

Abstract
An efficient route to substituted 1-aryl-1H-indazoles has been developed and optimized. The method involved the preparation of arylhydrazones from acetophenone or benzaldehyde substituted by fluorine at C2 and nitro at C5, followed by deprotonation and nucleophilic aromatic substitution (SNAr) ring closure in 45–90%. Modification of this procedure to a one-pot domino process was successful in the acetophenone series (73–96%), while the benzaldehyde series (63–73%) required a step-wise addition of reagents. A general one-pot protocol for 1-aryl-1H-indazole formation without the limiting substitution patterns required for the SNAr cyclization has also been achieved in 62–78% yields. A selection of 1-aryl-1H-indazoles was prepared in high yield by a procedure that requires only a single laboratory operation.
Keywords: 1-aryl-5-nitro-1H-indazole, 1-aryl-1H-indazole, arylhydrazone, SNAr reaction, Ullmann reaction
1. Introduction
Our synthetic methods program has recently been focused on the use of nucleophilic aromatic substitution (SNAr)-terminated domino reactions as a means to prepare heterocyclic compounds [1,2,3,4]. The current project has developed a synthesis of 1-aryl-1H-indazoles, which are core ring structures in a broad range of biologically active compounds (Figure 1). Among the compounds pictured, benzydamine (1) is a well-known NSAID drug [5], while alcohol 2 has likewise shown promise as an anti-inflammatory agent [6]. The urea-based structure 3 has recently been judged to possess significant tumor antiproliferative properties [7]. Similarly, amide 4 has also demonstrated anticancer activity [8,9].
Figure 1.

Drugs incorporating 1-aryl-1H-indazoles.
Several strategies have previously been disclosed as routes to 1-aryl-1H-indazoles. All but one required multiple steps. The closest work to that presented here utilized a two-step sequence involving acid catalyzed formation of the arylhydrazone, followed by a copper(I)-catalyzed Ullmann-type coupling to close the ring [10]. A related investigation described a similar cyclization of arylhydrazones from 2-(2-haloaryl)-2-oxoacetic esters [11]. A complementary approach utilized a copper(I)-promoted coupling of N-aroyl-N′-arylhydrazines with 2-bromoaryl ketones or aldehydes [12], followed by deacylation and condensative ring closure. Two additional accounts examined (1) an SNAr displacement of nitro by anions derived from arylhydrazones of 2-nitrobenzaldehydes [13] and (2) a one-step conversion from salicylaldehydes and arylhydrazine hydrochlorides [14]. Other reports have described a one-pot condensation-SNAr protocol using 2-fluorobenzaldehyde to prepare 1H-indazoles, but these utilized more reactive hydrazines and did not yield 1-aryl-substituted derivatives [15,16]. Along a different line, another study delineated a novel [3 + 2]-type annulation between arynes and arylhydrazones in the presence of fluoride under aerobic conditions [17]. Finally, syntheses promoted by palladium- [18] and rhodium-based [19] catalysts have also furnished these targets. A full listing of methods to prepare 1H-indazoles, with and without the N1 aryl group, have been cataloged in three published reviews [20,21,22].
The number of steps and the waste generated are two important considerations in planning synthetic procedures. Our aim, in the current study, was to develop a one-pot domino or consecutive process [23,24,25] to accomplish the synthesis of 1-aryl-1H-indazoles. Utilizing these techniques, a single operation—one set-up and one work-up—would generate members of this emerging class of heterocyclic building blocks rapidly and with a minimum of chemical waste. The details of our efforts towards this goal are given below.
2. Results and Discussion
A logical route to 1H-indazoles would involve the ring closure of an arylhydrazone anion derived from an appropriately substituted aromatic ketone or aldehyde. Although the arylhydrazones would form as two isomers, with the E isomer (less favorable for cyclization) predominating, these should readily interconvert with heat [26] and allow access to the Z isomer required for ring closure. Furthermore, the pKa of the phenylhydrazone proton (Ar = Ph) is 21.5 [27], and with slight variations due to substituents on the aromatic ring, should permit the use of K2CO3 in a polar aprotic solvent as the deprotonating agent [28,29]. Optimally, these annulations would proceed in one pot, but we initially sought to determine the feasibility of the sequence by evaluating each transformation separately. Once each step was optimized, attempts would then be made to merge the two reactions into a single laboratory operation. Finally, since this would generate structures with limited substitution patterns, we also aspired to generalize the process to include structures without SNAr activating groups.
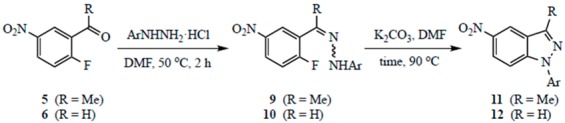
Although there are numerous examples of SNAr-type reactions to generate five-membered rings fused to a benzene, some strain would be expected in ring formations involving closure of a fragment incorporating three sp2 atoms onto an aromatic nucleus [30,31]. Thus, 2′-fluoro-5′-nitroacetophenone (5) and 2-fluoro-5-nitrobenzaldehyde (6) were initially reacted with hydrazine hydrate (NH2NH2·H2O) to assess the feasibility of forming indazoles in this manner. Due to the alpha-effect [32], hydrazine is an exceptionally potent nucleophile and might be expected to react without added base. Indeed, treatment of 5 and 6 with NH2NH2·H2O (3.0 equivalents for 5 and 2.0 equivalents for 6) in N,N-dimethylformamide (DMF) at 23 °C for two hours resulted in high yields of targets 7 and 8, respectively (see Scheme 1). Encouraged by this result, we forged ahead to prepare 1-aryl-5-nitro-1H-indazoles.
Scheme 1.

Reaction of 5 and 6 with hydrazine hydrate.
Formation of the intermediate arylhydrazones 9 and 10 was initially addressed. This involved warming a DMF solution of the carbonyl compound with an arylhydrazine hydrochloride (ArNHNH2·HCl) at 50 °C for two to three hours. DMF was used as a solvent for this process since we anticipated that a one-pot procedure where the hydrazone was prepared, deprotonated, and cyclized would likely require the use of a polar aprotic solvent. Optimization studies indicated that DMF was a suitable medium and the use of 3.0 equivalents of ArNHNH2·HCl with acetophenone 5 and 2.0 equivalents with benzaldehyde 6 afforded the highest yields of 9 and 10. In all cases, the intermediate hydrazones were isolated in ≥70% yield from 5 and ≥50% yield from 6. Interestingly, compounds arising from direct SNAr displacement of fluoride by the arylhydrazine were not observed to any significant extent under these conditions. While the product of addition–elimination by hydrazine is known to cyclize to a 1H-indazole [33,34], ring closure of the analogous product from arylhydrazine would give a 2-aryl-2H-indazole. Neither the arylhydrazine SNAr product nor the 2H-indazole were detected.
Cyclization of hydrazones 9 and 10 required base, higher temperature and occasionally extended reaction times. This reaction was performed in DMF at 90 °C and found to work best using 3.0 equivalents of K2CO3. Yields were slightly lower from the benzaldehyde since this substrate led to a product that was unhindered at C3 and possibly susceptible to nucleophilic attack by various species in the reaction. A summary of optimization experiments is given in the Electronic Supplemental Inofrmation, and the results of our two-step synthesis of 1-aryl-5-nitro-1H-indazoles are summarized in Table 1.
Table 1.
Yields for the two-step synthesis of 1-aryl-5-nitro-1H-indazoles.
| Substrate | Ar | Time (h) a | Pdt | % Yield b |
|---|---|---|---|---|
| 5 | a Ph- | 2 | 11a | 95 |
| 5 | b 2-MeOPh- | 2 | 11b | 82 |
| 5 | c 3-MeOPh- | 2 | 11c | 81 |
| 5 | d 4-MeOPh- | 2 | 11d | 94 |
| 5 | e 4-BrPh- | 2 | 11e | 95 |
| 5 | f 3-ClPh- | 2 | 11f | 87 |
| 5 | g 4-ClPh- | 2 | 11g | 93 |
| 5 | h 2,4-Cl2Ph | 36 | 11h | 80 |
| 5 | I 3-CF3Ph- | 2 | 11i | 88 |
| 5 | j 4-CF3Ph- | 2 | 11j | 70 |
| 5 | k 4-CNPh- | 2 | 11k | 80 |
| 5 | l 4-H2NSO2Ph- | 10 | 11l | 70 |
| 5 | m 4-HO2CPh- | 24 | 11m | 75 |
| 6 | a Ph- | 2 | 12a | 72 |
| 6 | b 2-MeOPh- | 2 | 12b | 0 c |
| 6 | c 3-MeOPh- | 2 | 12c | 67 |
| 6 | d 4-MeOPh- | 2 | 12d | 70 |
| 6 | e 4-BrPh- | 2 | 12e | 70 |
| 6 | f 3-ClPh- | 2 | 12f | 60 |
| 6 | g 4-ClPh- | 2 | 12g | 70 |
| 6 | h 2,4-Cl2Ph- | 2 | 12h | 60 |
| 6 | I 3-CF3Ph- | 2 | 12i | 62 |
| 6 | j 4-CF3Ph- | 2 | 12j | 68 |
| 6 | k 4-CNPh- | 3 | 12k | 60 |
| 6 | l 4-H2NSO2Ph- | 2 | 12l | 50 |
| 6 | m 4-HO2CPh- | 2 | 12m | 50 |
a All hydrazones were generated in two hours. Times given are for the final cyclization. b Isolated yield of 1H-indazole for the two-step sequence. c Only the hydrazone was isolated.
