

Abstract
Exiguobacterium antarcticum strain B7 is a psychrophilic Gram-positive bacterium that possesses enzymes that can be used for several biotechnological applications. However, many proteins from its genome are considered hypothetical proteins (HPs). These functionally unknown proteins may indicate important functions regarding the biological role of this bacterium, and the use of bioinformatics tools can assist in the biological understanding of this organism through functional annotation analysis. Thus, our study aimed to assign functions to proteins previously described as HPs, present in the genome of E. antarcticum B7. We used an extensive in silico workflow combining several bioinformatics tools for function annotation, sub-cellular localization and physicochemical characterization, three-dimensional structure determination, and protein-protein interactions. This genome contains 2772 genes, of which 765 CDS were annotated as HPs. The amino acid sequences of all HPs were submitted to our workflow and we successfully attributed function to 132 HPs. We identified 11 proteins that play important roles in the mechanisms of adaptation to adverse environments, such as flagellar biosynthesis, biofilm formation, carotenoids biosynthesis, and others. In addition, three predicted HPs are possibly related to arsenic tolerance. Through an in vitro assay, we verified that E. antarcticum B7 can grow at high concentrations of this metal. The approach used was important to precisely assign function to proteins from diverse classes and to infer relationships with proteins with functions already described in the literature. This approach aims to produce a better understanding of the mechanism by which this bacterium adapts to extreme environments and to the finding of targets with biotechnological interest.
Introduction
Exiguobacterium are Gram-positive mobile bacteria that have psychrophilic and thermophilic adaptations according to the environment they live in. Isolates from this genus can be found in the most variable environments, from glacial ice to temperate soils, and have the capacity to survive in a range of extreme temperatures and in effluents contaminated with heavy metals such as arsenic and chrome [1]. Most of the species, such as Exiguobacterium antarcticum, are extremophile microorganisms that produce several enzymes that are stable at a broad range of temperatures, with numerous industrial applications such as for biosensors, environmental bioremediation and pharmaceutical applications [2–9].
These characteristics have triggered biotechnological interest in these bacteria and have aroused the interest of researchers in the past few years to investigate the different proteins involved in cold-adaptation. As an example, the recent identification and structural and biochemical characterization of a novel esterase, EaEST, from E. antarcticum B7, was due to the great commercial potential of cold-adapted esterases for industrial applications [10]. Another study has revealed the different quaternary structure of GH1 β-glucosidase from the E. antarcticum B7 structural basis for cold adaptation [11]. Additionally, Baraúna and colleagues (2016) have investigated the role of the FapR regulator of E. antarcticum B7 as the main protein responsible for the regulation of fatty acid synthesis during cold adaptation [12].
E. antarcticum strain B7 was the second species of the genus to have its genome completely sequenced and published, allowing genomics, transcriptomics and proteomics studies to have better understanding of microbial adaptation mechanisms [9,13–15]. However, there are still challenges to improve the understanding of these mechanisms. Thus, bioinformatics approaches can play an important role in improving the understanding of biological processes, the gene repertoire and gene regulation, including protein-protein interactions (PPI) [16,17].
Functional annotation is crucial for determining the function of proteins during proteome analysis. Meanwhile, the function of a considerable number of coding sequences still cannot be predicted. For this reason, these molecules are labelled hypothetical proteins (HPs). Most of these proteins are believed to play an important role in the cell, and their annotation can lead to knowledge about new structures, functions and pathways. Proteins with unknown function can be assigned by homology-based gene annotation due to the correlation with known proteins [18–22].
Several recent bioinformatics tools, such as the Conserved Domain Architecture Retrieval Tool (CDART), the Simple Modular Architecture Research Tool (SMART), CATH, Pfam, SUPERFAMILY and SVMProt, have been developed to assign functions to HPs from many species [21,23–27]. These tools are associated with all the data available in many databases using domain, family and ontology information to support protein function characterizations. In addition, the study of PPI using software for protein interaction searches, such as the STRING database [28], is essential for understanding the role of a protein in a biological network [21,29]. These interactions play an important role in cellular processes, and by studying them, an understanding of HP function and inferences about biological functions for these non-elucidated proteins can be reached [30,31]. Furthermore, three-dimensional modeling is important to associate structural information with the function of unknown proteins, through homology searches at the Protein Data Bank (PDB) [32].
The utilization of in silico approaches to the functional prediction of HPs has been successfully used in several bacterial species, such as Vibrio cholerae, Neisseria gonorrhoeae, Clostridium difficile, and Staphylococcus aureus [22,33–35]. Due to the relevance of E. antarcticum B7, the purpose of this work was to assign function to the hypothetical proteins present on the genome of this species for the identification of new proteins that may contribute to an improved understanding of the adaptation of this bacterium to the extreme environment and for new biotechnological targets, adopting an integrated workflow, containing conventional annotation programs allied to PPI analysis, and three-dimensional protein modeling.
Materials and methods
Retrieval of genome data
In this work, we used the Exiguobacterium antarcticum B7 genome. This strain was isolated from biofilms in Ginger Lake, King George Island, Antarctica. Its genome has 2,815,863 bp and 2772 genes. It was retrieved from the National Center for Biotechnology Information database—NCBI (https://www.ncbi.nlm.nih.gov/genome/) under accession number CP003063.1 [9]. Subsequently, a total of 765 coding sequences (CDS) annotated as hypothetical proteins were extracted from this genome, using Artemis software [36].
Functional annotation of hypothetical proteins
To unveil the function of the HPs using the programs and databases described in S1 Table, we first submitted these proteins to annotation using the GO FEAT tool 1.0 to a preliminary prediction [37], using an e-value 1e-03. GO FEAT is a new, free, online platform for functional annotation based on the homology search analysis on multiple databases, such as protein (Uniprot) [38], genome annotation, domain and family (InterPro and Pfam) [25,39] databases and, a cross reference using NCBI (https://www.ncbi.nlm.nih.gov/) and EMBL (http://www.ebi.ac.uk/ena/) databases.
All HP-presenting products that described family and/or protein domains, according to the GO FEAT results, were then selected for further analysis using a variety of publicly available bioinformatics tools for domain and function assignment (Fig 1). CDART [24] and SMART 8.0 [26] were used to search conserved domains using the Conserved Domain Database (CDD), and protein function based on domain architecture, respectively. CATH 4.2 was used to classify the domains within structural hierarchy [40]. Pfam 31.0 [25], SUPERFAMILY 1.75 [23] and SVMProt [27] were used to classify the HPs into functional families to predict the function based on similarity. We also used InterPro 66.0 for motif detection, which uses the integration of numerous available databases for functional prediction. For all databases, we used default parameters.
Fig 1. Workflow used for the annotation of the hypothetical proteins from E. antarcticum B7.
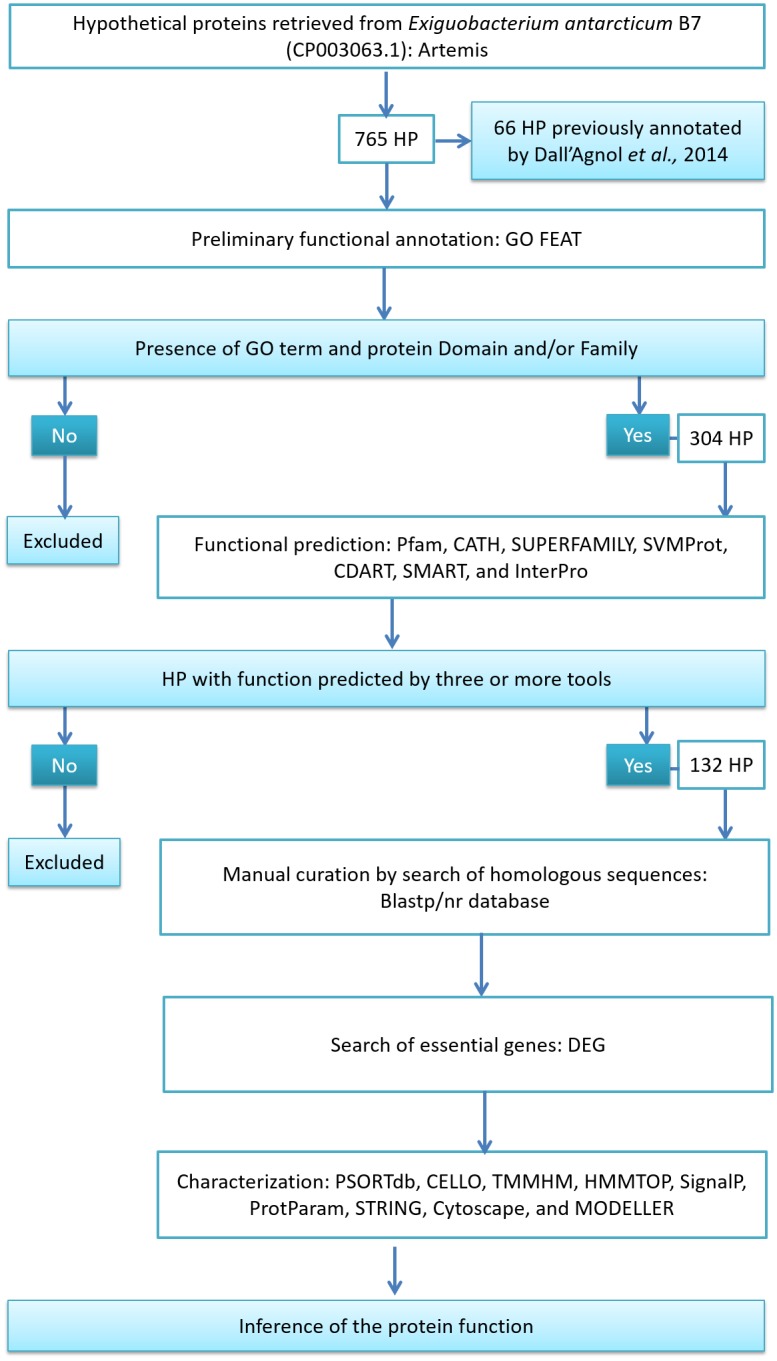
Then, CDS were manually annotated by searching for homologous proteins from related organisms using the Basic Local Alignment Search Tool (BLAST) against the NCBI non-redundant (nr) database, considering as parameter just hits with an identity ≥ 90%. The e-value, query cover and score parameters of every hits were described in supplemental material (S5 Table) [41].
To verify the presence of essential genes among our dataset, the DEG 15.2 database was used [42]. We adopted the BLOSUM62 matrix, score 100 and e-value 1e-05, using as a query a multifasta file containing the amino acid sequences with the 132 HPs versus the following available genomes belonging to the filo Firmicutes: Bacillus subtilis 168, Bacillus thuringiensis BMB171, Staphylococcus aureus N315 and Staphylococcus aureus NCTC 8325. On DEG, each gene has an identification from the single access number assignment, in addition to a reference number, sequence and function.
Determination of the sub-cellular localization
We used PSORTdb 3.0 [43] and CELLO 2.5 [44,45] to assign the location of the HPs in the cell. PSORTdb is a database that contains information through the association of both laboratory experimentations and computational prediction [46]. CELLO uses a two-level support vector machine (SVM), which involves a number of SVM classifiers. The second one uses a jury SVM to evaluate the outputs from the first level and generate the final probable sub-cellular localization [45]. The prediction of transmembrane helices and protein topology was performed through TMHMM 2.0 [47] and HMMTOP 2.0 [48], and SignalP 4.1 was used to verify the presence of signal peptide cleavage sites [49].
