

Abstract
A fundamental challenge in the design of bioresponsive (or bioactivated) GdIII-based magnetic resonance (MR) imaging probes is the considerable background signal present in the “preactivated” state that arises from outer-sphere relaxation processes. When sufficient concentrations of a bioresponsive agent are present (i.e., a detectable signal in the image), the inner- and outer-sphere contributions to r1 may be misinterpreted to conclude that the agent has been activated, when it has not. Of the several parameters that determine the observed MR signal of an agent, only the electron relaxation time (T1e) impacts both the inner- and outer-sphere relaxation. Therefore, strategies to minimize this background signal must be developed to create a near zero-background (or truly “off” state) of the agent. Here, we demonstrate that intramolecular magnetic exchange coupling when GdIII is coupled to a paramagnetic transition metal provides a means to overcome the contribution of second- and outer-sphere contributions to the observed relaxivity. We have prepared a series of complexes with the general formula LMLn(μ-O2CCH3)(O2CCH3)2 (M = Co, Cu, Zn). Solid-state magnetic susceptibility measurements reveal significant magnetic coupling between GdIII and the transition metal ion. Nuclear magnetic relaxation dispersion (NMRD) analysis confirms that the observed differences in relaxivity are associated with the modulation of T1e at GdIII. These results clearly demonstrate that magnetic exchange coupling between GdIII and a transition metal ion can provide a significant decrease in T1e (and therefore the relaxivity of GdIII). This design strategy is being exploited to prepare new generations of preclinical bioresponsive MR imaging probes with near zero-background.
Graphical Abstract
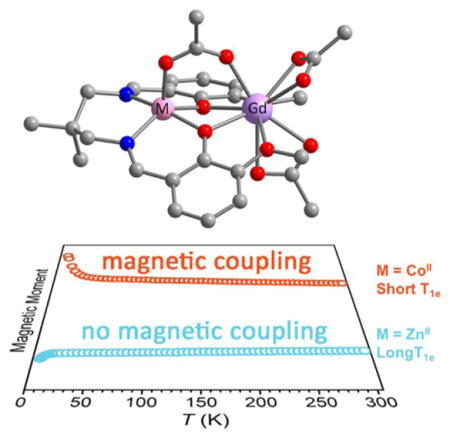
INTRODUCTION
Magnetic resonance imaging (MRI) is a staple of preclinical and clinical diagnostic radiology due to its tunable soft tissue contrast and high spatial resolution. The need to differentiate regions of tissues or organs that are magnetically similar but histologically distinct has been a major impetus for the development of contrast enhancement agents. Paramagnetic agents such as gadolinium [GdIII] complexes are often used (>40% of the 50 million clinical procedures performed annually) to decrease the T1 of adjacent protons, resulting in increased signal intensity (brightness) in the vicinity of the agent.1
The vast majority of GdIII agents are anatomical reporters and are designed to highlight regions of interest such as vasculature and tumors. Our laboratory has developed conditionally activated (or bioresponsive) probes that report on the in vivo physiological and biochemical status of whole animals in the form of an acquired magnetic resonance (MR) image.2 The use of these probes in intact organisms (combined with a modality that provides high spatiotemporal resolution) has resulted in a deeper understanding of the specific roles of in vivo gene expression and fluctuations in ion concentration in normal and pathological physiology.3–5
Current efforts toward developing responsive or bioactivated GdIII agents focus on optimizing several molecular parameters associated with paramagnetic GdIII chelates. These parameters include the number of inner-sphere water molecules (q), the rotational correlation time of the complex (τr), and the mean residence lifetime of the aquo ligand (τm).6–8 To date, numerous examples of MR molecular probes that modulate q in response to enzymatic activities,4,9–11 ion binding,12–16 and pH17–19 have been reported. Further, τr enhancement has been explored through molecular interactions with endogenous proteins20,21 and nanoparticle platforms22,23 to create high signal probes.
In order to maximize the signal enhancement in the acquired MR image using bioresponsive agents, the second- and outer-coordination sphere contributions of GdIII (which represent background signal in the image) must be minimized. For example, small-molecule agents at clinical field strengths (1–3 T), the outer-sphere contribution to the observed relaxation enhancement of water protons is ca. 45%.6,24 Therefore, when employing bioresponsive agents in vivo the background signal from outer-sphere water can be misinterpreted for the agent having been activated by an enzyme, or other physiologically relevant ions. In other words, we designed this new class of probes with low-background signal to minimize the ambiguity in MR images where an observed signal enhancement (brighter) can be the result of either high GdIII complex concentrations, or the activation of the bioresponsive agent being turned “on.”
The electronic relaxation time (T1e) of a contrast agent represents a key physical property that governs both the innersphere (described by Solomon–Bloembergen–Morgan (SBM) theory) and outer-sphere (described by the Freed and Bryant equations) relaxation enhancement of bulk water. Modulation of q, τr, or τm has created numerous bioresponsive probes by tuning the inner-sphere contribution to relaxivity. However, manipulating the value of T1e of a contrast agent can provide a unique opportunity to exploit both the inner- and outer-sphere contributions. Therefore, a mechanism that can modulate T1e in response to a biochemical event is desirable, as it would enable researchers to suppress undesired signals for developing highly responsive MR probes.
According to SBM theory, T1e of a paramagnetic center contributes to the overall correlation time (τc) of the molecule according to eq 1:
| (1) |
At clinical magnetic field strengths of 1.5 and 3 T, the optimal relationship for maximum contrast is (τm)−1 < (τr)−1 ≤ (T1e)−1, such that τc ≈ 10−9 s.25–28 For small molecular GdIII complexes, such as the clinically employed contrast agent [(DOTA)GdIII]− (H4DOTA = 1,4,7,10-tetraazacyclododecane- 1,4,7,10-tetraacetic acid), values for τm, τr, and T1e are typically measured to be on the order of ca. 10−7, 10−10, and 10−9 s, respectively.29 Accordingly, reduction in T1e in a contrast agent will result in deviation of the optimal correlation times and will thus lead to ineffective 1H relaxation of the molecule and low signal.
One method to decrease T1e in a molecular complex is through the introduction of magnetic exchange coupling between multiple paramagnetic metal centers. Here, the presence of magnetic exchange coupling gives rise to low-lying excited states through which spins can relax.30 Indeed, a number of multinuclear complexes that feature magnetic coupling between paramagnetic metal centers have been shown to exhibit smaller values of T1e relative to the constituent metal ions,31–33 and we recently employed this strategy to develop a CuII2-based chemical exchange saturation transfer (CEST) MR agent.34
Similarly, the introduction of magnetic coupling into Gd-containing multinuclear complexes should give rise to a shortening of T1e and a subsequent decrease in relaxivity. Merbach and co-workers explored this concept in trinuclear GdIII complexes where they found the weak magnetic coupling (0.033 cm−1) between unpaired electrons in the 4f orbitals to be insufficient to engender measurable effects on proton relaxivity.35 Other reported examples of multinuclear contrast agents with no magnetic coupling likewise likewise exhibited negligible reductions in measured T1e values.36,37
In contrast to the magnetic superexchange between lanthanides, a transition metal-lanthanide superexchange coupling can be much stronger (up to |J| = 7.0 cm−1) owing to the radially diffuse 3d orbitals in first-row transition metals.38–41 In principle, for a complex that features magnetic exchange between GdIII and a paramagnetic transition metal ion, the decrease in T1e of the GdIII ion induced by exchange coupling varies with the spin quantum number of the transition metal (STM), the exchange constant (J) of the coupling, and the electronic relaxation time of the transition metal (T1e′).28,30,42 Accordingly, the coupling constant (J) and the electronic structure of the transition metal (STM and T1e′) can be synthetically tuned to modulate T1e of GdIII. To achieve this, we proposed to employ dinuclear complexes where the GdIII center is magnetically coupled to a divalent first row transition metal, MII, as a strategy to reduce T1e of GdIII for the potential application of low-background GdIII CAs.
Bimetallic complexes of dinucleating salen derivatives (2,2′-ethylenebis(nitrilomethylidene)diphenol N,N′ethylenebis-(salicylimine)) have been shown to display significant magnetic coupling. Therefore, we selected a series of salen-based dinuclear complexes of the general formula LMGd(μ-O2CCH3)(O2CCH3)241,43 (Figure 1) to investigate the correlation between M and the resulting T1e of GdIII. Here, we should note that these complexes are intended to demonstrate a proof-of-concept and are not suitable for direct use in a biological system. Three first-row transition metals were chosen for this study: CoII (T1e ≈ 10−12 s), CuII (T1e ≈ 10−9 s), and ZnII as a diamagnetic control.30 We hypothesized that CoII, with a T1e value typically over 3 orders of magnitude faster than GdIII, would reduce the T1e of GdIII to the greatest extent. In addition, while GdIII complexes are our primary focus, the analogous complexes LMDy(μ-O2CCH3)-(O2CCH3)2 (M = Co, Cu, Zn) were prepared for solutionstate determination of q, and the complex LZnEu(μ-O2CCH3)-(O2CCH3)2 was prepared to elucidate the solution-state structure.
