

Abstract
MicroRNAs (miRNAs) are small, non-coding RNAs of ~18-25 nucleotides that have gained extensive attention as critical regulators in complex gene networks including immune cell lineage commitment, differentiation, maturation, and maintenance of immune homeostasis and function. Many viruses encode miRNAs that directly downregulate the expression of factors of the innate immune system, which includes proteins involved in promoting apoptosis and recruitment. In this study, we examined the expression profiles of three previously identified viral miRNAs (v-miRs) from the human herpesvirus (HHV) family, HSV-1 (miR-H1), KSHV (miR-K12-3-3p), and HCMV (miR-US4) in healthy and diseased periodontal tissues and observed increased levels of v-miRs in diseased tissues. To understand the significance of this increase, we overexpressed v-miRs in human oral keratinocytes (HOK), a common target for various HHV, and analyzed the impact of miR-H1 and miR-K12-3-3p on the host transcriptome. More than 1300 genes were altered in HOK overexpressing miR-H1 and miR-K12-3-3p. Global pathway analysis of deregulated genes identified several key cellular pathways that may favor viral persistence. Using bioinformatic analysis, we predicted hundreds of potential v-miR binding sites on genes downregulated by miR-H1 and miR-K12-3-3p and validated three novel target v-miR sites suggesting widespread direct and indirect modualtion of numerous host genes/pathways by a single v-miR. Finally, in vitro HSV-1 infection assays showed that miR-H1 can regulate viral entry and infection in human oral keratinocytes (HOK). Overall, our results demonstrate clinical and functional relevance of pathogenic viral molecules viz., v-miRs that regulate both host and viral functions and may contribute to the pathogenesis of inflammatory oral diseases.
Keywords: Herpesvirus, viral microRNA, transcriptome, inflammation, gingival keratinocytes, HSV-1
Introduction
Human Herpesviruses (HHV) are enveloped, linear, double stranded DNA (~140-250 kb) viruses that are highly prevalent across the world [1,2]. Based on geographical location and socioeconomic status, some populations are up to 100% seropositive for one or more members of the HHV family [3–5]. A unique feature of HHV is their ability to infect and persist for a lifetime. This is attributed to several viral proteins that efficiently subvert adaptive immune responses and modulate cellular homeostasis including interfering with antigen processing/presentation and preventing immune-mediated removal of virus-infected cells [6–8]. Further, it is well documented that viruses are capable of reprogramming cellular functions by altering expression of various host genes [9,10].
The discovery of viral miRNAs (v-miRs) has revealed yet another layer of intricate host-pathogen interactions [11,12]. MicroRNAs (miRs) are small, non-coding RNAs that fine tune translational output and can regulate cellular homeostasis, immune activation, cell differentiation, apoptosis, pathogen responses, cancer, etc., [13–19]. Interestingly, HHV and polyoma viruses are predominant viruses that exhibit v-miR encoding potential. The presence of multiple v-miRs in the HHV genome may impart an evolutionary benefit that confers the ability to persist inside the host and evade the immune responses. However, the role of v-miRs in the context of oral inflammatory diseases is poorly explored.
The oral cavity is a highly populated niche for various microbes including bacteria, yeast, and viruses. Consistent with this, oral infections are predominantly of polymicrobial etiology. Although the bacterial component is believed to serve as the major etiological factor of microbial-induced diseases such as periodontal disease, emerging evidence suggests the presence of multiple viruses (primarily HHV) which may augment the progression and pathogenesis of oral disorders [20–23]. HHV have been isolated from various regions of the oral cavity further supporting this notion. Similarly, co-infection of HHV and bacteria in periodontal and surrounding tissues (endodontic lesions, periapical tissues, etc.) is well-reported [24–27]. High prevalence of HCMV, EBV, and HSV-1 was observed in chronic and aggressive periodontitis lesions [28,29], while HCMV and EBV have been isolated from apical periodontitis clinical samples [16,30]. Indeed, multiple studies have detected viruses in more than 90% of granulomas of symptomatic and large periapical lesions [31]. Their presence has been suggested to contribute to pathogenesis whereby HHV may trigger the release of tissue destructive cytokines, upregulating the growth of pathogenic bacteria, and initiate cytotoxic or immunopathologic events [11,13]. The evidence supporting a periodontopathologic role of herpesviruses is derived from association studies and immunology-based research, but the specific molecular mechanisms of how HHV may instigate or exacerbate periodontitis has not been identified.
The ability of v-miRs to simultaneously regulate both viral and host genes allows the pathogen to adapt according to cellular microenvironment. In regards to viral infection, HHV employs v-miRs to establish latency after initial infection, switching the lytic-latency cycle as well as viral tropism [32,33]. This is achieved by silencing viral genes involved in the process. For instance, multiple v-miRs (miR-H2, -H4 and -H6) encoded by HSV-1 suppress expression of viral transcripts required for establishing infection including ICP0, ICP34.5, ICP4, thus promoting viral latency [33–35]. KSHV encoded miR-K12-7 and miR-K12-9 regulate expression of ORF50 encoded Replication and Transcription Activator (RTA) which leads to suppression of early genes and prevents inappropriate entry into the lytic phase [36,37]. Similarly, diverse cellular targets of v-miRs have been identified. By regulating these genes, v-miRs can control autophagy, apoptosis, cell cycle regulation, innate and adaptive immune responses, and cellular reprogramming. EBV-miR-BART5-5p has been shown to regulate PUMA. Either depletion of this v-miR or induction of PUMA expression is adequate to prompt apoptosis [39]. HCMV-miR-UL112-3p and HCMV-miR-UL148D target toll-like receptor 2 (TLR2) and chemokine (C-C Motif) ligand 5 (CCL5) genes thereby inhibiting the secretion of proinflammatory cytokines and suppressing Natural Killer (NK) activation and proliferation [40; functions of v-miRs described here are listed in Supplementary Table 1]. Together, these v-miRs significantly contribute to immune subversion and thus play key role in viral persistence.
The role of v-miRs in augmenting disease severity and pathogenesis is becoming more apparent. Moreover, v-miRs can be utilized as potential biomarkers for disease progression. For example, patients with nasopharyngeal carcinoma show increased EBV miR-BART7-3p and miR-BART13 levels in plasma samples compared with control patients [41,42]. Lisboa et al., detected significantly higher levels of HCMV-miR-UL22A-5p in a cohort of solid organ transplant patients with CMV disease [43]. However, the role of v-miRs in oral diseases in not well understood and poorly studied. The only study identifying v-miRs in diseased oral tissues was recently reported from our lab [44].
In this study, we examined the expression of four candidate miRNAs viz., HSV1-miR-H1, HCMV-miR-US4, HCMV miR-UL70-3p and KSHV-miR-K12-3-3p-3p in gingival biopsies derived from subjects with periodontitis compared with healthy subjects. We observed significantly higher expression of v-miRs in diseased periodontal tissues. To characterize potential v-miR functions, we assessed the impact of miR-H1 and miR-K12-3-3p-3p on transcriptome-wide changes in HOKs. Using a combinatorial in silico and functional approach, we identified novel targets of v-miRs which may facilitate viral persistence and immune evasion. We also examined the impact of miR-H1 on HSV-1 entry and infection in HOK. Our results provide the first report that shed light on the clinical detection of v-miRs in periodontal tissues and their potential role in pathogenic mechanisms.
Materials and Methods
Study population and sample collection
This study was approved by the Ethics Committee at the University Autónoma de Nuevo León Facultad de Odontología and The University of Illinois at Chicago, College of Dentistry. Two cohorts of subjects presenting to the Postgraduate Periodontics Clinic at the Dental School of the Universidad Autonóma de Nuevo León were recruited for this study. Inclusion criteria included male and female patients ages 18 to 65 years and in good systemic health. Exclusion criteria included chronic disease (e.g. diabetes, hepatitis, renal failure, clotting disorders, HIV, etc.)., antibiotics therapy for any medical or dental condition within a month before screening, and subjects taking medications known to affect periodontal status (e.g. phenytoin, calcium channel blockers, cyclosporine). Gingival biopsy samples were collected from subjects (n=6/group) with healthy periodontal tissues or from subjects diagnosed with chronic periodontitis, immediately placed in RNALater (Qiagen, Gaithersburg, MD) and stored at −70 °C. Subjects with chronic periodontitis displayed probing depth ≥ 6mm with bleeding on probing and radiographic evidence of bone loss [45]. Healthy periodontal patients displayed probing depths ≤ 3mm, with no bleeding on probing and no radiographic evidence of bone loss.
Total RNA Isolation and Quantitative Real-Time PCR
Tissue samples were lysed using the TissueLyzer (Qiagen) and total RNA isolated using the miRNeasy (Qiagen) kit as we previously reported [44,47,48]. The miRNeasy kit was also used to lyse cell cultures. For mature miRNA quantification, miScript primers and miScript II RT Kit were purchased from Qiagen. 100ng total RNA was reverse transcribed according to manufacturer’s instruction. The reactions were run using miRNA specific primer and universal primer (both from Qiagen) in the PCR mix buffer containing SYBR Green (Roche, Indianapolis, IN). RNU6B was used as endogenous control. The Ct values of replicates were analyzed to calculate relative fold change using the delta-delta Ct method and the normalized values plotted as histograms with standard deviations (SD).
Primary gingival human oral keratinocyte (HOK) culture
Primary HOK were purchased from ScienCell Research Laboratories (human gingival epithelial cells, Carlsbad, CA). Cultures were tested for HOK markers by immunofluorescent methods using antibodies to cytokeratine-8, -18 and -19 and were negative for HIV-1, HBV, HCV, mycoplasma, bacteria, yeast and fungi. Cells were cultured using DermaLife K Keratinocyte Medium Complete Kit (Lifeline Cell Technology, Frederick, MD). Immortalized human gingival epithelial cells (kindly provided by Dr. Richard Lamont, University of Louisville, Kentucky) were cultured in flasks containing keratinocyte growth medium (Lifeline Cell Technology).
Detection of virus in tissue samples and cell cultures
The presence of viruses was assessed by PCR based genome detection method using HSV-1 and EBV detection kits (Norgen Biotek Corp, Ontario, Canada). PCR was performed using virus specific primers provided in the kit. Amplicons were electrophoresed on 2% agarose gel. A positive control, isolation control, and a PCR control were included in the reaction.
Transient miRNA transfections
Transient transfections were performed using Lipofectaine 2000 reagent (Life Technologies, San Diego, CA) according to manufacturer’s instructions. Red siGLO oligos (ThermoScientific, Waltham, MA) were used to determine transfection efficiency. After 36 hours, cells were harvested for RNA isolation. Cells were transfected with viral miRNA mimics at increasing concentrations of 10, 20 and 50 nM (to determine dose effects) for 36 hours. We noticed robust (~90%) transfection at 20 nM, therefore, we used the same concentration for most of the experiments unless specified. Controls consisted of mock transfected and control mimic (Qiagen) transfected cultures.
