

Abstract
The present work aimed to synthesise promising antioxidant compounds as a valuable alternative to the currently expensive and easily degradable molecules that are employed as stabilizers in industrial preparation. Taking into account our experience concerning domino Friedel-Crafts/lactonization reactions, we successfully improved and extended the previously reported methodology toward the synthesis of 3,3-disubstituted-3H-benzofuran-2-one derivatives 9–20 starting from polyphenols 1–6 as substrates and either diethylketomalonate (7) or 3,3,3-trifluoromethyl pyruvate (8) as electrophilic counterpart. The antioxidant capacity of the most stable compounds (9–11 and 15–20) was evaluated by both DPPH assay and Cyclic Voltammetry analyses performed in alcoholic media (methanol) as well as in aprotic solvent (acetonitrile). By comparing the recorded experimental data, a remarkable activity can be attributed to few of the tested lactones.
Keywords: antioxidant activity, cyclic voltammetry, DPPH, domino reaction, benzofuran-2-ones
1. Introduction
The interplay between free radicals and antioxidants represents a crucial point in the clinical and nutritional research field [1,2,3,4,5]. Free radicals are responsible for oxidative stress, which is balanced by endogenous antioxidant defense mechanisms as well as by the ingestion of exogenous antioxidants [6,7]. In the human body, the overproduction of radical species can cause oxidative damage to fundamental biomolecules (i.e., DNA, lipids, proteins, carbohydrates, etc.), favoring cell apoptosis [8] and becoming a primary cause of several both chronic and degenerative diseases (i.e., aging related pathogenesis, neurological and cardiovascular diseases, skin disorders, cancer etc.) [9,10,11]. In order to prevent the undesirable outbreaks of such issues, various natural or synthetic antioxidants are nowadays used as dietary supplements (Smart Food), and are also considered candidate drugs for the reduction of the oxidative damage [12,13]. Among others, tocopherols and tocotrienols, different forms of Vitamin E [14], flavonoids (quercetin) [15,16], hydroxytyrosol (HT) [17], gallic acid [18,19], and their derivatives are noteworthy phenolic antioxidants (Figure 1) which have even shown fascinating antithrombotic activities (i.e., inhibition of LDL oxidation, platelet aggregation, and endothelial cell activation) [20]. Such outstanding actions probably rely on the catechol system, which is a common feature in the above bioactive compounds [21]. Unfortunately, the currently employed products are seldomly used not only as dietary supplements but also as stabilizers in foods, cosmetics, or industrial preparations, mainly due to their frequent degradation. Consequently, the research of more stable antioxidants with a broad application scope remains a challenging endeavor of great interest.
Figure 1.

Examples of natural phenolic antioxidants.
Prompted by these considerations, we planned the synthesis of new generation antioxidant compounds and the evaluation of their antioxidant activity through a deep comparison between DPPH assay and Cyclic Voltammetry analyses. Specifically, we design potential antioxidant molecules characterized by the 3,3-disubstituted-3H-benzofuran-2-one framework decorated with one or more hydroxyl groups on the aromatic ring. Such a distinguishing scaffold not only is found in key intermediates towards the synthesis of biologically active molecules [22,23], but occurs also as a common feature of many natural medicinal products, among which the antioxidant species play a critical role [24,25,26,27]. Indeed, the 3H-benzofuran-2-ones, or 2-coumaranones, are a significant class of heterocyclic molecules highly widespread in nature, consisting of a benzene fused with a furan-2-one ring [27,28,29,30]. For example, the 3,3-disubstitued 2-coumaronone scaffold is a prominent structural motif in many natural compounds, such as in yuccaol A–E, isolated from Yucca Schidigera, which have exhibited antioxidant, radical scavenging, and inflammatory properties, as well as inducible NO synthase (iNOS) expression and platelet aggregation inhibition [31,32,33,34,35].
2. Results and Discussion
2.1. Synthesis
Within this context, although several elegant strategies for the construction of the 3H-benzofuran-2-one scaffold [23,36,37,38] have been reported, the number of approaches to the corresponding 3-hydroxy derivatives is much more limited. Nevertheless, following our recent development of alternative, short, and practical route to 3-hydroxy-3H-benzofuran-2-one [39,40], we have figured out the possibility of obtaining various phenolic derivatives by domino reaction involving a first Friedel–Crafts alkylation and a subsequent intramolecular lactonization. Specifically, the reaction was performed employing the polyphenols 1–6 as substrates and either ketomalonate 7 or 3,3,3-trifluoromethyl pyruvate 8 as the electrophilic counterpart in the presence of TiCl4 (10 mol%) as catalyst, which should activate the alkylating agent as well as the postulated intermediate A (Table 1).
Table 1.