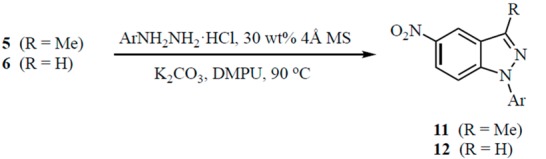
Modification of the protocol to allow for a one-pot procedure had to overcome several problems that could lead to unwanted impurities. Since water is produced during arylhydrazone formation, some conversion of the K2CO3 to KOH would be expected. Furthermore, KOH could hydrolyze the DMF solvent to form Me2NH. Both of these nucleophiles (OH– and Me2NH) could potentially undergo competitive SNAr addition to the carbonyl containing substrate. While no significant product resulting from OH– addition was observed, some of the Me2NH addition product was noted when prolonged heating was required. These side reactions were suppressed by adding 30 wt % (relative to 5 or 6) of powdered 4 Å molecular sieves to scavenge water produced during the initial condensation and using anhydrous 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (DMPU) as the solvent to minimize hydrolysis.
The one-step domino process was successful for the conversion of ketone 5 to indazoles 11, and in most cases, the yield was superior to the two-step sequence (see Table 2). Reactions of aldehyde 6 to give 12, however, were more limited and several of the substrates bearing acidic functions on the aromatic ring failed to react. For aldehydes that did generate a product, sequential treatment with ArNHNH2·HCl and base afforded yields that also surpassed the two-step process. In both one-pot procedures, it was often noted that the cyclization required a longer reaction time when the aryl group of the hydrazone was substituted by an electron-withdrawing group. This would be expected since the arylhydrazone anion would be less nucleophilic with a stabilizing group at C2 or C4 of the hydrazone aromatic ring [35]. Conversely, this same anion with an electron-donating substituent at C2 or C4 should be more reactive with potentially shorter reaction times.
Table 2.
Yields for the modified one-pot synthesis of 1-aryl-5-nitro-1H-indazoles.
| Substrate | Ar | Time (h) a | Pdt | % Yield b |
|---|---|---|---|---|
| 5 | a Ph- | 1.5 | 11a | 96 |
| 5 | d 4-MeOPh- | 1.5 | 11d | 95 |
| 5 | g 4-ClPh- | 1.5 | 11g | 94 |
| 5 | h 2,4-Cl2Ph- | 72 | 11h | 85 |
| 5 | l H2NSO2Ph- | 16 | 11m | 85 |
| 5 | m HO2CPh- | 45 | 11n | 73 |
| 6 | a Ph- | 3.5 | 12a | 73 |
| 6 | d 4-MeOPh- | 3.5 | 12d | 74 |
| 6 | g 4-ClPh- | 3.5 | 12g | 71 |
| 6 | k 4-NCPh- | 6.5 | 12k | 63 |
| 6 | l H2NSO2Ph- | 8.5 | 12l | 0 |
| 6 | m HO2CPh- | 8.5 | 12m | 0 |
a For 5, time is the total reaction time. For 6, the hydrazone was allowed to form for 1.5 h at 90 °C before base was added; time is for the final cyclization. b Isolated yield. DMPU: 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone.
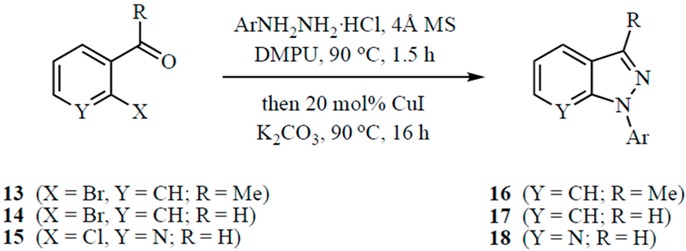
Finally, we sought to develop a general approach to 1H-indazoles unrestricted by the substitution patterns needed for the SNAr ring closure (see Table 3). Protocol development efforts indicated that this variant of the reaction required sequential addition of the reagents. The hydrazone was initially generated using 1.5 equivalents of the ArNHNH2·HCl and then cyclized by addition of 20 mol % of copper(I) iodide (CuI) and 2.5 equivalents of K2CO3, all in the same flask at 90 °C. Again, the key to success in this transformation was the inclusion of 30 wt % of powdered 4 Å molecular sieves and the use of DMPU as the solvent. Without activating groups on the ring, Ullmann conditions (CuI and base) for the final ring closure were essential. An earlier paper noted that this copper(I)-catalyzed ring closure was facilitated by the addition of 20 mol % of l-proline or 4-hydroxy-l-proline [8,9]. In the current procedure, however, addition of these ligands had no impact on the yield of the reaction. The cyclization using 2′-bromoacetophenone (13), 2-bromobenzaldehyde (14) and 2-chloronicotinaldehyde (15) was performed using phenylhydrazine as well as electron-rich and electron-poor arylhydrazines in relatively uniform yields. Though it was expected that the nicotinaldehyde derivatives would not need a copper catalyst, since it could directly cyclize by a SNAr reaction, this ring closure failed to proceed to completion without this additive. Overall, our procedure permits the rapid construction of 1-aryl-1H-indazoles in a single reaction vessel without isolation or purification of intermediates.
Table 3.
Yields for the general one-pot synthesis of 1-aryl-1H-indazoles.
| Substrate | Ar | Product | % Yield a |
|---|---|---|---|
| 13 | a Ph- | 16a | 81 |
| 13 | d 4-MeOPh- | 16d | 83 |
| 13 | k 4-CNPh- | 16l | 87 |
| 14 | a Ph- | 17a | 77 |
| 14 | d 4-MeOPh- | 17d | 72 |
| 14 | k 4-CNPh- | 17l | 79 |
| 15 | a Ph- | 18a | 68 |
| 15 | d 4-MeOPh- | 18d | 62 |
| 15 | k 4-CNPh- | 18l | 78 |
a Isolated yield.
3. Experimental Section
3.1. General Methods
Unless otherwise indicated, all reactions were carried out under dry N2 in oven-dried glassware. All reagents and solvents were used as received. Reactions were monitored by thin layer chromatography on silica gel GF plates (Analtech No. 21521, Newark, DE, USA). Preparative separations were performed by flash chromatography on silica gel (Davisil®, grade 62, 60–200 mesh, Sorbent Technologies, Norcross, GA, USA) containing UV-active phosphor (Sorbent Technologies No. UV-05) slurry packed into quartz columns. Band elution for all chromatographic separations was monitored using a hand-held UV lamp (Fisher Scientific, Pittsburgh, PA, USA). Melting points were obtained using a MEL-TEMP apparatus (Laboratory Devices, Cambridge, MA, USA) and are uncorrected. FT-IR spectra were run using a Varian Scimitar FTS 800 spectrophotometer (Varian Instruments, Randolph, MA, USA) as thin films or nujol mulls on NaCl disks. 1H- and 13C-NMR spectra were measured using a Bruker Avance 400 system (Bruker Corporation, Billerica, MA, USA) in the indicated solvents at 400 MHz and 101 MHz, respectively, with (CH3)4Si as the internal standard; coupling constants (J) are given in Hz. Unit resolution mass spectra were obtained using a Shimadzu LCMS-2010EV system (Shimadzu Corporation, Columbia, MD, USA). Elemental analyses (±0.4%) were determined by Atlantic Microlabs (Norcross, GA, USA).
3.2. Indazoles from 2′-Fluoro-5′-nitroacetophenone (5) or 2-Fluoro-5-nitrobenzaldehyde (6) with NH2NH2·H2O
To a stirred solution of 5 or 6 (1.0 mmol) in DMF (5 mL) at 23 °C was added NH2NH2·H2O (3.0 mmol for 5, 2.0 mmol for 6). The solution was stirred at 23 °C for 2 h at which time thin layer chromatography (TLC, 20% EtOAc in hexanes) indicated complete consumption of the starting carbonyl compound. The crude mixture was added to water and extracted with EtOAc (2 × 15 mL). The combined organic layers were washed with water and saturated aq NaCl, dried (MgSO4), filtered, and concentrated under vacuum to give the pure indazole products.
