (s, 3H, COOCH3), 4.93 (spt, 1H, 3J = 6.0 Hz, CH(CH3)2), 7.64 (dd, 1H, 3J = 8.4 Hz, 4J = 1.6 Hz, Ar-H-6), 7.78 (d, 1H, 4J= 1.2 Hz, Ar-H-2), 7.95 (d, 1H, 3J = 8.0 Hz, Ar-H-5).
4.8.3. General procedure C. Synthesis of Compounds 4a-b and 19a-c (with 4a as an Example)
To the solution of compound 3a (3.50 g, 14.6 mmol) in methanol (30 mL) 1 M NaOH (28 mL, 29.3 mmol) was added. The mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure and the residue acidified with 1 M HCl (10 mL). Water phase was extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed with water (3 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4, filtered and the solvent removed under reduced pressure to afford 4a (3.00 g) as yellow solid.
4.8.3.1. 3-Isopropoxy-4-nitrobenzoic acid (4a)
Yellow solid; yield 91% (3.00 g); mp 170–173 °C (173–175 °C, lit [41]). IR (ATR) ν = 1987, 2838, 2603, 1689, 1522, 1427, 1296, 110, 937, 837, 742 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 0.86 (d, 6H, 3J = 6.0 Hz, CH(CH3)2), 4.46 (spt, 1H, 3J = 6.0 Hz, CH(CH3)2), 7.17 (dd, 1H, 3J = 8.0 Hz, 4J = 1.6 Hz, Ar-H-6), 7.32 (d, 1H, 4J = 1.6 Hz, Ar-H-2), 7.48 (d, 1H, 3J = 8.4 Hz, Ar-H-5), 13.21 (s, 1H, COOH).
4.8.4. General procedure D. Synthesis of Compounds 5a-g, 13a-c and 20a-b (with 5a as an Example)
To the suspension of 4a (800 mg, 3.55 mmol) and TBTU (1.48 g, 4.62 mmol) in dichloromethane (40 mL) N-methylmorpholine (1.2 mL, 10.7 mmol) was added. The reaction mixture was stirred at rt for 30 min upon which a clear solution formed. l-valine methyl ester hydrochloride (715 mg, 4.26 mmol) was added and the mixture was stirred at rt for 15 h. The solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate (30 mL) and water (30 mL). The organic phase was washed with saturated aqueous NaHCO3 solution (3 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4, filtered and the solvent evaporated under reduced pressure to give 5a (1.11 g) as yellow oil.
4.8.4.1. Methyl (3-isopropoxy-4-nitrobenzoyl)-l-valinate (5a)
Yellow oil; yield 92% (1.11 g); -8.32 (c 0.263, MeOH). IR (ATR) ν = 3279, 2974, 1744, 1640, 1533, 1317, 1256, 1103, 1021, 842, 748 cm−1. 1H NMR (400 MHz, CDCl3) δ 1.00–1.04 (m, 6H, CHCH(CH3)2), 1.42 (d, 6H, 3J = 6 Hz, OCH(CH3)2), 2.27–2.35 (m, 1H, CHCH(CH3)2), 3.81 (s, 3H, COOCH3), 4.75–4.82 (m, 2H, CHCH(CH3)2, OCH(CH3)2), 6.71 (d, 1H, 3J = 8.4 Hz, NH), 7.31 (dd, 1H, 3J = 8.4 Hz, 4J = 1.6 Hz, Ar-H-6), 7.62 (d, 1H, 4J = 1.2 Hz, Ar-H-2), 7.81 (d, 1H, 3J = 8.0 Hz, Ar-H-5). 13C NMR (100 MHz, DMSO-d6) δ 19.01 (CH3), 19.10 (CH3), 21.50 (CH(CH3)2), 21.56 (CH(CH3)2), 29.60 (CH), 51.75 (COOCH3), 58.77 (CH), 72.43 (CH), 115.32, 119.76, 124.62, 138.42, 142.29, 149.61, 165.28 (CONH), 171.97 (COOCH3).
4.8.5. General procedure E. Synthesis of Compounds 6a-g, 14b-c and 21a-b (with 6a as an Example)
Compound 5a (1.11 g, 3.28 mmol) was dissolved in methanol (40 mL), Pd/C (111 mg) was added and the reaction mixture was stirred under hydrogen atmosphere for 3 h. The catalyst was filtered off and the solvent removed under reduced pressure.
4.8.5.1. Methyl (4-amino-3-isopropoxybenzoyl)-l-valinate (6a)
The crude product was purified with flash column chromatography using ethyl acetate/petroleum ether (1/1) as an eluent to obtain 6a (522 mg) as yellow oil. Yield: 55% (522 mg). -5.53 (c 0.378, MeOH). 1H NMR (400 MHz, CDCl3) δ 0.99–1.03 (m, 6H, CHCH(CH3)2), 1.38 (d, 6H, 3J = 6.4 Hz, OCH(CH3)2), 2.23–2.31 (m, 1H, CHCH(CH3)2), 3.79 (s, 1H, COOCH3), 4.11–4.17 (m, 2H NH2), 4.66 (spt, 1H, 3J = 6.0 Hz, OCH(CH3)2), 4.76–4.79 (m, 1H, CHCH(CH3)2), 6.50 (d, 1H, 3J = 8.8 Hz, NH), 6.70 (d, 1H, 3J = 8.0 Hz, Ar-H-5), 7.20 (dd, 1H, 3J = 8.0 Hz, 4J = 2.0 Hz, Ar-H-6), 7.40 (d, 1H, 4J = 2.0 Hz, Ar-H-2). MS (ESI) m/z = 331.37 ([M+Na]+).
4.8.6. General procedure F. Synthesis of Compounds 7a-i, 15a-d and 22a-b (with 7a as an Example)
To a solution of 4,5-dibromo-1H-pyrrole-2-carboxylic acid (157 mg, 0.584 mmol) in anhydrous dichloromethane (4 mL) oxalyl chloride (2 M solution in dichloromethane, 1.46 mL, 2.92 mmol) was added dropwise and the solution stirred at rt for 15 h under argon atmosphere. The solvent was evaporated under reduced pressure, fresh anhydrous dichloromethane (4 mL), 6a (150 mg, 0.487 mmol) and pyridine (2 mL) were added and the reaction mixture stirred under argon atmosphere at rt for 15 h. Solvent was removed under reduced pressure, the residue dissolved in ethyl acetate (20 mL) and washed with water (20 mL), saturated aqueous NaHCO3 solution (3 × 20 mL) and brine (2 × 20 mL). The organic phase was dried over Na2SO4, filtered and the solvent removed under reduced pressure.