Prediction of physicochemical parameters
Molecular mass, theoretical isoelectric point (pI), amino acid composition, atomic composition, extinction coefficient, instability index, aliphatic index and high average hydrotherapy were predicted using the ProtParam tool [50], which allows for the calculation of several physicochemical parameters of the proteins.
Protein-protein interaction network building
For the prediction of PPIs the STRING 10.5 database [51] was used, in which the amino acid sequences of E. antarcticum B7 were submitted to identify, by similarity, the described PPIs. This database includes direct (physical) and indirect (functional) associations through a computational forecast. To guarantee the reliability of the PPIs, we selected only the most reliable and experimental interactions with score values above 0.700.
Subsequently, the identified interactions were transferred to E. antarcticum B7 by the interolog mapping method, described in previous studies such as Yu et al. (2004) [44] and Folador et al. (2016) [52]. This method is based on the assumption that if two proteins interact, the orthologous pairs also interact [53,54]. To identify the homology between the proteins, the BLAST against STRING proteins was performed [55], in which we considered only reciprocal hits with a conserved interaction score (IS) greater than 0.5625, corresponding to a 75% identity and 75% coverage, as described by Folador et al. (2016) [52]. The IS value is calculated by multiplying the identity and coverage of the BLAST alignment. Thus, for an interaction, four alignments are performed (two reciprocal alignments among STRING and E. antarcticum), with IS being the smallest value of these alignments: IS = lowest (lowest (lower (A,a), lower (a, A)), lowest (lower (B, b) lower (b, B)), respectively, representing the "A" and "B" the proteins of STRING and the "a" and "b" proteins of E. antarcticum B7.
The validation of PPI networks was performed in the Cytoscape 3.6.1 program [56] with the Network Analyzer plugin [57]. This program is a large-scale general-purpose modeling platform for network integration, allowing the visualization and analysis of PPI networks, where protein molecules and molecular interactions are assigned to nodes and edges, respectively. The Network Analyzer computes several topological parameters from networks, such as the node degree distribution and the shortest path, both of which were used in this work.
Determination and validation of three-dimensional structures
Three-dimensional homology modeling of the target proteins was performed by the MODELLER 9.13 program [58]. To construct the target structures obtained from PDB, we considered only templates with an identity ≥ 30% through BLAST alignment. The models were evaluated according to their stereochemical qualities by PROCHECK 3.5.4 [59], using a range of resolutions from 1.96 Å to 2.70 Å according to the template selected for each protein. The output files in .pdb format were visualized in the UCSF Chimera 1.1.2 [60].
Performance assessment
To verify the accuracy of the predicted functions for the HPs from the E. antarcticum B7 genome, a receiver operating characteristic (ROC) was performed [61]. ROC has been extensively used to the analyze the accuracy of the prediction [21]. We randomly selected 100 proteins with known functions and gene names from E. antarcticum (S2 Table) to be carried by the ROC. These proteins were annotated using the same pipeline described above for the prediction of HPs. The diagnostic efficacy was evaluated on six levels. To classify the prediction, the binary numerals “1” and “0” were used, where “1” denotes a true positive and “0” denotes a true negative. For confidence rating, the integers “2”, “3”, “4” and “5” were used [21,62]. The classification data were submitted to online software ROC Analysis: Web-based Calculator for ROC Curves, which calculates the accuracy, sensitivity, specificity and the ROC area of the functional prediction of the HPs [63]. The average accuracy obtained by the used pipeline was 95.7% (S3 Table). The results from the ROC analysis indicated the high reliability of the set of bioinformatics tools used in our study.
Arsenic tolerance assay
Since proteins involved in arsenic (As) tolerance were predicted among the HPs analyzed in this study, the strain tolerance to this compound was evaluated. The arsenic stock solutions were prepared in distilled water and sterilized. TSA plates (Tryptic Soy Agar, Merck, Germany) supplemented with arsenic in different concentrations (As; 50, 100, 300, 600, 1000, 1200, 1500 and 2500 μg/ml As Na2AsO4) were spot-inoculated (3 spots of 10 μL) with 104, 105 and 106 cells mL-1 (prepared from an exponential growth phase culture). Triplicate plates were prepared for each metal concentration. The cultures were incubated at 25°C. Each plate was checked for growth after five days, and positives were recorded by the appearance of colonies on at least one spot at the plate surface. The lowest concentration that prevented growth was considered the minimal inhibitory concentration (MIC). The reference strain Escherichia coli ATCC 25922 was included for quality control.
Results and discussion
Analysis of the hypothetical proteins from the E. antarcticum B7 genome
The first complete genome to be sequenced for the species, E. antarcticum B7, was deposited in the NCBI database in 2012 by Carneiro and colleagues. On that date, a total of 2.772 protein-coding genes were predicted on this genome. Of these, 765 (27.59%) were termed hypothetical proteins. In a previous study to investigate the gene expression of E. antarcticum B7 during cold adaptation, 66 of these HPs were functionally annotated [13]. This indicates that nearly one-quarter of the proteins encoded by this genome still need to be functionally characterized. Therefore, in this work, we have assigned functional information to HPs by the association of several bioinformatics resources.
For this analysis, we first performed a preliminary prediction using the GO FEAT platform. After GO FEAT analysis, 304 HPs of known protein domain and/or families, and their GO terms were selected. Domains are structural, functional and evolutionary units of a protein that are usually responsible for a particular function of a protein; therefore, the knowledge of a protein domain is helpful in understanding its role within a cellular context. This pool of 304 proteins was extensively analyzed using CDART, SMART, Pfam, SUPERFAMILY, SVMProt, and InterPro. The results obtained from the prediction tools are presented in the S4 Table and were analyzed aiming to assign functions to HPs, as described. Functional annotation was assigned with strong confidence to the proteins that exhibited similar function predictions from three or more programs. Thus, we inferred the function of 132 HPs with high confidence (Table 1), where 36 have homologous sequences in the NCBI database with no product function described (S5 Table). Domain identification is important to determine the function of a protein because it is a distinct, functional, and stable structural unit of the protein that is highly conserved during the evolution process [64].
Table 1. Hypothetical proteins functionally annotated from E. antarcticum B7.
| No | HP ID | InterPro ID | Protein function |
|---|---|---|---|
| 1 | Eab7_0026 | IPR010375 | Cyclic-di-AMP receptor |
| 2 | Eab7_0139 | IPR014046 | Diadenylate cyclase |
| 3 | Eab7_0143 | IPR028951 | Immunity protein 64 |
| 4 | Eab7_0150 | IPR016898 | Polyphosphate kinase 2 |
| 5 | Eab7_0152 | IPR006750 | Inner membrane exporter, YdcZ |
| 6 | Eab7_0154 | IPR006750 | Inner membrane exporter, YdcZ |
| 7 | Eab7_0255 | IPR000644 | Cystathionine-beta synthase associated with the CorC_HlyC domain |
| 8 | Eab7_0278 | IPR027417 | P-loop containing Nucleoside Triphosphate Hydrolases |
| 9 | Eab7_0284 | IPR005524 | ArsP_1 Superfamily |
| 10 | Eab7_0378 | IPR006750 | Inner membrane exporter, YdcZ |
| 11 | Eab7_0379 | IPR006750 | Inner membrane exporter, YdcZ |
| 12 | Eab7_0392 | IPR007621 | TPM_phosphatase |
| 13 | Eab7_0417 | IPR025889 | Heat induced stress protein YflT |
| 14 | Eab7_0504 | IPR002696 | Haemolytic |
| 15 | Eab7_0509 | IPR000644 | Cystathionine-beta synthase associated with the CorC_HlyC domain |
| 16 | Eab7_0515 | IPR016047 | Peptidase family M23 |
| 17 | Eab7_0609 | IPR000620 | EamA-like transporter Family |
| 18 | Eab7_0622 | IPR001509 | NAD-dependent epimerase/dehydratase |
| 19 | Eab7_0636 | IPR029348 | Nucleotidyltransferase-like |
| 20 | Eab7_0655 | IPR010368 | Control of competence regulator ComK, YlbF/YmcA |
| 21 | Eab7_0707 | IPR029063 | S-adenosylmethionine-dependent methyltransferases (AdoMet_Mtases) |
| 22 | Eab7_0714 | IPR007354 | Carotene biosynthesis associated membrane protein |
| 23 | Eab7_0741 | IPR004394 | Ribosomal silencing factor RsfS |
| 24 | Eab7_0770 | IPR029063 | Phosphotransferase system, EIIC |
| 25 | Eab7_0774 | IPR029063 | S-adenosylmethionine-dependent methyltransferases (AdoMet_Mtases) |
| 26 | Eab7_0783 | IPR025945 | SGNH hydrolase-like domain, acetyltransferase AlgX |
| 27 | Eab7_0789 | IPR002810 | NfeD-like C-terminal, partner-binding |
| 28 | Eab7_0806 | IPR006901 | tRNA (adenine-N1-)-methyltransferase |
| 29 | Eab7_0807 | IPR002678 | GTP cyclohydrolase 1 type 2/Nif3 |
| 30 | Eab7_0823 | IPR010708 | 5' nucleotidase, deoxy (Pyrimidine), cytosolic type C protein |
| 31 | Eab7_0861 | IPR001763 | Rhodanese-like domain |
| 32 | Eab7_0909 | IPR009474 | Disulphide isomerase |
| 33 | Eab7_0918 | IPR010343 | Aromatic acid exporter family member 1 |
| 34 | Eab7_0931 | IPR000620 | EamA-like transporter Family |
| 35 | Eab7_1015 | IPR010368 | Control of competence regulator ComK, YlbF/YmcA |
| 36 | Eab7_1031 | IPR010343 | Aromatic acid exporter family member 1 |
| 37 | Eab7_1052 | IPR011010 | DNA breaking-rejoining enzymes/ phage integrase |
| 38 | Eab7_1085 | IPR025889 | Heat induced stress protein YflT |
| 39 | Eab7_1094 | IPR018958 | SMI1 / KNR4 family protein |
| 40 | Eab7_1169 | IPR014719 | Ribosomal protein L7/12, C-terminal domain |
| 41 | Eab7_1187 | IPR007344 | GrpB protein |
| 42 | Eab7_1213 | IPR000620 | EamA-like transporter Family |
| 43 | Eab7_1218 | IPR018958 | SMI1 / KNR4 family protein |
| 44 | Eab7_1252 | IPR004360 | Glyoxalase I (lactoylglutathione lyase) |
| 45 | Eab7_1290 | IPR000160 | Diguanylate cyclase (GGDEF domain) |
| 46 | Eab7_1307 | IPR032713 | Multidrug transporter EmrE and related cation transporters [Defense mechanisms] |
| 47 | Eab7_1311 | IPR000160 | Diguanylate cyclase (GGDEF domain) |
| 48 | Eab7_1319 | IPR004446 | D,D-heptose 1,7-bisphosphate phosphatase |
| 49 | Eab7_1321 | IPR002549 | Transmembrane protein TqsA-like |
| 50 | Eab7_1322 | IPR010162 | Zinc peptidase like protein |