3-Methyl-5-nitro-1H-indazole (7). Yield: 173 mg (0.98 mmol, 98%) as a yellow solid, m.p. 213–214 °C; IR: 1517, 1332 cm−1; 1H-NMR (DMSO-d6): δ 13.1 (br s, 1H), 8.79 (s, 1H), 8.18 (d, J = 9.1 Hz, 1H), 7.64 (d, J = 9.1 Hz, 1H), 2.59 (s, 3H); 13C-NMR (DMSO-d6): δ 145.4, 143.1, 141.3, 121.9, 121.3, 118.8, 111.2, 12.1; MS: m/z 177 (M+), calculated m/z 177.05. Anal. Calcd. for C8H7N3O2: C, 54.24; H, 3.98; N, 23.72. Found: C, 54.29; H, 4.01; N, 23.59.
5-Nitro-1H-indazole (8). Yield: 147 mg (0.90 mmol, 90%) as a tan solid, m.p. 207–208 °C; IR: 1534, 1341 cm−1; 1H-NMR (DMSO-d6): δ 13.7 (br s, 1H), 8.84 (s, 1H), 8.41 (s, 1H), 8.19 (d, J = 9.0 Hz, 1H), 7.74 (d, J = 9.0 Hz, 1H); 13C-NMR (DMSO-d6): δ 142.3, 142.0, 137.2, 122.5, 121.3, 119.3, 111.5; MS: m/z 163 (M+), calculated m/z 163.04. Anal. Calcd. for C7H5N3O2: C, 51.54; H, 3.09; N, 25.76. Found: C, 51.85; H, 3.15; N, 25.54.
3.3. Indazoles from 5 or 6 with ArNHNH2·HCl. Method A. Representative Two-Step Procedure
To a stirred solution of the carbonyl compound (1.0 mmol) in DMF (5 mL) at 50 °C (oil bath) was added ArNHNH2·HCl (3.0 mmol for 5, 2.0 mmol for 6) and the solution was stirred until TLC (20% EtOAc in hexanes) indicated complete conversion (roughly 2 h). The crude reaction mixture was added to water and extracted with EtOAc (2 × 15 mL). The combined organic layers were washed with water and saturated aq NaCl, dried (MgSO4), filtered, and concentrated under vacuum to give the crude hydrazones. The crude products were stirred with 20% ether/pentane for 1 h, filtered and dried to give arylhydrazones 9 and 10. Characterization data for these materials are given in the ESI.
To a stirred solution of the arylhydrazone (1.00 mmol) in DMF (5 mL) at 90 °C (oil bath) was added anhydrous K2CO3 (3.0 mmol) and the mixture was stirred for the time indicated in Table 1. The reaction mixture was added to water, extracted with EtOAc, dried (MgSO4), filtered, and concentrated under vacuum to afford the crude product. Indazoles from 5 were stirred with 20% ether in pentane, filtered and dried, while those from 6 were purified by silica gel chromatography on a 20 cm × 2 cm column eluted with increasing concentrations of EtOAc in hexanes. Yields as well as physical and spectral data are given below.
3.4. Indazoles from 5 or 6 with ArNHNH2·HCl. Method B. Representative One-Pot Procedure
To a stirred solution of the carbonyl com.p.ound (1.0 mmol) in DMPU (5 mL) were added powdered 4 Å molecular sieves (30 wt % relative to the carbonyl substrate), ArNHNH2·HCl (3.0 mmol for 5, 2.0 mmol for 6), and K2CO3 (3.0 mmol for 5, 2.0 mmol for 6). For 5, all reagents were placed in the flask and heated to 90 °C; for 6, the hydrazone was allowed to form at 90 °C (1.5 h) before K2CO3 was added. The reaction was stirred at 90 °C for the time indicated in Table 2. Workup was performed as described in Method A.
3-Methyl-1-phenyl-5-nitro-1H-indazole (11a). Yield: 240 mg, (0.95 mmol, 95%) as a brown solid, m.p. 115–117 °C; IR: 1516, 1336 cm−1; 1H-NMR (DMSO-d6): δ 8.89 (d, J = 2.2 Hz, 1H), 8.28 (dd, J = 9.3, 2.2 Hz, 1H), 7.93 (d, J = 9.3 Hz, 1H), 7.77 (d, J = 7.8 Hz, 2H), 7.62 (t, J = 7.7 Hz, 2H), 7.46 (t, J = 7.4 Hz, 1H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 147.3, 142.2, 141.1, 139.2, 130.3, 127.8, 124.4, 122.9, 122.8, 119.3, 111.7, 12.1; MS: m/z 253 (M+), calculated m/z 253.09. Anal. Calcd. for C14H11N3O2: C, 66.40; H, 4.38; N, 16.59. Found: C, 66.33; H, 4.34; N, 16.42.
1-(2-Methoxyphenyl)-3-methyl-5-nitro-1H-indazole (11b). Yield: 232 mg (0.82 mmol, 82%) as a light brown solid, m.p. 128–130 °C; IR: 2838, 1519, 1338 cm−1; 1H-NMR (DMSO-d6): δ 8.86 (s, 1H), 8.21 (d, J = 9.3 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.38–7.28 (overlapping signals, 2H), 7.16 (t, J = 7.7 Hz, 1H), 3.78 (s, 3H), 2.66 (s, 3H); 13C-NMR (DMSO-d6): δ 154.0, 146.6, 143.0, 141.9, 130.8, 128.6, 127.1, 123.2, 122.0, 121.4, 119.0, 113.4, 112.2, 56.2, 12.1; MS: m/z 283 (M+), calculated m/z 283.10. Anal. Calcd. for C15H13N3O3: C, 63.60; H, 4.63; N, 14.83. Found: C, 63.49; H, 4.61; N, 14.88.
1-(3-Methoxyphenyl)-3-methyl-5-nitro-1H-indazole (11c). Yield: 229 mg (0.81 mmol, 81%) as a light brown solid, m.p. 105–106 °C; IR: 2836, 1518, 1338 cm−1; 1H-NMR (DMSO-d6): δ 8.90 (s, 1H), 8.29 (d, J = 9.3 Hz, 1H), 7.96 (d, J = 9.3 Hz, 1H), 7.52 (t, J = 8.1 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.28 (s, 1H), 7.04 (d, J = 8.3 Hz, 1H); 13C-NMR (DMSO-d6): δ 160.6, 147.3, 142.3, 141.2, 140.3, 131.2, 124.4, 122.9, 119.3, 114.9, 113.7, 111.9, 108.6, 56.0, 12.1; MS: m/z 283 (M+), calculated m/z 283.10. Anal. Calcd. for C15H13N3O3: C, 63.60; H, 4.63; N, 14.83. Found: C, 63.52; H, 4.55; N, 14.71.
1-(4-Methoxyphenyl)-3-methyl-5-nitro-1H-indazole (11d). Yield: 266 mg (0.94 mmol, 94%) as a tan solid, m.p. 159–160 °C. IR: 2836, 1514, 1346 c cm−1; 1H-NMR (DMSO-d6): δ 8.88 (d, J = 2.2 Hz, 1H), 8.26 (dd, J = 9.3, 2.2 Hz, 1H), 7.79 (d, J = 9.3 Hz, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.15 (d, J = 8.1 Hz, 2H), 3.85 (s, 3H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 158.9, 146.7, 142.0, 141.2, 132.2, 124.8, 123.9, 122.5, 119.3, 1153, 111.4, 56.0, 12.1; MS: m/z 283 (M+), calculated m/z 283.10. Anal. Calcd. for C15H13N3O3: C, 63.60; H, 4.63; N, 14.83. Found: C, 63.57; H, 4.60; N, 14.76.
1-(4-Bromophenyl)-3-methyl-5-nitro-1H-indazole (11e). Yield: 314 mg, (0.95 mmol, 95%) as a tan solid, m.p. 202–203 °C; IR: 1512, 1345 cm−1; 1H-NMR (DMSO-d6): δ 8.90 (d, J = 2.2 Hz, 1H), 8.31 (dd, J = 9.2, 2.2 Hz, 1H), 7.96 (d, J = 9.2 Hz, 1H), 7.80 (d, J = 8.8 Hz, 2H), 7.75 (d, J = 8.8 Hz, 2H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 147.7, 142.4, 141.1, 138.5, 133.1, 124.7, 124.6, 123.0, 120.1, 119.3, 111.8, 12.1; MS: m/z 331 (M+), calculated m/z 331.00. Anal. Calcd. for C14H10BrN3O2: C, 50.62; H, 3.03; N, 12.65. Found: C, 50.44; H, 2.99; N, 12.73.