Microarray Analysis
We used Affymetrix HTA-2_0 arrays (Santa Clara, CA) for transcriptome profiling. RNA samples were labeled and hybridized according to standard WT (whole transcript) PLUS target labeling protocol recommended by the manufacturer. After hybridization, each image was analyzed for the following quality metrics: average signal present, signal intensity of species-specific house-keeping genes, relative signal intensities of labeling controls, and absolute signal intensities of hybridization controls.
Data was processed using Genomics Suite 6.6 Statistical Package (Partek, Inc., St. Louis, MO). The following parameters were applied for hybridization signal processing: Algorithm: RMA; Background Correction: RMA Background Correction; Normalization: Quantile Normalization; Log Probes using Base: 2; Probeset Summarization: Median Polish. ANOVA test was used to calculate significance of the differential expression in each comparison (miR-H1 vs control, miR-K12-3-3p vs control and control vs mock transfected). Differentially expressed transcripts were annotated according to Affymetrix ‘NetAffx Analysis Center’. The significance cut-off was at 0.05. Array data were in compliance with Minimum Information About a Microarray Experiment (MIAME) guidelines and deposited in the Gene Expression Omnibus public database under Accession Number GSE107005.
Pathway Analysis Using Ingenuity Pathways Analysis Software
Ingenuity pathway analysis software (IPA, Ingenuity® Systems, Redwood City, CA) was used to predict the networks and canonical pathways that are associated with the significantly expressed genes. This software collects information from a database built from published relationships between genes and their biological functions, mechanisms, canonical pathways, and networks. Significantly over- or under-expressed genes (p values <0.05) with relative expression values of ±1.25-fold change and their corresponding gene identifiers were imported into IPA and analyzed with the IPA Core Analysis Tool which identifies relationships, mechanisms, functions, and pathways relevant to a dataset. ‘Canonical pathways’, and ‘Networks’ were mainly used in this analysis. Figures were generated using the IPA Path Designer Graphical Representation. Gene symbols were mapped to their corresponding gene objects in the Ingenuity Pathways Knowledge Base.
Molecules are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). All edges are supported by at least one reference from the literature, from a textbook, or from canonical information stored in the Ingenuity Knowledge Base. The intensity of the node color indicates the degree of up- (red) or down- (green) regulation. Nodes are displayed using various shapes that represent the functional class of the gene product. Edges are displayed with various labels that describe the nature of the relationship between the nodes (see figure legends for details).
Luciferase reporter constructs and dual luciferase reporter assays
Genomic DNA were isolated from freshly prepared PBMCs using QIAamp DNA mini kit (Qiagen) according to manufacturer’s instructions. The 3′UTRs of predicted miRNA target genes were PCR amplified using Phusion Taq polymerase (NEB, Ipswich, MA). The amplified products were digested with restriction enzymes (Xho I and Not I) and ligated downstream to the luciferase reporter gene in psiCHECK™-2 vector (Promega Inc., Madison WI). The colonies were screened by restriction digestion and three positive clones for each gene were verified by DNA sequencing. Dual luciferase experiments were performed as previously described [16,17,49]. In brief, HEK293 and HOK cells were seeded at the density of 3 × 104 in DMEM supplemented with 10% fetal bovine serum. All transfections were performed in quadruplicate using 0.5 μL Lipofectamine 2000 (Invitrogen), 120 ng dual luciferase reporter plasmids, and a final concentration of 2 pmol of synthetic miRNA mimics (Thermo Fisher Scientific). For experimental controls, we used: 1) Empty vectors + viral miRNA mimics, and 2) reporter constructs + control mimic (Thermo Fisher Scientific). After 36 h post-transfection, cells were lysed in passive lysis buffer (Promega) and dual luciferase assays performed using the Lumat (Turner BioSystems, Sunnyvale, CA) luminometer. For each reporter 3′UTR construct, the Rluc/Fluc value obtained was normalized to the value obtained for psiCHECK™-2 no-insert control (EV) co-transfected with the same miRNA mimic. The values obtained were plotted as histograms, where EV is set at one.
HSV-1 Viral Entry and Infection Assay
HOK were grown in 96 or 48 well-plates to subconfluence and transfected with HSV-1 miR-H1 or control mimic. After 24 hours, cells were infected with HSV-1 (gL86) virus (β-galactosidase expressing recombinant HSV-1) at 5 or 10 MOI. Uninfected cells in the presence and absence of lipofectamine were used as additional controls. Six hours post-infection (hpi), β-galactosidase assays were performed using o-nitrophenyl-D-galactopyranoside (ImmunoPure ONPG; Pierce, Rockford, IL). For the soluble substrate, the enzymatic activity was measured at 410 nm using a microplate reader (Molecular Devices spectra MAX 190, Sunnyvale, CA).
For infection experiments, cells were infected with dual color recombinant HSV-1 strain KOS (expressing early protein ICP0-GFP and late protein gD-GFP) at 0.1 MOI. After 4 hours, media was replaced with fresh media and incubated for additional 36 hours. Cells were fixed with 2% PFA and nuclei were stained DAPI. Entire cell populations were used for mean fluorescence intensity calculations. Flow cytometry was performed on a CyAn ADP Analyzer (Beckman Coulter, Brea, CA) and data was analyzed using FlowJo analysis software. For florescence imaging, images were captured on EVOS florescence microscope (Thermo Fisher Scientific).
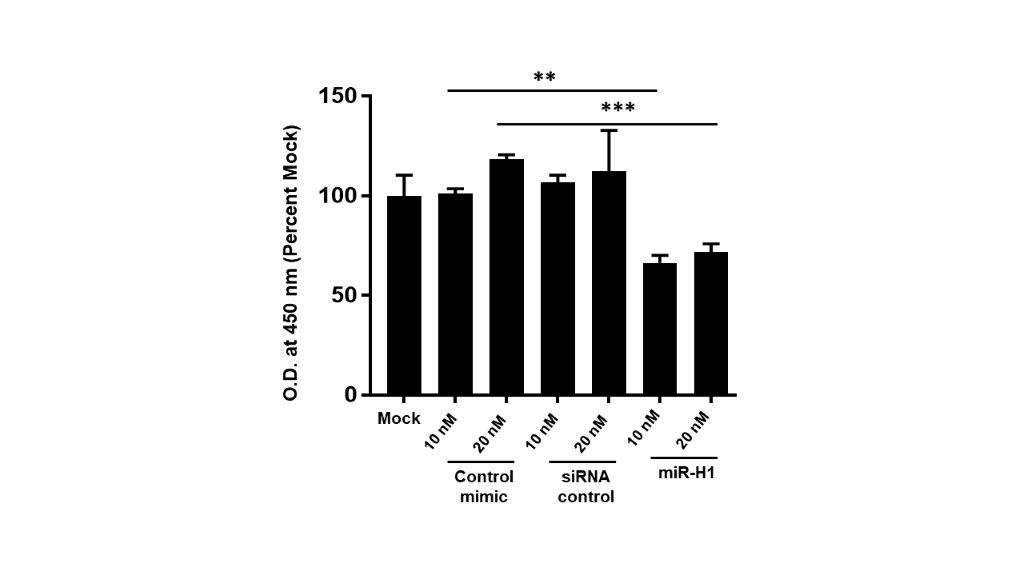
The impact of miRNA transfection on HOK viability was determined using the CellTiter 96 AQueous Cell Proliferation Assay Kit (Promega) according to manufacturer’s instructions. A total of 20 μl of MTS reagent was added to each well and incubated for two hours. Absorbance at 490 nm was monitored using the SpectraMax® M2 (Molecular Devices, Sunnyvale, CA, USA) plate reader.
Immunoblotting
Virally infected cell lysates were denatured in NuPAGE LDS Sample Buffer (Invitrogen, NP00007) and heated to 80°C for 10 min. Equal amounts of protein were added to 4%–12% SDS-PAGE gel and transferred to a nitrocellulose membrane. Nitrocellulose membrane was blocked in 5% nonfat milk in Tris-buffered saline (TBS) for 2 hr at room temperature. After the nonspecific binding blocking step was complete, membranes were incubated with mouse anti-gD monoclonal antibody (Abeam) at 1:1,000 dilution for overnight at 4°C. The following day, the blots were washed multiple times with 0.1 % TTBS (0.1% Tween 20 in TBS) before the addition of horseradish peroxidase conjugated anti-mouse IgG at dilutions of 1:25,000 at room temperature. Protein bands were visualized on an ImageQuant LAS 4000 imager (GE Healthcare) after the addition of SuperSignal West Pico maximum sensitivity substrate (Pierce, 34080). The density of the bands was quantified using ImageQuant TL image analysis software (version: 7). GAPDH was measured as a loading control.
Plaque assays
Monolayers of HOK cells were transfected with miR-H1 or control mimic. After overnight incubation, cells were infected with HSV-1 (KOS MOI 0.1) in Opti-MEM (Thermo Fisher Scientific). After 2 h incubation at 37°C, 5% CO2, inoculation solution was aspirated, cells were washed once with PBS, and complete media was added for 24 h. Culture supernatants were then collected, centrifuged at 13,000 rpm for 1 min, serially diluted in Opti-MEM, and overlayed on confluent monolayers of Vero cells in 24-well plates. After 2 h incubation, cells were washed, and complete DMEM containing 0.5% methylcellulose (Fisher Scientific) was added to cells for 48 to 72 h. To visualize and count plaques, cells were fixed with 100% methanol and stained with crystal violet solution.
Statistical Analysis
Data were analyzed on GraphPad Prism (Graphpad Software Inc., La Jolla, CA). The results were represented as standard deviation or ±SEM from three independent replicates and experiments were conducted at least thrice. P-values were calculated using Students t-test and p<0.05 were considered significant.
Results
Increased expression of herpes viral miRNAs in periodontitis
In our previous microarray study using tooth pulp tissue samples, we observed increased levels of four different v-miRs viz., miR-H1 (HSV-1), miR-K12-3-3p (KSHV), miR-UL70-3p and miR-US4 (both HCMV) in inflamed tooth pulps compared to corresponding healthy tissues [44]. Similar to dental pulps, HHV have been detected in tissue biopsies derived from periapical endodontic lesions, periodontitis and gingivitis lesions, as well as peri-implantitis lesions [24–27,49]. These observations indicate the likely presence of v-miRs in inflamed oral tissues. We therefore determined if miR-H1 (HSV-1), miR-K12-3-3p (KSHV) and miR-US4 (HCMV) are also present in gingival biopsies derived from healthy and periodontitis subjects. Our quantitative real-time PCR results confirm that miR-H1 was detected in all periodontitis lesions (Ct= 31.6±0.55), while none of the healthy tissue samples were positive (Ct cut off ≥35; Figure 1A). On the other hand, the expression values of miR-US4 were significantly lower in diseased (Ct=28.2±1.88) compared to healthy tissues (Ct=31.48±1.43; Figure 1B). Similarly, periodontitis lesions showed a higher prevalence for miR-K12-3-3p detection (Ct=31.35±1.7) compared to respective healthy controls (Ct=34.6±0.34) tissues indicating high expression of the viral miRNA (Figure 1C). Among all the v-miRs tested, miR-US4 expression levels were highest followed by miR-K12-3-3p and miR-H1.