| 51 | Eab7_1333 | IPR001845 | HTH ArsR-type DNA-binding domain |
| 52 | Eab7_1355 | IPR003010 | Carbon-nitrogen hydrolase |
| 53 | Eab7_1362 | IPR012259 | Dihydrofolate reductase (DHFR) |
| 54 | Eab7_1372 | IPR001845 | Bacterial regulatory protein/Helix-turn-helix, arsR Family |
| 55 | Eab7_1390 | IPR005545 | YCII-related domain |
| 56 | Eab7_1403 | IPR009267 | Nucleotidyltransferase |
| 57 | Eab7_1406 | IPR029068 | Glyoxalase/Bleomycin resistance protein/Dihydroxybiphenyl dioxygenase |
| 58 | Eab7_1409 | IPR034660 | DinB superfamily protein |
| 59 | Eab7_1438 | IPR016040 | Rossmann-fold NAD(P)(+)-binding proteins |
| 60 | Eab7_1462 | IPR000330 | SNF2 family N-terminal domain |
| 61 | Eab7_1478 | IPR026881 | WYL domain |
| 62 | Eab7_1498 | IPR005182 | Bacterial PH domain |
| 63 | Eab7_1499 | IPR005182 | Bacterial PH domain |
| 64 | Eab7_1514 | IPR012544 | Bacterial PH domain |
| 65 | Eab7_1515 | IPR005545 | YCII-related domain |
| 66 | Eab7_1541 | IPR005532 | Sulfatase-modifying factor enzyme 1 |
| 67 | Eab7_1554 | IPR029072 | Transcriptional regulator, YebC-like |
| 68 | Eab7_1581 | IPR000160 | Diguanylate cyclase (GGDEF/EAL domain) |
| 69 | Eab7_1590 | IPR003744 | Vitamin uptake transporter |
| 70 | Eab7_1596 | IPR000644 | Cystathionine-beta synthase associated with the CorC_HlyC domain |
| 71 | Eab7_1603 | IPR021027 | Transposase DNA-binding domain |
| 72 | Eab7_1639 | IPR017941 | Rieske [2Fe-2S] iron-sulfur protein |
| 73 | Eab7_1666 | IPR006158 | Cobalamin (vitamin B12)-binding domain/Transcritional regulator MerR |
| 74 | Eab7_1668 | IPR004589 | ATP-dependent DNA helicase RecQ |
| 75 | Eab7_1669 | IPR029491 | Helix-turn-helix domain |
| 76 | Eab7_1675 | IPR000524 | GntR family transcriptional regulator |
| 77 | Eab7_1682 | IPR011765 | Peptidase M16 |
| 78 | Eab7_1689 | IPR004038 | Ribosomal protein L7Ae/L30e/S12e/Gadd45 family |
| 79 | Eab7_1718 | IPR012826 | Flagellar motor switch FliN |
| 80 | Eab7_1743 | IPR029025 | Flagellar biosynthetic protein FlhB |
| 81 | Eab7_1766 | IPR004007 | Dihydroxyacetone kinase family |
| 82 | Eab7_1781 | IPR008532 | Fibronectin-binding protein A N-terminus (FbpA) |
| 83 | Eab7_1829 | IPR010368 | Control of competence regulator ComK, YlbF/YmcA |
| 84 | Eab7_1847 | IPR003714 | Phosphate starvation-inducible protein PhoH, predicted ATPase [Signal transduction mechanisms] |
| 85 | Eab7_1870 | IPR005119 | LysR substrate binding domain |
| 86 | Eab7_1875 | IPR004155 | PBS lyase HEAT-like repeat |
| 87 | Eab7_1922 | IPR006345 | ATP-dependent RecD-like DNA helicase |
| 88 | Eab7_1928 | IPR021886 | MgsA AAA+ ATPase C terminal |
| 89 | Eab7_1950 | IPR013196 | Helix-turn-helix transcriptional regulator/ 3H domain |
| 90 | Eab7_1971 | IPR001482 | Type II secretion system (T2SS), protein E, N-terminal domain |
| 91 | Eab7_1974 | IPR012902 | Prokaryotic N-terminal methylation site |
| 92 | Eab7_2007 | IPR021722 | YvbH-like oligomerisation region |
| 93 | Eab7_2029 | IPR003838 | FtsX-like permease Family |
| 94 | Eab7_2032 | IPR001537 | SpoU rRNA Methylase Family |
| 95 | Eab7_2049 | IPR005467 | Histidine Kinase A |
| 96 | Eab7_2065 | IPR025618 | YtpI-like protein |
| 97 | Eab7_2066 | IPR000644 | Cystathionine-beta synthase |
| 98 | Eab7_2073 | IPR003356 | N-6 DNA Methylase |
| 99 | Eab7_2103 | IPR025711 | PepSY domain |
| 100 | Eab7_2119 | IPR023753 | Oxidoreductase |
| 101 | Eab7_2173 | IPR025889 | Heat induced stress protein YflT |
| 102 | Eab7_2195 | IPR024775 | DinB superfamily protein |
| 103 | Eab7_2221 | IPR027417 | AAA ATPase domain |
| 104 | Eab7_2247 | IPR002882 | 2-phospho-L-lactate transferase |
| 105 | Eab7_2260 | IPR001478 | PDZ domain |
| 106 | Eab7_2266 | IPR003740 | YitT_membrane Superfamily protein |
| 107 | Eab7_2301 | IPR003740 | YitT_membrane Superfamily protein |
| 108 | Eab7_2309 | IPR007809 | Flagellar biosynthesis protein FlgN |
| 109 | Eab7_2341 | IPR003838 | FtsX-like permease Family |
| 110 | Eab7_2372 | IPR000361 | Iron-sulphur cluster biosynthesis |
| 111 | Eab7_2389 | IPR012336 | Thioredoxin |
| 112 | Eab7_2406 | IPR016181 | Acetyltransferase |
| 113 | Eab7_2419 | IPR01176 | ATP-grasp family |
| 114 | Eab7_2464 | IPR024596 | DNA-directed RNA polymerase subunit beta |
| 115 | Eab7_2498 | IPR003838 | FtsX-like permease Family |
| 116 | Eab7_2499 | IPR005467 | Two-component sensor histidine kinase |
| 117 | Eab7_2500 | IPR001789 | DNA-binding response regulator |
| 118 | Eab7_2518 | IPR029046 | Lipoprotein localisation LolA/LolB/LppX |
| 119 | Eab7_2549 | IPR005624 | Haem_degrading Superfamily protein |
| 120 | Eab7_2581 | IPR028974 | TSP type-3 repeat |
| 121 | Eab7_2599 | IPR002563 | Flavin reductase like domain |
| 122 | Eab7_2611 | IPR003442 | tRNA A37 threonylcarbamoyladenosine biosynthesis protein TsaE |
| 123 | Eab7_2638 | IPR019660 | Bacterial sensory transduction regulator, YbjN |
| 124 | Eab7_2641 | IPR022791 | Phosphatidylglycerol lysyltransferase |
| 125 | Eab7_2678 | IPR002781 | Sulfite exporter TauE/SafE |
| 126 | Eab7_2712 | IPR018445 | Phosphate transport regulator |
| 127 | Eab7_2737 | IPR029058 | Alpha/beta hydrolase family |
| 128 | Eab7_2789 | IPR005122 | Uracil-DNA glycosylase-like |
| 129 | Eab7_2816 | IPR011701 | Major facilitator superfamily |
| 130 | Eab7_2819 | IPR007612 | LURP-one-related |
| 131 | Eab7_2832 | IPR018604 | Regulatory protein YycI |
| 132 | Eab7_2855 | IPR009366 | Biofilm formation stimulator VEG |
In addition, we predicted essential genes using DEG, a database that accommodates in vivo and in vitro experiments to identify essential genes in eukaryotes and prokaryotes. These genes are fundamental for the cellular machinery acting in the essential processes of the cell [42]. Through the DEG results, it was possible to identify 26 homologous genes, as shown in S6 Table.
There were identified GO term predictions for 85 proteins. Fig 2 shows the distribution of the proteins that presented two or more HP in each category, within the three GO categories: biological process, molecular function and cellular component. The GO terms that were represented by only one protein and 47 proteins with no GO terms can be seen in the S7 and S8 Tables, respectively. For molecular function, we identified 71 different GO terminologies designating protein functions; most of these referred to protein binding such as DNA, ATP, and metal binding (Fig 2B and S7 Table). The cellular component category contained 62 different GO terminologies, including 53 that were involved in membrane function and 43 that were integral components of the membrane (Fig 2C). The importance of bacterial membrane proteins in the physiology of Gram-positive bacteria is well-established [65]; however, several membrane proteins are difficult to characterize due to challenges in the preparation of stable membrane proteins with the preservation of their native structure for further studies. The cell membranes act as the front line in the interaction between the cell and the environment [66], so the identification of these membrane proteins, once known as hypothetical, may be the key of understanding the E. antarcticum B7 mechanisms that enable this psychotropic bacterium to survive under inhospitable temperatures [9].
Fig 2. Distribution of the hypothetical proteins of E. antarcticum B7 among the GO categories.

(A) The figure shows how the HPs were distributed among the three gene ontology categories determined by the GoFeat software. (B) Graph of the molecular functions. (C) Graph of the cellular components. (D) Graph of the biological processes.
According to the GO annotation, the biological process category revealed 44 GO terminologies with more representative terms related to transcription, transport, DNA repair, and DNA recognition (Fig 2D). The interactions between the DNA molecule and proteins are in the center of many biological processes [67]. The regulation of transcription represents a vital process for any living organisms, as the control of transcription allows the cell to respond to intra- and extracellular signals, such as environmental stimuli or nutrient scarcity. Among these proteins, Eab7_1675 was functionally annotated as a GntR family transcriptional regulator. This family of transcriptional factors, named gluconate-operon repressors, was first described in Bacillus subtilis in 1991 and is a large group of proteins involved in the regulation of several biological processes.
The sequence of Eab7_2372 was predicted as a protein belonging to the iron-sulfur cluster (ISC) biosynthesis process. This cluster of gene functions in eukaryotic and prokaryotic organisms and is required in some biological functions, such as in DNA synthesis for the repair of regulatory processes and redox and non-redox system catalysis [68]. In Gram-negative bacteria, its main role is related to the capture of sulfur and iron atoms for storage and mounting for Fe/S cluster formation; these are used as the final protein receptors [69].
Additionally, Eab7_2641 was annotated as phosphatidylglycerol lysyltransferase (mprF), an enzyme known to protect bacteria from cationic antimicrobial peptides that adds L-lysine to phosphatidylglycerol and in this way increases the net positive charge of the bacterial surface and decreases the binding of daptomycin and some cationic antimicrobial peptides, thus contributing to bacterial virulence. This mechanism has been described as a resistance system in Staphylococcus aureus [70].
Another protein function identified was the LysR substrate-binding domain (Eab7_1870). LysR regulators are global transcriptional regulators that can act either as activators or repressors of a single gene or an operon [67,71]. The classification of the proteins with no GO terms can be observed in the S8 Table; they are distributed mostly between enzymes, binding proteins and regulatory proteins.
Prediction of physicochemical properties and sub-cellular localization
In our study, the amino acid sequences of all 132 HPs were analyzed to assess their physico-chemical parameters, and the results can be observed in S9 Table. However, we paid close attention to the proteins that revealed functions related to adaptation and biotechnological interest. The proteins Eab7_0284, Eab7_0655, Eab7_1015, Eab7_1666 and Eab7_2855 all had molecular weight values between 14577.4 and 33132.9. The isoelectric point is the point at which the amino acid of the protein does not tolerate liquid charge, and therefore does not move in an electric field of a direct current. This parameter is used to determine the protein load [22,50,72]. For this group of proteins, it ranged from 4.27 to 10.0. In combination these two parameters help in the visualization of two-dimensional electrophoresis gels (2D), contributing to the laboratorial investigation of these proteins [34].