1-(3-Chlorophenyl)-3-methyl-5-nitro-1H-indazole (11f). Yield: 250 mg, (0.87 mmol, 87%) as a tan solid, m.p. 135–136 °C; IR: 1517, 1339 cm−1; 1H-NMR (DMSO-d6): δ 8.89 (d, J = 2.2 Hz, 1H), 8.31 (dd, J = 9.3, 2.2 Hz, 1H), 7.98 (d, J = 9.3 Hz, 1H), 7.83 (t, J = 2.2 Hz, 1H), 7.78 (d, J = 8.4 Hz, 1H), 7.64 (t, J = 8.1 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 146.8, 141.1, 139.3, 133.4, 130.8, 126.4, 123.6, 122.0, 121.4, 120.1, 118.2, 110.9; MS: m/z 287 (M+), calculated m/z 287.05. Anal. Calcd. for C14H10ClN3O2: C, 58.45; H, 3.50; N, 14.61. Found: C, 58.58; H, 3.59; N, 14.53.
1-(4-Chlorophenyl)-3-methyl-5-nitro-1H-indazole (11g). Yield: 267 mg (0.93 mmol, 93%) as a tan solid, m.p. 217–218 °C; IR: 1511, 1339 cm−1; 1H-NMR (DMSO-d6): δ 8.91 (d, J = 2.2 Hz, 1H), 8.31 (dd, J = 9.3, 2.2 Hz, 1H), 7.95 (d, J = 9.3 Hz, 1H), 7.82 (d, J = 8.7 Hz, 2H), 7.67 (d, J = 8.7 Hz, 2H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 147.7, 1424, 141.1, 138.1, 131.8, 130.2, 124.6, 124.5, 123.0, 119.4, 111.8, 12.1; MS: m/z 287 (M+), calculated m/z 287.05. Anal. Calcd. for C14H10ClN3O2: C, 58.45; H, 3.50; N, 14.61. Found: C, 58.42; H, 3.49; N, 14.48.
1-(2,4-Dichlorophenyl)-3-methyl-5-nitro-1H-indazole (11h). Yield: 257 mg (0.80 mmol, 80%), m.p. 144–145 °C; IR: 1516, 1338 cm−1; 1H-NMR (DMSO-d6): δ 8.92 (d, J = 2.1 Hz, 1H), 8.27 (dd, J = 9.2, 2.1 Hz, 1H), 8.01 (d, J = 2.2 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 7.69 (dd, J = 8.5, 2.2 Hz, 1H), 7.44 (d, J = 9.2 Hz, 1H), 2.68 (s, 3H); 13C-NMR (DMSO-d6): δ 147.5, 143.1, 142.4, 135.3, 135.0, 132.0, 131.5, 130.7, 129.3, 123.5, 122.8, 119.3, 111.6, 12.1; MS: m/z 321 (M+), calculated m/z 321.01. Anal. Calcd. for C14H9Cl2N3O2: C, 52.20; H, 2.82; N, 13.04. Found: C, 52.11; H, 2.91; N, 13.09.
3-Methyl-5-nitro-1-(3-(trifluoromethyl)phenyl)-1H-indazole (11i). Yield: 282 mg (0.88 mmol, 88%) as a white solid, m.p. 112–113 °C; IR: 1520, 1339 cm−1; 1H-NMR (DMSO-d6): δ 8.90 (s, 1H), 8.32 (d, J = 9.3 Hz, 1H), 8.13 (d, J = 7.8 Hz, 1H), 8.07 (s, 1H), 7.99 (d, J = 9.3 Hz, 1H), 7.90–7.77 (complex, 2H), 2.70 (s, 3H); 13C-NMR (DMSO-d6): δ 148.0, 142.5, 141.1, 139.8, 131.6, 130.9 (q, J = 32.6 Hz), 126.2, 124.8, 124.2 (q, J = 272.6 Hz), 124.0 (q, J = 6.7 Hz), 123.1, 119.2 (2C), 111.8, 12.0; MS: m/z 321 (M+), calculated m/z 321.07. Anal. Calcd. for C15H10F3N3O2: C, 56.08; H, 3.14; N, 13.08. Found: C, 56.02; H, 3.18; N, 13.11.
3-Methyl-5-nitro-1-(4-(trifluoromethyl)phenyl)-1H-indazole (11j). Yield: 225 mg, (0.70 mmol, 70%) as a white solid, mp 167–168 °C; IR: 1525, 1331 cm−1; 1H-NMR (DMSO-d6): δ 8.89 (d, J = 2.2 Hz, 1H), 8.31 (dd, J = 9.3, 2.2 Hz, 1H), 8.05 (d, J = 9.3 Hz, 1H), 8.02 (d, J = 8.6 Hz, 2H), 7.95 (d, J = 8.6 Hz, 2H), 2.69 (s, 3H); 13C-NMR (DMSO-d6): δ 148.3, 142.6, 142.3, 141.1, 127.4 (q, J = 4.0 Hz), 127.1, 125.1, 124.5 (q, J = 269.1 Hz), 123.2, 122.6, 119.2, 112.0, 12.1; MS: m/z 321 (M+). calculated m/z 321.07. Anal. Calcd. for C15H10F3N3O2: C, 56.08; H, 3.14; N, 13.08. Found: C, 55.98; H, 3.19; N, 13.04.
1-(4-Cyanophenyl)-3-methyl-5-nitro-1H-indazole (11k). Yield: 222 mg (0.80 mmol, 80%) as a brown solid, m.p. ≥ 194 °C (sub), ≥ 260 °C (dec); IR: 2226, 1516, 1338 cm−1; 1H-NMR (DMSO-d6): δ 8.93 (d, J = 2.2 Hz, 1H), 8.35 (dd, J = 9.1, 2.2 Hz, 1H), 8.11 (d, J = 9.1 Hz, 1H), 8.08 (d, J = 8.9 Hz, 2H), 8.04 (d, J = 8.9 Hz, 2H), 2.71 (s, 3H); 13C-NMR (DMSO-d6): δ 148.7, 142.8, 142.7, 141.1, 134.5, 125.3, 123.3, 122.6, 119.3, 118.9, 112.3, 109.4, 12.1; MS: m/z 278 (M+), calculated m/z 278.08. Anal. Calcd. for C15H10N4O2: C, 64.74; H, 3.62; N, 20.13. Found: C, 64.69; H, 3.67; N, 20.06.
4-(3-Methyl-5-nitro-1H-indazol-1-yl)benzenesulfonamide (11l). Yield: 249 mg (0.75 mmol, 75%), m.p. 265–266 °C; IR: 3422, 1515, 1331, 1320, 1123 cm−1; 1H-NMR (DMSO-d6): δ 8.92 (d, J = 2.2 Hz, 1H), 8.34 (dd, J = 9.3, 2.2 Hz, 1H), 8.09 (d, J = 9.3 Hz, 1H), 8.04 (d, J = 8.4 Hz, 2H), 8.01 (d, J = 8.4 Hz, 2H), 7.51 (s, 2H), 2.71 (s, 3H); 13C-NMR (DMSO-d6): δ 148.2, 142.54, 145.50, 141.6, 141.0, 127.9, 125.0, 123.1, 122.3, 119.2, 112.0, 12.0; MS: m/z 332 (M+), calculated m/z 332.06. Anal. Calcd. for C14H12N4O4S: C, 50.60; H, 3.64; N, 16.86. Found: C, 50.70; H, 3.63; N, 16.78.
4-(3-Methyl-5-nitro-1H-indazol-1-yl)benzoic acid (11m). Yield: 208 mg (0.70 mmol, 70%) as a tan solid, m.p. ≥ 240 °C (sub), ≥ 320 °C (dec). IR: 3441–2352, 1697, 1514, 1346 cm−1; 1H-NMR (DMSO-d6): δ 12.8 (br s, 1H), 8.93 (d, J = 2.2 Hz, 1H), 8.34 (dd, J = 9.3, 2.2 Hz, 1H), 8.16 (d, J = 8.6 Hz, 2H), 8.09 (d, J = 9.3, Hz, 1H), 7.95 (d, J = 8.6 Hz, 2H), 2.71 (s, 3H); 13C-NMR (DMSO-d6): δ 167.1, 148.2, 142.6, 142.5, 141.1, 131.5, 125.0, 123.1, 122.0, 119.4, 119.3, 112.2, 12.1; MS: m/z 297 (M+), calculated m/z 297.07. Anal. Calcd. for C15H11N3O4: C, 60.61; H, 3.73; N, 14.14. Found: C, 60.53; H, 3.68; N, 14.22.