4.8.6.1. Methyl (4-(4,5-dibromo-1H-pyrrole-2-carboxamido)-3-isopropoxybenzoyl)-l-valinate (7a)
To the crude product ethyl acetate and petroleum ether (1:2, 15 mL) were added and the precipitate was filtered off to obtain 7a (40 mg) as light brown solid. The mother liquid was concentrated and purified with flash column chromatography using ethyl acetate/petroleum ether (1/2) as an eluent to give another 61 mg of 7a as light brown solid. Yield 37% (101 mg); mp 145–148 °C +8.94 (c 0.273, MeOH). IR (ATR) ν = 3417, 3304, 3238, 2964, 1741, 1633, 1515, 1418, 1311, 1202, 1124, 978, 831, 743 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 0.94–1.00 (m, 6H, CHCH(CH3)2), 1.32–1.35 (m, 6H, OCH(CH3)2), 2.15–2.23 (m, 1H, CHCH(CH3)2), 3.67 (s, 3H, COOCH3), 4.30 (t, 1H, 3J = 8.0 Hz, CHCH(CH3)2), 4.72 (spt, 1H, 3J = 6.0 Hz, OCH(CH3)2), 7.18 (d, 1H, 4J = 2.4 Hz, Pyrr-CH), 7.53–7.55 (m, 2H, Ar-H-2,6), 7.92 (d, 1H, 3J = 8.4 Hz, Ar-H-5), 8.59 (d, 1H, 3J = 8.0 Hz, CONHCH), 9.06 (s, 1H, CONHAr), 13.02 (d, 1H, 4J = 2.8 Hz, Pyrr-NH). 13C NMR (100 MHz, DMSO-d6) δ 19.06 (CHCH(CH3)2), 19.09 (CHCH(CH3)2), 21.59 (OCH(CH3)2), 21.67 (OCH(CH3)2), 29.45 (CHCH(CH3)2), 51.55 (COOCH3), 58.59 (CHCH(CH3)2), 71.29 (OCH(CH3)2), 98.35, 106.22, 113.18, 113.77, 120.11, 122.44, 127.50, 130.06, 130.50, 148.15, 156.95, 166.15 (Ar-CONH), 172.26 (COOCH3). MS (ESI) m/z = 556.0 ([M-H]-), HRMS for C21H24Br2N3O5: calculated 556.0083, found 556.0076. HPLC: tr 14.108 min (97.2% at 280 nm, method A).
4.8.7. General procedure G. Synthesis of Compounds 8a-e, 8g-i and 23a-d (with 8a as an Example)
To the solution of 7a (65 mg, 0.116 mmol) in tetrahydrofuran (8 mL) 1 M NaOH (348 μL, 348 μmol) was added and the mixture was stirred at rt for 15 h. The solvent was evaporated under reduced pressure, the residue acidified with 1 M HCl (5 mL) to pH 1 and the water phase was extracted with ethyl acetate (3 × 10 mL). The combined organic phases were washed with water (3 × 10 mL) and brine (10 mL), dried over Na2SO4, filtered and the solvent removed under reduced pressure to obtain 8a (53 mg) as white solid.
4.8.7.1. (4-(4,5-Dibromo-1H-pyrrole-2-carboxamido)-3-isopropoxybenzoyl)-l-valine (8a)
White solid; yield 87% (53 mg); mp 116–120 °C; +18.6 (c 0.288, MeOH). IR (ATR) ν = 3402, 3183, 2970, 1716, 1648, 1507, 1389, 1265, 1179, 1109, 977, 805, 751 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 0.96–1.00 (m, 6H, CHCH(CH3)2), 1.32–1.36 (m, 6H, OCH(CH3)2), 2.15–2.22 (m, 1H, CHCH(CH3)2), 4.30 (t, 1H, 3J = 8.0 Hz, CHCH(CH3)2), 4.73 (spt, 1H, 3J = 5.6 Hz, OCH(CH3)2), 7.18 (d, 1H, 4J = 2.4 Hz, Pyrr-CH), 7.53–7.57 (m, 2H, Ar-H-2,6), 7.92 (d, 1H, 3J = 8.0 Hz, Ar-H-5), 8.42 (d, 1H, 3J = 8.0 Hz, CONHCH), 9.05 (s, 1H, CONHAr), 12.62 (br s, 1H, COOH), 13.02 (s, 1H, Pyrr-NH). 13C NMR (100 MHz, DMSO-d6) δ 18.89 (CHCH(CH3)2), 19.34 (CHCH(CH3)2), 21.67 (OCH(CH3)2), 21.76 (OCH(CH3)2), 29.52 (CHCH(CH3)2), 58.38 (CHCH(CH3)2), 71.36 (OCH(CH3)2), 98.41, 106.28, 113.28, 113.82, 120.16, 122.53, 127.60, 130.43, 130.46, 148.22, 157.03, 166.13 (Ar-CONH), 173.22 (COOH). MS (ESI) m/z = 542.0 ([M-H]-), HRMS for C20H22Br2N3O5: calculated 541.9926, found 541.9935. HPLC: tr 12.049 min (98.1% at 280 nm, method A).
4.8.8. General procedure H. Synthesis of Compounds 9a-e (with 9a as an Example)
The starting compound (7h, 40 mg, 0.057 mmol) was dissolved in 4M HCl in 1,4-dioxane (4 mL) and tetrahydrofuran (1 mL) and the reaction mixture stirred at rt for 2 h. The solvent was removed under reduced pressure, to the residue diethyl ether (10 mL) was added, the obtained suspension was sonicated, filtered, washed with diethyl ether (2 × 2 mL) and dried to give 9a (36 mg) as white solid.