The aliphatic index is directly related to the molecular fraction of some amino acids and is associated with protein thermostability; that is, the higher its value, the higher the temperature range for which this protein will be stable [22,73,35]. Protein Eab7_2855, which is involved in biofilm formation, had one of the highest aliphatic index values, of 133.14. The grand average of hydropathy (GRAVY) reveals the protein interaction with water, which occurs better with low GRAVYs [22,73]. In Eab7_0655, Eab7_1015 and Eab7_1666, the GRAVY values are between -0.111, -0.490 and -0.678. For the instability investigation, the instability index was applied. This parameter offers an assumption of protein stability in a test tube. To discriminate between stable and unstable proteins, we use as cutoff values >40 and <40, respectively [50,74]. From the proteins of interest, Eab7_0655 (52.91), Eab7_1015 (42.24) and Eab7_1666 (40.20) were considered to be stable. The physicochemical characterization and sub-cellular localization analysis contribute to the elucidation of proteins predicted as proteins of unknown function [19]; this knowledge corroborates the findings of in vitro experiments with bacteria that exhibit biotechnological interest, such as E. antarcticum B7.
The sub-cellular localization of a protein plays an important role in the determination of its function, mainly because protein function is typically correlated to its location [45,62]. Therefore, the knowledge of a protein’s localization in the cellular space is helpful to unveil proteins with unknown function [22,75,76]. The sub-cellular localization can be inferred from the composition in amino acids due to evolutionary adaptation to different sub-cellular sites [75,77]. The proteins Eab7_0284, Eab7_0655, Eab7_1015, Eab7_1666 and Eab7_2588 were predicted to be in the cytoplasm. Proteins in this cellular localization are involved in functional processes such as biosynthesis and transport, which contributes to the secretion of substrates or even other proteins. In cases of environmental bacteria, this process can help in the competition between bacteria inhabiting the same ecological niche [78].
These types of analyses have already been carried out in studies of pathogenic bacteria. In this study 132 HPs were analyzed for their physicochemical parameters and sub-cellular localization. In environmental bacteria, studies with this theme have not yet been reported in the literature, making this study a pioneer.
Predicted proteins with adaptational functions to extreme environments
Among the 132 proteins functionally annotated in our study, we identified 10 that play an important role for the E. antarcticum B7 adaptation that allows theses bacteria to survive in extreme environmental conditions.
The protein Eab7_0284 was annotated as the ArsP_1 superfamily, which is a permease encoded by the arsenic resistance operon already identified in Campylobacter jejuni [79,80] and that has been recently identified as an organic arsenic transporter [79]. Microorganisms have developed mechanisms to survive in arsenic-contaminated environments, usually lowering arsenic concentrations in the cell by regulating the arsenic uptake, expulsing, or metabolizing arsenic to less toxic compounds [80,81]. Studies have revealed the presence of numerous genes involved in metal and metalloid response, including arsenic resistance, in the genome of species from the Exiguobacterium genus [82]. However, arsP was not yet identified. The permease ArsP together with ArsB, already described in E. antarcticum B7 [83], are commonly used to eliminate arsenic toxins in some bacteria through resistance pathways [84]. In general, these pathways act in three ways: reducing the concentrations of arsenic in the cytoplasm that can lead to the metabolism of arsenic into less toxic compounds, limiting its absorption and/or causing its expulsion [81]. A previous study revealed that most of the genes described for other strains of this genus that were considered highly resistant to arsenic, were absent in E. antarcticum B7 [82]. This indicates that arsP might be of great importance to this strain. Two other proteins were functionally predicted for this strain as related to arsenic resistance regulation (Eab7_1333 and Eab7_1372).
The protein Eab7_2599 was predicted to contain a Flavin reductase like domain. Flavin (oxy)reductases stimulate the reduction of flavin by NA(P)H. Among the functions of this protein family is the reduction of cobalamins (III), ferrisiderophores and chromate (III). The latter is a highly toxic compound, with carcinogenicity and mutagenicity properties, that has been described in studies related to bioremediation and which can be harmful in mineral and industrial processes that lead to serious environmental problems and health. E. antarcticum B7 resistance to chromium confers advantages to its adaptation and may be a future target in bioremediation trials against this toxic metal [85,86].
The protein Eab7_0714 was identified as related to the carotene function biosynthesis associated membrane protein. In non-photosynthetic bacteria this protein protects against ultraviolet (UV) radiation and may act as a regulator of membrane fluidity in these types of environments, interacting with the cell membrane in a similar way to cholesterol. This interaction is dependent on the degree of desaturation and on the length of the lipid chains [87–89].
Through the HPs studied, we identified the proteins that are functionally involved in flagella formation (Eab7_1718, Eab7_1743 e Eab7_2309). The E. antarcticum B7 flagella operon has already been identified, and its relationship with the adaptation at low temperatures has been inferred. In a study of the gene expression of E. antarcticum B7, Dall’Agnol et al. (2014) reported the differential expression in genes related to flagella synthesis regulation. They suggested that bacterial motility might be substantial under low temperature conditions.
Proteins involved in biofilm formation were also identified. The proteins Eab7_1015, Eab7_0655 and Eab7_1822 were annotated as the Control of competence regulator ComK and YlbF/YmcA. This family of proteins includes YlbF and YmcA, which are involved in the formation of biofilms and are necessary for correct biofilm formation [90].
The protein Eab7_2855 was annotated as Biofilm formation stimulator VEG. This protein stimulates the formation of the biofilm by inducing the transcription of the tapA-sipW-tasA operon, which formats a component of the biofilm, the amyloid fiber (TasA) [91]. Biofilm formation occurs when bacteria switches its state from a free-living form to a surface-associated multicellular state, producing a three-dimensional growth community [91,92]. This structure is supported by a highly hydrated extracellular matrix that is responsible for the adhesion of the cells in the biofilm and to solid surfaces, and serves as a source of nutrient providing carbon, nitrogen, and phosphorus [91]. Biofilm formation of a multicellular bacterial community in a dense barrier is responsible for the high tolerance of these microbial cells to environmental stresses [93,94]. Thus, the biofilm plays a great role for the adaptation of E. antarcticum in extreme environments.
We found protein Eab7_0741 as Ribosomal silencing factor RsfS. This protein acts by slowing cell growth by inhibiting protein synthesis when the nutrient availability is reduced [95]. RsfS was characterized by Escherichia coli and has been demonstrated to slow down or block translation when necessary [96]. The studies conducted in E. coli propose that RsfS works by binding the large 50S ribosomal subunit [97] so that it prevents 50S and 30S form forming a functional 70S complex [96]. Therefore, it seems to have great importance in the preservation of energy levels during nutritional absences, proving the importance of this protein for the adaptation of E. antarcticum B7 to the deprivation of nutrient resources in the environment it leaves in.
Arsenic tolerance in E. antarcticum B7
Previous research has described the absence in E. antarcticum B7 of several arsenic resistance genes necessary for arsenic detoxification [82]. However, in our study, we annotated proteins functionally related with arsenic resistance, such as ArsP1 Superfamily (Eab7_0284), together with two other (Eab7_1333 and Eab7_1372) presenting protein domain and family of the transcriptional repressor ArsR, respectively.
Our experimental results indicate that E. antarcticum B7 is resistant to arsenic, with the ability to grow in all the concentrations tested, up to 2500 μg/mL or 33.5 mM of As. This tolerance level is similar to the ones described for other Exiguobacterium strains that possess the genes included in the arsenic resistance operon, reported to grow in arsenic concentrations as high as 10 mM of arsenite (As[III]) and 150 mM of arsenate (As[V]) [82,83,98]. This demonstrates that even though this strain does not contain all of the genes included in the arsenic resistance operon in its genome, it is still highly resistant to arsenic.
Interestingly, we obtained better growth rates in concentrations above 300 μg/mL when compared with the lower concentrations tested. The effect of arsenic in stimulating bacterial growth has been previously reported [99] and has been related to a positive effect on bacteria metabolism, resulting in a shorter generation time and higher cell yield. On the other hand, the observed effect may be related to the addition of sodium (since arsenic was added as Na2AsO4), which may promote bacterial growth [100].
In nature, bacteria responses to arsenic are different and are usually mediated by genes present in the ars operons [101,102]. The most common configuration of this operon includes genes encoding a transcriptional repressor ArsR, an arsenate reductase ArsC, and an arsenite efflux pump [83,103]. Castro-Severyn and colleagues (2017) [82] investigated the presence of the genes responsible for arsenic resistance in 34 genomes within the Exiguobacterium genus, using Exiguobacterium sp. S17 as a reference due to its confirmed resistance to arsenic [83,98]. According to their study, E. antarcticum B7 detains only the gene arsR, for the arsenical resistance operon repressor, and arsB, for the arsenical pump membrane protein, with 50–69% and 85–94% of gene identity compared to the reference, respectively [82]. Thus, the mechanisms that allow E. antarcticum B7 to tolerate arsenic are still unclear, and proteins such as ArsP, predicted in our study, might be important for this phenotype.
Although arsenic is a natural element, it is a genotoxic component when present at high levels. Indeed, even low concentrations are detrimental to human health. The presence of arsenic detoxifying genes in bacteria is indicative of a potential application in bioremediation processes, for instance, in for the depollution of water effluents [104]. Nevertheless, studies with gene expression and proteomics are still necessary to produce a better understanding of the E. antarcticum B7 mechanism of arsenic resistance.
Probable targets with biotechnological interest
We identified some proteins that are functionally involved in processes that can have biotechnological applications, as Eab7_1666 (Cobalamin (vitamin B12)-binding domain), which is related to vitamin B12 synthesis. Vitamin B12 belongs to the cobalamin family and its biosynthesis is restricted to prokaryotes via a complex pathway [105,106]. This vitamin is essential and has been extensively used in the medical and food industries [106]. Its industrial production relies on microbial biosynthetic fermentation, specially using Pseudomonas denitrificans and Propionibacterium shermanii. Nonetheless, these bacteria present several limitations that make the production of vitamin B12 challenging [105,106].
Researchers have conducted several studies on vitamin B12 engineering. For the industrial production of cobalamin, it is crucial the use of efficient genetic tools and the knowledge of the metabolic pathways in order to improve the production of vitamin B12 [106,107]. Advances in metabolic engineering have been allowed to construct microbial chemical factories; however, the number of microbes that can be used efficiently to produce cobalamin is still small. In this way, the investigation of the E. antarcticum capability for industrial production of vitamin B12 may be conducted.
Furthermore, given its potential for biotechnology use, studies have suggested that cobalamin also plays an important role in bacterial adaptation to extreme environments, increasing the competitiveness during biofilm formation. However, the protective role of cobalamin has not yet been completely understood [108,109].
The proteins Eab7_0774 and Eab7_0707 were both predicted to be S-adenosylmethionine-dependent methyltransferases, known as AdoMet_Mtases (EC 2.1.1). The Eab7_0806 protein was identified as a tRNA (adenine-N1-)-methyltransferase (EC 2.1.1.217). Methyltransferases are a class of enzymes very present in nature acting in the methylation of biopolymers, as proteins and small metabolites in the three domains of life [110]. These enzymes act by catalyzing a methyl donor group for a receptor molecule, which in turn generates S-adenosylmethionine (SAM-MT) and a modified methylated molecule. The first SAM-MT discovered was catechol, a substance used in the pharmaceutical industry as an anti-cancer and anti-microbial compound [110,111].