Figure 1.

Synthetic scheme for the preparation and metalation of LH2 to give the complexes LMLn(μ-O2CCH3)(O2CCH3)2, where M = CoII, CuII, or ZnII and Ln = EuIII, GdIII, or DyIII.
EXPERIMENTAL SECTION
General Considerations
Unless otherwise noted, syntheses and manipulations were carried out in an MBraun LABstar glovebox operated under a humid dinitrogen atmosphere. The ligand LH2 was synthesized following a modified literature procedure.44 Dioxygen was removed from H2O through at least three successive freeze–pump–thaw cycles. Anhydrous organic solvents were dried using a solvent purification system from Pure Process Technology. The solvents H2O (10% 17O) and CD3OD were purchased from Cambridge Isotope Laboratories, Inc. The compound Gd(O2CCH3)3·4H2O was purchased from Strem Chemical Inc. All other reagents and solvents were purchased from Sigma-Aldrich and used without further purification.
Synthetic Procedures
6,6′-((1E,1′E)-((2,2-Dimethylpropane 1,3-diyl)bis(azanylylidene))bis(methanylylidene))bis(2-methoxyphenol) (LH2). To a stirring solution of o-vanillin (15 g, 98 mmol) in 200 mL of anhydrous MeOH under an inert atmosphere, 2,2-dimethyl-1,3-propanediamine (5.0 g, 49 mmol) in 30 mL of dry MeOH was added dropwise via a syringe. The reaction was stirred at room temperature for 24 h, producing a yellow/orange solution. The MeOH was removed in vacuo, and the resulting yellow residue was recrystallized from hot toluene to yield the product as yellow block-like crystals in 87% yield (Figure 1). Full 1H and 13C NMR spectra can be found in Figure S1. 1H NMR (500 MHz, chloroform-d) δ 14.12 (s, 2H), 8.29 (d, J = 1.4 Hz, 2H), 6.89 (dd, J = 7.8, 1.6 Hz, 2H), 6.85 (dd, J = 7.9, 1.5 Hz, 2H), 6.77 (t, J = 7.8 Hz, 2H), 3.88 (s, 6H), 3.46 (d, J = 1.2 Hz, 4H), 1.04 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 165.80, 152.05, 148.47, 122.92, 118.40, 117.90, 113.81, 67.31, 56.03, 36.17, 24.25. 1H NMR and 13C NMR spectra were obtained at 25 °C on a Bruker Avance III 500 MHz NMR spectrometer.
LCoGd(μ-O2CCH3)(O2CCH3)2·0.7MeCN (1)
LH2 (100 mg, 0.27 mmol) and Co(O2CCH3)2·4H2O (67 mg, 0.27 mmol) were dissolved in MeOH (6 mL) in a 20 mL scintillation vial fit with a stir bar. The reaction was sealed with a Teflon cap and heated to 30 °C with stirring for 10 min. The reaction was then stirred at ambient temperature for 1 h to produce an amber solution. Gd(O2CCH3)3·4H2O (90 mg, 0.27 mmol) was then added to the solution, and after stirring for 16 h, the solvent was removed under reduced pressure to give a red-orange residue. The residue was dissolved in MeCN (1 mL), and the resulting solution was filtered through a column of diatomaceous earth. Vapor diffusion of Et2O into the filtrate afforded an amber crystalline solid, which was collected by vacuum filtration and dried under reduced pressure to give 1 (190 mg, 87%). FT-IR (ATR, cm−1): 3016 (w), 2898 (w), 1652 (m), 1586 (m), 1475 (s), 1444 (s), 1343 (m), 1255 (s), 1102 (m), 765 (s) (see Figure S2). Anal. Calcd C28.4H35.1GdN2.7O10Co; C, 43.14; H, 4.47; N, 4.77%. Found: C, 43.03; H, 4.40; N, 4.77%. ESI-MS (positive mode): m/z = 703.058 g/mol (M-O2CCH3). Vapor diffusion of Et2O into a dilute MeCN (4 mL) solution of the red-orange residue from above gave amber rod-shaped crystals of LCoGd(μ-O2CCH3)(O2CCH3)2·0.6MeCN that were suitable for single-crystal X-ray diffraction analysis.
LCuGd(μ-O2CCH3)(O2CCH3)2·0.8MeCN (2)
LH2 (100 mg, 0.27 mmol) and Cu(O2CCH3)2·H2O (54 mg, 0.27 mmol) were dissolved in MeOH (6 mL) in a 20 mL scintillation vial fit with a stir bar. The reaction was sealed with a Teflon cap and heated to 30 °C with stirring for 10 min. The reaction was then stirred at ambient temperature for 1 h to produce a blue-green precipitate. Gd(O2CCH3)3·4H2O (90 mg, 0.27 mmol) was then added to the reaction mixture, which was stirred for 16 h to yield a blue-green solution. The solvent was removed under reduced pressure to give a blue-green residue. The residue was dissolved in MeCN (1 mL), and the resulting solution was filtered through a column of diatomaceous earth. Vapor diffusion of Et2O into the filtrate afforded a blue-green crystalline solid, which was collected by vacuum filtration and dried under reduced pressure to give 2 (180 mg, 83%). FT-IR (ATR, cm−1): 2983 (w), 2886 (w), 1661 (m), 1588 (m), 1479 (s), 1444 (m), 1345 (m), 1259 (s), 1102 (s), 1004 (m), 767(s) (see Figure S3). Anal. Calcd C28.9H36.2GdN2.8O10Cu; C, 43.05; H, 4.52; N, 4.93%. Found: C, 43.03; H, 4.53; N, 4.94%. Vapor diffusion of Et2O into a dilute MeCN (4 mL) solution of the blue-green residue from above gave blue-green rod-shaped crystals of LCuGd(μ-O2CCH3)(O2CCH3)2 that were suitable for single-crystal X-ray diffraction analysis.
LZnGd(μ-O2CCH3)(O2CCH3)2·0.1MeCN (3)
This complex was prepared in air. LH2 (100 mg, 0.27 mmol) and Zn(O2CCH3)2·H2O (54 mg, 0.27 mmol) were dissolved in MeOH (6 mL) in a 25 mL round-bottom flask fit with a stir bar. The reaction was heated to 30 °C for 10 min and stirred at ambient temperature for 1 h to produce a yellow solution. Gd(O2CCH3)3·4H2O (90 mg, 0.27 mmol) was added to the solution, and after stirring for 12 h, the solvent was removed under reduced pressure to give a yellow residue. The residue was dissolved in MeCN (1 mL), and the resulting solution was filtered through a column of diatomaceous earth. Vapor diffusion of Et2O into the filtrate afforded a colorless crystalline solid, which was collected by vacuum filtration and dried under reduced pressure to give 3 (190 mg, 88%). FT-IR (ATR, cm−1): 3028 (w), 3001 (w), 1659 (m), 1591 (m), 1473 (s), 1448 (s), 1343 (m), 1257 (s), 1100 (m), 773 (s) (see Figure S4). Anal. Calcd C27.5H34GdN2.1O10Zn; C, 42.42; H, 4.40; N, 3.93%. Found: C, 42.43; H, 4.41; N, 3.83%. ESI-MS (positive mode): m/z = 708.051 g/mol (M-O2CCH3). Vapor diffusion of Et2O into a dilute MeCN (4 mL) solution of the colorless residue from above gave colorless rod-shaped crystals of LZnGd(μ-O2CCH3)(O2CCH3)2·0.9 MeCN that were suitable for single-crystal X-ray diffraction analysis.
LCoDy(μ-O2CCH3)(O2CCH3)2·0.7MeCN (4)
LH2 (100 mg, 0.27 mmol) and Co(O2CCH3)2·H2O (67 mg, 0.27 mmol) were dissolved in MeOH (6 mL) in a 20 mL scintillation vial fit with a stir bar. The reaction was sealed with a Teflon cap and heated to 30 °C with stirring for 10 min. The reaction was then stirred at ambient temperature for 1 h to produce an amber solution. Dy(O2CCH3)3·xH2O (46 mg, 0.27 mmol) was added to the solution, and after stirring for 16 h, the solvent was removed under reduced pressure to give an amber residue. The residue was dissolved in MeCN (1 mL), and the resulting solution was filtered through a column of diatomaceous earth. Vapor diffusion of Et2O into the filtrate afforded an orange crystalline solid, which was collected by vacuum filtration and dried under reduced pressure to give 4 (190 mg, 86%). FT-IR (ATR, cm−1): 2983 (w), 2886 (w), 1661 (s), 1558 (m), 1479 (s), 1345 (m), 1259 (s), 1102 (m), 767 (s) (see Figure S5). Anal. Calcd C28.9H36.9DyN2.8O10Co; C, 43.05; H, 4.53; N, 4.94%. Found: C, 43.05; H, 4.53; N, 4.94%. ESI-MS (positive mode): m/z = 709.064 g/mol (M-O2CCH3).