Figure 1. Increased expression of viral miRNAs in diseased periodontal tissues.
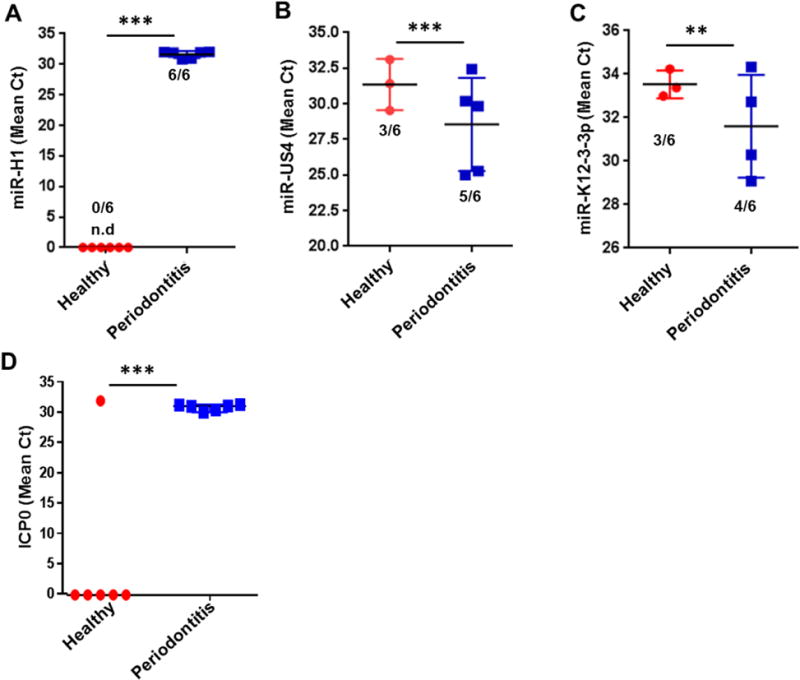
Expression analysis of candidate viral miRNAs in healthy and diseased gingival samples. Total RNA was isolated from healthy and diseased tissues (n=6 subjects/group). Expression of three viral miRNAs (A) miR-H1, (B) miR-K12-3-3p and (C) miR-US4 were detected by quantitative RT-PCR. (D) Quantification of HSV-1 encoded ICP0 transcript was assessed by RT-PCR in healthy and diseased gingival biopsies. Numbers of positive samples are mentioned for each group. Student’s t-test was used to calculate p-values. **p<0.01; ***p<0.001.
To examine if other viral components are also altered in diseased gingiva compared to healthy tissues, we quantified HSV-1 encoded early protein ICP0 transcript levels. Our results show that ICP0 was detected in all the periodontitis samples (Ct=30.53±0.49) while, only one healthy control (Ct=31.68) showed presence of transcript but other samples did not show any detectable level of ICP0 (Ct cut off ≥35; Figure 1D). This corroborates with the miR-H1 expression data (Figure 1A).
These results clearly indicate that induced expression of v-miRs (and other viral transcripts) is associated with inflammation. The detection of v-miRs in periodontitis subjects confirm our previous data and support our hypothesis that v-miRs are associated with oral inflammatory diseases.
Differential expression of genes in HSV miR-H1 and KSHV miR-K12-3-3p transfected HOK
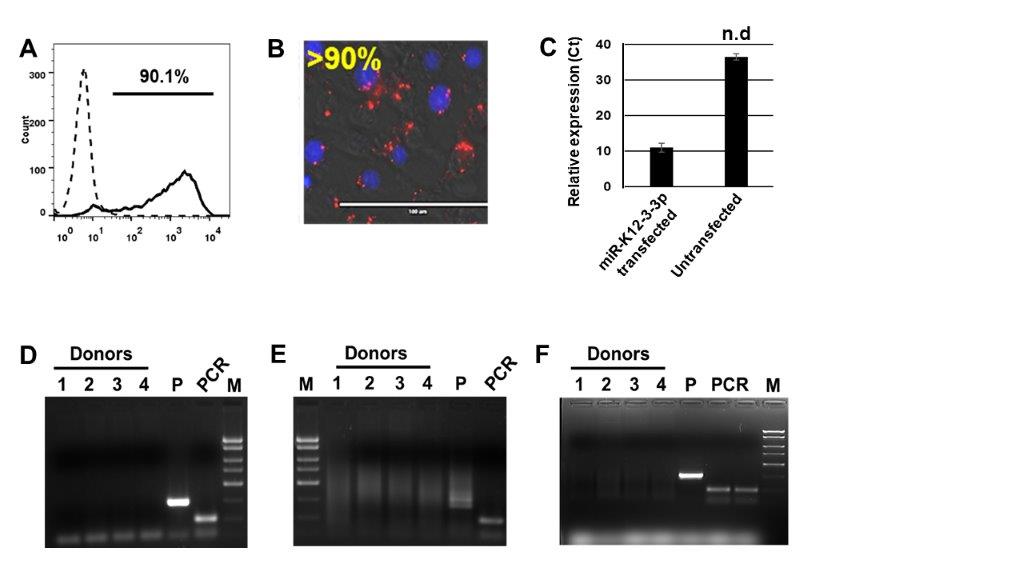
Before examining the impact of v-miRs on the HOK transcriptome, we standardized the transfection efficiency of miRNA mimics in HOK cells using a DY-547 dye labeled siRNA (siGLO) which localizes to the nucleus. Cells were transfected and analyzed for transfection efficiency using flow cytometry and florescent microscopy after 24 hours. Qualitative estimation using flow cytometry showed ~90% HOK positive for siGLO (Supplementary Figure 1A). This corroborates well with the florescent images and qRT-PCR data (Supplementary Figure 1B–C) demonstrating the presence of labelled siRNA and v-miR. We therefore achieved high miRNA transfection efficiency to perform subsequent experiments.
HOK are common target cells for various HHV. To rule out prior contamination with HHV, we screened commercially procured primary HOK cultures for HSV-1, HCMV and KSHV. Using virus specific primers, no amplification of HSV-1, HCMV or KSHV DNA was detected among the four donors examined (Supplementary Figure 1D–F). As expected, the positive control viral template demonstrated amplification (Lane 5; P). Similarly, PCR control also showed amplification (PCR; Supplementary Figure 1D–F). Overall, these results clearly show that our HOK cultures were free of HSV-1, HCMV and KSHV contamination.
Having established that miR-H1 and K12-3-3p expression are induced in oral inflammatory disease, we next examined the functional significance of these v-miRs in HOK. Total RNA isolated from v-miRs or control mimic transfected HOK were analyzed for differentially expressed genes using microarrays. Compared to control mimic, our results show expression of many genes significantly (p<0.05) altered in both miR-H1 and miR-K12-3-3p transfected HOK. In the miR-H1 dataset, we found 1376 genes that were differentially expressed (cut-off 1.25-fold), of which 618 were upregulated while, 758 were downregulated (Figure 2A). For miR-K12-3-3p, 1377 transcripts displayed differential expression. Among these 613 and 764 were up- and down-regulated, respectively (Figure 2B). We also examined the convergent and divergent expression of genes in miR-H1 and miR-K12-3-3p expressing HOK. The Venn diagram in Figure 2C shows that 408 genes were common to both v-miRs while, 968 and 969 transcripts were unique to miR-H1 and miR-K12-3-3p, respectively.
Figure 2. Differential expression of host genes in miR-H1 and miR-K12-3-3p transfected HOK.
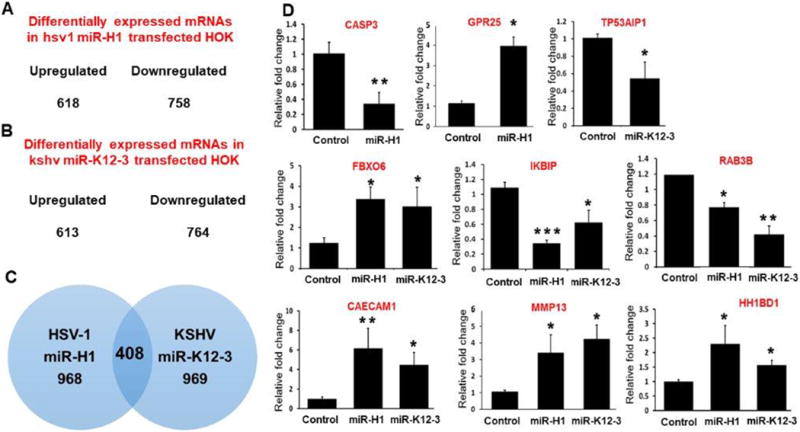
Viral miRNA transfected HOK cells were assessed for whole transcriptome using microarray analysis. The number of upregulated and downregulated genes in (A) miR-H1 and (B) miR-K12-3-3p transfected cells (n=4). (C) Venn diagram showing list of unique and commonly expressed genes in miR-H1 and miR-K12-3-3p overexpressing HOK. (D) Validation of selected differentially expressed genes by quantitative RT-PCR in a second cohort of HOK cultures. Total RNA isolated from miR-H1, miR-K12-3-3p and control transfected HOK were analyzed for the expression of differentially expressed genes by quantitative RT-PCR. Data is presented as ±SEM of four independent experiments. Student’s t-test was conducted to calculate p-values. *p<0.05, **p < 0.01
To confirm the microarray data, we examined the levels of 9 differentially expressed genes (unique and common) from miR-H1 and miR-K12-3-3p datasets by quantitative PCR in a separate cohort of v-miR transfected HOK. Caspase 3 and GPR25 were unique to miR-H1 while TP53AIP1 was deregulated only in miR-K12-3-3p transfected cells. Six other genes (FBXO6, IKBIP, RAB3B, CAECAM1, MMP13 and HH1BD1) common to both miR-H1 and miR-K12-3-3p datasets were also evaluated. Compared to control mimic, caspase 3 was significantly downregulated and GPR25 was upregulated in miR-H1 transfected cells (Figure 2D). Expression analysis of miR-K12-3-3p regulated genes show reduced expression of TP53AIP1 (Figure 2D). Similarly, commonly downregulated (IKBIP and RAB3B) and upregulated (FBXO6, CAECAM1, MMP13 and HH1BD1) genes from microarray exhibit similar gene expression profiles in RT-PCR analysis (Figure 2D). Thus, expression profiles of genes assessed by RT-PCR analysis corroborates with our microarray data. Overall, these results show that v-miRs identified in clinical samples are functionally relevant as they can alter expression of various host genes in a key target cell, HOK.