1-Phenyl-5-nitro-1H-indazole (12a). Yield: 172 mg (0.72 mmol, 72%) as a tan solid, m.p. 178–180 °C (lit m.p. 178–180 °C [18]); IR: 1534, 1341 cm−1; 1H-NMR (DMSO-d6): δ 8.95 (s, 1H), 8.70 (s, 1H), 8.30 (d, J = 9.3 Hz, 1H), 7.98 (d, J = 9.3 Hz, 1H), 7.81 (d, J = 7.9 Hz, 2H), 7.65 (d, J = 7.7 Hz, 2H), 7.51 (t, J = 7.4 Hz, 1H); 13C-NMR (DMSO-d6): δ 142.8, 140.5, 139.1, 138.9, 130.3, 128.2, 124.8, 123.3, 122.7, 119.9, 111.9; MS: m/z 239 (M+), calculated m/z 239.07. Anal. Calcd. for C13H9N3O2: C, 65.27; H, 3.79; N, 17.56. Found: C, 65.16; H, 3.77; N, 17.57.
1-(2-Methoxyphenyl)-5-nitro-1H-indazole (12b). This reaction stopped at the hydrazone stage and did not give an indazole.
1-(3-Methoxyphenyl)-5-nitro-1H-indazole (12c): Yield: 180 mg (0.67 mmol, 67%) as a yellow solid, m.p. 132–133 °C (lit [36] m.p. 135 °C); IR: 1516, 1344 cm−1; 1H-NMR (DMSO-d6): δ 8.94 (s, 1H), 8.69 (s, 1H), 8.30 (d, J = 9.2 Hz, 1H), 8.01 (d, J = 9.3 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.32 (s, 1H), 7.08 (d, J = 8.2 Hz, 1H), 3.87 (s, 3H); 13C-NMR (DMSO-d6): δ 160.6, 142.8, 140.5, 140.2, 130.8, 131.2, 124.8, 122.7, 119.8, 115.3, 114.1, 112.0, 109.0, 56.0; MS: m/z 269 (M+), calculated m/z 269.08. Anal. Calcd. for C14H11N3O3: C, 62.45; H, 4.12; N, 15.61. Found: C, 62.37; H, 4.06; N, 15.74.
1-(4-Methoxyphenyl)-5-nitro-1H-indazole (12d). Yield: 199 mg (0.74 mmol, 74%) as a light yellow solid, m.p. 181–182 °C; IR: 1516, 1342 cm−1; 1H-NMR (DMSO-d6): δ 8.92 (d, J = 2.2 Hz, 1H), 8.64 (s, 1H), 8.27 (d, J = 9.2, 2.2 Hz, 1H), 7.84 (d, J = 9.2 Hz, 1H), 7.68 (d, J = 8.7 Hz, 2H), 7.18 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H); 13C-NMR (DMSO-d6): δ 159.2, 142.6, 141.6, 138.3, 132.1, 125.2, 124.3, 122.4, 119.8, 115.6, 111.6, 56.2; MS: m/z 269 (M+), calculated m/z 269.08. Anal. Calcd. for C14H11N3O3: C, 62.45; H, 4.12; N, 15.61. Found: C, 62.41; H, 4.07; N, 15.56.
1-(4-Bromophenyl)-5-nitro-1H-indazole (12e). Yield: 222 mg (0.70 mmol, 70%) as a tan solid, m.p. 169–170 °C; IR: 1511, 1348 cm−1; 1H-NMR (DMSO-d6): δ 8.95 (s, 1H), 8.71 (s, 1H), 8.32 (d, J = 9.3 Hz, 1H), 8.01 (d, J = 9.2 Hz, 1H), 7.84 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H); 13C-NMR (DMSO-d6): δ 142.9, 140.4, 139.2, 138.4, 133.2, 125.2, 125.0, 122.8, 120.7, 119.9, 111.9; MS: m/z 317 (M+), calculated m/z 316.98. Anal. Calcd. for C13H8BrN3O2: C, 49.08; H, 2.53; N, 13.21. Found: C, 48.97; H, 2.59; N, 13.25.
1-(3-Chlorophenyl)-5-nitro-1H-indazole (12f). Yield: 164 mg (0.60 mmol, 60%) as an off-white solid, m.p. 143–144 °C; IR: 1514, 1346 cm−1; 1H-NMR (DMSO-d6): δ 8.95 (s, 1H), 8.72 (s, 1H), 8.32 (d, J = 9.2 Hz, 1H), 8.03 (d, J = 9.2 Hz, 1H), 7.88 (s, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.67 (t, J = 8.1 Hz, 1H), 7.57 (d, J = 8.2 Hz, 1H); 13C-NMR (DMSO-d6): δ 142.9, 140.5, 140.3, 139.4. 134.6, 132.0, 128.0, 125.0, 123.0, 122.9, 121.7, 119.8, 112.0; MS: m/z 273 (M+), calculated m/z 273.03. Anal. Calcd. for C13H8ClN3O2: C, 57.05; H, 2.95; N, 15.35. Found: C, 56.96; H, 3.00; N, 15.29.
1-(4-Chlorophenyl)-5-nitro-1H-indazole (12g). Yield: 191 mg (0.70 mmol, 70%) as a light yellow solid, m.p. 179–180 °C; IR: 1509, 1343 cm−1; 1H-NMR (DMSO-d6): δ 8.95 (d, J = 2.2 Hz, 1H), 8.71 (s, 1H), 8.31 (dd, J = 9.3 , 2.2 Hz, 1H), 7.99 (d, J = 9.3 Hz, 1H), 7.85 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.6 Hz, 2H); 13C-NMR (DMSO-d6): δ 142.9, 140.5, 139.2, 138.0, 132.4, 130.3, 124.9, 122.8, 119.9, 111.9 (one C not observed); MS: m/z 273 (M+), calculated m/z 273.03. Anal. Calcd. for C13H8ClN3O2: C, 57.05; H, 2.95; N, 15.35. Found: C, 57.12; H, 2.97; N, 15.33.
1-(2,4-Dichlorophenyl)-5-nitro-1H-indazole (12h). Yield: 215 mg (0.70 mmol, 70%) as a light pink solid, m.p. 101–102 °C. IR: 1517, 1344 cm−1; 1H-NMR (DMSO-d6): δ 8.96 (s, 1H), 8.72 (s, 1H), 8.29 (d, J = 9.2 Hz, 1H), 8.03 (s, 1H), 7.78 (d, J = 8.3 Hz, 1H), 7.73 (d, J = 8.3 Hz, 1H), 7.52 (d, J = 9.2 Hz, 1H); 13C-NMR (DMSO-d6): δ 143.0, 142.3, 139.2, 135.7, 135.0, 132.2, 131.6, 130.8, 129.3, 123.9, 122.7, 119.8, 111.7; MS: m/z 307 (M+), calculated m/z 306.99. Anal. Calcd. for C13H7Cl2N3O2: C, 50.68; H, 2.29; N, 13.64. Found: C, 50.67; H, 2.34; N, 13.53.
1-(3-(Trifluoromethyl)phenyl)-5-nitro-1H-indazole (12i). Yield: 190 mg (0.62 mmol, 62%) as a light yellow solid, m.p. 118–119 °C. IR: 1513, 1347 cm−1; 1H-NMR (DMSO-d6): δ 8.97 (s, 1H), 8.75 (s, 1H), 8.35 (d, J = 9.2 Hz, 1H), 8.18 (d, J = 7.2 Hz, 1H), 8.13 (s, 1H), 8.05 (d, J = 9.3 Hz, 1H), 7.94–7.85 (complex, 2H); 13C-NMR (DMSO-d6): δ 143.0, 140.6, 139.8, 139.6, 131.7, 131.0 (q, J = 32.7 Hz), 126.9, 125.2, 124.6 (q, J = 4.3 Hz), 124.1 (q, J = 272.6 Hz), 123.0, 119.8 (q, J = 4.9 Hz), 112.0 (one C not observed); MS: m/z 307 (M+), calculated m/z 307.06. Anal. Calcd. for C14H8F3N3O2: C, 54.73; H, 2.62; N, 13.68. Found: C, 54.66; H, 2.64; N, 13.57.