LCuDy(μ-O2CCH3)(O2CCH3)2·2.6H2O (5)
LH2 (100 mg, 0.27 mmol) and Cu(O2CCH3)2·4H2O (67 mg, 0.27 mmol) were dissolved in MeOH (6 mL) in a 20 mL scintillation vial fit with a stir bar. The reaction was sealed with a Teflon cap and heated to 30 °C with stirring for 10 min. The reaction was then stirred at ambient temperature for 1 h to produce a blue-green precipitate. Dy(O2CCH3)3·xH2O (46 mg, 0.27 mmol) was then added to the reaction mixture, which was stirred for 16 h to yield a blue-green solution. The solvent was removed under reduced pressure to give a blue-green residue. The residue was dissolved in MeCN (1 mL), and the resulting solution was filtered through a column of diatomaceous earth. Vapor diffusion of Et2O into the filtrate afforded a blue-green crystalline solid, which was collected by vacuum filtration and dried under reduced pressure to afford 5 (180 mg, 80%). FT-IR (ATR, cm−1): 2979 (w), 2942 (w), 1619 (m), 1556 (m), 1437 (s), 1415 (s), 1309 (m), 1223 (s), 1066 (m), and 744 (s) (see Figure S6). Anal. Calcd C27H38.1DyN2O10Cu; C, 39.64; H, 4.70; N, 3.43%. Found: C, 39.53; H, 4.03; N, 3.49%.
LZnDy(μ-O2CCH3)(O2CCH3)2·0.21MeCN·0.38H2O·0.22MeOH (6)
This compound was prepared in air. LH2 (100 mg, 0.27 mmol) and Zn(O2CCH3)2·H2O (54 mg, 0.27 mmol) were dissolved in MeOH (6 mL) in a 25 mL round-bottom flask fit with a stir bar. The reaction was heated to 30 °C for 10 min and stirred at ambient temperature for 1 h to produce a yellow solution. Dy(O2CCH3)3·xH2O (46 mg, 0.27 mmol) was added to the solution, and after stirring for 12 h, the solvent was removed under reduced pressure to give a yellow residue. The residue was dissolved in MeCN (1 mL), and the resulting solution was filtered through a column of diatomaceous earth. Vapor diffusion of Et2O into the filtrate afforded a colorless crystalline solid, which was collected by vacuum filtration and dried under reduced pressure to give 6 (180 mg, 82%). FT-IR (ATR, cm−1): 2979 (w), 2942 (w), 1619 (m), 1556 (m), 1437 (s), 1415 (m), 1309 (m), 1223 (s), 1066 (m), 744 (s) (see Figure S7). Anal. Calcd C27.78H35.6DyN2.21O10Zn; C, 41.78; H, 4.49; N, 3.88%. Found: C, 41.78; H, 4.49; N, 3.89%. ESI-MS (positive mode): m/z = 714.058 g/mol (M-O2CCH3).
LZnEu(μ-O2CCH3)(O2CCH3)2·0.1MeCN·0.17H2O (7)
This compound was prepared in air. LH2 (100 mg, 0.27 mmol) and Zn(O2CCH3)2·H2O (54 mg, 0.27 mmol) were dissolved in MeOH (6 mL) in a 25 mL round-bottom flask fit with a stir bar. The reaction was heated to 30 °C for 10 min, and then stirred at ambient temperature for 1 h to produce a yellow solution. Eu(O2CCH3)3· xH2O (90 mg, 0.27 mmol) was added to the solution, and after stirring for 16 h, the solvent was removed under reduced pressure to give a yellow residue. The residue was dissolved in MeCN (1 mL), and the resulting solution was filtered through a column of diatomaceous earth. Vapor diffusion of Et2O into the filtrate afforded a colorless crystalline solid, which was collected by vacuum filtration and dried under reduced pressure to give 7 (180 mg, 86%). FT-IR (ATR, cm−1): 3032 (w), 2960 (w), 1677 (m), 1659 (m), 1471 (s), 1448 (m), 1343 (m), 1259 (s), 1100 (m), 773 (s) (see Figure S8). Anal. Calcd C27.21H33.67DyN2.12O10Zn; C, 41.78; H, 4.49; N, 3.88%. Found: C, 44.23; H, 4.41; N, 3.83%. ESI-MS (positive mode): m/z = 703.048 g/mol (M-O2CCH3). Vapor diffusion of Et2O into a dilute MeCN (4 mL) solution of the colorless residue from above gave colorless rod-shaped crystals of LZnEu(μ-O2CCH3)(O2CCH3)2·1.5MeCN that were suitable for single-crystal X-ray diffraction analysis.
NMR Spectroscopy
Total NMR structural elucidation of 7 (see Figure S9) was completed to verify that no lanthanide dissociated from the O4 (2-phenoxo and 2-methoxo) binding pocket. 7 was the only complex with line widths acceptable for 2D NMR analysis. Two-dimensional experiments were performed in methanol-d4 on a 400 MHz Agilent DD MR-400 system equipped with autoX probe (COSY, TOCSY, NOESY) (see Figures S9 and S10) and an Agilent DD2 500 MHz for HSQC and HMBC data (see Figure S11). The chemical shifts of 7 dissolved in D2O were inferred based on the assignment made in methanol-d4. Two-dimensional NMR experiments were not successful in D2O due to rapid relaxation times. Fully assigned spectra in D2O can be found for in Figure S12.
The number of bound H2O molecules (q) was measured by VT 17O NMR of aqueous solutions of 4, 5, and 6. DyIII analogues were utilized to measure q due to suitable line shape (in the range of 30–40 Hz) of the 17O peak. In the N2 glovebox, saturated solutions of the complexes were prepared with freeze–pump–thawed D2O, doped with 1% 17O H2O. The solutions were filtered using a 5 μm syringe filter and transferred to J. Young NMR tubes. VT-NMR experiments were performed using an Agilent DD MR-400 system equipped with an autoX probe. Each sample was heated to 80 °C and allowed to equilibrate for 10 min to ensure that H2O molecules were in the fast-exchanging regime. 17O NMR spectra were collected for each sample.
The 17O chemical shifts (in ppm) for diamagnetic control were −2.74 (4), −4.701 (5), and −3.53 (6). The molar ratio of compound (Pm) was determined using ICP-MS and <SZ> used for DyIII was 28.565. q was calculated using equations found in Figure S13; calculated values for q are reported in Table 1.
Table 1.
T1e Values at 1 T Obtained from Fits of the NMRD Profiles, for q Ranging from 5 to 6 for 1 and 3 and from 6 to 7 for 2a
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| T (°C) | 25 | 37 | 25 | 37 | 25 | 37 |
| T1e (ns) | 0.065 (±0.025) | 0.065 (±0.025) | 0.135 (±0.025) | 0.145 (±0.025) | 2.6 (±0.7) | 2.5 (±0.7) |
| q | 6.0 (±0.2) | 5.0 (±0.2) | 5.6 (±0.2) | |||
The number of water molecules coordinated to GdIII, q, is reported for each complex based on measurements of the DyIII analogue.
Relaxivity Measurements at 14 T
Saturated aqueous solutions of 1, 2, and 3 were prepared in an MBraun LABstar glovebox and filtered through 5 μm syringe filters. Serial dilutions were prepared four times to produce 500 μL of five sample concentrations. The sample tubes were sealed under inert atmosphere and removed from the glovebox. Analogous solutions of 1:1 M(O2CCH3)2/Gd-(O2CCH3)3 were prepared; M = Co, Cu, and Zn. The samples were then incubated at 37 °C for 30 min, and the T1 and T2 relaxation times were measured on a Bruker mq60 NMR analyzer equipped with Minispec V2.51 Rev.00/NT software (Billerica, MA, U.S.A.) operating at 1.41 T (60 MHz) and at 37 °C. T1 was measured by an inversion recovery pulse sequence (t1_ir_mb) with a final pulse separation ≥5T1. T2 relaxation was measured by the Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence (t2_cpmb). The T1 and T2 relaxation rates were plotted as a function of the GdIII concentration determined by ICP-MS. r1 and r2 were determined from the slopes of the linear fits of three independent replicates. r1 and r2 values can be found in Figure S14.