4.8.8.1. (S)-2-(2-(4,5-Dibromo-1H-pyrrole-2-carboxamido)-5-((1-methoxy-1-oxo-3-phenylpropan-2-yl)carbamoyl)phenoxy)ethan-1-aminium chloride (9a)
White solid; yield 99% (36 mg); mp 146–148 °C; -48.7 (c 0.252, MeOH). IR (ATR) ν = 3394, 3326, 3222, 3029, 2951, 2859, 1723, 1649, 1509, 1414, 1277, 1179, 1021, 972, 816, 746, 700 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 3.09–3.21 (m, 2H, CH2Ph), 3.38–3.43 (m, 2H, OCH2CH2NH, overlapping with the signal for water), 3.65 (s, 3H, CH3), 4.30 (t, 2H, 3J = 4.4 Hz, OCH2CH2NH), 4.64–4.70 (m, 1H, CH), 7.18–7.36 (m, 6H, 5 × Ar-H, Pyrr-CH), 7.48 (d, 1H, 4J = 1.6 Hz, Ar-H), 7.51 (dd, 1H, 3J = 8.4 Hz, 4J = 1.6 Hz, Ar-H), 8.03 (d, 1H, 3J = 8.4 Hz, Ar-H), 8.25 (s, 3H, ), 8.89 (d, 1H, 3J = 8.0 Hz, CONHCH), 9.44 (s, 1H, CONHAr), 13.10 (d, 1H, 4J = 2.4 Hz, Pyrr-NH). 13C NMR (100 MHz, DMSO-d6) δ 36.25 (CH2), 38.35 (CH2), 51.95 (CH3), 54.33 (CH), 64.84 (CH2), 98.41, 106.29, 110.57, 115.03, 120.24, 122.13, 126.49, 127.47, 128.24, 129.07, 129.51, 129.53, 137.66, 147.98, 157.29, 165.54 (Ar-CONH), 172.21 (COOH). MS (ESI) m/z = 605.0 ([M-H]-). HRMS for C24H23Br2N4O5: calculated 605.0035, found 605.0018. HPLC: tr 8.015 min (95.0% at 220 nm, 95.3% at 280 nm, method A).
4.8.9. General procedure I. Synthesis of Compounds 10a-b (with 10a as an Example)
To the solution of 7e (180 mg, 0.395 mmol) in anhydrous methanol and anhydrous tetrahydrofurane (3:1, 16 mL) hydrazine monohydrate (240 μL, 3.95 mmol) was added and the reaction mixture stirred at 65 °C for 15 h. The obtained suspension was cooled on ice bath, the precipitate filtered off and dried to give 10a (104 mg) as white solid.
4.8.9.1. (S)-3,4-Dichloro-N-(4-((1-hydrazineyl-1-oxopropan-2-yl) carbamoyl)-2-isopropoxyphenyl)-5-methyl-1H-pyrrole-2-carboxamide (10a)
White solid; yield 58% (104 mg); mp 235–237 °C +87.9 (c 0.220, DMF). IR (ATR) ν = 3397, 3365, 3310, 3252, 3134, 2984, 2940, 1733, 1648, 1518, 1441, 1324, 1261, 1173, 1042, 990, 831, 747, 702 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.32–1.38 (m, 9H, CH3, CH(CH3)2), 2.24 (s, 3H, Pyrr-CH3), 4.23 (s, 2H, NH2), 4.47 (quint, 1H, 3J = 7.2 Hz, CONHCH), 4.88 (spt, 1H, 3J = 6.0 Hz, CH(CH3)2), 7.56 (dd, 1H, 3J = 8.4 Hz, 4J = 1.6 Hz, Ar-H-6), 7.64 (d, 1H, 4J = 1.6 Hz, Ar-H-2), 8.44–8.48 (m, 2H, Ar-H-5, CONHCH), 9.18 (s, 1H, NHNH2), 9.25 (s, 1H, CONHAr), 12.45 (s, 1H, Pyrr-NH); 13C NMR (100 MHz, DMSO-d6) δ 10.75 (Pyrr-CH3), 18.16 (CH3), 21.78 (CH(CH3)2), 21.84 (CH(CH3)2), 47.67 (CH), 71.28 (CH(CH3)2), 108.58, 109.67, 111.89, 117.87, 118.66, 120.65, 128.92, 129.75, 131.01, 145.05, 156.19, 165.22 (Ar-CONH), 171.77 (CONHNH2). MS (ESI) m/z = 454.1 ([M-H]-). HRMS for C19H22Cl2N5O4: calculated 454.1049, found 454.1041. HPLC: tr 9.111 min (95.8% at 254 nm, method A).
4.8.10. General procedure J. Synthesis of Compounds 11a-b (with 11a as an Example)
To the solution of 10a (83 mg, 0.172 mmol) in 1,4-dioxane and anhydrous dimethylformamide (2:1, 7.5 mL) 1,1′-carbonyldiimidazole (CDI, 55.8 mg, 0.344 mmol) was added and the reaction mixture was stirred at 101 °C for 15 h. Additional 0.5 equivalents of 1,1′-carbonyldiimidazole (14 mg, 0.0863 mmol) were added and the mixture was stirred at 101 °C for 1 h. The solvent was removed under reduced pressure.
4.8.10.1. (S)-3,4-Dichloro-N-(2-isopropoxy-4-((1-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)ethyl)carbamoyl)phenyl)-5-methyl-1H-pyrrole-2-carboxamide (11a)
To the residue acetonitrile (10 mL) was added, the obtained suspension was sonicated and filtered. To the precipitate water (10 mL) was added, the suspension was sonciated, filtered, washed with water (2 × 5 mL) and dried to obtain 11a (48 mg) as an off-white solid. Yield 58% (48 mg); mp 244–246°C; +37.9 (c 0.463, DMF). IR (ATR) ν = 3397, 3361, 3232, 3136, 2981, 2923, 2852, 1775, 1649, 1514, 1415, 1322, 1260, 1175, 1110, 917, 831, 759, 702 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.36–1.37 (m, 6H, CH(CH3)2), 1.49 (d, 3H, 3J = 6.8 Hz, CH3), 2.24 (s, 3H, Pyrr-CH3), 4.86 (spt, 1H, 3J = 6.0 Hz, CH(CH3)2), 5.11 (quint, 1H, 3J = 7.2 Hz, Hz, CONHCH), 7.55–7.61 (m, 2H, 2 × Ar-H), 8.47 (d, 1H, 3J = 8.4 Hz, Ar-H-5), 8.88 (d, 1H, 3J = 7.6 Hz, CONHCH), 9.26 (s, 1H, CONHAr), 12.27 (br s, 1H, oxadiazolone-NH/Pyrr-NH), 12.45 (s, 1H, oxadiazolone-NH/Pyrr-NH). 13C NMR (100 MHz, DMSO-d6) δ 10.76 (Pyrr-CH3), 16.87 (CH3), 21.77 (CH(CH3)2), 21.80 (CH(CH3)2), 41.73 (CH), 71.36 (CH(CH3)2), 108.60, 109.74, 111.65, 117.99, 118.63, 120.55, 128.28, 129.82, 131.35, 145.20, 154.89, 156.22, 157.23, 165.10 (Ar-CONH). MS (ESI) m/z = 480.1 ([M-H]-). HRMS for C20H20Cl2N5O5: calculated 480.0841, found 480.0848. HPLC: tr 12.275 min (95.2% at 220 nm, method A).