Peptidases present biotechnological importance due to their applicability in the industries, acting in the composition of detergents, pharmaceuticals and foods [112]. In this study, proteins comprising the zinc metallopeptidases family were identified. Eab7_0515 belongs to the Peptidase M23 subfamily and has glycylglycine endopeptidase activity. This group also includes some bacterial lipoproteins [113]. Eab7_1322 corresponds to peptidase T, which is a metalloenzyme belonging to the M20 family. This enzyme, under anaerobic conditions, hydrolyzes the tripeptides at their N-termini region. Studies report that the regulation of this gene may contribute to the use of amino acids as energy sources. Håkansson and colleagues demonstrated, that the peptidase T (PepT) is involved in amino acid utilization in Salmonella typhimurium [114]. The occurrence of free amino acids is related to the nutritional value, bioactivity, and organoleptic characteristics of the hydrolysates. A protease from Exiguobacterium sp. SWJS2 compared with two commercial proteases (papain and alcalase 2.4L), has been reported to have the potential to produce hydrolysates containing peptides or amino acids of nutritional and sensorial importance [115].
The enzyme Eab7_1682 was predicted as an M16 peptidase, present in most prokaryotic and eukaryotic organisms [116]. Frias and colleagues analyzed the membrane vesicles in the extracellular matter, and identified this peptidase was superinduced at 4°C in Shewanella livingstonensis NF22T, the psychrotolerant bacterium, suggesting that it could be involved in bacterial survival in the Antarctic environment [117].
Protein-protein interaction network
PPI network analysis was performed for the 11 proteins for cold adaptation and biotechnological interest (Eab7_1743, Eab7_2309, Eab7_0284, Eab7_2599, Eab7_0714, Eab7_1015, Eab7_0655, Eab7_1829, Eab7_2855, Eab7_0741 and Eab7_1666) to evaluate the interactions between them and the other proteins of E. antarcticum B7. We obtained the network for these proteins individually and in clusters. The interactions within this group of proteins can be observed in the S1 Fig.
The degree of interaction is evaluated according to the color of the nodes on the network; the darker the green, the greater the interaction. The classification in physical and non-physical (regulatory) interactions is represented by the solid and discontinuous lines, respectively. The line thickness represents the IS value, such that thin lines have lower IS and thick lines have greater IS. However, the smaller IS in this network is 0.5625, meaning that all the interactions were mapped with a minimum of 75% identity and 75% coverage. The color of the lines represents the confidence of the interaction, with the yellow/green lines representing a high level of confidence (70% to 90%) and the blue lines representing experimental confidence (>90%) [118].
The protein Eab7_0284, related with arsenic resistance, showed interaction with only 12 proteins, and these interactions were mostly hypothetical. It also presented high confidence interaction with the transcriptional regulator, PadR-like protein (S1 Fig). PadR transcriptional regulators are frequently related with control of detoxification genes [119,120], acting in behaviors such as repressing the phenolic acid decarboxylase gene in Lactobacillus plantarum and Pediococcus pentosaceus [121,122] and the phenol acid decarboxylase gene in Bacillus subtilis [123].
The proteins Eab7_0714 and Eab7_1666 presented interactions with 10 and 12 proteins, respectively (S1 Fig). Eab7_0714 interacted with three phytoene desaturase (Eab7_0708, Eab7_0709 and Eab7_0711) and one phytoene synthase (Eab7_0710), which are proteins involved in the carotenoid biosynthesis pathway [124,125]. Carotenoids are natural organic pigments that are produced by plants, algae, fungi and bacteria in response to various environmental stresses [126]. The interaction revealed between our protein (once known as hypothetical and now assigned as a protein related to carotene biosynthesis) and these proteins reinforces the accurate function attribution performed by this study. The protein Eab7_1666 also showed a high confidence interaction with a protein producer of phytoene synthase (Eab7_0710). Studies have shown that in prokaryotes, such as the extremophilic bacteria from the genera Deinococcus and Thermus, colalamin, in addition to its role as an enzyme cofactor, is involved in the regulation of gene transcription related to the biosynthesis of carotenoids by binding to CarH repressor [109,127,128]. Carotenoids protect against oxidative damage. Therefore, the presence of cobalamin might provide an advantage in environments that are extremely acidic or highly loaded with metal [109].
The flagella formation proteins Eab7_2309 and Eab7_1743 have been demonstrated to interact with 14 and 61 proteins, respectively (S1 Fig). Both revealed strong interactions with other flagellar synthesis proteins. Eab7_1743 was shown to interact with 16 flagellar proteins that were annotated as part of an operon in the E. antarcticum B7 genome. Eab7_1743 also exhibited a high-confidence relationship with the gene sigD that encoded a sigma factor, which was shown to regulate genes involved in flagella biosynthesis. The expression of the gene sigD was induced under cold conditions, suggesting that bacterial motility might be substantial at a low temperature [13].
The proteins related to biofilm formation, Eab7_1829, Eab7_2855, Eab7_0655 and Eab7_1015, interact with 35, 36, 48 and 57 proteins, respectively. Some of these proteins were closely related to the biofilm formation or regulation. For instance, Eab7_1829 exhibited a high degree interaction with the protein encoded by the gene clpP that has been described to be involved in biofilm formation. Protein Eab7_0655 showed an interaction with the ctsR gene, which is a regulator of the genes clpC and clpP and is involved in biofilm development in Staphylococcus aureus [129]. Eab7_0655 also exhibited a great degree of interaction with murB, an up-regulated gene in biofilm formation and maintenance, responsible for the synthesis of cell wall structures [130]. Both Eab7_1829 and Eab7_2855 have demonstrated a high degree of interaction with coaD, included in the three-gene operon, and waaAE-coaD in Yersinia pestis, where waaA is a key determinant in biofilm formation [131].
The protein with the wider interaction network was Eab7_0741 assigned to the transcriptional regulator RsfS. It is involved in 156 interactions, the great majority of which present a high degree of interaction and confidence. Between these interactions, we identified many proteins related to transcription, such as the 50S and 30S ribosomal proteins, ribosomal RNA large subunit methyltransferases, ribosomal RNA small subunit methyltransferases, tRNA ligases and tRNA synthetase subunits, and elongation factors (Fig 3). Many of the interactions identified support the correct functional prediction of this protein.
Fig 3. Protein-protein interaction network of protein Eab7_0741, the Ribosomal silencing factor RsfS.

The figure shows that the majority of the 156 proteins interacting with Eab7_0741 presents a great number of interactions, which is supported by the node color intensity (dark green), thickness and line color.
Protein cellular functions are determined through their interactions with other proteins and the knowledge of these relationships have led to the development of diverse experimental methods for measurement [132]. Thus, the study of protein-protein interactions are important to infer the function of a completely unidentified protein once its function can be inferred based on the evidence of their interactions with the known proteome of a given organism [64]. Consequently, the identification of the protein interaction is essential since the execution of one function is strongly dependent on the contact or regulatory interaction with another protein [54].
The cellular environment is congested, and the proteins communicate with each other in specific ways leading to cellular processes and biological functions. Therefore, the analysis of PPI networks is required to understand the protein function and the complexity of living systems [54,133]. Functional prediction is one of the main goals of the PPI network, and the availability of PPI networks have helped in the development of computational methods to predict protein functions [64].
Three-dimensional structures
We also constructed the three-dimensional structures for those proteins considered important for the adaptation or potential application in biotechnology. Models for five proteins were obtained (Table 2) with the rate of identity with the model from PDB ranging from 34.53% to 63.46%. The three-dimensional models and the Ramachandran values can be observed in the S2 Fig. Four of the models were constructed from homologous proteins derived from bacteria belonging to the Bacillus genus, closely related to the Exiguobacterium genus. Based on the value of identity and resolution, the best model obtained was of the protein Eab7_0741, which was annotated as Ribosomal silencing factor RsfS (Fig 4A).
Table 2. Three-dimensional structure characteristics for the proteins tested.
| HP ID | Protein Name | % Identity | PDB Template | Organism | Method | R-value free | R-value work | Resolution |
|---|---|---|---|---|---|---|---|---|
| Eab7_0741 | Ribosomal silencing factor RsfS | 63.46 | 2o5a | Bacillus halodurans | X-ray diffraction | 0.278 | 0.251 | 2.70 Å |
| Eab7_1666 | Cobalamin (vitamin B12)-binding domain | 34.53 | 5c8a | Thermus thermophilus | X-ray diffraction | 0.227 | 0.183 | 2.15 Å |
| Eab7_2599 | Flavin reductase like domain | 45.17 | 3bpk | Bacillus cereus | X-ray diffraction | 0.187 | 0.153 | 1.56 Å |
| Eab7_1015 | Control of competence regulator ComK, YlbF/YmcA | 56.19 | 2pih | Bacillus subtilis | X-ray diffraction | 0.254 | 0.207 | 2.10 Å |
| Eab7_0655 | Control of competence regulator ComK, YlbF/YmcA | 54.54 | 2oee | Bacillus subtilis | X-ray diffraction | 0.249 | 0.217 | 1.96 Å |
Fig 4. Structural model of protein Eab7_0741, predicted as a Ribosomal silencing factor RsfS.

(A) Three-dimensional model obtained from the Protein Data Bank. (B) Alignment between the RsfS protein structures from B. halodurans and E. antarcticum generated by MODELLER. (C) Ramachandran plot provided by the PROCHECK program for the RsfS protein from E. antarcticum B7.
The model structure obtained for the RsfS protein was previously determined by X-ray crystallography and refined with diffraction data to 2.1 Å resolution from Mycobacterium tuberculosis, which was solved by molecular replacement with a truncated poly-Ala model (Ala7-Ala103) derived from an ortholog isolated from B. halodurans (PDB 2o5a), the same model used in this study to the three-dimensional modeling by homology. RsfS contains the α1-β1-β2-α2-β3-β4-β5-α3 fold and ortholog structures from Chromobacterium violaceum (PDB 2id1), Zymomonas mobilis (PDB 3ups) and B. halodurans onto M. tuberculosis RsfS, demonstrating that the overall structure is well-conserved from the N terminus to the end of β5 [95]. The alignment between the structures from B. halodurans and E. antarcticum RsfS generated by MODELLER also shows a high structural similarity (Fig 4B). RsfS is known as component of the protein synthesis regulation system through the binding to the 50S ribosomal protein L14, impairing joining with the 30S ribosomal subunit. Haüser and colleagues reported that the RsfS-interaction epitope of L14 involves the highly conserved residues K114 and T97 as the most important ones, in addition to R98. The residues T97 and R98 are involved in bridge B8, which contacts the 30S ribosomal subunit. The authors performed a docking model to predict the binding of RsfA to these residues, showing that they sterically interfere with ribosome subunit joining, probably blocking translation [96].
To identify the stereochemical characteristics and possible mismatches in the molecule architecture, as well as to confirm the quality of the model, it is important to evaluate the quality of protein structures [134]. The PROCHECK program provided the Ramachandran plot for the Rsfs protein of E. antarcticum B7, and the results showed that 88% of the residues are in favorable regions, indicating that the generated model presents an excellent degree of reliability (Fig 4C).
The relationship between three-dimensional proteins structures and their biological functions is evident and the knowledge of the protein structure combined with functional annotation methods can lead to the elucidation of uncharacterized proteins [32]. Bioinformatics methods based on available databases have been used to understand protein structure and its function [54]. Therefore, the sequence-to-function method of functional prediction has identified the function of the proteins in this study. The association with the structural information performed for a few groups has confirmed the accurate function annotation.
The model obtained for the RsfS protein predicted from E. antarcticum B7 was already evaluated experimentally to M. tuberculosis [95], and our study has shown great results to the model obtained by computational methods of databases comparison of the amino acid sequence of this protein with the ones previously characterized. This kind of comparison is possible because amino acid sequences determine the structure, and the structures commend the biochemical function. In this way, proteins with shared similarity of their amino acid sequence usually perform similar functions [135].