Relaxivity Measurements at 7 T
Samples were prepared the same as the 1.4 T measurements but were performed on an 89 mm-boresize PharmaScan 7.05 T MRI spectrometer fitted with shielded gradient coils (Bruker BioSpin, Billerica, MA) using an RF RES 300 1H 089/023 quadrature transmit/receive mouse brain volume coil (Bruker BioSpin, Billerica, MA). Image acquisition was performed using Paravision 5.0.1 software (Bruker BioSpin, Billerica, MA). T1 and T2 weighted images were acquired using a rapid-acquisition rapid-echo variable repetition time (RAREVTR) and multislice multiecho pulse sequences, respectively. RARE scan = 13 factor = 1, echo time = 11 ms, averages = 3, matrix size (MTX) = 128 × 128, field of view = 25 mm × 25 mm, six slices, slice thickness = 1.5 mm, interslice distance = 2.0 mm, repetition times = 15000, 10000, 8000, 6000, 3000, 1500, 1000, 750, 500, 300, 200, and 150 ms, and a total scan time of ~1 h and 32 min. T1 values of selected regions of interest of five out of six slices were calculated using the T1 saturation recovery monoexponential curve fitting formula provided by the image sequence analysis tool in Paravision 5.0.1 software. The 7 T T1 and T2 relaxation rates were plotted as a function of the GdIII concentration determined by ICPMS. 7 T r1 and r2 values can be found in Figure S15.
MR Solution Phantom Imaging at 1.5 T
Samples of 1, 2, and 3 were prepared (1 mM) in 15 mL conical tubes for imaging in a 70 cmbore-size Aera 1.5 T MR spectrometer fitted with shielded gradient coils (Siemens Medical Solutions, Erlangen, DE) using a four-channel receive only head matrix coil (Siemens). Image acquisition was performed using advanced mapping sequences using Siemens Syngo software. T1 images were acquired using a saturation recovery sequence with variable TR’s ranging from 25 to 1500 ms. Selection of this range of TR’s was chosen to accurately sample the spin–lattice relaxation recovery curve with short T1 values. The echo time was set to ~2 ms for all scans. All images were acquired in 2D with in-plane resolution of 900 μm, slice thickness = 4 mm (n = 3 slices). Maps were generated using Xinapse JIM software and least-squares fitting using standard MR signal equations.
Nuclear Magnetic Relaxation Dispersion (NMRD)
NMRD profiles were obtained using a Stelar Spinmaster fast field cycling FFC 200-1T relaxometer by measuring the water 1H longitudinal relaxation rates of aqueous solutions containing 1 (2.31 mM), 2 (0.64 mM), or 3 (0.58 mM) as a function of the applied magnetic field, in a range corresponding to proton Larmor frequencies from 0.01 to 40 MHz (ca. 2 × 10−4 − 1 T). The relaxivity profiles were obtained by normalization of the relaxation rate data, subtracted by the diamagnetic contribution, to the GdIII concentration determined by ICP-MS. The measurements were performed at 25 and 37 °C. The measurement error was below 1%.
UV–vis Spectroscopy
UV–vis spectra were obtained for aqueous solutions of LH2, 1, 2, and 3 on an Agilent 8453 UV–vis in an air-free cuvette dissolved in freeze pump thawed Milli-Q water (see Figure S16). Air stability of 2 and 3 was evaluated over 2 weeks after exposing the solutions to air (see Figure S17).
Inductively Coupled Plasma – Mass Spectrometry (ICP-MS)
ICP-MS samples were prepared by dissolving 10 μL aliquots of sample in concentrated HNO3 (300 μL) in a 15 mL conical tube and digested at 60 °C for 1 h. Samples were diluted to a total volume of 10 mL with Milli-Q water and analyzed using a computer-controlled Thermo iCAP Q ICP-MS. Data were acquired using one survey run (10 sweeps) and three main (peak jumping) runs (100 sweeps). Isotopes selected for analysis were 157,158Gd, 163Dy, 153Eu, 59Co, 68Zn, 65Cu, 89Y, 115In, and 159Tb; the latter three elements were used for internal calibration.
W-band Electron Paramagnetic Resonance Spectroscopy (EPR)
1 mM aqueous stock solutions of each complex (1, 2, and 3) were prepared using Milli-Q water in an inert atmosphere; the solutions were diluted to a final concentration of 400 μM and freeze–pump–thawed three times. The solutions were transferred to quartz EPR tubes and sealed with Teflon. Continuous wave EPR (cw-EPR) spectra were collected at room temperature with a Bruker E-680X/W outfitted with a cylindrical resonator (EN-680-1021H), the experimental parameters used were 0.05 mW of power; 4 G magnetic field modulation amplitude at 100 kHz; and a time constant of 40.96 ms and time constant of 163.84 ms. The cw-EPR lineshapes were modeled in Easyspin using a single isotropic S = 1/2 electron spin convoluted with a Lorentzian line shape, where the line width parameter is defined as the full width at half-max.45
Magnetic Measurements
Magnetic measurements were performed using a Quantum Design MPMS-XL SQUID magnetometer. Polycrystalline samples were sealed in a polyethylene bag under a dinitrogen atmosphere. Approximately 10 mg of each compound was pulverized and weighed into a small polyethylene bag; exact weights were obtained to the nearest milligram. Variable temperature dc susceptibility data were measured from 1.8 to 300 K. Magnetic data were simulated using MagPro software46 to obtain the coupling constant J. The Hamiltonians used were Ĥ = −2JŜGdŜM, where M is CoII or CuII. Experimental errors were determined by averaging simulations of two independently prepared samples.
X-ray Structure Determination
Single crystals of reported compounds were directly coated with Paratone-N oil and mounted on a MicroMounts rod. The crystallographic data were collected at 100 K on a Bruker APEX II diffractometer equipped with MoKα or CuKα sealed tube source. Raw data were integrated and corrected for Lorentz and polarization effects using Bruker APEX2 v. 2009.1.47 SADABS48 was used to apply multiscan absorption correction. Space group assignments were achieved by examining systematic absences, Estatistics, and successive refinement of the structure. Structures were solved by SHELXT49 direct method and refined by SHELXL within the OLEX interface. In cases where solvent molecules were severely disordered, the solvent mask function was applied in the OLEX interface to calculate the void space for solvent molecules as well as averaged electron density found in such void. For the complexes LCoGd(μ-O2CCH3)(O2CCH3)2, LZnGd(μ-O2CCH3)(O2CCH3)2, and LZnEu(μ-O2CCH3)(O2CCH3)2, there were determined to be 112.8, 156.6, and 129.1 electrons/unit cell (Z = 8 for all three complexes), corresponding to approximately 0.6, 0.9, and 1.5 molecule of CH3CN/complex, respectively (see Tables S1–S4).
RESULTS AND DISCUSSION
Synthesis and Structures
The ligand LH2 was synthesized according to a modified literature procedure.44 All dinuclear complexes were synthesized through a one-pot stepwise metalation by addition of transition metal(II) then lanthanide(III) acetate salts to H2L. Here, the acetate anion acts as an internal base to deprotonate the two phenol protons on H2L. Following metalation, slow diffusion of Et2O vapor in concentrated MeCN solutions gave crystalline solids that were dried under reduced pressure to give the compounds 1–7. In the cases of 1, 2, 3, and 7, Et2O diffusion into a dilute MeCN solution of the compound gave rod-shaped crystals, suitable for single-crystal X-ray diffraction analysis, of LCoGd-(μ-O2CCH3)(O2CCH3)2·0.6MeCN, LCuGd(μ-O2CCH3)-(O2CCH3)2, LZnGd(μ-O2CCH3)(O2CCH3)2·0.9MeCN, and LZnEu(μ-O2CCH3)(O2CCH3)2·1.5MeCN, respectively. The CoGd, ZnGd, and ZnEu compounds crystallized in the space group P21/n, while the CuGd compound crystallized in the space group P1̄.
The neutral complex in all four compounds features a GdIII ion that is coordinated to nine O atoms (see Figures 2 and S18). Four coordination sites of GdIII are occupied by O atoms from two phenoxo bridges and two phenyl methyl ether groups of L−, while the remaining five sites are occupied by one O atom from a bridging μ-O2CCH3− ion and two κ2 O2CCH3− ions.
Figure 2.

Crystal structures of LMGd(μ-O2CCH3)(O2CCH3)2 complexes, where M = Co (left), Cu (middle), and Zn (right), as observed in single crystals grown from concentrated solutions of 1, 2, and 3, respectively. Orange, green, cyan, magenta, red, blue, and gray spheres represent Gd, Zn, Cu, Co, O, N, and C atoms, respectively; H atoms are omitted for clarity.