4.8.10.2. (S)-2-(2-(3,4-Dichloro-5-methyl-1H-pyrrole-2-carboxamido)-5-((1-(5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)ethyl) carbamoyl)phenoxy)ethan-1-aminium chloride (12)
Synthesized according to General procedure H with stirring the reaction mixture for 5 h. The product was additionally purified with reverse-phase flash chromatography on a Biotage Isolera One System using a Biotage SNAP Cartridge KP-C18-HS 12 g column and 15–60% acetonitrile in TFA (0.1%) as a mobile phase to afford 12 (5 mg) as an off white solid. Yield 16% (5 mg); mp 234–236 °C; +29.6 (c 0.180, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 1.50 (d, 3H, 3J = 7.6 Hz, CH3), 2.25 (s, 3H, Pyrr-CH3), 3.34 (2H, OCH2CH2, overlapping with the signal for water), 4.39 (t, 2H, 3J = 4.4 Hz, OCH2CH2), 5.11 (quint, 1H, 3J = 7.2 Hz, CONHCH), 7.61–7.62 (m, 2H, Ar-H-4,6), 8.24 (s, 3H, ), 8.41 (d, 1H, 3J = 8.8 Hz, Ar-H-3), 8.98 (d, 1H, 3J = 7.6 Hz, CONHCH), 9.26 (s, 1H, CONHAr), 12.31 (s, 1H, oxadiazole-NH/Pyrr-NH), 12.51 (s, 1H, oxadiazole-NH/Pyrr-NH). MS (ESI) m/z = 484.1 ([M-H]+). HRMS for C19H20Cl2N5O6: calculated 484.0791, found 484.0798. HPLC: tr 3.697 (97.8% pri 280 nm, method A).
4.8.10.3. 3-Isopropoxy-4-nitro-N-(pyridin-2-ylmethyl)benzamide (13a)
Synthesized according to General procedure D with pyridin-2-ylmethanamine (420 μL, 3.73 mmol) as a reagent. The crude product was purified with flash column chromatography using dichloromethane/methanol (20/1) as an eluent to give 13a (612 mg) as pale yellow solid. Yield 66% (612 mg); mp 73–75 °C. IR (ATR) ν = 3192, 2991, 2937, 1656, 1517, 1412, 1357, 1301, 1253, 1103, 1008, 960, 837, 751 cm−1. 1H NMR (400 MHz, CDCl3) δ 1.44 (d, 6H, 3J = 6.4 Hz, CH(CH3)2), 4.78 (d, 2H, 3J = 4.4 Hz, CH2), 4.82 (spt, 1H, 3J = 6.4 Hz, CH(CH3)2), 7.26–7.27 (m, 1H, NH/Ar-H-2), 7.29–7.30 (m, 1H, NH/Ar-H-2), 7.35 (d, 1H, 3J = 8.0 Hz, Ar-H-5), 7.40 (dd, 1H, 3J = 8.4 Hz, 4J = 1.6 Hz, Ar-H-6), 7.70–7.84 (m, 4H, 4 × Pyridine-H), 8.59–8.61 (d, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 21.58 (CH(CH3)2), 44.86 (CH2), 72.38 (CH(CH3)2), 114.91, 119.40, 121.12, 122.21, 124.76, 136.78, 138.76, 142.15, 148.91, 149.76, 158.28, 164.60 (CONH). MS (ESI) m/z = 314.38 ([M-H]-).
4.8.10.4. 4-Amino-3-isopropoxy-N-(pyridin-2-ylmethyl)benzamide (14a)
Synthesized according to General procedure E with stirring the reaction mixture for 4 h. Yellow oil; yield 99% (230 mg). IR (ATR) ν = 3464, 3289, 3170, 2978, 1622, 1546, 1506, 1303, 1225, 1110, 956, 825, 754, 608 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.29 (d, 6H, 3J = 6.0 Hz, CH(CH3)2), 4.52 (d, 2H, 3J = 6 Hz, CH2), 4.56 (spt, 1H, 3J = 6 Hz, CH(CH3)2), 5.22 (s, 2H, NH2), 6.64 (d, 1H, 3J = 8.0 Hz, Ar-H-5), 7.24–7.29 (m, 2H, 2 × Pyridine-H), 7.35 (dd, 1H, 3J = 8.0Hz, 4J = 2.0 Hz, Ar-H-6), 7.40 (d, 1H, 4J = 2.0 Hz, Ar-H-2), 7.73–7.77 (td, 1H, Pyridine-H), 8.49–8.51 (dq, 1H, Pyridine-H), 8.72 (t, 1H, 3J = 6.0 Hz, NH). 13C NMR (100 MHz, DMSO-d6) δ 21.96 (CH(CH3)2), 44.57 (CH2), 70.28 (CH(CH3)2), 112.55, 112.91, 120.83, 121.24, 121.33, 121.91, 136.61, 142.39, 143.09, 148.70, 159.43, 166.27 (CONH). MS (ESI) m/z = 284.39 ([M-H]-).
4.8.10.5. 4,5-Dibromo-N-(2-isopropoxy-4-((pyridin-2-ylmethyl)carbamoyl)phenyl)-1H-pyrrole-2-carboxamide (15a)
Synthesized according to General procedure F but a few seconds after the addition of pyridine a precipitate started to form which was filtered off and washed with methanol (10 mL) to give 15a (37 mg) as white solid. Yield 13% (37 mg); mp 253–255 °C. IR (ATR) ν = 3383, 2976, 2633, 1652, 1521, 1402, 1310, 1274, 1201, 1127, 967, 874, 753 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 1.34 (d, 6H, 3J = 6.0 Hz, CH(CH3)2), 4.62 (d, 2H, 3J = 5.6 Hz, CH2), 4.71 (spt, 1H, 3J = 6.0 Hz, CH(CH3)2), 7.18 (d, 1H, 4J = 2.4 Hz, Pyrr-CH), 7.36–7.39 (m, 1H, Pyridine-H), 7.43 (d, 1H, 3J = 7.6 Hz, Pyridine-H), 7.56 (dd, 1H, 3J = 8.0 Hz, 4J = 1.6 Hz, Ar-H-6), 7.62 (d, 1H, 4J = 1.6 Hz, Ar-H-2), 7.86–7.90 (td, 1H, Pyridine-H), 7.94 (d, 1H, 3J = 8.0 Hz, Ar-H-5), 8.57–8.58 (m, 1H, Pyridine-H), 9.05 (s, 1H, CONHAr), 9.16 (t, 1H, 3J = 5.6 Hz, CONHCH2), 13.03 (d, 1H, 4J = 2.0 Hz, Pyrr-NH). 13C NMR (100 MHz, DMSO-d6) δ 21.75 (CH(CH3)2), 44.72 (CH2), 71.29 (CH(CH3)2), 98.43, 106.30, 112.77, 113.83, 119.75, 120.96, 122.08, 122.60, 127.60, 130.39, 130.51, 136.72, 148.31, 148.82, 157.02, 158.84, 165.68 (Ar-CONH). MS (ESI) m/z = 533.0 ([M-H]-), HRMS for C21H19Br2N4O3: calculated 532.9824, found 532.9834. HPLC: tr 8.831 min (98.9% at 280 nm, method A).