Conclusions
Proteins are versatile macromolecules that play crucial role in biological processes. The identification of protein functions is fundamental for the understanding of these processes. We used an in silico approach to predict the function of hypothetical proteins from the E. antarcticum B7 genome. We attributed a function to 132 HPs with high confidence. The prediction of sub-cellular localization and physicochemical parameters were useful to reinforce the understanding of the particular characteristics of the proteins annotated. Those proteins were further investigated for their interactions and three-dimensional structures. PPI investigation is important to determine the relationship between these proteins and the known proteome of a given organism, helping to infer correctly its function. We identified the presence of proteins that play important roles in the mechanisms of adaptation to adverse environments, such as biofilm formation, flagellar biosynthesis, transcription regulation, carotenoid biosynthesis, and others. The pipeline used in this study allowed us to obtain excellent results and can be used to assign protein function to hypothetical proteins. We also demonstrated E. antarcticum B7 resistance to arsenic. Our findings open possibilities for better investigation of this bacterium for application in the biotechnology field.
Supporting information
This image shows how each of the proteins tested interacts with each other and with other proteins form the E. antarcticum B7.
(TIFF)
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
We thank Dr. Rommel T. J. Ramos for the support in the GO FEAT plataform analysis.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Authors thank the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – CAPES (88881.068052/2014-01) and PROPESP/UFPA for the financial support on this study. We also acknowledge FCT (Fundação para a Ciência e Tecnologia, Portugal) financing to CESAM (UID/AMB/50017/2013 - POCI-01-0145-FEDER-007638) and Isabel Henriques (FCT Investigator Programme – IF/00492/2013). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Vishnivetskaya TA, Kathariou S, Tiedje JM. The Exiguobacterium genus: biodiversity and biogeography. Extremophiles. 2009;13: 541–555. doi: 10.1007/s00792-009-0243-5 [DOI] [PubMed] [Google Scholar]
- 2.Usuda Y, Kawasaki H, Shimaoka M, Utagawa T. Molecular characterization of guanosine kinase gene from a facultative alkalophile, Exiguobacterium aurantiacum ATCC 35652. Biochim Biophys Acta. 1998;1442: 373–379. doi: 10.1016/S0167-4781(98)00166-3 [DOI] [PubMed] [Google Scholar]
- 3.Suga S, Koyama N. Purification and properties of a novel azide-sensitive ATPase of Exiguobacterium aurantiacum. Arch Microbiol. 2000;173: 200–205. doi: 10.1007/s002039900129 [DOI] [PubMed] [Google Scholar]
- 4.Früling A, Schumann P, Hippe H, Stra B. Exiguobacterium undae sp. nov. and Exiguobacterium antarticum sp. nov. Int J Syst Evol Microbiol. 2002; 1171–1176. doi: 10.1099/00207713-52-4-1171 [DOI] [PubMed] [Google Scholar]
- 5.Wada M, Yoshizumi A, Furukawa Y, Kawabata H, Ueda M. Cloning and overexpression of the Exiguobacterium sp. F42 gene encoding a new short chain dehydrogenase, which catalyzes the stereoselective reduction of ethyl 3-Oxo-3- (2-thienyl) propanoate to ethyl (S)-3-hydroxy-3-(2-thienyl) propanoate. Biosci Biotechnol Biochem. 2004;68: 1481–1488. doi: 10.1271/bbb.68.1481 [DOI] [PubMed] [Google Scholar]
- 6.Hwang B-Y, Kim J-H, Kim B-G. Screening of Exiguobacterium acetylicum from soil samples showing enantioselective and alkatolerant esterase activity. Biotechnol Bioprocess Eng. 2005;10: 367–371. doi: 10.1007/BF02931857 [Google Scholar]
- 7.Kasana RC, Yadav SK. Isolation of a psychrotrophic Exiguobacterium sp. SKPB5 (MTCC 7803) and characterization of its alkaline protease. Curr Microbiol. 2007;54: 224–229. doi: 10.1007/s00284-006-0402-1 [DOI] [PubMed] [Google Scholar]
- 8.Hara I, Ichise N, Kojima K, Kondo H, Ohgiya S, Matsuyama H, et al. Relationship between the size of the bottleneck 15 Å from iron in the main channel and the reactivity of catalase corresponding to the molecular size of substrates. Biochemistry. 2007;46: 11–22. doi: 10.1021/bi061519w [DOI] [PubMed] [Google Scholar]
- 9.Carneiro AR, Ramos RTJ, Dall’Agnol H, Pinto AC, Soares S de C, Santos AR, et al. Genome sequence of Exiguobacterium antarcticum B7, isolated from a biofilm in Ginger Lake, King George Island, Antarctica. J Bacteriol. 2012;194: 6689–6690. doi: 10.1128/JB.01791-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee CW, Kwon S, Park SH, Kim BY, Yoo W, Ryu BH, et al. Crystal structure and functional characterization of an esterase (EaEST) from Exiguobacterium antarcticum. PLoS One. 2017;12: 1–19. doi: 10.1371/journal.pone.0169540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zanphorlin LM, De Giuseppe PO, Honorato RV, Tonoli CCC, Fattori J, Crespim E, et al. Oligomerization as a strategy for cold adaptation: structure and dynamics of the GH1 β-glucosidase from Exiguobacterium antarcticum B7. Sci Rep. Nature Publishing Group; 2016;6: 1–14. doi: 10.1038/srep23776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baraúna RA, das Graças DA, Nunes CIP, Schneider MPC, Silva A, Carepo MSP. De novo synthesis of fatty acids is regulated by FapR protein in Exiguobacterium antarcticum B7, a psychrotrophic bacterium isolated from Antarctica. BMC Res Notes. 2016;9: 447 doi: 10.1186/s13104-016-2250-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dall’Agnol HPMB, Baraúna RA, de Sá PHCG, Ramos RTJ, Nóbrega F, Nunes CIP, et al. Omics profiles used to evaluate the gene expression of Exiguobacterium antarcticum B7 during cold adaptation. BMC Genomics. 2014;15: 986 doi: 10.1186/1471-2164-15-986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawasaki R, Baraúna RA, Silva A, Carepo MSP, Oliveira R, Marques R, et al. Reconstruction of the fatty acid biosynthetic pathway of Exiguobacterium antarcticum B7 based on genomic and bibliomic data. Biomed Res Int. 2016;2016:7863706 doi: 10.1155/2016/7863706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baraúna R, Freitas D, Pinheiro J, Folador A, Silva A. A proteomic perspective on the bacterial adaptation to cold: integrating OMICs data of the psychrotrophic bacterium Exiguobacterium antarcticum B7. Proteomes. 2017;5: 9 doi: 10.3390/proteomes5010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosca R, Pons T, Céol A, Valencia A, Aloy P. Towards a detailed atlas of protein-protein interactions. Curr Opin Struct Biol. 2013;23: 929–940. doi: 10.1016/j.sbi.2013.07.005 [DOI] [PubMed] [Google Scholar]
- 17.Nourani E, Khunjush F, Durmus S. Computational approaches for prediction of pathogen-host protein-protein interactions. Front Microbiol. 2015;6: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doerks T, Von Mering C, Bork P. Functional clues for hypothetical proteins based on genomic context analysis in prokaryotes. 2004;32: 6321–6326. doi: 10.1093/nar/gkh973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawkins T, Kihara D. Function prediction of uncharacterized proteins. J Bioinform Comput Biol. 2007;5: 1–30. doi: 10.1142/S0219720007002503 [DOI] [PubMed] [Google Scholar]
- 20.Nimrod G, Schushan M, Steinberg DM, Ben-Tal N. Detection of functionally important regions in “hypothetical proteins” of known structure. Struct Des. 2008;16: 1755–1763. doi: 10.1016/j.str.2008.10.017 [DOI] [PubMed] [Google Scholar]
- 21.Shahbaaz M, Hassan I, Ahmad F. Functional annotation of conserved hypothetical proteins from Haemophilus influenzae Rd KW20. 2013;8 doi: 10.1371/journal.pone.0084263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Islam SS, Sohel S, Patwary NIA, Hasan A. In Silico structural and functional annotation of hypotetical proteins of Vibrio cholerae O139. Genomics Inform. 2015;13: 53–59. doi: 10.5808/GI.2015.13.2.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313: 903–919. doi: 10.1006/jmbi.2001.5080 [DOI] [PubMed] [Google Scholar]
- 24.Geer LY, Domrachev M, Lipman DJ, Bryant SH. CDART: protein homology by domain architecture. Genome Res. 2002;12: 1619–1623. doi: 10.1101/gr.278202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42: 222–230. doi: 10.1093/nar/gkt1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40: 302–305. doi: 10.1093/nar/gkr931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai CZ, Han LY, Ji ZL, Chen X, Chen YZ. SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003;31: 3692–3697. doi: 10.1093/nar/gkg600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39: 561–568. doi: 10.1093/nar/gkq973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeong H, Qian X, Yoon B-J. Effective comparative analysis of protein-protein interaction networks by measuring the steady-state network flow using a Markov model. BMC Bioinformatics. 2016;17: 395 doi: 10.1186/s12859-016-1215-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snider J, Kotlyar M, Saraon P, Yao Z, Jurisica I, Stagljar I. Fundamentals of protein interaction network mapping. Mol Syst Biol. 2015;11: 848 doi: 10.15252/msb.20156351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. Part I. Experimental techniques and databases. PLoS Comput Biol. 2007;3: 0337–0344. doi: 10.1371/journal.pcbi.0030042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jez JM. Revisiting protein structure, function, and evolution in the genomic era. J Invertebr Pathol. Elsevier Inc.; 2017;142: 11–15. doi: 10.1016/j.jip.2016.07.013 [DOI] [PubMed] [Google Scholar]
- 33.Singh A, Singal B, Nath O, Singh IK. Functional annotation and classification of the hypothetical proteins of Neisseria meningitidis H44 / 76. 2015;3: 57–64. doi: 10.11648/j.bio.20150305.16 [Google Scholar]
- 34.Malhotra H, Kaur H. A bioinformatics approach for functional and structural analysis of hypothetical proteins of Clostridium difficile. Imp J Interdiscip Res. 2016;2: 1601–1609. [Google Scholar]
- 35.School K, Marklevitz J, Schram WK, Harris LK. Predictive characterization of hypothetical proteins in Staphylococcus aureus NCTC 8325. Bioinformation. 2016;12: 209–220. doi: 10.6026/97320630012209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M a, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16: 944–945. doi: 10.1093/bioinformatics/16.10.944 [DOI] [PubMed] [Google Scholar]
- 37.Araujo FA, Barh D, Silva A, Guimarães L, Ramos RTJ. GO FEAT: a rapid web-based functional annotation tool for genomic and transcriptomic data. Sci Rep. 2018;8: 1794 doi: 10.1038/s41598-018-20211-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C, Huang H, Wu C. Protein Bioinformatics. Methods Mol Biol. 2017;1558: 28150231 doi: 10.1007/978-1-4939-6783-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017;45: D190–D199. doi: 10.1093/nar/gkw1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sillitoe I, Lewis TE, Cuff A, Das S, Ashford P, Dawson NL, et al. CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015;43: D376–D381. doi: 10.1093/nar/gku947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36: 5–9. doi: 10.1093/nar/gkn201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo H, Lin Y, Gao F, Zhang CT, Zhang R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014;42: 574–580. doi: 10.1093/nar/gkt1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26: 1608–1615. doi: 10.1093/bioinformatics/btq249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu C, Lin C-J, Hwang J-K. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004;13: 1402–1406. doi: 10.1110/ps.03479604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu C-S, Chen Y-C, Lu C-H, Hwang J-K. Prediction of protein subcellular localization. proteins struct funct bioinforma. 2006;64: 643–651. doi: 10.1002/prot.21018 [DOI] [PubMed] [Google Scholar]
- 46.Peabody MA, Laird MR, Vlasschaert C, Lo R, Brinkman FSL. PSORTdb: expanding the bacteria and archaea protein subcellular localization database to better reflect diversity in cell envelope structures. Nucleic Acids Res. 2016;44: D663–D668. doi: 10.1093/nar/gkv1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krogh A, Larsson B, Von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305: 567–580. doi: 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 48.Tusnády GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17: 849–850. doi: 10.1093/bioinformatics/17.9.849 [DOI] [PubMed] [Google Scholar]
- 49.Petersen TN, Brunak S, Von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8: 785–786. doi: 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 50.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A, et al. ExPASy: the proteomics server for in-depth protein knowledge and analysis. 2003;31: 3784–3788. doi: 10.1093/nar/gkg563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41: 808–815. doi: 10.1093/nar/gks1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Folador EL, Vinícius P, Daltro S, Silva WM, Ferreira RS, Silva A, et al. In silico identification of essential proteins in Corynebacterium pseudotuberculosis based on protein-protein interaction networks. BMC Syst Biol. BMC Systems Biology; 2016; 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, Uetz P, et al. Conserved patterns of protein interaction in multiple species. Proc Natl Acad Sci U S A. 2005;102: 1974–9. doi: 10.1073/pnas.0409522102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wetie AG, Sokolowska I, Woods AG, Roy U, Deinhardt K, Darie CC. Protein-protein interactions: switch from classical methods to proteomics and bioinformatics-based approaches. Cell Mol Life Sci. 2014;71: 205–228. doi: 10.1007/s00018-013-1333-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10: 421 doi: 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shannon P, Markiel A, Ozier Owen, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 2498–2504. doi: 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Assenov Y, Ramírez F, Schelhorn SESE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24: 282–284. doi: 10.1093/bioinformatics/btm554 [DOI] [PubMed] [Google Scholar]
- 58.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen M, et al. Comparative protein structure modeling using Modeller. Current Protocols in Bioinformatics. 2006. doi: 10.1002/0471250953.bi0506s15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. International Union of Crystallography; 1993;26: 283–291. doi: 10.1107/S0021889892009944 [Google Scholar]
- 60.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25: 1605–1612. doi: 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 61.Swets JA, Dawes RM, Monahan J. Better decisions through science. Sci Am. 2000; 82–87. [DOI] [PubMed] [Google Scholar]
- 62.Naqvi AAT, Shahbaaz M, Ahmad F, Hassan MI. Identification of functional candidates amongst hypothetical proteins of Treponema pallidum ssp. pallidum. PLoS One. 2015;10: 1–17. doi: 10.1371/journal.pone.0124177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eng J. ROC analysis: web-based calculator for ROC curves. Baltimore: Johns Hopkins University [updated 2014 March 19; cited <19/08/2017>]. http://www.jrocfit.org.