Each divalent transition metal center resides in a pseudosquare pyramidal coordination geometry with the equatorial sites occupied by two imide N atoms and two bridging phenoxo O atoms afforded by L−, and the apical site occupied by an O atom from a μ-O2CCH3 ion. In the case of the CoGd, ZnGd, and ZnEu complexes, the transition metal ion is significantly displaced out of the N2O2 plane, with M–Oacetate distances of 1.996(2), 1.978(4), and 1.980(2) Å, respectively. This distortion leads to the two N-bound C atoms of the propylene group to bend away from the square pyramid. By contrast, the CuII center in the CuGd complex is not nearly as displaced from the N2O2 pocket with a significant Jahn–Teller distortion giving a Cu–Oacetate distance of 2.224(2) Å. Consequently, one N-bound C atom of the propylene group is bent away from the square pyramid, while the other is bent toward the pyramid. This conformational difference likely results in the lower-symmetry space group for the CuGd complex relative to the others.
The MLn complexes feature M–O–Ln angles of 103.27(7)° (CoGd), 103.44(9)° (CuGd), 103.3(2)° (ZnGd), and 103.34(9)° (ZnEu), and corresponding M···Gd distances of 3.4528(7), 3.3911(4), 3.463(2), and 3.4787(5) Å. These structural parameters are consistent with related transition metal-lanthanide complexes.41,43 Moreover, the similar structural features across the family of complexes supports the validity of comparing values of relaxivity and q between different metal compositions.
Magnetic Behavior
In order to probe the magnetic interactions in 1 and 2, dc magnetic susceptibility data were collected for solid-state samples (see Figure 3). The high temperature value of χMT for 1 and 2 are 10.27 and 8.06 cm3 K/mol, respectively, consistent with noninteracting spins of S = 3/2 (CoII) and S = 7/2 (GdIII) for 1 and S = 1/2 (CuII) and S = 7/2 (GdIII) for 2. For both compounds, χMT increases with decreasing temperature, albeit more rapidly for 2, eventually reaching maxima of 11.27 cm3 K/mol (1) and 9.57 cm3 K/mol (2) at 10 K. Upon decreasing the temperature further, χMT undergoes a sharp downturn, reaching minima of 7.30 cm3 K/mol (1) and 9.87 cm3 K/mol (2) at 1.8 K.
Figure 3.

Variable temperature dc susceptibility data of 1 (red) and 2 (blue) measured at an applied field of 10 000 Oe. The black lines represent fits to the data.
The gradual increase of χMT with decreasing temperature indicates the presence of ferromagnetic coupling between GdIII and MII centers. These interactions were modeled with the spin Hamiltonian Ĥ = −2J(ŜGd·ŜM)46,50 to give exchange coupling constants of J = +0.22 cm−1 for 1 and J = +2.6 cm−1 for 2. The much stronger coupling observed in 2 can likely be attributed to the large degree of orbital overlap between the p orbital on the phenoxo O atom and the singly occupied d(x2−y2) on the Cu center.
While not explicitly modeled, the sharp downturn in χMT observed for both compounds likely stems from weak intermolecular antiferromagnetic coupling and zero-field splitting. The values of J obtained for 1 and 2 are comparable to those previously obtained for related dinuclear complexes.38 Most importantly, the presence of significant exchange coupling for these complexes, particularly when paired with the inherently short T1e of CoII in 1, may lead to a shortening of T1e and thus decrease in relaxivity for GdIII in solution.
Solution Properties
Before determining T1e and the resulting H2O relaxation rate in 1, 2, and 3, the complex compositions in aqueous solution were examined by solution spectroscopic techniques. Compounds 1, 2, and 3 exhibited modest solubility in aqueous solution, leading us to suspect the coordinated acetates exchange with water to yield a tricationic complex of form [LMGd(H2O)5–6]3+.
To examine this hypothesis, the solution structure of each complex was investigated by 2D NMR techniques to ensure the lanthanide was stable in the O4 binding pocket afforded by L−. Total NMR structural assignment was made in methanol-d4 due to inadequate line shape in D2O. 1H assignments were made based on the correlations in the NOESY and TOCSY spectra coupled with the heteronuclear HSQC and HMBC. The rigidity of the structure results in integration doubling of chemically equivalent resonances. Three 1H resonances at 6.2, 9.4, and 18.3 ppm in the methanol-d4 spectrum were assigned to the bridging and capping acetates in 7, consistent with three coordinating inequivalent acetates. In contrast, only one acetate resonance near 1 ppm was found in the D2O spectrum of 7, resembling a dissociated diamagnetic acetate resonance (see Figures S9 and S12). Additionally, none of the integration doubling observed in the methanol-d4 spectra was observed in D2O. Thus, the rigidity of the complex is reduced, and we attribute this result to the dissociation of O2CCH3− ions.
The precise number of bound water molecules in aqueous solution was determined by measuring the 17O NMR chemical shifts of 4, 5, and 6, which are the DyIII analogues of 1, 2, and 3, respectively. The choice of DyIII in place of GdIII is to take advantage of the short T1e of DyIII, such that the 17O resonances of bound water molecules can be observed. Saturated solutions of 4, 5, and 6 were found to exhibit values of q = 6, 5, and 5.6, respectively, with an inherent error of 20% per the limitations of the technique (see Experimental Section and Table 1). Additional support for the aqueous stability of [LCoGd(H2O)6]3+ and [LCuGd(H2O)5]3+ was evidenced by no significant changes in the UV–vis spectra in aqueous solution over time (see Figure S17).
After confirming the number of bound H2O molecules in 1, 2, and 3 in aqueous solution, the ability of these complexes to increase the relaxation of bulk H2O protons was evaluated. The relaxivities (r1) for samples containing 1, 2, and 3 were found to be 5.2, 9.8, and 11.4 mM−1 s−1, respectively (see Figure 4, top). These values are high compared to conventional GdIII contrast agents where q ≈ 1–2, due to the high q values (~5–6) present in here. Nevertheless, the value of r1 for 1 is remarkably less than half that for 3, despite the structural similarities between the two compounds. By comparison, the r1 for 2 is lower than that for 3, but still considerably higher than r1 for 1.
Figure 4.

Relaxivity values of 1 mM aqueous solutions 1, 2, and 3 at 1.4 T and 37 °C. Normalized r1 to the number of inner-sphere waters, q (top) and measured r1 values (bottom). Error bars represent the propagated error (top) or the standard deviation of three independent measurements (top).
The reduction in r1 for 1 is even more apparent when the values are normalized for q, with values of 0.9, 1.9, and 2.0 mM−1 s−1 for 1, 2, and 3, respectively. Moreover, such a small value of r1/q is remarkable when compared to the clinically employed agent [Gd(DOTA)]−, which features r1/q = 3.2 mM−1 s−1.51
Relaxivities for aqueous solutions containing 1:1 M-(O2CCH3)2/Gd(O2CCH3)3, where M = Co, Cu and Zn, were measured to verify that the observed modulation of r1 for 1, 2, and 3 is not an intrinsic property of the free ions interacting in solution (see Figure S14). Importantly, the r1 values for these mixtures show no correlation to the identities of M, in contrast to the r1 comparison among 1, 2, and 3.52 In addition, MR solution phantom images at 1.5 T offer visual evidence that 1 features the smallest relaxation enhancement of the three compounds (see Figure 5). The T1 values were measured at ambient temperature and as such cannot be directly compared to the relaxivity measurements at 37 °C.
Figure 5.
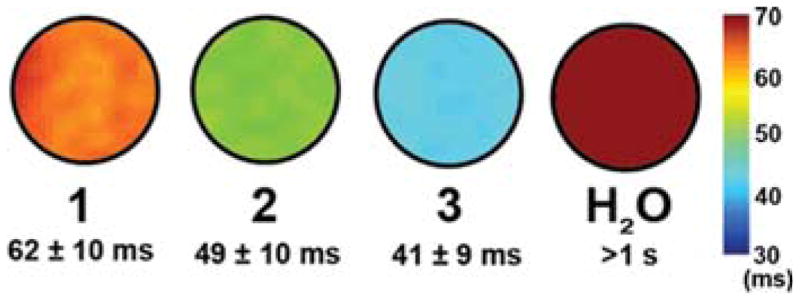
T1 MR solution phantom images of 1, 2, and 3 at 1.5 T and ambient temperature. T1 times represent averages over three slices with the associated standard deviation.