4.8.10.6. Methyl 2-hydroxy-4-nitrobenzoate (17)
Synthesized according to General procedure A without purification with petroleum ether. The starting compound (16, 5.00 g, 25.7 mmol) was dissolved from the begining. Yellow solid; yield 89% (4.80 g); mp 97–99 °C (101–102 °C, lit [42]). IR (ATR) ν = 3114, 2961, 2868, 1676, 1519, 1434, 1339, 1250, 1197, 1070, 957, 902, 812, 786, 732 cm−1. 1H NMR (400 MHz, CDCl3) δ 4.05 (s, 3H, COOCH3), 7.73 (dd, 1H, 3J = 8.8 Hz, 4J = 2.4 Hz, Ar-H-5), 7.85 (d, 1H, 4J = 2.4 Hz, Ar-H-3), 8.05 (d, 1H, 3J = 8.4 Hz, Ar-H-6), 11.01 (s, 1H, OH).
4.8.10.7. Methyl 2-(benzyloxy)-4-nitrobenzoate (18a) [43]
To a solution of methyl 2-hydroxy-4-nitrobenzoate (17, 1.00 g, 5.06 mmol) and potassium carbonate (1.40 g, 10.1 mmol) in acetonitrile (40 mL) benzyl bromide (0.60 mL, 5.06 mmol) was added and the reaction mixture was stirred at 60°C for 15 h. The solvent was evaporated under reduced pressure, the residue was dissolved in ethyl acetate (40 mL) and water (40 mL), the organic phase was washed with brine (2 × 30 mL), dried over Na2SO4, filtered and the solvent removed under reduced pressure to give 18a (1.24 g) as yellow solid. Yield 85% (1.24g); mp 74–76°C; 1H NMR (400 MHz, CDCl3) δ 3.97 (s, 3H, COOCH3), 5.30 (s, 2H, CH2), 7.35–7.54 (m, 5H, 5 × Ar-H), 7.86–7.96 (m, 2H, 3 × Ar-H).
4.8.10.8. 2-(Benzyloxy)-4-nitrobenzoic acid (19a) [43]
Synthesized according to General procedure C. Yellow solid; yield 87% (1.02 g); mp 151–153 °C. IR (ATR) ν = 3061, 2941, 2865, 2648, 2537, 1678, 1522, 1345, 1244, 1082, 988, 949, 875, 811, 734 cm−1. 1H NMR(400 MHz, CDCl3) δ 5.42 (s, 2H, CH2), 7.45–7.52 (m, 5H, 5 × Ar-H′), 8.00 (dd, 1H, 3J = 8.4 Hz, 4J = 2.0 Hz, Ar-H-5), 8.03 (d, 1H, 4J = 2.0 Hz, Ar-H-3), 8.38 (d, 1H, 3J = 8.4 Hz, Ar-H-6), 10.56 (br s, 1H, COOH).
4.8.10.9. Methyl (2-(benzyloxy)-4-nitrobenzoyl)glycinate (20a)
Synthesized according to General procedure D with 2-(benzyloxy)-4-nitrobenzoic acid (19a, 1.00 g, 3.64 mmol) and glycine methyl ester hydrochloride (505 mg, 4.03 mmol) as reagents. The crude product was purified by adding petroleum ether, the obtained suspension was sonicated, filtered and washed with petroleum ether to give 20a (1.18 g) as yellow solid. Yield 85% (1.18 g); mp 119–122 °C. IR (ATR) ν = 3367, 3105, 2950, 2855, 1750, 1648, 1518, 1442, 1342, 1202, 1101, 989, 923, 838, 743 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 3.66 (s, 1H, COOCH3), 4.08 (d, 2H, 3J = 6.0 Hz, CONHCH2), 5.44 (s, 2H, OCH2Ph), 7.32–7.54 (m, 5H, 5 × Ar-H′), 7.85 (d, 1H, 3J = 8.4 Hz, Ar-H-6), 7.91 (dd, 1H, 3J = 8.4 Hz, 4J = 2.0 Hz, Ar-H-5), 7.98 (d, 1H, 4J = 2.0 Hz, Ar-H-3), 8.90 (t, 1H, 3J = 6.0 Hz, CONHCH2). 13C NMR (100 MHz, DMSO-d6) δ 41.25 (CH2), 51.81 (COOCH3), 70.35 (CH2), 108.46, 115.68, 127.33, 128.01, 128.52, 130.03, 131.09, 135.90, 149.35, 155.98, 164.44 (CONH), 169.93 (COOCH3). MS (ESI) m/z = 367.32 ([M+Na]+).
4.8.10.10. Methyl (4-amino-2-(benzyloxy)benzoyl)glycinate (21a)
A mixture of compound 20a (675 mg, 1.98 mmol) and SnCl2 (1.85 g, 16.9 mmol) in ethyl acetate (20 mL) and methanol (20 mL) was stirred at 55 °C for 15 h. The solvent was removed under reduced pressure and the residue dissolved in ethyl acetate (20 mL) and water (20 mL). The aqueous phase was basified with 1 M NaOH to pH 9 and extracted with ethyl acetate (2 × 20 mL). The combined organic phases were dried over Na2SO4, filtered and the solvent evaporated under reduced pressure. The crude product was purified with flash column chromatography using ethyl acetate/petroleum ether (2/1) as an eluent to give compound 21a (356 mg) as yellow oil. Yield 58% (356 mg). IR (ATR) ν = 3378, 3335, 3232, 2953, 1737, 1596, 1539, 1440, 1281, 1198, 1118, 1005, 825, 732 cm−1. 1H NMR (400 MHz, CDCl3) δ 3.70 (s, 1H, COOCH3), 4.16 (d, 2H, 3J = 5.6 Hz, CONHCH2), 4.33 (br s, 2H, NH2), 5.17 (s, 2H, CH2Ph), 6.31 (d, 1H, 4J = 2.0 Hz, Ar-H-3), 6.38 (dd, 1H, 3J = 8.4 Hz, 4J = 2.0 Hz, Ar-H-5), 7.35–7.48 (m, 5H, 5 × Ar-H′), 8.04 (d, 1H, 3J = 8.8 Hz, Ar-H-6), 8.29 (t, 1H, 3J = 4.4 Hz, CONHCH2). 13C NMR (100 MHz, DMSO-d6) δ 41.20 (CH2), 51.64 (COOCH3), 69.39 (CH2), 97.31, 106.47, 108.42, 127.34, 127.88, 128.51, 132.73, 136.61, 153.44, 158.06, 165.05 (CONH), 170.69 (COOCH3). MS (ESI) m/z = 337.33 ([M+Na]+).