Multiple pathways relevant to HHV infection are associated with differentially expressed genes
To translate the microarray data into functional biological information, we utilized Ingenuity Pathway Analysis (IPA) to gain insights into networks, functions and pathways associated with the v-miR-mediated transcriptomic response. Our results show that miR-H1 and miR-K12-3-3p target various pathways that can be beneficial for virus survival, replication, immune evasion and persistence in HOK. For miR-H1 the most relevant pathways identified include endocytosis, cell cycle regulation, cell survival and apoptosis, NFκB signaling, actin cytoskeleton rearrangement, and p38 kinase pathway, among others. Supplementary Table 2 lists the top ten pathways and the p-values identified in our analysis. miR-H1 targets various genes involved in integrin signaling pathway. Figure 3A shows the integrin pathway associated genes with miR-H1 downregulated genes highlighted in green.
Figure 3. Pathway analysis identified key cellular functions targeted by viral miRNAs.
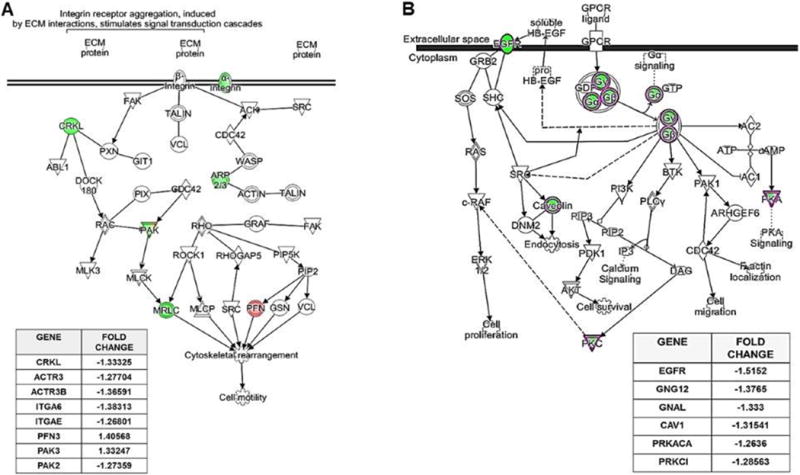
(A) miR-H1 targets genes involved in the integrin signaling pathway. Pathway depicting multiple differentially expressed genes regulated by miR-H1 using Ingenuity Pathway Analysis (IPA). Table lists miR-H1 altered genes involved in integrin signaling with fold change. (B) miR-K12-3-3p targets genes involved in the endocytic pathway. Pathway depicting miR-K12-3-3p downregulated genes involved in GPCR signaling assessed by IPA. Table lists genes involved in GPCR signaling with fold change.
Analysis of miR-K12-3-3p regulated genes reveal various affected pathways. These include cell proliferation and migration, viral replication, NFκB signaling, cell cycle regulation, inflammation, etc. Supplementary Table 3 lists the top ten pathways and the p-values identified in our analysis. GPCR signaling is among the miR-K12-3-3p targeted pathways affected with downregulation of multiple genes in this network (Figure 3B, highlighted in green).
Comparison of pathways related to miR-H1 and miR-K12-3-3p data revealed unique pathways of each v-miRs. For instance, insulin/IGF pathway-protein kinase B signaling and N-acetylglucosamine metabolism were specifically identified in miR-H1 pathway analysis while JAK/STAT signaling, synaptic vesicle trafficking, and p38 MAPK pathway were uniquely regulated by miR-K12-3-3p. It can be noted that pathways listed in Supplementary Tables 2 and 3 have large number of differentially expressed genes. Therefore, for representation we used pathways with fewer number of genes in the network. These findings suggest v-miRs modulate specific pathways and hence function of host cells thereby contributing to viral infection and pathogenesis.
Identification of potential direct gene targets of miR-K-12-3 and miR-H1
In general, miRNAs and their direct target genes exhibit antagonistic expression patterns. We therefore selected all the downregulated genes from miR-H1 and miR-K12-3-3p datasets and scanned for potential v-miR binding sites [50].
miR-K12-3-3p target prediction analysis of downregulated genes revealed hundreds of potential binding sites. Using stringent parameters, we obtained 110 different interactions. While most of the genes exhibit one miR-K12-3-3p binding site, some genes showed two or rarely three different binding sites. The less stringent analysis yielded 207 different interactions that satisfy our potential binding site criterion. In this case, we noted that most of the genes possessed single miR-K12-3-3p binding sites and only a few genes were identified with multiple binding sites. However, this number was higher than observed with the stringent analysis. For instance, SLC9A6 exhibit two miR-K12-3-3p binding sites at position 322 and 2257 while, GPAUCH2L also harbor two different predicted miR-K12-3-3p sites at nucleotide positions 33 and 69.
MiRNA-target prediction analysis was also performed using miR-H1 and downregulated genes. Using highly stringent parameters, 163 target interactions were obtained. With relaxed stringency, we obtained 197 different target interactions. As expected, more target binding sites were observed with relaxed parameters. However, compared to miR-K12-3-3p where stringent and relaxed parameters yielded 110 and 210 interactions, respectively, we did not notice profound differences in the miR-H1 analysis.
miR-H1 directly regulates multiple host genes involved in immune response and autophagy
To validate the in silico predicted v-miR binding sites on various host genes, we focused on miR-H1 targeted genes with known functions in immune response and autophagy. The 3′UTR of four genes including Leukemia Inhibitory Factor Receptor Alpha (LIFR), Transforming Growth Factor Beta Receptor 1 (TGFBR1), Interleukin 1 Receptor Accessory Protein (IL1RAP), Autophagy Related 16 Like 1 (ATG16L1) encompassing miR-H1 binding sites were cloned in psiCHECK2 vector expressing both renilla and firefly luciferase. The cloned 3′UTR of TGFBR, ATG16L1, LIFR, IL1RAP regions were 567, 1000, 1582 and 976 nucleotides long, respectively. Figure 4 (A–D) shows the sequence alignment of miR-H1 and its predicted gene targets. We noted two highly complementary miR-H1 binding sites on the 3′UTR of LIFR (nucleotide position 1045 and 2175) and ATG16L1 (nucleotide position 1965 and 2609). We cloned the region that encompasses both the binding sites for these predicted targets.
Figure 4. miR-H1 directly regulate host antiviral genes.
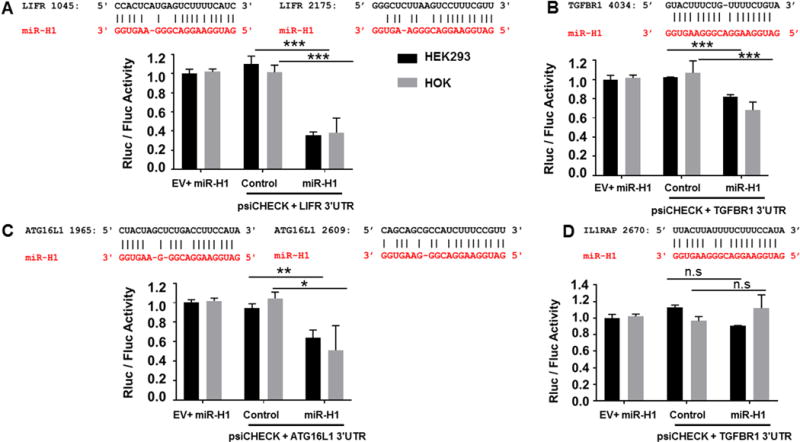
Luciferase reporter constructs were generated by cloning the 3′UTR of LIFR, ATG16L1, TGFBR and IL1RAP downstream of the renilla luciferase in psiCHECK2. (A–D) Top panel shows the sequence alignment of the predicted miR-H1 (in red) binding site on the target and gene (in black). HEK293 and HOK cells were transiently co-transfected with reporter construct and either miR-H1 or control mimics. Constitutively expressed firefly luciferase readings were used for normalization. Histograms shows impact of miR-H1 on normalized renilla activity compared to experimental controls. Error bars represent mean ± SD from three biological replicates. P-values were calculated using Student’s t-test. *p<0.05; **p<0.01; ***p<0.0001
HEK293 cells were co-transfected with 3′UTR cloned or empty vector and miR-H1 or control miRNA mimics. Dual luciferase assays show that compared with control mimic, significantly reduced renilla luciferase activity (normalized to firefly luciferase) for LIFR, TGFBR1 and ATG16L1 but not IL1RAP was observed in miR-H1 transfected cells (Figure 4A–D). Based on the fold reduction, marked suppression of LIFR (~3 fold) and ATG16L1 (~1.5 fold) constructs indicates a stronger interaction of miR-H1 with their UTRs. Interestingly, we noticed two sites for miR-H1 on the 3′UTR of both LIFR and ATG16L1, as mentioned above. The presence of multiple miRNA binding sites is known to exert more functional silencing of target genes [51,52]. To further confirm v-miR and target gene interaction, we performed dual luciferase assays in HOK, as the transcriptome changes were assessed in these cells. Our results show that compared to control mimic, miR-H1 targeted LIFR, TGFBR1 and ATG16L1 3′UTR as observed by reduction in renilla luciferase activity (Figure 4A–D). Similar to our HEK data, luciferase assays did not show significant reduction in IL1RAP construct suggesting that IL1RAP is not a bona-fide target of miR-H1.
Together, these results confirm that v-miR can directly bind and modulate the activity of target host genes identified in our microarray data and further supports their widespread functional impact on host-virus interactions.
miR-H1 regulates viral entry and productive infection in oral keratinocytes
Viral miRNAs can regulate host cellular and viral life cycle by targeting host and viral genes. Having established that disease associated v-miRs can modulate a large array of host genes, we next wanted to examine their impact on viral infection. To this end, we evaluated the role of HSV-1 encoded miR-H1 on viral entry and infection in oral keratinocytes. For entry assays, HOK were transfected with miR-H1 (1 and 2.5 pmol), or control mimic. After 36 hours, cells were infected with 5 MOI of HSV-1 gL86 (engineered with β-galactosidase gene) strain for 6 hours. Subsequently, cells were assayed for β-galactosidase activity in the presence of substrate ortho-Nitrophenyl-β-galactoside (ONPG). Figure 5A shows that compared with control miRNA or mock, reduced β-gal activity was observed for miR-H1 expressing epithelial cells. This is neither due to enhanced cell death (Supplementary Figure 2) or production of antiviral response in miR-H1 expressing cells that may restrict viral infection (data not shown). Similar results were observed in miR-H1 transfected U937 differentiated macrophages (Mφ), key regulators of mucosal immunity, that exhibit reduced HSV-1 entry compared to control mimic (Supplementary Figure 3).
Figure 5.