- 64.Rao VS, Srinivas K, Sujini GN, Kumar GNS. Protein-protein interaction detection: methods and analysis. Int J Proteomics. 2014;2014: 1–12. doi: 10.1155/2014/147648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee BY, Hefta SA, Brennan PJ. Characterization of the major membrane protein of virulent Mycobacterium tuberculosis. Infect Immun. 1992;60: 2066–2074. 0019-9567/92/052066-09$02.00/0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walian PJ, Allen S, Shatsky M, Zeng L, Szakal ED, Liu H, et al. High-throughput isolation and characterization of untagged membrane protein complexes: outer membrane complexes of Desulfovibrio vulgaris. J Proteome Res. 2012;11: 5720–5735. doi: 10.1021/pr300548d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suvorova IA, Korostelev YD, Gelfand MS. GntR Family of bacterial transcription factors and their DNA binding motifs: structure, positioning and co-evolution. 2015; 1–21. doi: 10.1371/journal.pone.0132618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beinert Holm, Münck; Iron-sulfur clusters : nature’s modular, multipurpose structures. Science (80-). 2007;277: 653 doi: 10.1126/science.277.5326.653 [DOI] [PubMed] [Google Scholar]
- 69.Ayala-Castro C, Saini A, Outten FW. Fe-S cluster assembly pathways in bacteria. Microbiol Mol Biol Rev. 2008;72: 110–125. doi: 10.1128/MMBR.00034-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peschel BA, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with L -Lysine. 2001;193: 1067–1076. doi: 10.1084/jem.193.9.1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng M, Yu M, Hung L, Cieslik M, Derewenda U, Scott A, et al. Research papers structure of Thermotoga maritima TM0439 : implications for the mechanism of bacterial GntR transcription regulators with Zn 2 + -binding FCD domains research papers. 2009; 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Enany S. Structural and functional analysis of hypothetical and conserved proteins of Clostridium tetani. J Infect Public Health. King Saud Bin Abdulaziz University for Health Sciences; 2014;7: 296–307. doi: 10.1016/j.jiph.2014.02.002 [DOI] [PubMed] [Google Scholar]
- 73.Ikai A. Thermostability and Aliphatic Index of Globular Proteins. J Biochem. 1980;88: 1895–1898. [PubMed] [Google Scholar]
- 74.Varma PBS, Adimulam YB, Kodukula S. In silico functional annotation of a hypothetical protein from Staphylococcus aureus. J Infect Public Health. King Saud Bin Abdulaziz University for Health Sciences; 2015;8: 526–532. doi: 10.1016/j.jiph.2015.03.007 [DOI] [PubMed] [Google Scholar]
- 75.Hua S, Sun Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics. 2001;17: 721–728. doi: 10.1093/bioinformatics/17.8.721 [DOI] [PubMed] [Google Scholar]
- 76.Naqvi AAT, Anjum F, Khan FI, Islam A, Ahmad F, Hassan MI. Sequence analysis of hypothetical proteins from Helicobacter pylori 26695 to identify potential virulence factors. Genomics Inform. 2016;14: 125 doi: 10.5808/GI.2016.14.3.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andrade MA, O’Donoghue SI, Rost B. Adaptation of protein surfaces to subcellular location. Edited by Cohen F. E. J Mol Biol. 1998;276: 517–525. doi: 10.1006/jmbi.1997.1498 [DOI] [PubMed] [Google Scholar]
- 78.Nakashima H. Discrimination of intracelullar and extracellular proteins using amino acid compostion and residue-pair frequences. J Mol Biol. 1994;238: 54–61. doi: 10.1006/jmbi.1994.1267 [DOI] [PubMed] [Google Scholar]
- 79.Shen Z, Luangtongkum T, Qiang Z, Jeon B, Wang L, Zhang Q. Identification of a novel membrane transporter mediating resistance to organic arsenic in Campylobacter jejuni. Antimicrob Agents Chemother. 2014;58: 2021–2029. doi: 10.1128/AAC.02137-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang Y, Wu S, Lilley RM, Zhang R. The diversity of membrane transporters encoded in bacterial arsenic-resistance operons. PeerJ. 2015; 3:e943; doi: 10.7717/peerj.943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu Y-G, Yoshinaga M, Zhao F-J, Rosen BP. Earth abides arsenic biotransformations. Annu Rev Earth Planet Sci. 2014;42: 443–467. doi: 10.1146/annurev-earth-060313-054942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Castro-Severyn J, Remonsellez F, Valenzuela SL, Salinas C, Fortt J, Aguilar P, et al. Comparative genomics analysis of a new Exiguobacterium strain from salar de huasco reveals a repertoire of stress-related genes and arsenic resistance. Front Microbiol. 2017;8: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ordoñez OF, Lanzarotti E, Kurth D, Cortez N, Farías ME, Turjanski AG. Genome comparison of two Exiguobacterium strains from high altitude andean lakes with different arsenic resistance: identification and 3D modeling of the Acr3 efflux pump. Front Environ Sci. 2015;3: 1–12. doi: 10.3389/fenvs.2015.00050 [Google Scholar]
- 84.Tawfik DS, Viola RE. Arsenate replacing phosphate: alternative life chemistries and ion promiscuity. Biochemistry. 2011;50: 1128–1134. doi: 10.1021/bi200002a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Puzon GJ, Petersen JN, Roberts AG, Kramer DM, Xun L. A bacterial flavin reductase system reduces chromate to a soluble chromium(III)-NAD+ complex. Biochem Biophys Res Commun. 2002;294: 76–81. doi: 10.1016/S0006-291X(02)00438-2 [DOI] [PubMed] [Google Scholar]
- 86.Pradhan SK, Singh NR, Rath BP, Thatoi H. Bacterial chromate reduction: a review of important genomic, proteomic, and bioinformatic analysis. Crit Rev Environ Sci Technol. Taylor & Francis; 2016;46: 1659–1703. doi: 10.1080/10643389.2016.1258912 [Google Scholar]
- 87.Jagannadham M V., Rao VJ, Shivaji S. The major carotenoid pigment of a psychotrophic Micrococcus roseus strain: purification, structure, and interaction with synthetic membranes. J Bacteriol. 1991;173: 7911–7917. doi: 10.1006/bbrc.1996.0471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Subczynski WK, Markowska E, Sielewiesiuk J. Spin-label studies on phosphatidylcholine-polar carotenoid membranes: effects of alkyl-chain length and unsaturation. BBA—Biomembr. 1993;1150: 173–181. doi: 10.1016/0005-2736(93)90087-G [DOI] [PubMed] [Google Scholar]
- 89.Jagannadham M V., Chattopadhyay MK, Subbalakshmi C, Vairamani M, Narayanan K, Rao CM, et al. Carotenoids of an Antarctic psychrotolerant bacterium, Sphingobacterium antarcticus, and a mesophilic bacterium, Sphingobacterium multivorum. Arch Microbiol. 2000;173: 418–424. doi: 10.1007/s002030000163 [DOI] [PubMed] [Google Scholar]
- 90.Carabetta VJ, Tanner AW, Greco TM, Defrancesco M, Cristea IM, Dubnau David. A complex of YlbF, YmcA and YaaT regulates sporulation, competence and biofilm formation by accelerating the phosphorylation of Spo0A i. NIH Public Access. 2013;88: 283–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lei Y, Oshima T, Ogasawara N, Ishikawa S. Functional analysis of the protein veg, which stimulates biofilm formation in Bacillus subtilis. J Bacteriol. 2013;195: 1697–1705. doi: 10.1128/JB.02201-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Branda SS, Vik Å, Friedman L, Kolter R. Biofilms: The matrix revisited. Trends Microbiol. 2005;13: 20–26. doi: 10.1016/j.tim.2004.11.006 [DOI] [PubMed] [Google Scholar]
- 93.Feng G, Cheng Y, Wang S-Y, Borca-Tasciuc DA, Worobo RW, Moraru CI. Bacterial attachment and biofilm formation on surfaces are reduced by small-diameter nanoscale pores: how small is small enough? npj Biofilms Microbiomes. Nature Publishing Group; 2015;1: 15022 doi: 10.1038/npjbiofilms.2015.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Limoli DH, Jones CJ, Wozniak DJ, Cruz S. Bacterial extracellular polysaccharides in biofilm formation and function. Microbiol Spectr. 2015;3: 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li X, Sun Q, Jiang C, Yang K, Hung L, Zhang J, et al. Structure of ribosomal silencing factor bound to Mycobacterium tuberculosis ribosome. Structure. 2016;23: 1858–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Häuser R, Pech M, Kijek J, Yamamoto H, Titz B, Naeve F, et al. RsFA (YbeB) proteins are conserved ribosomal silencing factors. PLoS Genet. 2012;8: 1–12. doi: 10.1371/journal.pgen.1002815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jiang M, Sullivan SM, Walker AK, Strahler JR, Andrews PC, Maddock JR. Identification of novel Escherichia coli ribosome-associated proteins using isobaric tags and multidimensional protein identification techniques. J Bacteriol. 2007;189: 3434–3444. doi: 10.1128/JB.00090-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Belfiore C, Ordoñez OF, Farías ME. Proteomic approach of adaptive response to arsenic stress in Exiguobacterium sp. S17, an extremophile strain isolated from a high-altitude Andean Lake stromatolite. Extremophiles. 2013;17: 421–431. doi: 10.1007/s00792-013-0523-y [DOI] [PubMed] [Google Scholar]
- 99.Sarkar A, Kazy SK, Sar P. Characterization of arsenic resistant bacteria from arsenic rich groundwater of West Bengal, India. Ecotoxicology. 2013;22: 363–376. doi: 10.1007/s10646-012-1031-z [DOI] [PubMed] [Google Scholar]
- 100.Dastager SG, Mawlankar R, Sonalkar V V., Thorat MN, Mual P, Verma A, et al. Exiguobacterium enclense sp. nov., isolated from sediment. Int J Syst Evol Microbiol. 2015;65: 1611–1616. doi: 10.1099/ijs.0.000149 [DOI] [PubMed] [Google Scholar]