At high magnetic field, the proton Larmor frequency increases while T1e slows dramatically. If the difference in r1 between complexes 1, 2, and 3 arises from the difference in T1e of GdIII, then this difference should be less prominent at higher field. To examine this, r1 values were measured on a 7 T MR scanner. Figures S15 and S20 show the relaxivity data from the 7 T relaxivity measurements and image intensities, respectively, where each complex exhibits a value of r1 ≈ 10 mM−1 s−1. These data further support the contention that the mechanism of relaxivity decrease in 1 arises from the presence of magnetic coupling to CoII and not through other parameters such as τr or τm, which are field-independent.
EPR Spectroscopy
In order to determine whether magnetic coupling between MII and GdIII leads to a decrease in T1e of GdIII, electron paramagnetic resonance (EPR) spectroscopy of each complex was measured to probe the electronic relaxation. Precise quantitation of T1e is not possible for these complexes as the T1e was found to be too short for the instrument acquisition time scale (ns). Nevertheless, the transverse electron relaxation time T2e is expected to follow a similar trend as T1e in the presence of magnetic coupling,42 and therefore W-band (95 GHz) continuous-wave EPR spectra were collected for aqueous solutions of 1–3 at 25 °C (see Figure 6).
Figure 6.

W-band EPR spectra of 1 (top, red), 2 (middle, blue), and 3 (bottom, green) in aqueous solution at room temperature. Crosses and solid lines represent experimental data and simulation, respectively.
Each spectrum shows only a single feature corresponding to S = 7/2 GdIII, with g values of 1.9928, 1.9934, and 1.9922 for 1–3, respectively. Importantly, 1 and 2 exhibit an increased Lorentzian line width of 14.3 mT and 13.5 mT, respectively, relative to that of 5.8 mT observed for 3. For a homogeneously broadened line, the full width at half-maximum (Γ) is inversely related to T2e (T2e ∝ Γγe−1).53 As such, the significantly larger line widths for 1 and 2 indicate smaller T2e values of Gd compared to that of 3. In sum, while EPR measurements here cannot provide direct determination of T1e, acceleration of the electronic relaxation of GdIII in 1 and 2 is evidenced by the observed reduction in T2e.
Nuclear Magnetic Relaxation Dispersion
To verify that the observed differences in relaxivity arise from changes in the T1e of GdIII, nuclear magnetic relaxation dispersion (NMRD) data were obtained at 25 and 37 °C (see Figure 7) for 1–3. This analysis enables the estimation of T1e from fitting the parameters of the SBM model for paramagnetic relaxation enhancement, namely, T1e, τr, and τm (eq 1). The resulting NMRD profiles verify that the 1H relaxivities decrease from 3 to 2 to 1 in the range 20–40 MHz, in accord with the value above obtained at 60 MHz. In addition, all profiles exhibit a single dispersion occurring near 10 MHz. However, the ratio between the relativities at 40 MHz and low field (<1 MHz) differ between the complexes, indicating that both the field dependent T1e and τr contribute to the dipolar relaxation correlation time (τc).
Figure 7.

Best fit NMRD profiles obtained in the assumption of anisotropic (top) or isotropic (bottom) reorientation time. Aqueous solutions of 1 (red), 2 (blue), and 3 (green). The corresponding red, blue, and green stars represent the relaxivity measured at 1.4 T reported above. Solid and hollow spheres represent data obtained at 25 and 37 °C respectively. Lines represent the best fits obtained from the minimization.
The temperature dependence of the profiles shows that r1 decreases upon heating for all complexes, thus indicating that the water molecules are in fast exchange. This observation indicates that τm is smaller than the nuclear relaxation time of the protons on coordinated H2O molecules, in agreement with the observed value of 60 ns at 37 °C (see Table S5). The outersphere contribution is plotted independently for the 25 °C data in Supplemental Figure S23.
The NMRD profiles were fit using the SBM model assuming isotropic motion for inner-sphere dipolar relaxation (see eq 1, eqs 4–6 in the Supporting Information, and Figures S21–S22), including a minor outer-sphere contribution due to freely diffusing water. This second contribution was calculated according to Freed equation,54 as described by the distance of closest approach d (fixed to 3.6 Å) and the diffusion coefficient D (2.5 × 10−9 m2/s and 3.5 × 10−9 m2/s at 25 and 37 °C, respectively). Application of the Bloembergen–Morgan equation to describe the field dependence of electron relaxation in a magnetically coupled system may be debatable. Indeed, we found this model does heuristically describes the field dependence of T1e in 1 (CoII analogue), but does not fully characterize 2 (CuII analogue). The field dependence of T1e of GdIII in 2 is parametrized by including a field independent contribution for electronic relaxation (Figure S21, eq 8). This difference in behavior is ascribed to the fact T1e of CoII is always smaller than T1e of GdIII, whereas T1e for CuII is longer at low field and shorter at high field than GdIII. In the presence of magnetic coupling between two metal ions, as measured between GdIII and CoII or CuII, the SBM equation was rescaled by a constant to account for the coupling in the high-temperature limit, as defined in Figure S21 and eqs 9–11.
In the minimization, rGd–H was fixed to 3.0 Å, q was fixed to either 5 or 6, and τm to 100 and 60 ns at 25 and 37 °C, respectively. The best-fit parameters included the electronic parameters Dt (transient zero-field splitting), τv (correlation time for the zero-field splitting modulation), and τr. Here, the values of Dt and τv should change depending on the identity of the transition metal, while τr is expected to remain essentially constant, as shown in Table S5. A relatively good fit with constant τr is possible only if q, fixed to 5 for 1 and 3, is increased to 6 for 2 (Table S5 and Figure 7, bottom). The larger q obtained for 2 can account for the contribution to the water proton relaxation from the dipole–dipole interactions between a water molecule coordinated to the CuII center. Different from CoII, this contribution may not be negligible due to the long T1e of CuII. If in fast exchange, it can provide a contribution to the low-field relaxivity up to ~1 s−1 (corresponding to an apparent increase in q of ~0.2).
Upon closer inspection, the best-fit profiles (see Figure 7, bottom) are suboptimal, with dispersion occurring with a frequency dependence less steep than what is obtained from a single Lorentzian function. Moreover, the nonspherical, planar structure of the complexes suggests that the reorientation should be anisotropic, such that two rotational correlation times should be considered with a weighting factor described by the parameter SLS.25 The NMRD profiles were therefore fit using an anisotropic rotation model (see Figure S21 and eq 7) that accounts for two separate rotational correlation times (τr1 and τr2). In this case, the NMRD fits were excellent (see Figure 7, top) for q ranging between 5 and 6 for 1 and 3 and between 6 and 7 for 2. In addition, values for SLS2 = 0.39, τr1 = 150 ps, and τr2 = 30 ps were obtained at 25 °C (see Table S6). Most importantly, the fits reveal that T1e at high fields is much shorter in the complexes with magnetic exchange coupling. For instance, at 25 °C and 1 T, values of T1e = 0.065 ± 0.025 and 0.135 ± 0.025 ns were obtained for 1 and 2, respectively, considerably shorter than that of T1e = 2.6 ± 0.7 ns for 3.
Conclusion and Outlook
The foregoing results demonstrate that magnetic exchange coupling between GdIII and transition metal ions provides a route toward low-background MR imaging contrast agents. This approach is exemplified by a study of the complexes LMLn(μ-O2CCH3)(O2CCH3)2 (M = Co, Cu, Zn). Solid-state magnetic susceptibility measurements reveal the presence of magnetic coupling between GdIII and the paramagnetic transition metal ions, with J = +0.22 cm−1 for the LCoGd(μ-O2CCH3)(O2CCH3)2 (1) complex and J = +2.6 cm−1 for the LCuGd(μ-O2CCH3)(O2CCH3)2 (2) complex. The three complexes are shown by 1H and 17O NMR spectroscopy to be [LCoGd(H2O)6]3+, [LCoGd(H2O)5]3+, and [LCoGd(H2O)5.6]3+ in aqueous solution.
Further, NMR experiments demonstrate that relaxivity values are loweered by half for LCoGd(μ-O2CCH3)(O2CCH3)2 in comparison to LZnGd(μ-O2CCH3)(O2CCH3)2 by virtue of the short T1e of CoII. Finally, NMRD analysis confirms that these differences in relaxivity are associated with differences in modulation of T1e of GdIII, with fits to the profiles giving values of T1e = 0.065 ± 0.025, 0.135 ± 0.025, and 2.6 ± 0.7 ns for LCoGd(μ-O2CCH3)(O2CCH3)2, LCuGd(μ-O2CCH3)-(O2CCH3)2, and LZnGd(μ-O2CCH3)(O2CCH3)2, respectively. These dinuclear complexes provide the proof-of-concept to develop biologically applicable complexes.