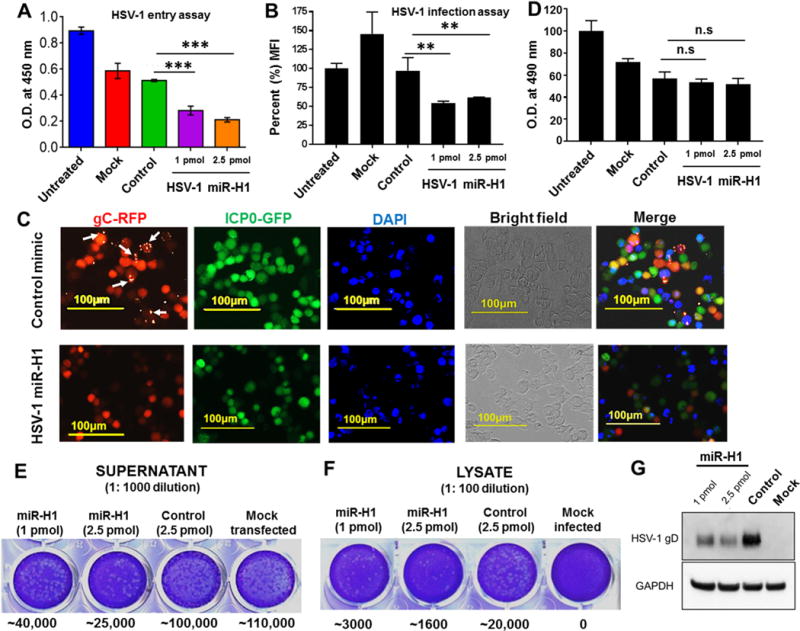
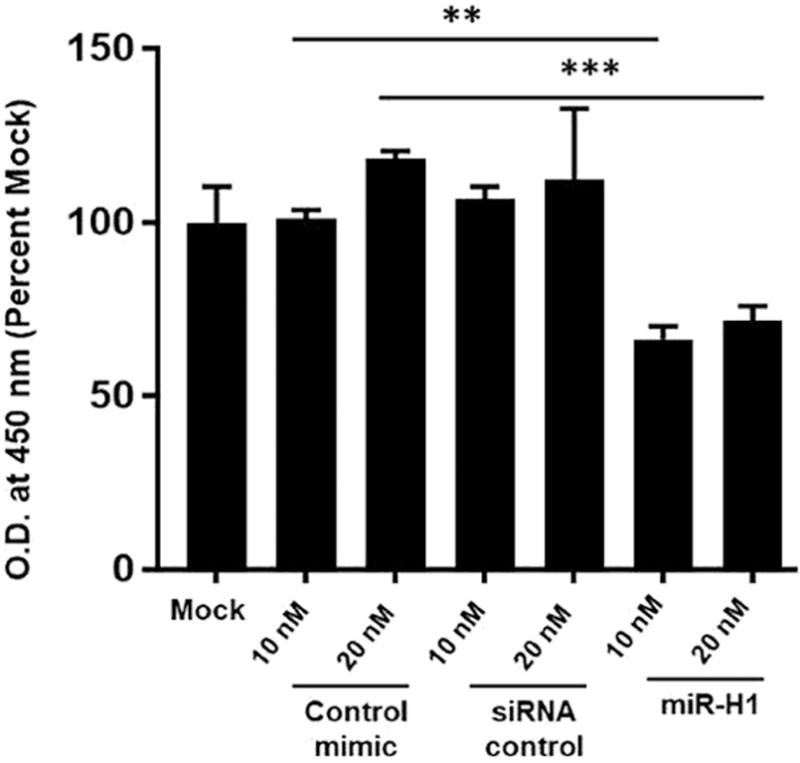
miR-H1 control HSV-1 entry and infection in host cell. (A) HOK were transfected with miR-H1 mimic, control mimics or mock transfected. To examine the impact of miR-H1 on viral entry, cells were incubated HSV-1 (gL86) at 10 MOI. β-galactosidase activity was assayed by providing substrate (ONPG) 6 h.p.i. Histograms show absorbance (yellow color) at 450 nm quantified on a plate reader. (B) miR-H1 or control infected cells were assessed for productive infection using dual reporter expressing HSV-1 (KOS). After 24 hours, GFP (ICP0) florescence was measured by flow cytometric analysis. Geometric MFI (of GFP) were normalized with respect to untransfected cells and the percent values are shown as histograms. (C) Representative images showing florescent-tagged HSV-1 early (ICP0;GFP) and late (gC;RFP) proteins in miR-H1 or control mimic transfected cells. Scale bar:100μm. (D) Impact of v-miR or control mimic and HSV-1 (gL86) infection on the viability of HOK was evaluated by MTS assays. Histograms showing percent absorbance (at 490 nm) for untransfected, mock, miR-H1 or control mimics. Plaque assays show virion formation in the (E) supernatant and (F) lysates of HSV-1 infected HOK cells expressing miR-H1 or control mimic. Colony numbers are mentioned for each sample below corresponding well. (G) Immunoblot showing accumulation of late protein gD in the lysates of the same samples. GAPDH was used a loading control. Error bars represent mean ± SD from three biological replicates. P-values were calculated using Student’s t-test. **p<0.01; ***p<0.0001
We further examined the impact of miR-H1 on productive viral infection. Epithelial cells overexpressing miR-H1 or control mimic were infected with HSV-1 KOS strain (0.1 MOI). This strain encodes green florescent protein-tagged (GFP) immediate early protein ICP0 and red florescent protein-tagged (RFP) viral gene gC. Both flow cytometry (GFP MFI) and florescent microscopy data demonstrate reduced infection in miR-H1 overexpressing cells compared with control (Figure 5B, C). However, we did not notice any delay in the visualization of GFP (ICP0) or RFP (gC) reporter signals indicating that viral life cycle is progressing at a normal pace. Interestingly, florescent imaging showed that unlike control miRNA transfected cells which showed massive egress of HSV-1 as observed by the red blebs on the cell surface (marked with white arrows; Figure 5C; upper panel), miR-H1 transfected cells may likely control release of virions. Given that miR-H1is associated with both the latent and productive phases, it may be highly relevant to fine-tune virion release to minimize the immune response. These differences were not due to the cytotoxic effect of miR-H1 as assessed by MTS viability assay (Figure 5D).
To further confirm the impact of miR-H1 on viral release, we assessed the virion formation in the supernatant and lysates of HSV-1 infected HOK transfected with miR-H1 or control mimic. As shown in Figure 5 (E, F), we noticed significantly reduced number of plaques in miR-H1 transfected cells compared with control mimic after 48 hpi. The reduction in plaque number correlates with the increased dose of miR-H1 (compare 1 and 2.5 pmol) suggesting a negative regulation of virion formation by miR-H1. Mock infected cells showed no plaques while, mock transfected cells were used as positive control and displayed plaque numbers similar to control mimics (Figure 5 E, F). We also checked the levels of gD, a key virion structure protein, in the lysates of miR-H1 or control HOK infected with virus. Compared to control mimic, we observed reduced level of gD in miR-H1 transfected cells. Similar to plaque assays, higher dose (2.5 pmol) of miR-H1 further reduced the gD protein (Figure 5G). Together, these results suggest a critical role of miR-H1 in controlling viral load in infected host cells.
DISCUSSION
Detection of HHV and their association with different oral inflammatory diseases has been reported in numerous clinical studies [24–27]. However, viral pathogenicity factors that can contribute to the disease remains largely unknown. In the present study, we showed detection and increased prevalence of three v-miRs encoded by HHV in the gingival tissues of periodontitis subjects compared to healthy controls. These findings corroborate with our previous observation that showed increased v-miR expression in diseased human tooth pulp [44].
The question arises as to identification of cellular reservoirs in these tissues. While we did not detect miR-H1 in healthy tissues, both miR-US4 and miR-K12-3-3p were detected in both healthy and diseased periodontal tissues, albeit with higher prevalence in diseased samples. Herpesviruses are known to exhibit broad tropism for human cells. Epithelial cells are the primary target of HSV-1 and establish latent infection in sensory ganglia [53], while HCMV primarily target epithelial cells but also exhibit tropism for monocytes, T cells and NK cells [54,55]. Latent HCMV infection is established in monocyte precursors and kidney epithelial cells. In vivo, KSHV detection is reported in lymphoid cells, epithelial cells, monocytes, keratinocytes as well as oral epithelial cells [56,57]. Thus, epithelial cells and monocytes are common target cells that can potentially serve as direct viral reservoirs expressing v-miRs. Moreover, indirect sources of v-miRs can also contribute to the detection of these viral biomolecules. Similar to cellular miRNAs, v-miRs can be packaged into exosomes and delivered to various cell types [58,59]. Exosomes are ubiquitously secreted membrane bound nano-vesicles (20-100 nm in diameter) capable of transporting RNA, miRNAs, proteins, and DNA molecules to distant cell types through membrane fusion [60]. This may permit viruses to modulate key host cell functions distally, including immunity without directly infecting these cells. Indeed, multiple studies have shown the presence of v-miR derived exosomes in-vivo and in-vitro signifying the functional relevance of v-miRs in viral immunoevasion [58,61,62]. For instance, EBV miR-BART-15 is secreted in exosomes of EBV infected B cells as well as Mφ ectopically expressing v-miRs [62]. This v-miR shares the same seed region as that of cellular miR-223 for its target NLRP3. Functionally, B cell derived exosome-mediated delivery of BART-15 into recipient Mφ suppress inflammasome formation by silencing NLRP3 [62]. Such interactions provide an evolutionary benefit as these sequences are less likely to undergo mutation to avoid v-miR regulation.
It is important to note that the expression of v-miRs is dependent on not only the presence of the virus, but also the specific stage of its life cycle. For example, miR-H1 is detected primarily during the lytic stage, while it is not similarly expressed during viral latency [34,63]. This is relevant because the detection of v-miRs should not be utilized to evaluate the presence or absence of the virus in tissue biopsies. For instance, HSV-1 genome was detected in tears and saliva of healthy human subjects [64]; however, in our study HSV-1 encoded miR-H1 could not be detected in gingival tissues of healthy individuals. Three possible reasons may be attributed to this observed discrepancy. Firstly, herpesviruses are shed into biological fluids like tears or saliva that contains secreted biomolecules of various cells and tissues. However, our study used gingival tissues that may lack latent infected cells in healthy individuals. This is supported by the multiple studies showing extremely low detection rate of HSV-1 (and other herpesviruses) genome in gingival biopsies [20,21,28,29]. Secondly, the sero-prevalence of herpesviruses varies geographically, thus the rate of viral genome detection may also vary among different cohorts. Finally, our small sample size (n=6) may further reduce the probability of viral miRNA detection. Nonetheless, these observations cumulatively suggest that determining the clinical presence of viruses may require detection of multiple viral derived biomolecules.