- 101.de Azevedo JSN, Silva-Rocha R, Silva A, Peixe Carepo MS, Cruz Schneider MP. Gene expression of the arsenic resistance operon in Chromobacterium violaceum ATCC 12472. Can J Microbiol. 2008;54: 137–142. doi: 10.1139/w07-123 [DOI] [PubMed] [Google Scholar]
- 102.Ciprandi A, Baraúna RA, Santos AV, Gonçalves EC, Carepo MSP, Schneider MPC, et al. Proteomic response to arsenic stress in Chromobacterium violaceum. J Integr OMICS. 2012;2: 69–73. doi: 10.5584/jiomics.v2i1.84 [Google Scholar]
- 103.Páez-Espino D, Tamames J, De Lorenzo V, Cánovas D. Microbial responses to environmental arsenic. BioMetals. 2009;22: 117–130. doi: 10.1007/s10534-008-9195-y [DOI] [PubMed] [Google Scholar]
- 104.Bloom MS, Surdu S, Neamtiu IA, Garzau ES. Maternal arsenic exposure and birth outcomes: a comprehensive review of the epidemiologic literature focused on drinking water. Int J Hyg Env Heal. 2014;217: 709–719. doi: 10.1016/j.ijheh.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Martens JH, Barg H, Warren M, Jahn D. Microbial production of vitamin B12. Appl Microbiol Biotechnol. 2002;58: 275–285. doi: 10.1007/s00253-001-0902-7 [DOI] [PubMed] [Google Scholar]
- 106.Fang H, Kang J, Zhang D. Microbial production of vitamin B12: a review and future perspectives. Microb Cell Fact. BioMed Central; 2017;16: 15 doi: 10.1186/s12934-017-0631-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Biedendieck R, Malten M, Barg H, Bunk B, Martens JH, Deery E, et al. Metabolic engineering of cobalamin (vitamin B12) production in Bacillus megaterium. Microb Biotechnol. 2010;3: 24–37. doi: 10.1111/j.1751-7915.2009.00125.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Denef VJ, Kalnejais LH, Mueller RS, Wilmes P, Baker BJ, Thomas BC, et al. Proteogenomic basis for ecological divergence of closely related bacteria in natural acidophilic microbial communities. Proc Natl Acad Sci. 2010;107: 2383–2390. doi: 10.1073/pnas.0907041107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ferrer A, Rivera J, Zapata C, Norambuena J, Sandoval Á, Chávez R, et al. Cobalamin protection against oxidative stress in the acidophilic iron-oxidizing bacterium Leptospirillum group II CF-1. Front Microbiol. 2016;7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Banco MT, Mishra V, Greeley SC, Ronning DR. Direct cetection of products from S-adenosylmethionine-dependent enzymes using a competitive fluorescence polarization assay. Anal Chem. 2018;90: 1740–1747. doi: 10.1021/acs.analchem.7b03556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Martin JL, McMillan FM. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 2002;12: 783–93. doi: 10.1016/S0959-440X(02)00391-3 [DOI] [PubMed] [Google Scholar]
- 112.Harada J, Takaku S, Watanabe K. An On-demand metalloprotease from psychro-tolerant Exiguobacterium undae Su-1, the activity and stability of which are controlled by the Ca 2+ Concentration. Biosci Biotechnol Biochem. 2012;76: 986–992. doi: 10.1271/bbb.110997 [DOI] [PubMed] [Google Scholar]
- 113.Grabowska M, Jagielska E, Czapinska H, Bochtler M, Sabala I. High resolution structure of an M23 peptidase with a substrate analogue. Sci Rep. Nature Publishing Group; 2015;5: 1–8. doi: 10.1038/srep14833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Håkansson K, Miller CG. Structure of peptidase T from Salmonella typhimurium. Eur J Biochem. 2002;269: 443–450. doi: 10.1046/j.0014-2956.2001.02665.x [DOI] [PubMed] [Google Scholar]
- 115.Lei F, Cui C, Zhao Q, Sun-Waterhouse D, Zhao M. Evaluation of the hydrolysis specificity of protease from marine Exiguobacterium sp. SWJS2 via free amino acid analysis. Appl Biochem Biotechnol. 2014;174: 1260–1271. doi: 10.1007/s12010-014-1088-7 [DOI] [PubMed] [Google Scholar]
- 116.Ohtsuka J, Ichihara Y, Ebihara A, Nagata K, Tanokura M. Crystal structure of TTHA1264, a putative M16-family zinc peptidase from Thermus thermophilus HB8 that is homologous to the β subunit of mitochondrial processing peptidase. Proteins Struct Funct Bioinforma. 2009;75: 774–780. doi: 10.1002/prot.22365 [DOI] [PubMed] [Google Scholar]
- 117.Frias A, Manresa A, de Oliveira E, López-Iglesias C, Mercade E. Membrane vesicles: a common feature in the extracellular matter of cold-adapted antarctic bacteria. Microb Ecol. 2010;59: 476–486. doi: 10.1007/s00248-009-9622-9 [DOI] [PubMed] [Google Scholar]
- 118.Folador EL, Hassan SS, Lemke N, Barh D, Silva A, Ferreira RS, et al. An improved interolog mapping-based computational prediction of protein-protein interactions with increased network coverage. Integr Biol (Camb). Royal Society of Chemistry; 2014;6: 1080–7. doi: 10.1039/c4ib00136b [DOI] [PubMed] [Google Scholar]
- 119.Agustiandari H, Lubelski J, Van Den Berg Van Saparoea HB, Kuipers OP, Driessen AJM. LmrR is a transcriptional repressor of expression of the multidrug ABC transporter LmrCD in Lactococcus lactis. J Bacteriol. 2008;190: 759–763. doi: 10.1128/JB.01151-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kaval KG, Hahn B, Tusamda N, Albrecht D, Halbedel S. The PadR-like transcriptional regulator LftR ensures efficient invasion of Listeria monocytogenes into human host cells. Front Microbiol. 2015;6: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Barthelmebs L, Lecomte B, Divies C, Cavin JF. Inducible metabolism of phenolic acids in Pediococcus pentosaceus is encoded by an autoregulated operon which involves a new class of negative transcriptional regulator. J Bacteriol. 2000;182: 6724–6731. doi: 10.1128/JB.182.23.6724-6731.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gury J, Barthelmebs L, Tran NP, Diviès C, Cavin JF. Cloning, deletion, and characterization of PadR, the transcriptional repressor of the phenolic acid decarboxylase-encoding padA gene of Lactobacillus plantarum. Appl Environ Microbiol. 2004;70: 2146–2153. doi: 10.1128/AEM.70.4.2146-2153.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tran NP, Jerôme G, Véronique D, Chi NTK, Hélène S, Lise B, et al. Phenolic acid-mediated regulation of the padC gene, encoding the phenolic acid decarboxylase of Bacillus subtilisa. J Bacteriol. 2008;190: 3213–3224. doi: 10.1128/JB.01936-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Welsch R, Beyer P, Hugueney P, Kleinig H, von Lintig J. Regulation and activation of phytoene synthase, a key enzyme in carotenoid biosynthesis, during photomorphogenesis. Planta. 2000;211: 846–54. doi: 10.1007/s004250000352 [DOI] [PubMed] [Google Scholar]
- 125.Harada J, Nagashima K V, Takaichi S, Misawa N, Matsuura K, Shimada K. Phytoene desaturase, CrtI, of the purple photosynthetic bacterium, Rubrivivax gelatinosus, produces both neurosporene and lycopene. Plant Cell Physiol. 2001;42: 1112–1118. doi: 10.1093/pcp/pce140 [DOI] [PubMed] [Google Scholar]
- 126.Kirti K, Amita S, Priti S, Kumar AM, Jyoti S. Colorful world of Mmicrobes: carotenoids and their applications. Adv Biol. 2014;2014: 1–13. http://dx.doi.org/10.1155/2014/837891. [Google Scholar]
- 127.Tian B, Hua Y. Carotenoid biosynthesis in extremophilic Deinococcus-Thermus bacteria. Trends Microbiol. Elsevier Ltd; 2010;18: 512–520. doi: 10.1016/j.tim.2010.07.007 [DOI] [PubMed] [Google Scholar]
- 128.Ortiz-Guerrero JM, Polanco MC, Murillo FJ, Padmanabhan S, Elias-Arnanz M. Light-dependent gene regulation by a coenzyme B12-based photoreceptor. Proc Natl Acad Sci. 2011;108: 7565–7570. doi: 10.1073/pnas.1018972108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Frees D, Chastanet A, Qazi S, Sørensen K, Hill P, Msadek T, et al. Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus. Mol Microbiol. 2004;54: 1445–1462. doi: 10.1111/j.1365-2958.2004.04368.x [DOI] [PubMed] [Google Scholar]
- 130.Moreno-Paz M, Gomez MJ, Arcas A, Parro V, Gómez MJ, Arcas A, et al. Environmental transcriptome analysis reveals physiological differences between biofilm and planktonic modes of life of the iron oxidizing bacteria Leptospirillum spp. in their natural microbial community. BMC Genomics. 2010;11: 404 doi: 10.1186/1471-2164-11-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liu L, Fang H, Yang H, Zhang Y, Han Y, Zhou D, et al. CRP is an activator of Yersinia pestis biofilm formation that operates via a mechanism involving gmhA and waaAE-coaD. Front Microbiol. 2016;7: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zahiri J, Bozorgmehr JH, Masoudi-Nejad A. Computational prediction of protein–protein interaction networks: algorithms and resources. Curr Genomics. 2013;14: 397–414. doi: 10.2174/1389202911314060004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Andreani J, Guerois R. Evolution of protein interactions: from interactomes to interfaces. Arch Biochem Biophys. Elsevier Inc.; 2014;554: 65–75. doi: 10.1016/j.abb.2014.05.010 [DOI] [PubMed] [Google Scholar]
- 134.Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7: 95–99. doi: 10.1016/S0022-2836(63)80023-6 [DOI] [PubMed] [Google Scholar]
- 135.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4: 1–13. doi: 10.1038/nprot.2008.197 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This image shows how each of the proteins tested interacts with each other and with other proteins form the E. antarcticum B7.
(TIFF)
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.