4.8.10.11. Methyl (2-(benzyloxy)-4-(4,5-dibromo-1H-pyrrole-2-carboxamido)benzoyl)glycinate (22a)
Synthesized according to General procedure F. The crude product was purified by adding diethyl ether (15 mL) and methanol (5 mL), the obtained suspension was sonicated, filtered and washed with diethyl ether (2 × 10 mL) to obtain 22a (192 mg) as white solid. Yield 34% (192 mg); mp 103–106°C. IR (ATR) ν = 3421, 3370, 3306, 3129, 1736, 1627, 1593, 1520, 1332, 1217, 1076, 979, 836, 740 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 3.65 (s, 3H, COOCH3), 4.08 (d, 2H, 3J = 6.0 Hz, CONHCH2), 5.34 (s, 2H, OCH2Ph), 7.27 (d, 1H, 4J = 2.8 Hz, Pyrr-CH), 7.32–7.42 (m, 4H, 4 × Ar-H), 7.52–7.54 (m, 2H, 2 × Ar-H), 7.77–7.86 (m, 2H, 2 × Ar-H), 8.58 (t, 1H, 3J = 6.0 Hz, CONHCH2), 10.05 (s, 1H, Pyrr-CONH-Ar), 13.01 (d, 1H, 4J = 2.4 Hz, Pyrr-NH); 13C NMR (100 MHz, DMSO-d6) δ 41.26 (CH2), 51.65 (COOCH3), 69.74 (CH2), 98.19, 104.16, 106.53, 111.74, 114.17, 116.57, 127.34, 127.46, 127.90, 128.44, 131.58, 136.14, 142.86, 156.49, 157.39, 164.48 (Ar-CONH), 170.29 (COOCH3). MS (ESI) m/z = 562.0 ([M-H]-), HRMS for C22H18Br2N3O5: calculated 561.9613, found 561.9606. HPLC: tr 12.252 min (95.1% at 280 nm, method A).
4.8.10.12. (2-(Benzyloxy)-4-(4,5-dibromo-1H-pyrrole-2-carboxamido)benzoyl)glycine (23a)
Synthesized according to General procedure G with 2 equivalents of 1 M NaOH (140 μL, 140 μmol). White solid; yield 46% (18 mg); mp 195–198 °C. IR (ATR) ν = 3369, 3222, 2930, 1714, 1618, 1510, 1408, 1294, 1205, 1119, 1019, 842, 737 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 4.00 (d, 2H, 3J = 5.6 Hz, CONHCH2), 5.33 (s, 2H, CH2Ph), 7.26 (s, 1H, Pyrr-CH), 7.30–7.55 (m, 6H, 5 × Ar-H′, Ar-H-5), 7.77 (d, 1H, 4J = 2.0 Hz, Ar-H-3), 7.87 (d, 1H, 3J = 8.4 Hz, Ar-H-6), 8.49 (t, 1H, 3J = 5.6 Hz, CONHCH2), 10.03 (s, 1H, Pyrr-CONH-Ar), 12.68 (br s, 1H, COOH/Pyrr-NH), 13.00 (br s, 1H, COOH/Pyrr-NH); 13C NMR (100 MHz, DMSO-d6) δ 41.32 (CH2), 69.84 (CH2), 98.18, 104.16, 106.53, 111.75, 114.17, 116.62, 127.44, 127.48, 127.93, 128.43, 131.63, 136.07, 142.82, 156.51, 157.40, 164.23 (Ar-CONH), 171.21 (COOH). MS (ESI) m/z = 547.9 ([M-H]-), HRMS for C21H16Br2N3O5: calculated 547.9457, found 547.9454. HPLC: tr 10.497 min (97.5% at 280 nm, method A).
Supplementary Material
Supplementary Information. Full results of antibacterial activity of compounds, detailed experimental procedures, analytical data and NMR spectra of the representative compounds. This material is available free of charge via the internet.
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ejmech.2018.05.011.
Scheme 2.

Reagents and conditions: (a) hydrazine monohydrate, MeOH/THF, 65 °C, 15 h (for the synthesis of 10a) or 4 d (for the synthesis of 10b); (b) CDI, 1,4-dioxane/DMF, 100 °C, 15 h; (c) 4 M HCl in 1,4-dioxane, THF, rt, 5 h.
Acknowledgment
This work was supported by the Slovenian Research Agency (Grant No. P1-0208), Academy of Finland (Grant No. 277001, 304697 and 312503), grants from the European Research Council H2020-ERC-2014-CoG 648364 - Resistance Evolution (to C.P.), the Wellcome Trust (to C.P.), and GINOP (MolMedEx TUMORDNS) GINOP-2.3.2-15-2016-00020 and GINOP (EVOMER) GINOP-2.3.2-15-2016-00014 (to C.P.), the 'Lendület' Program of the Hungarian Academy of Sciences (to C.P.) and the PhD fellowship from the Boehringer Ingelheim Fonds (to Á.N.). We thank Dr. Dušan Žigon (Mass Spectrometry Center, Jožef Stefan Institute, Ljubljana, Slovenia) for recording mass spectra, Michaela Barančoková for the help with biochemical evaluation, and Heli Parviainen, Heidi Mäkkylä and Cristina Carbonell Duacastella for their technical assistance in the antibacterial assays. The authors thank Prof. Roger Pain for proofreading the manuscript.
Funding sources
The work was funded by the Slovenian Research Agency (Grant No. P1-0208), Academy of Finland (Grant No. 277001, 304697 and 312503), grants from the European Research Council (to C.P.), the Wellcome Trust (to C.P.), and GINOP (MolMedEx TUMORDNS) GINOP-2.3.2-15-2016-00020 and GINOP (EVOMER) GINOP-2.3.2-15-2016-00014 (to C.P.), the 'Lendület' Program of the Hungarian Academy of Sciences (to C.P.) and the PhD fellowship from the Boehringer Ingelheim Fonds (to Á.N.).