KSHV is a known oncovirus that encodes multipe v-miRs [11,12]. Chugh et al., tested the impact of exosomes from control and KSHC infected primary effusion lymphoma (PEL) subjects [61]. Compared to control human plasma (CHP) derived exosomes, HUVEC cells treated with exosomes derived from PEL patients showed increased cell migration capacity. These observations demonstrate a key role of v-miRs in moduating paracrine signaling leading to aberrant cellular functions. Considering that exosomal-derived v-miRs can theoretically be delivered to any cell type, v-miRs can be considered as broad spectrum virulence factors. While copious amounts of life cycle associated v-miRs are produced in both the latent as well as lytic phases of viral infection, the functional relevance of v-miRs as virulence factors is plausible in latency where they have ample time to downmodulate target genes in a wide range of host cells, in particular immune cells. This provides a microenvironment that may favor viral persistence or spread. Overall, non-immunogenic v-miRs are employed as tools by certain viruses to modulate functions of a wide range of non-permissive cells.
Viral miRNAs perform unique biological functions that can regulate virus life cycle switch, cell tropism, persistence and immune evasion [65]. While several studies focus on identifying direct targets of viral miRNAs, we used a novel approach of profiling global host transcripts under potential regulation of our candidate v-miRs. Using this strategy, pathway analysis of these altered genes highlighted various networks modulated by v-miRs that can alter key host pathogen defense mechanisms. For example, miR-K12-3-3p inhibits endocytosis by downregulating multiple genes involved in the pathway. Given the significance of this pathway in pathogen clearance, antibody-mediated antigen uptake, antigen processing, etc., this may be highly detrimental for host cells. Consistent with this notion, we previously showed that primary Mφ transfected with miR-K12-3-3p showed reduced uptake of whole E. coli [66]. Similarly, other pathways relevant to HHV persistence were also inhibited. These include cell replication, GPCR signaling, cell death, cell adhesion, etc. Moreover, employing in silico target prediction analysis, we identified hundreds of potential v-miR binding sites on the 3′UTRs of downregulated genes, even with stringent parameters [50; see Tables 1, 2 and supplementary data]. These genes are potentially regulated by v-miRs as our microarray data shows reduced mRNA levels and in silico analysis identifies single or in some cases, multiple v-miR binding sites suggesting that v-miR binding to host mRNA leads to transcript degradation. We have validated three novel v-miR binding sites on downregulated mRNAs, further validation of other predicted sites is required. Nonetheless, this information is highly valuable in focusing on the genes/pathways that are under direct regulation of viral miRNAs. Together, our results highlight findings that HSV v-miRs can modulate multiple cellular functions thereby potentially compromising host defense mechanisms aimed at countering viral infection and survival. These results also strengthen the plausible role of v-miRs as key pathogenic factors that can contribute to oral inflammatory disease progression.
Our microarray data coupled with in silico and dual luciferase assays identified three novel host genes viz., LIFR, ATG16L1 and TGFBR1 that are directly regulated by miR-H1. While LIFR and TGFBR1 are important for activation of immune cells, cell proliferation and differentiation, the role of ATG16L1 in autophagy and antiviral response has been reported. Both LIFR and TGFBR1 are key receptors for two pleiotropic cytokines LIF and TGFB, respectively. LIF is a member of IL-6 cytokine family and is known to modulate cell proliferation, differentiation, survival, immune cell chemotaxis, immune tolerance, etc., [67]. These properties suggest an antiviral function of LIF receptor (LIFR). Indeed, LIFR alpha (or CD118) deficient mice exhibit increased susceptibility to HSV-2 which was attributed to reduced infiltration of cytotoxic (CD8) T cells and reduced IFN gamma (IFNγ) production [68]. LIF has both pro- and anti-inflammatory properties depending on the tissue and the form of inflammatory stimuli [69,70]. Using a complete Freund’s adjuvant (CFA)-induced cutaneous inflammation model, LIF was shown to regulate IL-1β, IL-6, IL-7, IL-2Rα, and IFN-γ expression [70]. It is thus likely that miR-H1 mediated suppression of LIFR observed in our study would likely attenuate LIF signaling causing reduced secretion of pro-inflammatory cytokines. Consistent with this, we observed decreased pro-inflammatory cytokine and chemokine levels in culture supernatants of miR-H1 transfected primary Mφ challenged with whole E. coli [66].
Another key cytokine target of miR-H1 validated in this study is TGFBR1. Similar to LIFR, this gene encodes for a receptor for the pleiotropic cytokine, TGFβ which is involved in maintaining self-tolerance via the regulation of lymphocyte proliferation, differentiation, and survival [71]. TGFβ interactions with TGFBR1 trigger SMAD dependent or independent signaling that activates immune-suppressive gene required during the resolution of inflammation or wound healing [71]. Periodontitis is a consequence of an overt, uncontrolled immune response triggered by microbial challenge. Thus, downregulation of TGFBR1 by miR-H1 noted in our microarray data suggest that this v-miR may attenuate resolution of inflammation thus exacerbating local inflammation. Further, it is possible that miR-H1-mediated interference with host immunosuppressive signaling will lead to increased pro-inflammatory cytokine levels thus directly contributing to periodontal inflammation.
Autophagy is an essential host antiviral pathway that rapidly clears endocytosed virus. Evidently, suppression of this pathway can favor long-term virus survival. In particular, HSV-1 have evolved various mechanisms to block autophagy. We show that miR-H1 downregulate ATG16L1 mRNA by directly interacting with its 3′UTR. This gene, through its interaction with ATG12-ATG5, facilitates production of an active component of autophagy viz., LC3-II [72,73]. Thus, miR-H1 reduction in ATG16L1 may favor virus persistence. ATG16L1 is also reported to participate in the autophagic clearance of bacteria. Intracellular sensors such as the Nod-like receptors Nod1 and Nod2, recruit ATG16L1 resulting in coalescence of bacteria-triggered autophagosomes thus triggering autophagic responses against various intracellular bacteria [74]. Given that both bacteria and viruses are major etiological factors in periodontal pathogenesis, the potential role of viral miRNA (miR-H1) in subverting cellular responses and restricting infection of both pathogens through silencing a key host factor (ATG16L1) is highly significant. Besides its role in autophagy, ATG16L1 is shown to regulate antiviral responses [75]. Post-transcriptional silencing of ATG16L1 by miR-H1 observed in our study suggest that this v-miR can benefit virus to overcome not only autophagy-mediated virus clearance but also by attenuating hosts’ antiviral responses.
Employing two different assays for viral infection in HOK cells, we observed reduced HSV-1 entry in miR-H1 transfected cells compared with control mimic. Global pathway analysis of miR-H1 modulated genes identified endocytosis, cell movement and cytoskeletal rearrangement pathways which may interfere with the uptake of HSV-1. This corroborates with the reduced uptake of whole E. coli by miR-H1 transfected primary Mφ [66]. Indeed, endocytosis is required for efficient uptake of HSV-1 and suppression of this pathway can attenuate viral entry. For instance, enhanced endosomal pH by bafilomycin, ammonium chloride and monensin blocked HSV-1 in keratinocytes [76]. Thus, miR-H1 mediated silencing of endocytosis pathway allow this v-miR to restrict virion entry in an HSV-1 infected cell. Further, using dual reporter recombinant HSV-1 virus, we also noted reduced expression of early (ICP0-GFP) as well as late (gD-RFP) proteins in miR-H1 expressing HOK. The lack of detectable miR-H1-mediated cell death and antiviral response (IFN-α/β) strongly suggests other operating mechanisms. Over millions of years, HSV-1 have coevolved to infect host cells, as evidenced by the widespread global seroprevalance, but with minimal damage to host cell/tissue except for neonates and immunocompromised individuals [77]. An important question is what molecular cues HSV-1 have evolved to maintain this successful interaction with its host. HSV-1 is known to encode miRNA genes that provides an ingenious way to fine tune both viral and host functions. miR-H1 expressing HOK show entry and infection, however, it appears more tightly regulated compared to control transfected cells. It is plausible that miR-H1 may regulate HSV-1 entry into previously infected cells and facilitate optimization of virion generation during the productive phase. High miR-H1 expression during productive as well as latency cycle further support this notion [78]. Moreover, controlled virion production will facilitate virus spread effectively without triggering an antiviral response. Interestingly, two newly identified HSV-1 miRNAs, viz., miR-H28 and miR-H29 are reported to regulate HSV-1 infection and spread [58]. Similar to miR-H1, both v-miRs are produced during productive infection, are exported into exosomes and prevent spread and infection of HSV-1. Importantly, miR-28 and miR-29 are expressed in reactivated murine ganglia but not during latent infection strongly suggesting their specific role in infection. Besides miRNAs, HSV-1 is also known to encode two other small RNAs viz., small non-coding RNA (sncRNA) 1 and 2 [79]. These sncRNAs are encoded by the LAT transcript and cooperates with RIG-I to consistently stimulate IFN-β promoter activity and NF-κB dependent transcription. Through activation of type I interferon, these sncRNAs reduce HSV-1 replication efficiency and thus maintain latency [80]. Concurrently, NF-κB dependent transcription of anti-apoptotic genes promote survival of virus-infected cells [81]. Given their crucial role in viral persistence, examination of these small RNAs in oral inflammatory disease will reveal novel insights on their role in the pathogenesis. Together, these findings reveal that HSV-1 has evolved an ingenious strategy in which multiple small RNAs namely miR-H1 reported in this study), miR-H28, miR-H29 [58] and sncRNA 1 and sncRNA 2 [79,80] are employed as non-immunogenic, paracrine regulators of host-virus interaction that allow successful persistence and spread of virus. One such mechanism involves controlled virus production.
Overall, our gene profiling, target prediction and validation data suggest that v-miRs can use multi-prong approaches to suppress immune activation, interfere with resolution of inflammation, a key component of wound healing, as well as facilitate viral persistence within the host. In combination with additional environmental factors and bacteria challenge with periodontal pathogens such as Porphyromonas gingvalis, Aggrebacter actinomycetemcomitans, etc., it is plausible that periodontitis induced herpesvirus derived miRNAs can exacerbate the severity of disease. Using anti-v-miR strategies in conjunction with conventional periodontal therapies may provide better treatment options. Interstingly, in support of this notion, Sunde et al., [82] showed that treating an EBV positive patient with recurent periodontitis with antiviral drug (Valtrex) remarkably reduced viral titres in subgingival tissue and improved periodontal health. This study, for the first time, reveal clinical as well as biological relevance of HHV derived pathogenic components (v-miRs) in oral infection. The findings will pave the way for future research to dissect key pathways/molecules targeted by v-miRs that can be of therapeutic value for the treatment oral inflammtory disease including periodontitis, pulpitis, and peri-implantitis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Figure 6.