1-(4-(Trifluoromethyl)phenyl)-5-nitro-1H-indazole (12j). Yield: 209 mg (0.68 mmol, 68%) as a light yellow solid, m.p. 151–152 °C. IR: 1519, 1327 cm−1; 1H-NMR (DMSO-d6): δ 8.95 (s, 1H), 8.76 (s, 1H), 8.34 (d, J = 9.5 Hz, 1H), 8.11 (obscured d, J = 9.5 Hz, 1H), 8.08 (d, J = 8.5 Hz, 2H), 8.00 (d, J = 8.5 Hz, 2H); 13C-NMR (DMSO-d6): δ 143.1, 142.3, 140.5, 139.8, 127.9, (q, J = 32.3 Hz), 127.5 (q, J = 3.5 Hz), 125.4, 124.4 (q, J = 273.7 Hz), 123.3, 123.1, 119.9, 112.1; MS: m/z 307 (M+), calculated m/z 307.06. Anal. Calcd. for C14H8F3N3O2: C, 54.73; H, 2.62; N, 13.68. Found: C, 54.68; H, 2.61; N, 13.64.
1-(4-Cyanophenyl)-5-nitro-1H-indazole (12k). Yield: 158 mg (0.60 mmol, 60%) as a light yellow solid, m.p. 250–251 °C. IR: 2227, 1512, 1344 cm−1; 1H-NMR (DMSO-d6): δ 8.97 (d, J = 2.2 Hz, 1H), 8.78 (s, 1H), 8.36 (dd, J = 9.3, 2.2 Hz, 1H), 8.15 (d, J = 9.3 Hz, 1H), 8.12 (d, J = 8.8 Hz, 2H), 8.07 (d, J = 8.8 Hz, 2H); 13C-NMR (DMSO-d6): δ 142.1, 141.6, 139.4, 139.1, 133.6, 124.5, 122.1, 120.0, 118.9, 117.7, 111.3, 109.0; MS: m/z 264 (M+), calculated m/z 264.06. Anal. Calcd. for C14H8N4O2: C, 63.64; H, 3.05; N, 21.20. Found: C, 63.55; H, 3.09; N, 21.08.
4-(5-Nitro-1H-indazol-1-yl)benzenesulfonamide (12l). Yield: 159 mg (0.50 mmol, 50%), m.p. 237–238 °C; IR: 3329, 3241, 1512, 1339 cm−1; 1H-NMR (DMSO-d6): δ 8.97 (d, J = 2.2 Hz, 1H), 8.76 (s, 1H), 8.35 (dd, J = 9.3, 2.2 Hz, 1H), 8.13 (d, J = 9.3 Hz, 1H), 8.06 (s, 4H), 7.53 (br s, 2H); 13C-NMR (DMSO-d6): δ 143.1, 141.60, 141.55, 140.5, 139.8, 128.0, 125.3,123.1, 122.2, 119.9, 112.2; MS: m/z 318 (M+), calculated m/z 318.04. Anal. Calcd. for C13H10N4O4S: C, 49.05; H, 3.17; N, 17.60. Found: C, 49.11; H, 3.16; N, 17.66.
4-(5-Nitro-1H-indol-1yl)benzoic acid (12m). This product was formed only using the two-step procedure. Yield: 142 mg (0.50 mmol, 50%) as a brown product, m.p. 158–159 °C; IR: 3395–2372, 1694, 1518, 1345 cm−1; 1H-NMR (DMSO-d6): δ 13.3 (br s, 1H), 8.97 (d, J = 2.2 Hz, 1H), 8.76 (s, 1H), 8.35 (dd, J = 9.3, 2.2 Hz , 1H), 8.18 (d, J = 8.4 Hz, 2H), 8.13 (d, J = 9.3 Hz, 1H), 7.98 (d, J = 8.4 Hz, 2H); 13C-NMR (DMSO-d6): δ 167.0, 143.0, 142.5, 140.4, 139.7, 131.5, 129.8, 125.3, 123.0, 122.5, 119.9, 112.2; MS: m/z 283 (M+), calculated m/z 283.06. Anal. Calcd. for C14H9N3O4: C, 59.37; H, 3.20; N, 14.84. Found: C, 59.32; H, 3.23; N, 14.75.
3.5. Representative Procedure for the General Indazole Synthesis
To a stirred solution of the carbonyl compound (13, 14 or 15, 1.0 mmol) in DMPU (5 mL) were added powdered 4 Å molecular sieves (30 wt% relative to the carbonyl substrate) and ArNHNH2·HCl (1.5 mmol). The mixture was heated at 90 °C (oil bath) for 1.5 h at which time CuI (0.2 mmol) and K2CO3 (2.5 mmol) were added and heating was continued at this temperature for 16 h. The crude reaction mixture was cooled to 23 °C and filtered through a Celite® pad (Fisher Scientific, Pittsburgh, PA, USA). The pad was rinsed with ether (2 × 20 mL) and the combined filtrate was washed with water (25 mL), saturated NaCl (25 mL), dried (MgSO4), filtered, and concentrated under vacuum. The products were purified by silica gel chromatography using increasing concentrations of EtOAc in hexanes. Yields as well as physical and spectral data are given below.
3-Methyl-1-phenyl-1H-indazole (16a). Yield: 169 mg (0.81 mmol, 81%) as tan solid, m.p. 73–74 °C (lit [18] m.p. 73–74 °C); IR: 1597, 1505 cm−1; 1H-NMR (CDCl3): δ 7.75–7.96 (complex, 4H), 7.52 (t, J = 7.3 Hz, 2H), 7.42 (t, J = 7.5 Hz, 1H), 7.32 (t, J = 7.3 Hz, 1H), 7.21 (t, J = 7.5 Hz, 1H), 2.65 (s, 3H); 13C-NMR (CDCl3): δ 144.0, 140.3, 139.5, 129.4, 127.1, 126.1, 124.9, 122.4, 120.8, 120.6, 110.3, 12.0; MS: m/z 208 (M+), calculated m/z 208.10. Anal. Calcd. for C14H12N2: C, 80.74; H, 5.81; N, 13.45. Found: C, 81.01; H, 6.08; N, 13.23.
1-(4-Methoxyphenyl)-3-methyl-1H-indazole (16d). Yield: 207 mg (0.87 mmol, 87%) as a yellow oil; IR: 2845, 1517 cm−1; 1H-NMR (CDCl3): δ 7.72 (d, J = 8.1 Hz, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.59 (d, J = 8.9 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.04 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H), 2.65 (s, 3H); 13C-NMR (CDCl3): δ 158.1, 143.8, 139.7, 133.5, 126.9, 124.4, 124.3, 120.50, 120.47, 114.6, 110.1, 56.6, 11.9; MS: m/z 238 (M+), calculated m/z 238.11. Anal. Calcd. for C15H14N2O: C, 75.61; H, 5.92; N, 11.76. Found: C, 75.35; H, 6.18; N, 11.25.
4-(3-Methyl-1H-indazol-1-yl)benzonitrile (16k). Yield: 198 mg (0.85 mmol, 85%) as tan solid, m.p. 124–126 °C; IR: 2224, 1604, 1517 cm−1; 1H-NMR (CDCl3): δ 7.91 (d, J = 8.6 Hz, 2H), 7.80 (d, J = 8.6 Hz, 2H), 7.78 (d, J = 8.4 Hz, 1H), 7.75 (d, J = 7.9 Hz, 1H), 7.50 (t, J = 8.3 Hz, 1H), 7.28 (t, J = 7.4 Hz, 1H), 2.65 (s, 3H); 13C-NMR (CDCl3): δ 146.1, 143.9, 139.1, 133.5, 128.0, 125.9, 121.9, 121.3, 121.1, 118.7, 110.5, 108.5, 12.0; MS: m/z 233 (M+), calculated m/z 233.10. Anal. Calcd. for C15H11N3: C, 77.23; H, 4.75; N, 18.01. Found: C, 77.35; H, 4.31; N, 18.32.
1-Phenyl-1H-indazole (17a). Yield: 149 mg (0.77 mmol, 77%) as off-white solid, m.p. 77–78 °C (lit [18] m.p. 76–78 °C); IR: 1595, 1500 cm−1; 1H-NMR (CDCl3): δ 8.21 (s, 1H), 7.81 (d, J = 8.1 Hz, 1H), 7.77 (d, J = 8.4 Hz, 1H), 7.75 (d, J = 8.7 Hz, 2H), 7.54 (t, J = 8.4 Hz, 2H), 7.43 (t, J = 8.3 Hz, 1H), 7.37 (t, J = 8.3 Hz, 1H), 7.23 (t, J = 8.1 Hz, 1H); 13C-NMR (CDCl3): δ 140.2, 138.8, 135.4, 129.5, 127.1, 126.6, 125.3, 122.8, 121.5, 121.3, 110.4; MS: m/z 194 (M+), calculated m/z 194.08. Anal. Calcd. for C13H10N2: C, 80.39; H, 5.19; N, 14.42. Found: C, 80.15; H, 5.31; N, 14.69.