Abbreviations
- ATCC
American type culture collection
- ATR
attenuated total reflectance
- CDI
1,1′-carbonyldiimidazole
- CFU
colony-forming unit
- CLSI
Clinical and Laboratory Standards Institute
- DIAD
diisopropyl azodicarboxylate
- DTT
dithiothreitol
- FBS
fetal bovine serum
- GyrA
DNA gyrase A
- GyrB
DNA gyrase B
- HepG2
human hepatocellular carcinoma cell line
- HUVEC
human umbilical vein endothelial cells
- MH
Mueller Hinton
- MRSA
methicillin-resistant Staphylococcus aureus
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NMM
N-methylmorpholine
- ParC
topoisomerase IV subunit A
- ParE
topoisomerase IV subunit B
- PAβN
phenylalanine-arginine β-naphthylamide
- RA
residual activity
- TBTU
N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate
- topo IV
topoisomerase IV
- VRE
vancomycin-resistant Enterococci
Footnotes
Author contributions
The manuscript was written using contributions from all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest including any financial, personal or other relationships with other people or organizations.
References
- [1].Klahn P, Bronstrup M. New structural templates for clinically validated and novel targets in antimicrobial drug research and development. Curr Top Microbiol Immunol. 2016;398:365–417. doi: 10.1007/82_2016_501. [DOI] [PubMed] [Google Scholar]
- [2].Chan PF, Germe T, Bax BD, Huang J, Thalji RK, Bacque E, Checchia A, Chen D, Cui H, Ding X, Ingraham K, et al. Thiophene antibacterials that allosterically stabilize DNA-cleavage complexes with DNA gyrase. Proc Natl Acad Sci U S A. 2017;114:4492–4500. doi: 10.1073/pnas.1700721114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! an update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- [4].World Health Organisation. Global priority list of antibiotic-resistant to guide research, discovery, and development of new antibiotics. [Accessed 30 September 2017];2017 Available at, http://www.who.int/medicines/publications/WHO-PPL-Short_Summary_ 25Feb-ET_NM_WHO.pdf.
- [5].Collin F, Karkare S, Maxwell A. Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl Microbiol Biotechnol. 2011;92:479–497. doi: 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tomašič T, Mašič LP. Prospects for developing new antibacterials targeting bacterial type IIA topoisomerases. Curr Top Med Chem. 2014;14:130–151. doi: 10.2174/1568026613666131113153251. [DOI] [PubMed] [Google Scholar]
- [7].Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- [8].Oblak M, Kotnik M, Solmajer T. Discovery and development of ATPase inhibitors of DNA gyrase as antibacterial agents. Curr Med Chem. 2007;14:2033–2047. doi: 10.2174/092986707781368414. [DOI] [PubMed] [Google Scholar]
- [9].Savage VJ, Charrier C, Salisbury AM, Moyo E, Forward H, Chaffer-Malam N, Metzger R, Huxley A, Kirk R, Uosis-Martin M, Noonan G, et al. Biological profiling of novel tricyclic inhibitors of bacterial DNA gyrase and topoisomerase IV. J Antimicrob Chemother. 2016;71:1905–1913. doi: 10.1093/jac/dkw061. [DOI] [PubMed] [Google Scholar]
- [10].Azam MA, Thathan J, Jubie S. Dual targeting DNA gyrase B (GyrB) and topoisomerse IV (ParE) inhibitors: a review. Bioorg Chem. 2015;62:41–63. doi: 10.1016/j.bioorg.2015.07.004. [DOI] [PubMed] [Google Scholar]
- [11].Bisacchi GS, Manchester JI. A new-class antibacterial-almost. Lessons in drug discovery and development: a critical analysis of more than 50 years of effort toward ATPase inhibitors of DNA gyrase and topoisomerase IV. ACS Infect Dis. 2015;1:4–41. doi: 10.1021/id500013t. [DOI] [PubMed] [Google Scholar]
- [12].Silver LL. A gestalt approach to Gram-negative entry. Bioorg Med Chem. 2016;24:6379–6389. doi: 10.1016/j.bmc.2016.06.044. [DOI] [PubMed] [Google Scholar]
- [13].Gjorgjieva M, Tomašič T, Barančoková M, Katsamakas S, Ilaš J, Tammela P, Peterlin Mašič L, Kikelj D. Discovery of benzothiazole scaffold-based DNA gyrase B inhibitors. J Med Chem. 2016;59:8941–8954. doi: 10.1021/acs.jmedchem.6b00864. [DOI] [PubMed] [Google Scholar]
- [14].Trzoss M, Bensen DC, Li X, Chen Z, Lam T, Zhang J, Creighton CJ, Cunningham ML, Kwan B, Stidham M, Nelson K, et al. Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE), Part II: development of inhibitors with broad spectrum, Gram-negative antibacterial activity. Bioorg Med Chem Lett. 2013;23:1537–1543. doi: 10.1016/j.bmcl.2012.11.073. [DOI] [PubMed] [Google Scholar]
- [15].Zhang J, Yang Q, Romero JA, Cross J, Wang B, Poutsiaka KM, Epie F, Bevan D, Wu Y, Moy T, Daniel A, et al. Discovery of indazole derivatives as a novel class of bacterial gyrase B inhibitors. ACS Med Chem Lett. 2015;6:1080–1085. doi: 10.1021/acsmedchemlett.5b00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Durcik M, Tammela P, Barančoková M, Tomašič T, Ilaš J, Kikelj D, Zidar N. Synthesis and evaluation of N-phenylpyrrolamides as DNA gyrase B inhibitors. ChemMedChem. 2017;5 doi: 10.1002/cmdc.201700549. 201700549. [DOI] [PubMed] [Google Scholar]
- [17].Tari LW, Li X, Trzoss M, Bensen DC, Chen Z, Lam T, Zhang J, Lee SJ, Hough G, Phillipson D, Akers-Rodriguez S, et al. Tricyclic GyrB/ParE (TriBE) inhibitors: a new class of broad-spectrum dual-targeting antibacterial agents. PLoS One. 2013;8:e84409. doi: 10.1371/journal.pone.0084409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang J, Yang Q, Cross JB, Romero JA, Poutsiaka KM, Epie F, Bevan D, Wang B, Zhang Y, Chavan A, Zhang X, et al. Discovery of azaindole ureas as a novel class of bacterial gyrase B inhibitors. J Med Chem. 2015;58:8503–8512. doi: 10.1021/acs.jmedchem.5b00961. [DOI] [PubMed] [Google Scholar]