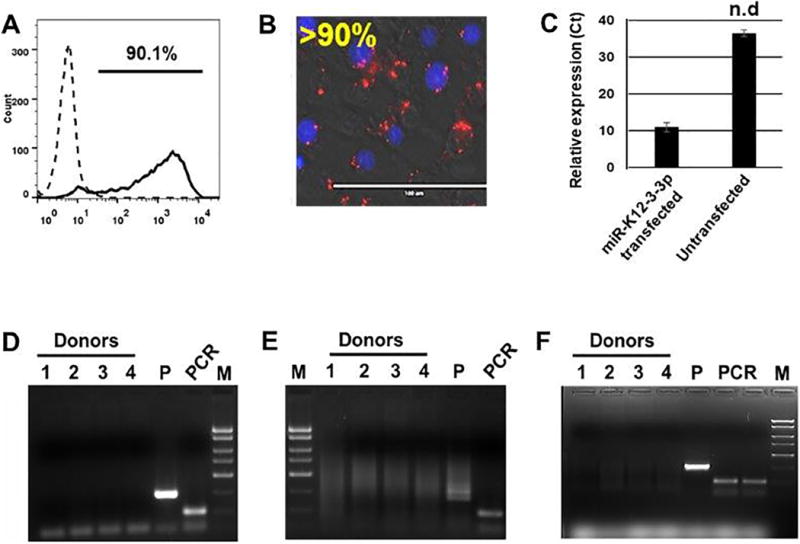
Figure 7.
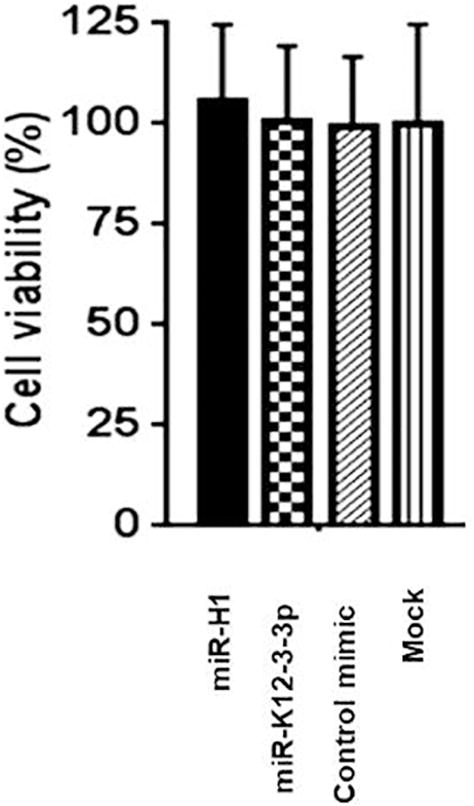
Figure 8.
Highlights.
The impact of three viral miRNAs (v-miRs) from the human herpesvirus family, HSV-1 (miR-H1), KSHV (miR-K12-3-3p), and HCMV (miR-US4) on the transcriptome of human oral keratinocytes and macrophages were investigated.
These v-miRs were previously identified in biopsies derived from healthy and diseased periodontal (gingival) tissues.
Functionally characterization of these viral microRNAs revealed their significant effects on cellular function.
Our results demonstrate that v-miRs regulate both host and viral functions and may contribute to the pathogenesis of inflammatory oral diseases.
Acknowledgments
We would like to thank Dr. Richard Lamont, University of Louisville, Kentucky for graciously providing the immortalized human gingival epithelial cell line used in this study.
Funding
Part of this work was funded by the NIH/NIDCR R21 DE026259-01A1 to ARN and NIH/NIDCR R01 DE02105201A1 to SN.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
ARN and SN conceptualized the study; MN, GM, GP recruited subjects and performed sample collection; ARN, JS, AS performed most of the experiments; SH helped with the transcriptome and IPA analysis; DJ, TY and DS provided reagents and helped perform HSV-1 related experiments; ARN and SN acquired and analyzed the data and drafted the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Roizman B, Sears AE. Herpes simplex viruses and their replication. In: Roizman B, Whitley RJ, Lopez C, editors. The Human Herpesviruses. 1st. New York: Raven Press; 1993. pp. 11–68. [Google Scholar]
- 2.Pellet P, Roizman B. The family herpesviridae: a brief introduction. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott Williams, Wilkins; 2007. pp. 2479–2499. [Google Scholar]
- 3.Alford CA, Stagno S, Pass RF, Huang ES. Epidemiology of cytomegalovirus. In: Nahmais A, Dowdle W, Schinazi R, editors. The Human Herpesviruses: An Interdisciplinary Perspective. New York: Elsevier; 1981. pp. 159–171. [Google Scholar]
- 4.Adland E, Klenerman P, Goulder P, Matthews PC. Ongoing burden of disease and mortality from HIV/CMV coinfection in Africa in the antiretroviral therapy era. Front Microbiol. 2015;6:1016. doi: 10.3389/fmicb.2015.01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaftenaar E, Verjans GM, Getu S, et al. High seroprevalence of human herpesviruses in HIV-infected individuals attending primary healthcare facilities in rural South Africa. PLoS One. 2014;9:e99243. doi: 10.1371/journal.pone.0099243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White DW, Beard WS, Barton ES. Immune modulation during latent herpesvirus infection. Immunol Rev. 2012;245:189–208. doi: 10.1111/j.1600-065X.2011.01074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Røder G, Geironson L, Bressendorff I, Paulsson K. Viral proteins interfering with antigen presentation target the major histocompatibility complex class I peptide-loading complex. J Virol. 2008;82:8246–8252. doi: 10.1128/JVI.00207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verweij MC, Horst D, Griffin BD, et al. Viral inhibition of the transporter associated with antigen processing (TAP): a striking example of functional convergent evolution. PLoS Pathog. 2015;11:e1004743. doi: 10.1371/journal.ppat.1004743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naranatt PP, Krishnan HH, Svojanovsky SR, Bloomer C, Mathur S, Chandran B. Host gene induction and transcriptional reprogramming in Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 2004;64:72–84. doi: 10.1158/0008-5472.can-03-2767. [DOI] [PubMed] [Google Scholar]
- 10.Collins-McMillen D, Kim JH, Nogalski MT, et al. Human Cytomegalovirus Promotes Survival of Infected Monocytes via a Distinct Temporal Regulation of Cellular Bcl-2 Family Proteins. J Virol. 2015;90:2356–2371. doi: 10.1128/JVI.01994-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfeffer S, Zavolan M, Grasser FA, et al. Identification of virus-encoded microRNAs. Science. 2004;304:734–736. doi: 10.1126/science.1096781. [DOI] [PubMed] [Google Scholar]
- 12.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci U S A. 2005;102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 14.Naqvi AR, Islam MN, Choudhury NR, Haq QM. The fascinating world of RNA interference. Int J Biol Sci. 2009;5:97–117. doi: 10.7150/ijbs.5.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fordham JB, Naqvi AR, Nares S. Regulation of miR-24, miR-30b, and miR-142-3p during macrophage and dendritic cell differentiation potentiates innate immunity. J Leukoc Biol. 2015;98:195–207. doi: 10.1189/jlb.1A1014-519RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naqvi AR, Fordham JB, Nares S. miR-24, miR-30b, and miR-142-3p regulate phagocytosis in myeloid inflammatory cells. J Immunol. 2015;194:1916–1927. doi: 10.4049/jimmunol.1401893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naqvi AR, Fordham JB, Nares S. MicroRNA target Fc receptors to regulate Abdependent Ag uptake in primary macrophages and dendritic cells. Innate Immun. 2016;22:510–521. doi: 10.1177/1753425916661042. [DOI] [PubMed] [Google Scholar]
- 18.Self-Fordham JB, Naqvi AR, Uttamani JR, Kulkarni V, Nares S. MicroRNA: Dynamic Regulators of Macrophage Polarization and Plasticity. Front Immunol. 2017;8:1062. doi: 10.3389/fimmu.2017.01062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–469. doi: 10.1016/j.molmed.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Saygun I, Kubar A, Sahin S, Sener K, Slots J. Quantitative analysis of association between herpesviruses and bacterial pathogens in periodontitis. J Periodontal Res. 2008;43:352–359. doi: 10.1111/j.1600-0765.2007.01043.x. [DOI] [PubMed] [Google Scholar]
- 21.Slots J. Herpesviral-bacterial interactions in periodontal diseases. Periodontol 2000. 2010;52:117–140. doi: 10.1111/j.1600-0757.2009.00308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vincent-Bugnas S, Vitale S, Mouline CC, et al. EBV infection is common in gingival epithelial cells of the periodontium and worsens during chronic periodontitis. PLoS One. 2013;8:e80336. doi: 10.1371/journal.pone.0080336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slots J. Periodontitis: facts, fallacies and the future. Periodontol 2000. 2017;75:7–23. doi: 10.1111/prd.12221. [DOI] [PubMed] [Google Scholar]
- 24.Contreras A, Nowzari H, Slots J. Herpesviruses in periodontal pocket and gingival tissue specimens. Oral Microbiol Immunol. 2000;15:15–18. doi: 10.1034/j.1399-302x.2000.150103.x. [DOI] [PubMed] [Google Scholar]
- 25.Sabeti M, Simon JH, Nowzari H, Slots J. Cytomegalovirus and Epstein-Barr virus active infection in periapical lesions of teeth with intact crowns. J Endod. 2003;29:321–323. doi: 10.1097/00004770-200305000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Saboia-Dantas CJ, Coutrin de Toledo LF, Sampaio-Filho HR, Siqueira JF., Jr Herpesviruses in asymptomatic apical periodontitis lesions: an immunohistochemical approach. Oral Microbiol Immunol. 2007;22:320–325. doi: 10.1111/j.1399-302X.2007.00362.x. [DOI] [PubMed] [Google Scholar]
- 27.Ferreira DC, Rocas IN, Paiva SS, et al. Viral-bacterial associations in acute apical abscesses. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112:264–271. doi: 10.1016/j.tripleo.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Das S, Krithiga GS, Gopalakrishnan S. Detection of human herpes viruses in patients with chronic and aggressive periodontitis and relationship between viruses and clinical parameters. J Oral Maxillofac Pathol. 2012;16:203–209. doi: 10.4103/0973-029X.98502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li F, Zhu C, Deng FY, Wong MCM, Lu HX, Feng XP. Herpesviruses in etiopathogenesis of aggressive periodontitis: A meta-analysis based on case-control studies. PLoS One. 2017;12:e0186373. doi: 10.1371/journal.pone.0186373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sunde PT, Olsen I, Enersen M, Beiske K, Grinde B. Human cytomegalovirus and Epstein-Barr virus in apical and marginal periodontitis: a role in pathology? J Med Virol. 2008;80:1007–1011. doi: 10.1002/jmv.21180. [DOI] [PubMed] [Google Scholar]
- 31.Slots J, Sabeti M, Simon JH. Herpesviruses in periapical pathosis: an etiopathogenic relationship? Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:327–331. doi: 10.1016/s1079-2104(03)00352-4. [DOI] [PubMed] [Google Scholar]
- 32.Nicoll MP, Proença JT, Efstathiou S. The molecular basis of herpes simplex virus latency. FEMS Microbiol Rev. 2012;36:684–705. doi: 10.1111/j.1574-6976.2011.00320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan C, Zhu D, Wang Y, et al. Human Cytomegalovirus miR-UL148D Facilitates Latent Viral Infection by Targeting Host Cell Immediate Early Response Gene 5. PLoS Pathog. 2016;12:e1006007. doi: 10.1371/journal.ppat.1006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang S, Bertke AS, Patel A, Wang K, Cohen JI, Krause PR. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc Natl Acad Sci U S A. 2008;105:10931–10936. doi: 10.1073/pnas.0801845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan D, Flores O, Umbach JL, et al. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe. 2014;15:446–456. doi: 10.1016/j.chom.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellare P, Ganem D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe. 2009;6:570–575. doi: 10.1016/j.chom.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin X, Liang D, He Z, Deng Q, Robertson ES, Lan K. miR-K12-7-5p encoded by Kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS One. 2011;6:e16224. doi: 10.1371/journal.pone.0016224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choy EY, Siu KL, Kok KH, et al. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med. 2008;205:2551–2560. doi: 10.1084/jem.20072581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landais I, Pelton C, Streblow D, DeFilippis V, McWeeney S, Nelson JA. Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFkappaB Signaling Pathway. PLoS Pathog. 2015;11:e1004881. doi: 10.1371/journal.ppat.1004881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gourzones C, Gelin A, Bombik I, et al. Extra-cellular release and blood diffusion of BART viral micro-RNAs produced by EBV-infected nasopharyngeal carcinoma cells. Virol J. 2010;7:271. doi: 10.1186/1743-422X-7-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang G, Zong J, Lin S, et al. Circulating Epstein-Barr virus microRNAs miR-BART7 and miR-BART13 as biomarkers for nasopharyngeal carcinoma diagnosis and treatment. Int J Cancer. 2015;136:E301–12. doi: 10.1002/ijc.29206. [DOI] [PubMed] [Google Scholar]