Future work will focus on alternative pathways to generate near zero-background probes through incorporation pathways for both better donor ligands and stronger magnetic coupling. Finally, we are using the critical lessons learned from this work to synthesize bioresponsive agents that are redox-sensitive where the T1e can be modulated via biochemical redox events. This can be conceived as starting from a zero-background, magnetically coupled (fast T1e) state being oxidized/reduced to break the coupling and restore the T1e of GdIII, thus subsequently increasing the relaxivity.
Supplementary Material
Acknowledgments
Work in the Meade laboratory was supported by the National Cancer Institute (U54CA151880) and the National Institute of General Medical Sciences (R01GM121518-01A1). Work in the Harris laboratory was supported by the Air Force Research Laboratory (FA8650-15-5518) and the National Science Foundation (DMR-1351959). T.D.H. thanks the Alfred P. Sloan Foundation. G.P. and C.L acknowledge the COST Action CA15209 “European Network on NMR Relaxometry”, Fondazione Cassa di Risparmio di Firenze, Consozio Interuniversitario CIRMMP, and Insruct-ERIC (ESFRI Core Centre CERM, Italy). We thank the Magnetic Resonance Center at the University of Florence, Drs. Yongbo Zhang for assistance with NMR, and Keith MacRenaris for assistance with ICP-MS, and Daniele Procissi for assistance with MR imaging.
Footnotes
Accession Codes
CCDC 1821082–1821085 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.8b00120.
1D-1H structural paramagnetic NMR assignment; equations for fitting q by 17O NMR; r1 and r2 values; UV-vis spectra; X-ray crystal structure of 7; DC-susceptibility measurement of 3; T1 and T2 solution phantom MR images; SBM equations for fitting NMR-dispersion profiles; best-fit NMRD profile; outer-sphere NMRD contribution; X-ray crystallographic data for 1, 2, 3, and 7; NMRD best-fit parameters for isotropic rotation; NMRD best-fit parameters for anisotropic rotation of 1, 2, and 3 (PDF)
References
- 1.Major JL, Meade TJ. Bioresponsive, Cell-Penetrating, and Multimeric MR Contrast Agents. Acc Chem Res. 2009;42:893–903. doi: 10.1021/ar800245h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heffern MC, Matosziuk LM, Meade TJ. Lanthanide Probes for Bioresponsive Imaging. Chem Rev. 2014;114:4496–4539. doi: 10.1021/cr400477t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louie AY, Hüber MM, Ahrens ET, Rothbächer U, Moats R, Jacobs RE, Fraser SE, Meade TJ. In vivo Visualization of Gene Expression Using Magnetic Resonance Imaging. Nat Biotechnol. 2000;18:321–325. doi: 10.1038/73780. [DOI] [PubMed] [Google Scholar]
- 4.Duimstra JA, Femia FJ, Meade TJ. A Gadolinium Chelate for Detection of β-Glucuronidase: A Self-Immolative Approach. J Am Chem Soc. 2005;127:12847–12855. doi: 10.1021/ja042162r. [DOI] [PubMed] [Google Scholar]
- 5.Li WH, Fraser SE, Meade TJ. A Calcium-Sensitive Magnetic Resonance Imaging Contrast Agent. J Am Chem Soc. 1999;121:1413–1414. [Google Scholar]
- 6.Caravan P. Strategies for Increasing the Sensitivity of Gadolinium Based MRI Contrast Agents. Chem Soc Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 7.Hingorani DV, Bernstein AS, Pagel MD. A Review of Responsive MRI Contrast Agents: 2005–2014. Contrast Media Mol Imaging. 2015;10:245–265. doi: 10.1002/cmmi.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powell DH, Ni Dhubhghaill OM, Pubanz D, Helm L, Lebedev YS, Schlaepfer W, Merbach AE. Structural and Dynamic Parameters Obtained from 17O NMR, EPR, and NMRD Studies of Monomeric and Dimeric Gd3+ Complexes of Interest in Magnetic Resonance Imaging: An Integrated and Theoretically Self-Consistent Approach. J Am Chem Soc. 1996;118:9333–9346. [Google Scholar]
- 9.Moats RA, Fraser SE, Meade TJ. A “Smart” Magnetic Resonance Imaging Agent That Reports on Specific Enzymetic Activity. Angew Chem, Int Ed Engl. 1997;36:726–728. [Google Scholar]
- 10.Giardiello M, Lowe MP, Botta M. An Esterase-Activated Magnetic Resonance Contrast Agent. Chem Commun. 2007;39:4044–4046. doi: 10.1039/b711989e. [DOI] [PubMed] [Google Scholar]
- 11.Keliris A, Mamedov I, Hagberg GE, Logothetis NK, Scheffler K, Engelmann J. A Smart 19F and 1H MRI Probe with Self-Immolative Linker as a Versatile Tool for Detection of Enzymes. Contrast Media Mol Imaging. 2012;7:478–483. doi: 10.1002/cmmi.1470. [DOI] [PubMed] [Google Scholar]
- 12.Que EL, Chang CJ. A Smart Magnetic Resonance Contrast Agent for Selective Copper Sensing. J Am Chem Soc. 2006;128:15942–15943. doi: 10.1021/ja065264l. [DOI] [PubMed] [Google Scholar]
- 13.MacRenaris KW, Ma Z, Krueger RL, Carney CE, Meade TJ. Cell-Permeable Esterase-Activated Ca(II)-Sensitive MRI Contrast Agent. Bioconjugate Chem. 2016;27:465–473. doi: 10.1021/acs.bioconjchem.5b00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matosziuk LM, Leibowitz JH, Heffern MC, MacRenaris KW, Ratner MA, Meade TJ. Structural Optimization of Zn(II)-Activated Magnetic Resonance Imaging Probes. Inorg Chem. 2013;52:12250–12261. doi: 10.1021/ic400681j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esqueda AC, López JA, Andreu-de-Riquer G, Alvarado-Monzòn JC, Ratnakar J, Lubag AJ, Sherry AD, De León-Rodríguez LM. A New Gadolinium-Based MRI Zinc Sensor. J Am Chem Soc. 2009;131:11387–11391. doi: 10.1021/ja901875v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Song R, Guo K, Meng Q, Zhang R, Kong X, Zhang Z. A Gadolinium(III) Complex Based Dual-Modal Probe for MRI and Fluorescence Sensing of Fluoride Ions in Aqueous Medium and In Vivo. Dalton Trans. 2016;45:17616–17623. doi: 10.1039/c6dt02229d. [DOI] [PubMed] [Google Scholar]
- 17.Woods M, Zhang S, Ebron VH, Sherry AD. pH-Sensitive Modulation of the Second Hydration Sphere in Lanthanide(III) Tetraamide-DOTA Complexes: A Novel Approach to Smart MR Contrast Media. Chem - Eur J. 2003;9:4634–4640. doi: 10.1002/chem.200305159. [DOI] [PubMed] [Google Scholar]
- 18.Kálmán FK, Woods M, Caravan P, Jurek P, Spiller M, Tircsó G, Király R, Brücher E, Sherry AD. Potentiometric and Relaxometric Properties of a Gadolinium-Based MRI Contrast Agent for Sensing Tissue pH. Inorg Chem. 2007;46:5260–5270. doi: 10.1021/ic0702926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tóth É, Bolskar RD, Borel A, González G, Helm L, Merbach AE, Sitharaman B, Wilson LJ. Water-Soluble Gadofullerenes: Towards High-Relaxivity pH-Responsive MRI Contrast Agents. J Am Chem Soc. 2005;127:799–805. doi: 10.1021/ja044688h. [DOI] [PubMed] [Google Scholar]
- 20.Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Albumin Binding, Relaxivity, and Water Exchange Kinetics of the Diastereoisomers of MS-325: a Gadolinium(III)-Based Magnetic Resonance Angiography Contrast Agent. Inorg Chem. 2007;46:6632–6639. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- 21.Overoye-Chan K, Koerner S, Looby RJ, Kolodziej AF, Zech SG, Deng Q, Chasse JM, McMurry TJ, Caravan P. EP-2104R: A Fibrin-Specfic Gadolinium-Based MRI Contrast Agent for Detection of Thrombus. J Am Chem Soc. 2008;130:6025–6039. doi: 10.1021/ja800834y. [DOI] [PubMed] [Google Scholar]