1-(4-Methoxyphenyl)-1H-indazole (17d). Yield: 177 mg (0.79 mmol, 79%) as a yellow oil; IR: 2836, 1515 cm−1; 1H-NMR (CDCl3): δ 8.17 (s, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.65 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 8.9 Hz, 2H), 7.41 (t, J = 8.3 Hz, 1H), 7.21 (t, J = 8.0 Hz, 1H), 7.06 (d, J = 8.9 Hz, 2H), 3.88 (s, 3H); 13C-NMR (CDCl3): δ 158.4, 139.0, 134.8, 133.4, 126.9, 124.9, 124.5, 121.23, 121.21, 114.6, 110.2, 55.6; MS: m/z 224 (M+), calculated m/z 224.09. Anal. Calcd. for C14H12N2O: C, 74.98; H, 5.39; N, 12.49. Found: C, 74.63; H, 5.52; N, 12.63.
4-(1H-Indazol-1-yl)benzonitrile (17k). Yield: 164 mg (0.75 mmol, 75%) as white solid, m.p. 104–106 °C (lit m.p. [18] 103–106 °C); IR: 2225, 1604, 1510 cm−1; 1H-NMR (CDCl3): δ 8.26 (s, 1H), 7.94 (d, J = 8.6 Hz, 2H), 7.84 (d, J = 8.6 Hz, 2H), 7.86–7.81 (complex, 2H), 7.51 (t, J = 8.4 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H); 13C-NMR (CDCl3): δ 143.8, 138.5, 137.3, 133.6, 128.1, 126.1, 122.5, 121.9, 121.8, 118.5, 110.4, 109.3; MS: m/z 219 (M+), calculated m/z 219.08. Anal. Calcd. for C14H9N3: C, 76.70; H, 4.14; N, 19.17. Found: C, 76.85; H, 4.31; N, 19.32.
1-Phenyl-1H-pyrazolo[3,4-b]pyridine (18a). Yield: 133 mg (0.68 mmol, 68%) as a white solid, m.p. 52–54 °C (lit [10] 53–55 °C); IR: 1595, 1499 cm−1; 1H-NMR (CDCl3): δ 8.65 (dd, J = 4.5, 1.7 Hz, 1H), 8.28 (dd, J = 7.7, 1.2 Hz, 2H), 8.21 (s, 1H), 8.14 (dd, J = 8.0, 1.7 Hz, 1H), 7.54 (t, J = 7.7 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H), 7.22 (dd, J = 8.0, 4.5 Hz, 1H); 13C-NMR (CDCl3): δ 150.1, 149.2, 139.5, 133.8, 130.2, 129.1, 126.1, 121.4, 117.6, 117.2; MS: m/z 195 (M+), calculated m/z 195.08. Anal. Calcd. for C12H9N3: C, 73.83; H, 4.65; N, 21.52. Found: C, 73.95; H, 4.87; N, 21.78.
1-(4-Methoxyphenyl)-1H-pyrazolo[3,4-b]pyridine (18d). Yield: 140 mg (0.62 mmol, 62%) as a purple solid, m.p. 204–205 °C; IR: 2833, 1513 cm−1; 1H-NMR (CDCl3): δ 9.00 (dd, J = 8.0, 1.7 Hz, 1H), 8.70 (dd, J = 4.5, 1.7 Hz, 1H), 8.28 (d, J = 9.0 Hz, 2H), 7.35 (dd, J = 8.0, 4.5 Hz, 1H), 7.26 (s, 1H), 7.14 (d, J = 9.0 Hz, 2H), 3.91 (s, 3H); 13C-NMR (CDCl3): δ 158.0, 150.6, 149.6, 138.7, 132.9, 132.6, 123.0, 118.3, 115.3, 114.4, 55.6; MS: m/z 225 (M+), calculated m/z 225.09. Anal. Calcd. for C13H11N3O: C, 69.32; H, 4.92; N, 18.66. Found: C, 69.68; H, 5.11; N, 19.82.
4-(1H-Pyrazolo[3,4-b]pyridine-1-yl)benzonitrile (18k). Yield: 172 mg (0.78 mmol, 78%) as a white solid, m.p. 125–127 °C; IR: 2222, 1603, 1450 cm−1; 1H-NMR (CDCl3): δ 8.68 (dd, J = 4.5, 1.6 Hz, 1H), 8.66 (d, J = 8.8 Hz, 2H), 8.25 (s, 1H), 8.17 (dd. J = 8.0, 1.6 Hz, 1H), 7.82 (d, J = 8.8 Hz, 2H), 7.30 (dd, J = 8.0, 4.5 Hz, 1H); 13C-NMR (CDCl3): δ 150.7, 149.4, 143.1, 135.4, 133.2, 130.6, 120.3, 11.89, 118.4, 117.9, 108.7; MS: m/z 220 (M+), calculated m/z 220.07. Anal. Calcd. for C13H8N4: C, 70.90; H, 3.66; N, 25.44. Found: C, 71.23; H, 3.82; N, 25.67.
4. Conclusions
We have developed and optimized one and two-pot syntheses of 1-aryl-5-nitro-1H-indazoles from 2′-fluoro-5′-nitroacetophenone (5) and 2-fluoro-5-nitrobenzaldehyde (6). The one-pot conversion of 5 to 11 (a domino reaction) and 6 to 12 (a consecutive reaction) proceeded in yields (70–96%) that surpassed those of the two-pot sequence. We have also developed a general one-pot approach to 1-aryl-1H-indazoles that is not limited by the substitution patterns normally required for the SNAr ring closure. Treatment of 2′-bromoacetophenone (13), 2-bromobenzaldehyde (14) and 2-chloronicotinaldehyde (15) with ArNHNH2·HCl and 30 wt % of powdered 4 Å molecular sieves in DMPU at 90 °C, followed by addition of CuI and K2CO3 afforded 1H-indazoles bearing phenyl as well as electron-rich and electron-poor aromatic groups at N1 in 62–87% yields. The efficiency of the one-pot process provides access to a selection of 1-aryl-1H-indazoles in a single laboratory operation—one set-up and one work-up—without isolation or purification of intermediates. The methodology described herein has streamlined the synthesis of 1-aryl-1H-indazoles, improved the yields, and minimized the waste generated in preparing these systems.
Acknowledgments
The authors wish to acknowledge support from the NSF (BIR-9512269), the Oklahoma State Regents for Higher Education, the W. M. Keck Foundation, and Conoco, Inc. for establishing the Oklahoma State-wide NMR facility. The authors are also grateful to the OSU College of Arts and Sciences for funds that allowed the Chemistry Department to purchase a new 400 MHz NMR for this facility.
Supplementary Materials
Electronic Supplementary Information (ESI) are available online: Tables summarizing optimization experiments, a listing of characterization data for the intermediate arylhydrazones, and 1H- and 13C-NMR spectra for all compounds, are available online.
Author Contributions
J.K.A.-G. and K.K.G. did the experimental work and developed the one pot protocols. R.A.B. wrote the paper. All of the authors read and approved the final version of the manuscript before submission.
Conflicts of Interest
The authors declare no conflicts of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors or MDPI.