- 43.Lisboa LF, Egli A, O’Shea D, et al. Hcmv-miR-UL22A-5p: A biomarker in transplantation with broad impact on host gene expression and potential immunological implications. Am J Transp. 2015;15:1893–1902. doi: 10.1111/ajt.13222. [DOI] [PubMed] [Google Scholar]
- 44.Zhong S, Naqvi A, Bair E, Nares S, Khan AA. Viral microRNAs identified in human dental pulp. J Endod. 2017;43:84–89. doi: 10.1016/j.joen.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The American Academy of Periodontology. Parameter on chronic periodontitis with advanced loss of periodontal support. J Periodontol. 2000;71:856–858. doi: 10.1902/jop.2000.71.5-S.856. [DOI] [PubMed] [Google Scholar]
- 46.Perri R, Nares S, Zhang S, Barros SP, Offenbacher S. MicroRNA modulation in obesity and periodontitis. J Dent Res. 2012;91:33–38. doi: 10.1177/0022034511425045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan LT, Zhong S, Naqvi AR, et al. MicroRNAs: new insights into the pathogenesis of endodontic periapical disease. J Endod. 2013;39:1498–1503. doi: 10.1016/j.joen.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naqvi AR, Fordham JB, Khan A, Nares S. MicroRNAs responsive to Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis LPS modulate expression of genes regulating innate immunity in human macrophages. Innate Immun. 2014;20:540–551. doi: 10.1177/1753425913501914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jankovic S, Aleksic Z, Dimitrijevic B, Lekovic V, Milinkovic I, Kenney B. Correlation between different genotypes of human cytomegalovirus and Epstein-Barr virus and peri-implant tissue status. Aust Dent J. 2011;56:382–388. doi: 10.1111/j.1834-7819.2011.01360.x. [DOI] [PubMed] [Google Scholar]
- 50.Naqvi AR, Shango J, Seal A, Shukla S, Nares S. In silico prediction of cellular targets of herpesvirus encoded microRNAs. Data in Brief. Submitted. 2018 doi: 10.1016/j.dib.2018.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hon LS, Zhang Z. The roles of binding site arrangement and combinatorial targeting in microRNA repression of gene expression. Genome Biol. 2007;8:R166. doi: 10.1186/gb-2007-8-8-r166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kulkarni V, Naqvi AR, Uttamani JR, Nares S. MiRNA-target interaction Reveals cell-specific post-transcriptional regulation in mammalian cell lines. Int J Mol Sci. 2016;17:pii: E72. doi: 10.3390/ijms17010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heldwein EE, Krummenacher C. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci. 2008;65:1653–1668. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schelhaas M, Jansen M, Haase I, Knebel-Morsdorf D. Herpes simplex virus type 1 exhibits a tropism for basal entry in polarized epithelial cells. J Gen Virol. 2003;84:2473–2484. doi: 10.1099/vir.0.19226-0. [DOI] [PubMed] [Google Scholar]
- 55.Sinzger C, Digel M, Jahn G. Cytomegalovirus cell tropism. Curr Top Microbiol Immunol. 2008;325:63–83. doi: 10.1007/978-3-540-77349-8_4. [DOI] [PubMed] [Google Scholar]
- 56.Chakraborty S, Veettil MV, Chandran B. Kaposi’s Sarcoma Associated Herpesvirus Entry into Target Cells. Front Microbiol. 2012;3:6. doi: 10.3389/fmicb.2012.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duus KM, Lentchitsky V, Wagenaar T, Grose C, Webster-Cyriaque J. Wild-type Kaposi’s sarcoma-associated herpesvirus isolated from the oropharynx of immune-competent individuals has tropism for cultured oral epithelial cells. J Virol. 2004;78:4074–4084. doi: 10.1128/JVI.78.8.4074-4084.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han Z, Liu X, Chen X, et al. miR-H28 and miR-H29 expressed late in productive infection are exported and restrict HSV-1 replication and spread in recipient cells. Proc Natl Acad Sci U S A. 2016;113:E894–901. doi: 10.1073/pnas.1525674113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A. 2010;107:6328–6333. doi: 10.1073/pnas.0914843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 61.Chugh PE, Sin SH, Ozgur S, et al. Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog. 2013;9:e1003484. doi: 10.1371/journal.ppat.1003484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haneklaus M, Gerlic M, Kurowska-Stolarska M, et al. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J Immunol. 2012;189:3795–3799. doi: 10.4049/jimmunol.1200312. [DOI] [PubMed] [Google Scholar]
- 63.Jurak I, Hackenberg M, Kim JY, et al. Expression of herpes simplex virus 1 microRNAs in cell culture models of quiescent and latent infection. J Virol. 2014;88:2337–2339. doi: 10.1128/jvi.03486-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaufman HE, Azcuy AM, Varnell ED, Sloop GD, Thompson HW, Hill JM. HSV-1 DNA in tears and saliva of normal adults. Invest Ophthalmol Vis Sci. 2005;46:241–247. doi: 10.1167/iovs.04-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Piedade D, Azevedo-Pereira JM. The Role of microRNAs in the pathogenesis of Herpesvirus infection. Viruses. 2016;8:pii: E156. doi: 10.3390/v8060156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naqvi AR, Shango J, Seal A, Shukla D, Nares S. Viral miRNAs alter host cell miRNAs profiles and modulate innate immune responses. Front Immunol. 2018 doi: 10.3389/fimmu.2018.00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nicola NA, Babon JJ. Leukemia inhibitory factor (LIF) Cytokine Growth Factor Rev. 2015;26:533–544. doi: 10.1016/j.cytogfr.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conrady CD, Halford WP, Carr DJ. Loss of the type I interferon pathway increases vulnerability of mice to genital herpes simplex virus 2 infection. J Virol. 2011;85:1625–1633. doi: 10.1128/JVI.01715-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gadient RA, Patterson PH. Leukemia inhibitory factor, Interleukin 6, and other cytokines using the GP130 transducing receptor: roles in inflammation and injury. Stem Cells. 1999;17:127–137. doi: 10.1002/stem.170127. [DOI] [PubMed] [Google Scholar]
- 70.Zhu M, Oishi K, Lee SC, Patterson PH. Studies using leukemia inhibitory factor (LIF) knockout mice and a LIF adenoviral vector demonstrate a key anti-inflammatory role for this cytokine in cutaneous inflammation. J Immunol. 2001;166:2049–2054. doi: 10.4049/jimmunol.166.3.2049. [DOI] [PubMed] [Google Scholar]
- 71.Yoshimura A, Muto G. TGF-β function in immune suppression. Curr Top Microbiol Immunol. 2011;350:127–147. doi: 10.1007/82_2010_87. [DOI] [PubMed] [Google Scholar]
- 72.Boada-Romero E, Letek M, Fleischer A, Pallauf K, Ramón-Barros C, Pimentel-Muiños FX. TMEM59 defines a novel ATG16L1-binding motif that promotes local activation of LC3. EMBO J. 2013;32:566–582. doi: 10.1038/emboj.2013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nishimura T, Kaizuka T, Cadwell K, et al. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep. 2013;14:284–291. doi: 10.1038/embor.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 75.Grimm WA, Messer JS, Murphy SF, et al. The Thr300Ala variant in ATG16L1 is associated with improved survival in human colorectal cancer and enhanced production of type I interferon. Gut. 2016;65:456–464. doi: 10.1136/gutjnl-2014-308735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nicola AV, Hou J, Major EO, Straus SE. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J Virol. 79:7609–7616. doi: 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roizman B. HSV gene functions: what have we learned that could be generally applicable to its near and distant cousins? Acta Virol. 1999;43:75–80. [PubMed] [Google Scholar]
- 78.Du T, Han Z, Zhou G, Roizman B. Patterns of accumulation of miRNAs encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc Natl Acad Sci U S A. 2015;112:E49–55. doi: 10.1073/pnas.1422657112. Erratum in: Proc Natl Acad Sci U S A. 113:E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Peng W, Vitvitskaia O, Carpenter D, Wechsler SL, Jones C. Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1-encoded latency-associated transcript. J Neurovirol. 2008;14:41–52. doi: 10.1080/13550280701793957. [DOI] [PubMed] [Google Scholar]
- 80.Shen W, Sa e Silva M, Jaber T, Vitvitskaia O, Li S, Henderson G, Jones C. Two small RNAs encoded within the first 1.5 kilobases of the herpes simplex virus type 1 latency-associated transcript can inhibit productive infection and cooperate to inhibit apoptosis. J Virol. 2009;83:9131–9139. doi: 10.1128/JVI.00871-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.da Silva LF, Jones C. Small non-coding RNAs encoded within the herpes simplex virus type 1 latency associated transcript (LAT) cooperate with the retinoic acid inducible gene I (RIG-I) to induce beta-interferon promoter activity and promote cell survival. Virus Res. 2013;175:101–109. doi: 10.1016/j.virusres.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sunde PT, Olsen I, Enersen M, Grinde B. Patient with severe periodontitis and subgingival Epstein-Barr virus treated with antiviral therapy. J Clin Virol. 2008;42:176–178. doi: 10.1016/j.jcv.2008.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.