- 22.Song Y, Kohlmeir EK, Meade TJ. Synthesis of Multimeric MR Contrast Agents for Cellular Imaging. J Am Chem Soc. 2008;130:6662–6663. doi: 10.1021/ja0777990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vistain LF, Rotz MW, Rathore R, Preslar AT, Meade TJ. Targeted Delivery of Gold Nanoparticle Contrast Agents for Reporting Gene Detection by Magnetic Resonance Imaging. Chem Commun. 2016;52:160–163. doi: 10.1039/c5cc06565h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahrens E, Rothbächer U, Jacobs R, Fraser S. A Model for MRI Contrast Enhancement Using T1 Agents. Proc Natl Acad Sci U S A. 1998;95:8443–8448. doi: 10.1073/pnas.95.15.8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipari G, Szabo A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 1. Theory and Range of Validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 26.Bloembergen N, Purcell EM, Pound RV. Relaxation Effects in Nuclear Magnetic Resonance Absorption. Phys Rev. 1948;73:679–712. [Google Scholar]
- 27.Solomon I. Relaxation Processes in a System of Two Spins. Phys Rev. 1955;99:559–565. [Google Scholar]
- 28.Bloembergen N, Morgan L. Proton Relaxation Times in Paramagnetic Solutions. Effects of Electron Spin Relaxation. J Chem Phys. 1961;34:842–850. [Google Scholar]
- 29.Rast S, Borel A, Helm L, Belorizky E, Fries PH, Merbach AE. EPR Spectroscopy of MRI-Related Gd(III) Complexes: Simultaneous Analysis of Multiple Frequency and Temperature Spectra, Including Static and Transient Crystal Field Effects. J Am Chem Soc. 2001;123:2637–2644. doi: 10.1021/ja003707u. [DOI] [PubMed] [Google Scholar]
- 30.Bertini I, Luchinat C, Parigi G, Ravera E. NMR of Paramagnetic Molecules in Biological Systems. 2. Elsevier; 2017. [Google Scholar]
- 31.Bertini I, Galas O, Luchinat C, Parigi G, Spina G. Nuclear and electron relaxation in magnetic exchange coupled dimers: implications for NMR spectroscopy. J Magn Reson. 1998;130:33–44. doi: 10.1006/jmre.1997.1266. [DOI] [PubMed] [Google Scholar]
- 32.Murthy NN, Karlin KD, Bertini L, Luchinat C. NMR and Electronic Relaxation in Paramagnetic Dicopper(II) Compounds. J Am Chem Soc. 1997;119:2156–2162. [Google Scholar]
- 33.Holz RC, Bennett B, Chen G, Ming LJ. Proton NMR Spectroscopy as a Probe of Dinuclear Copper(II) Active Sites in Metalloproteins. Characterization of the Hyperactive Copper(II)-Substituted Aminopeptidase from Aeromonas Proteolytica. J Am Chem Soc. 1998;120:6329–6335. [Google Scholar]
- 34.Du K, Harris TD. A CuII2 Paramagnetic Chemical Exchange Saturation Transfer Contrast Agent Enabled by Magnetic Exchange Coupling. J Am Chem Soc. 2016;138:7804–7807. doi: 10.1021/jacs.6b03060. [DOI] [PubMed] [Google Scholar]
- 35.Tóth É, Helm L, Merbach AE, Hedinger R, Hegetschweiler K, Jánossy A. Structure and Dynamics of a Trinuclear Gadolinium(III) Complex: The Effect of Intramolecular Electron Spin Relaxation on Its Proton Relaxivity. Inorg Chem. 1998;37:4104–4113. doi: 10.1021/ic980146n. [DOI] [PubMed] [Google Scholar]
- 36.Moriggi L, Aebischer A, Cannizzo C, Sour A, Borel A, Bünzli JCG, Helm L. A Ruthenium-Based Metallostar: Synthesis, Sensitized Luminescence and 1H Relaxivity. Dalton Trans. 2009;12:2088–2095. doi: 10.1039/b818599a. [DOI] [PubMed] [Google Scholar]
- 37.Koullourou T, Natrajan LS, Bhavsar H, Pope SJ, Feng J, Narvainen J, Shaw R, Scales E, Kauppinen R, Kenwright AM, Faulkner S. Synthesis and Spectroscopic Properties of a Prototype Single Molecule Dual Imaging Agent Comprimising a Heterobimetallic Rhenium-Gadolinium Complex. J Am Chem Soc. 2008;130:2178–2179. doi: 10.1021/ja710859p. [DOI] [PubMed] [Google Scholar]
- 38.Gómez V, Vendier L, Corbella M, Costes JP. Tetranuclear [Co-Gd]2 Complexes: Aiming at a Better Understanding of the 3d-Gd Magnetic Interaction. Inorg Chem. 2012;51:6396–6404. doi: 10.1021/ic300711h. [DOI] [PubMed] [Google Scholar]
- 39.Costes JP, Dahan F, Dupuis A, Laurent JP. A Genuine Example of a Discrete Bimetallic (Cu, Gd) Complex: Structural Determination and Magnetic Properties. Inorg Chem. 1996;35:2400–2402. doi: 10.1021/ic951382q. [DOI] [PubMed] [Google Scholar]
- 40.Costes JP, Dahan F, Dupuis A, Laurent JP. A General Route to Strictly Dinuclear Cu(II)/Ln(III) Complexes. Structural Determination and Magnetic Behavior of Two Cu(II)/Gd(III) Complexes. Inorg Chem. 1997;36:3429–3433. doi: 10.1021/ic970264v. [DOI] [PubMed] [Google Scholar]
- 41.Yamaguchi T, Costes JP, Kishima Y, Kojima M, Sunatsuki Y, Bréfuel N, Tuchagues JP, Vendier L, Wernsdorfer W. Face-Sharing Heterotrinuclear MII-LnIII-MII (M = Mn, Fe, Co, Zn; Ln = La, Gd, Tb, Dy) Complexes: Synthesis, Structures, and Magnetic Properties. Inorg Chem. 2010;49:9125–9135. doi: 10.1021/ic100460w. [DOI] [PubMed] [Google Scholar]
- 42.Bertini I, Galas O, Luchinat C, Parigi G, Spina G. Nuclear and Electron Relaxation in Magnetic Exchange Coupled Dimers: Implications for NMR Spectroscopy. J Magn Reson. 1998;130:33–34. doi: 10.1006/jmre.1997.1266. [DOI] [PubMed] [Google Scholar]
- 43.Costes JP, Donnadieu B, Gheorghe R, Novitchi G, Tuchagues JP, Vendier L. Di- or Trinuclear 3d-4f Schiff Base Complexes: the Role of Anions. Eur J Inorg Chem. 2008;2008:5235–5244. [Google Scholar]
- 44.Romain C, Bennington MS, White AJP, Williams CK, Brooker S. Macrocyclic Dizinc(II) Alkyl and Alkoxide Complexes: Reversiblel CO2 Uptake and Polymerization Catalysis Tesing. Inorg Chem. 2015;54:11842–11851. doi: 10.1021/acs.inorgchem.5b02038. [DOI] [PubMed] [Google Scholar]
- 45.Stoll S, Schweiger A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 46.Azuah RT, Kneller LR, Qiu Y, Tregenna-Piggott PL, Brown CM, Copley JR, Dimeo RM. DAVE: a Comprehensive Software Suite for the Reduction, Visualization, and Analysis of Low Energy Neutron Spectroscopic Data. J Res Natl Inst Stand Technol. 2009;114:341–358. doi: 10.6028/jres.114.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruker AV. Bruker AXS Inc; Madison, WI: 2008. [Google Scholar]
- 48.Sheldrick G. SADABS, Program for Empirical Absorption Correction of Area Detector Data. University of Göttingen; Germany: 1996. [Google Scholar]
- 49.Sheldrick GM. SHELXTL, Version 6.12. Bruker Analytical Xray Systems, Inc; Madison, WI: 2000. [Google Scholar]
- 50.Magnetic susceptibility data were simulated using the program MagProp. Tregenna-Piggott PLW. MagProp (part of the NIST DAVE software suite), version 2.0. 2008 http://www.ncnr.nist.gov/dave.
- 51.Meyer D, Schaefer M, Bonnemain B Gd-DOTA, a Potential MRI Contrast Agent. Current Status of Physicochemical Knowledge. Invest Radiol. 1988;23:S232–235. doi: 10.1097/00004424-198809001-00048. [DOI] [PubMed] [Google Scholar]
- 52.Micskei K, Helm L, Brucher E, Merbach AE. Oxygen-17 NMR Study of Water Exchange on Gadolinium Polyaminopolyacetates [Gd(DTPA)(H2O)]2- and [Gd(DOTA)(H2O)]-Related to NMR Imaging. Inorg Chem. 1993;32:3844–3850. [Google Scholar]
- 53.Weil JA, Bolton JR. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. John Wiley & Sons; 2007. [Google Scholar]
- 54.Freed JH, Bruno GV, Polnaszek CF. Electron Spin Resonance Line Shapes and Saturation in the Slow Motional Region. J Phys Chem. 1971;75:3385–3399. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.