References and Notes
- 1.Nammalwar B., Bunce R.A. Recent Syntheses of 1,2,3,4-tetrahydroquinolines, 2,3-dihydro-4(1H)-quinolinones and 4(1H)-quinolinones using a domino strategy. Molecules. 2014;19:204–232. doi: 10.3390/molecules19010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunce R.A., Nammalwar B., Gnanasekaran K.K., Cain N.R. Isoquinolin-1(2H)-ones and 1,6-naphthyridin-5(6H)-ones by an N-acylation-SNAr sequence. Tetrahedron. 2014;70:838–844. doi: 10.1016/j.tet.2013.12.033. [DOI] [Google Scholar]
- 3.Gnanasekaran K.K., Muddala N.P., Bunce R.A. Pyrazoloquinazolinones and pyrazolo-pyridopyrimidinones by a sequential N-acylation-SNAr reaction. Tetrahedron Lett. 2015;56:1367–1369. doi: 10.1016/j.tetlet.2015.01.146. [DOI] [Google Scholar]
- 4.Gnanasekaran K.K., Muddala N.P., Bunce R.A. Benzo[4,5]imidazo[2,1-b]quinazolin-12-ones and benzo[4,5]imidazo[1,2-a]pyrido[2,3-d]pyrimidin-5-ones by a sequential N-acylation-SNAr reaction. Tetrahedron Lett. 2015;56:7180–7183. [Google Scholar]
- 5.Palazzo G., Corsi G., Baiocchi L., Silvestrini B. Synthesis and pharmacological properties of 1-substituted 3-dimethylaminoalkoxy-1H-indazoles. J. Med. Chem. 1966;9:38–41. doi: 10.1021/jm00319a009. [DOI] [PubMed] [Google Scholar]
- 6.Bai M., Carr G., DeOrazio R.J., Friedrich T.D., Dobritsa S., Fitzpatrick K., Guzzo P.R., Kitchen D.B., Lynch M.A., Peace D., et al. 5-Funtionalized indazoles as glucocorticoid receptor agonists. Bioorg. Med. Chem. Lett. 2010;20:3017–3020. doi: 10.1016/j.bmcl.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 7.Zhao C., Wang R., Li G., Xue X., Sun C., Qu X., Li W. Synthesis of indazole based diarylurea derivatives and their antiproliferative activity against tumor cell lines. Bioorg. Med. Chem. Lett. 2013;23:1989–1992. doi: 10.1016/j.bmcl.2013.02.034. [DOI] [PubMed] [Google Scholar]
- 8.Dessole G., Branca D., Ferrigno F., Kinzel O., Muraglia E., Palumbi M.C., Rowley M., Serafini S., Steinkühler C., Jones P. Discovery of N-[(1-aryl-1H-indazol-5-yl)methyl]amide derivatives as smoothened antagonists for inhibition of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009;19:4191–4195. doi: 10.1016/j.bmcl.2009.05.112. [DOI] [PubMed] [Google Scholar]
- 9.Mahindroo N., Punchihewa C., Fujii N. Hedgehog-gli pathway inhibitors as anticancer agents. J. Med. Chem. 2009;52:3829–3845. doi: 10.1021/jm801420y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao M., Liu X., Wang X., Cai Q., Ding K. Synthesis of 1-aryl-1H-indazoles via a ligand-free copper-catalyzed intramolecular amination reaction. Chin. J. Chem. 2011;29:1199–1204. doi: 10.1002/cjoc.201190223. [DOI] [Google Scholar]
- 11.Esmaeili-Marandi F., Saeedi M., Mahdavi M., Yavari I., Foroumadi A., Shafiee A. Potassium tert-butoxide promoted intramolecular amination of 1-aryl-2-(nitrobenzylidene)hydrazines: Efficient synthesis of 1-aryl-1H-indazoles. Synlett. 2014;25:2605–2608. [Google Scholar]
- 12.Lokhande P.D., Raheem A., Sabale S.T., Chabukswar A.R., Jagdale S.C. An efficient synthesis of 1H-indazoles. Tetrahedron Lett. 2007;48:6890–6892. doi: 10.1016/j.tetlet.2007.07.162. [DOI] [Google Scholar]
- 13.Xiong X., Jiang Y., Ma D. Assembly of N,N-disubstituted hydrazines and 1-aryl-1H-indazoles via copper-catalyzed coupling reactions. Org. Lett. 2012;14:2552–2555. doi: 10.1021/ol300847v. [DOI] [PubMed] [Google Scholar]
- 14.Veerareddy A., Gogireddy S., Dubey P.K. Regioselective synthesis of 1-substituted indazole-3-carboxylic acids. J. Heterocycl. Chem. 2014;51:1311–1321. doi: 10.1002/jhet.1717. [DOI] [Google Scholar]
- 15.Wheeler R.C., Baxter E., Campbell I.B., Macdonald S.J.F. A General one-step synthesis of substituted indazoles using a flow reactor. Org. Process Res. Dev. 2011;15:565–569. doi: 10.1021/op100288t. [DOI] [Google Scholar]
- 16.Lu Y., Cole K.P., Fennell J.W., Maloney T.D., Mitchell D., Subbiah R., Ramadas B. An alternative indazole synthesis for merestinib. Org. Process Res. Dev. 2018;22 doi: 10.1021/acs.oprd.8b00016. [DOI] [Google Scholar]
- 17.Li P., Wu C., Zhao J., Rogness D.C., Shi F. Synthesis of substituted 1H-indazoles from arynes and hydrazones. J. Org. Chem. 2012;77:3149–3158. doi: 10.1021/jo202598e. [DOI] [PubMed] [Google Scholar]
- 18.Lebedev A.Y., Khartulyari A.S., Voskoboynikov A.Z. Synthesis of 1-aryl-1H-indazoles via a palladium-catalyzed intramolecular amination of aryl halides. J. Org. Chem. 2005;70:596–602. doi: 10.1021/jo048671t. [DOI] [PubMed] [Google Scholar]
- 19.Xu P., Wang G., Wu Z., Li S., Zhu C. Rh(III)-catalyzed double C-H activation of aldehyde hydrazones: A route for functionalized 1H-indazole synthesis. Chem. Sci. 2017;8:1303–1308. doi: 10.1039/C6SC03888C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt A., Beutler A., Snovydovych B. Recent advances in the chemistry of indazoles. Eur. J. Org. Chem. 2008:4073–4095. doi: 10.1002/ejoc.200800227. [DOI] [Google Scholar]
- 21.Gaikwad D.D., Chapolikar A.D., Devkate C.G., Warad K.D., Tyade A.P., Pawar R.P., Domb A.J. Synthesis of indazole motifs and their medicinal importance: An overview. Eur. J. Med. Chem. 2015;90:707–731. doi: 10.1016/j.ejmech.2014.11.029. [DOI] [PubMed] [Google Scholar]
- 22.Cankarová N., Hlavác J., Krchnák V. Recent synthetic approaches to 1H- and 2H-indazoles. A review. Org. Prep. Proced. Int. 2010;42:433–465. doi: 10.1080/00304948.2010.513898. [DOI] [Google Scholar]
- 23.Tietze L.F., Beifuss U. Sequential transformations in organic chemistry: A synthetic strategy with a future. Angew. Chem. Int. Ed. 1993;32:131–163. doi: 10.1002/anie.199301313. [DOI] [Google Scholar]
- 24.Tietze L.F. Domino reactions in organic chemistry. Chem. Rev. 1996;96:115–136. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]
- 25.Tietze L.F., Brasche G., Gericke K.M. Domino Reactions in Organic Synthesis. Wiley-VCH; Weinheim, Germany: 2006. [Google Scholar]
- 26.Kim S., Yoon J.-Y. In: Science of Synthesis Houben-Weyl Methods of Molecular Transformations. Padwa A., editor. Volume 27. Thieme; Stuttgart, Germany: 2004. pp. 671–722. [Google Scholar]
- 27.Bordwell F.G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988;21:456–463. doi: 10.1021/ar00156a004. [DOI] [Google Scholar]
- 28.Reich H.J., Rigby J.H. Handbook of Reagents for Organic Synthesis: Acidic and Basic Reagents. Volume 4. Wiley; New York, NY, USA: 1999. pp. 296–298. [Google Scholar]
- 29.Fieser M., Fieser L.F. Reagents for Organic Synthesis. Volume 5. Wiley; New York, NY, USA: 1975. pp. 552–553. [Google Scholar]
- 30.Gribble G.W. Indole Ring Synthesis: From Natural Products to Drug Discovery. Wiley; New York, NY, USA: 2016. p. 229. [Google Scholar]
- 31.Katritsky A.R., Ramsden C.A., Joule J.A., Zhankin V.V. Handbook of Heterocyclic Chemistry. 3rd ed. Elsevier; Amsterdam, The Netherlands: 2010. Most ring closures to produce 6,5-fused aromatic rings involve reaction of an aryl-bound nucleophile with a side chain electrophilic center. [Google Scholar]
- 32.Smith M.B., March J. March’s Advanced Organic Chemistry: Reactions, Mechanism and Structure. 6th ed. Wiley-Interscience; Hoboken, NJ, USA: 2007. p. 495. [Google Scholar]
- 33.Lukin K., Hsu M.C., Fernando D., Leanna M.R. New practical synthesis of indazoles via condensation of O-fluorobenzaldehydes and their O-methyloximes with hydrazine. J. Org. Chem. 2006;71:8166–8172. doi: 10.1021/jo0613784. [DOI] [PubMed] [Google Scholar]
- 34.Sagitullina G.P., Garkushenko A.K., Poendaev N.V., Sagitullin R.S. Simple and efficient synthesis of 1H-indazoles. Mendeleev Commun. 2012;22:167–168. doi: 10.1016/j.mencom.2012.05.020. [DOI] [Google Scholar]
- 35.This statement is further substantiated by the fact that hydrazones bearing nitro groups at C2 and C4 failed to cyclize under our optimized conditions
- 36.Anderson K.W., Tundel R.E., Ikawa T., Altman R.A., Buchwald S.L. Monodentate phosphines provide highly active catalysts for Pd-catalyzed C–N bond forming reactions of heteroaromatic halides/amines and (H)N-heterocycles. Angew. Chem. Int. Ed. 2006;45:6523–6527. doi: 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.