- [19].Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, Hergenrother PJ. Predictive compound accumulation rules yield a broad - spectrum antibiotic. Nature. 2017;545:299–304. doi: 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Basarab GS, Manchester JI, Bist S, Boriack-Sjodin PA, Dangel B, Illingworth R, Sherer BA, Sriram S, Uria-Nickelsen M, Eakin AE. Fragment-to-hit-to-lead discovery of a novel pyridylurea scaffold of ATP competitive dual targeting type II topoisomerase inhibiting antibacterial agents. J Med Chem. 2013;56:8712–8735. doi: 10.1021/jm401208b. [DOI] [PubMed] [Google Scholar]
- [21].Lassalas P, Gay B, Lasfargeas C, James MJ, Tran V, Vijayendran KG, Brunden KR, Kozlowski MC, Thomas CJ, Smith AB, 3rd, Huryn DM, et al. Structure property relationships of carboxylic acid isosteres. J Med Chem. 2016;59:3183–3203. doi: 10.1021/acs.jmedchem.5b01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brvar M, Perdih A, Renko M, Anderluh G, Turk D, Solmajer T. Structure-based discovery of substituted 4,5'-bithiazoles as novel DNA gyrase inhibitors. J Med Chem. 2012;55:6413–6426. doi: 10.1021/jm300395d. [DOI] [PubMed] [Google Scholar]
- [23].Barrish JC, Gordon E, Alam M, Lin PF, Bisacchi GS, Chen P, Cheng PTW, Fritz AW, Greytok JA, Hermsmeier MA, Humphreys WG, et al. Aminodiol HIV protease inhibitors .1. Design, synthesis, and preliminary SAR. J Med Chem. 1994;37:1758–1768. doi: 10.1021/jm00038a005. [DOI] [PubMed] [Google Scholar]
- [24].PyMOL. Delano Scientific LLC; San Francisco, CA: http://pymol.sourceforge.net. [Google Scholar]
- [25].Tomašič T, Katsamakas S, Hodnik Z, Ilaš J, Brvar M, Solmajer T, Montalvão S, Tammela P, Banjanac M, Ergović G, Anderluh M, et al. Discovery of 4,5,6,7-tetrahydrobenzo[1,2-d]thiazoles as novel DNA gyrase inhibitors targeting the ATP-binding site. J Med Chem. 2015;58:5501–5521. doi: 10.1021/acs.jmedchem.5b00489. [DOI] [PubMed] [Google Scholar]
- [26].Zidar N, Macut H, Tomašič T, Brvar M, Montalvão S, Tammela P, Solmajer T, Peterlin Mašić L, Ilaš J, Kikelj D. N-Phenyl-4,5-dibromopyrrolamides and N-phenylindolamides as ATP competitive DNA gyrase B inhibitors: design, synthesis, and evaluation. J Med Chem. 2015;58:6179–6194. doi: 10.1021/acs.jmedchem.5b00775. [DOI] [PubMed] [Google Scholar]
- [27].Baker KR, Jana B, Franzyk H, Guardabassi L. A high-throughput approach to identify compounds that impair envelope integrity in Escherichia coli. Antimicrob Agents Chemother. 2016;60:5995–6002. doi: 10.1128/AAC.00537-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tillotson G. A crucial list of pathogens. Lancet Infect Dis. 2017:30754–30755. doi: 10.1016/S1473-3099(17). [DOI] [PubMed] [Google Scholar]
- [29].Stokes JM, MacNair CR, Ilyas B, French S, Cote JP, Bouwman C, Farha MA, Sieron AO, Whitfield C, Coombes BK, Brown ED. Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat Microbiol. 2017;2 doi: 10.1038/nmicrobiol.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 inframe, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].ISO 20776–1:2006-Clinical laboratory testing and in vitro diagnostic test systems – Susceptibility testing of infectious agents and evaluation of performance of antimicrobial susceptibility test devices – Part 1: Reference method for testing the in vitro activity of antimicrobial agents against rapidly growing aerobic bacteria involved in infectious diseases. ISO; http://www.iso. org/iso/catalogue_detail.htm?csnumber=41630. [Google Scholar]
- [32].Garg G, Zhao H, Blagg BS. Design, synthesis, and biological evaluation of ring-constrained novobiocin analogues as Hsp90 C-terminal inhibitors. ACS Med Chem Lett. 2015;6:204–209. doi: 10.1021/ml5004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Promega Corporation; Madison, WI: https://worldwide.promega.com/ [Google Scholar]
- [34].Pingeaw R, Mandi P, Nantasenamat C, Prachayasittikul S, Ruchirawat S, Prachayasittikul V. Design, synthesis and molecular docking studies of novel N-benzenesulfonyl-1,2,3,4-tetrahydroisoquinoline-based triazoles with potent anticancer activity. Eur J Med Chem. 2014;81:192–203. doi: 10.1016/j.ejmech.2014.05.019. [DOI] [PubMed] [Google Scholar]
- [35].Budovská M, Pilátová M, Varinská L, Mojžiš J, Mezencev R. The synthesis and anticancer activity of analogs of the indole phytoalexins brassinin, 1-methoxyspirobrassinol methyl ether and cyclobrassinin. Bioorg Med Chem. 2013;21:6623–6633. doi: 10.1016/j.bmc.2013.08.020. [DOI] [PubMed] [Google Scholar]
- [36].GraphPad Software. San Diego, CA: https://www.graphpad.com/ [Google Scholar]
- [37].GAMESS interface, Chem3D 16.0, ChemOffice Professional 16.0 Suite, CambridgeSoft
- [38].Halgren TA. Merck molecular force field .1. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17:490–519. [Google Scholar]
- [39].Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- [40].Ekici OD, Li ZZ, Campbell AJ, James KE, Asgian JL, Mikolajczyk J, Salvesen GS, Ganesan R, Jelakovic S, Grutter MG, Powers JC. Design, synthesis, and evaluation of aza-peptide Michael acceptors as selective and potent inhibitors of caspases-2, -3, -6, -7, -8, -9, and -10. J Med Chem. 2006;49:5728–5749. doi: 10.1021/jm0601405. [DOI] [PubMed] [Google Scholar]
- [41].Plante J, Campbell F, Malkova B, Kilner C, Warriner SL, Wilson AJ. Synthesis of functionalised aromatic oligamide rods. Org Biomol Chem. 2008;6:138–146. doi: 10.1039/b712606a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wilder Smith AE, Frommel E. The preparation of some 1,3,4-oxadiazol-2-ols and oxadiazol-2-thiols active as tuberculostatics. Arzneimittelforschung. 1962;12:485–487. [PubMed] [Google Scholar]
- [43].Prabhakaran P, Azzarito V, Jacobs T, Hardie MJ, Kilner CA, Edwards TA, Warriner SL, Wilson AJ. Conformational properties of O-alkylated benzamides. Tetrahedron. 2012;68:4485–4491. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.