Friedel–Crafts alkylation/lactonization of polyphenols 1–6 performed with TiCl4 as catalyst a.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R2 | Product | Yield (%) b | ||
| 1 | 1 | 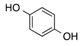 |
CO2Et | 9 | 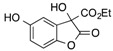 |
70 |
| 2 | 2 |  |
CO2Et | 10 |  |
62 |
| 3 | 3 | 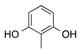 |
CO2Et | 11 | 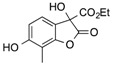 |
81 |
| 4 | 4 |  |
CO2Et | 12 | 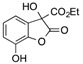 |
4 (22 c) |
| 5 | 5 |  |
CO2Et | 13 |  |
7 (18 c) |
| 6 | 6 | 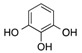 |
CO2Et | 14 | 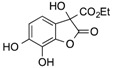 |
3 (19 c) |
| 7 | 1 | 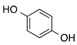 |
CF3 | 15 | 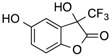 |
35 |
| 8 | 2 |  |
CF3 | 16 | 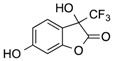 |
28 |
| 9 | 3 | 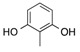 |
CF3 | 17 |  |
61 |
| 10 | 4 | 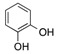 |
CF3 | 18 | 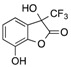 |
5 |
| 11 | 5 | 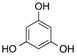 |
CF3 | 19 | 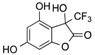 |
11 |
| 12 | 6 | 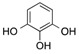 |
CF3 | 20 | 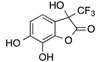 |
7 |
a Unless otherwise stated, the reactions were performed with indicated phenols (2.0 mmol) and alkylating agents (7 or 8; 2.2 mmol) in the presence of TiCl4 (10 mol%) in anhydrous CHCl3 (9 mL); b Yield of the isolated product; c Yield calculated at the 1H-NMR of the crude reaction mixture.
As depicted in Table 1, employing the previously optimized conditions, only the polyphenols 1–3 furnished the desired bicyclic compounds 9–11 with good to high yields (up to 81%, entries 1–3), when the ketomalonate 7 was used as alkylating agent; meanwhile substrates 4–5, owing the second hydroxyl group in the ortho position (4) or two OH substituents (5 and 6), led to a complex reaction mixture where the corresponding products 12 and 13, initially detected at the 1H-NMR, were isolated in very poor yields, mainly due to their instability (entry 4–6). Additionally, the more reactive 3,3,3-trifluoromethyl pyruvate 8 provided exclusively the benzofuran-2-one 17 in acceptable amount (61%, entry 9). On the contrary, the same electrophile 8 reacted slowly with hydroxyl phenols 2 and 3, which furnished the 3-hydroxy lactones 15 (entry 7) and 16 (entry 8) in moderate yields (35% and 28% respectively) and almost no conversion was detected starting from substrates 4–6 (entries 10–12).
In light of these outcomes, we supposed that the installation of a strong interaction between the employed Lewis acid (TiCl4) and the several hydroxyl groups bounded to the substrates should establish various stable complexes by prohibiting any possible nucleophilic attack of the alkylating agent on aromatic ring. For this reason, we carried out additional investigations in order to optimize the domino Friedel–Crafts/lactonization reaction between polyphenols 1–6 and 3,3,3-trifluoromethyl pyruvate 8 as alkylating agent. Specifically, we employed p-hydroxyphenol (hydroquinone) 1 as the model substrate and different reaction conditions. The observed results are summarized in Table 2.
Table 2.
Reaction of 3,3,3-trifluoromethyl pyruvate 8 with polyphenols 1–6 a.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Sub | Catalyst | mol% | Solvent | Temp (°C) | Time (h) | Product | Yield (%) b |
| 1 | 1 | BF3·Et2O | 30 | CH2Cl2 | r.t | 48 | 15 | 5 |
| 2 | 1 | BF3·Et2O | 30 | THF | 60 | 22 | 15 | n.d. |
| 3 | 1 | AlCl3 | 30 | CH2Cl2 | r.t. | 72 | 15 | n.d. |
| 4 | 1 | PhCO2H | 30 | CHCl3 | 60 | 72 | 15 | n.d. |
| 5 | 1 | PhCO2H | 100 | CHCl3 | 60 | 24 | 15 | n.d. |
| 6 | 1 | CSA | 30 | CHCl3 | 60 | 24 | 15 | n.d. |
| 7 | 1 | CSA | 100 | Toluene | 100 | 10 | 15 | n.d. |
| 8 | 1 | DBU | 30 | CH2Cl2 | r.t. | 72 | 15 | n.d. |
| 9 | 1 | AcOH | 30 | CH2Cl2 | r.t. | 72 | 15 | n.d. |
| 10 | 1 | - | - | AcOH | 120 | 12 | 15 | 45 |
| 11 c | 1 | - | - | AcOH | 120 | 10 | 15 | 55 |
| 12 d | 1 | - | - | AcOH | 120 | 4 | 15 | 80 |
| 13 d | 2 | - | - | AcOH | 120 | 4 | 16 | 72 |
| 14 d | 3 | - | - | AcOH | 120 | 6 | 17 | 86 |
| 15 d | 4 | - | - | AcOH | 120 | 4 | 18 | 78 |
| 16 d | 5 | - | - | AcOH | 120 | 5 | 19 | 66 |
| 17 d | 6 | - | - | AcOH | 120 | 4 | 20 | 76 |
a Unless otherwise stated, the reactions were performed under inert atmosphere with the indicated phenols (5.0 mmol) and 3,3,3-trifluoromethyl pyruvate (8; 5.5 mmol) in 8 mL of solvent; b Yield of the isolated product. (n.d. = not detected); c 5 mL of AcOH was employed; d 3 mL of AcOH was employed.
Firstly, we examined catalytic amounts of two different Lewis acids, precisely, BF3·Et2O (entries 1 and 2) and AlCl3 (entry 3), but both of them were neither effective nor efficient, leading to a complex reaction mixture without any trace of the target compound. Subsequently, benzoic acid (entries 4 and 5) and camphorsulfonic acid (CSA) (entries 6 and 7), were tested, varying both their amounts and the reaction temperature, but unfortunately the starting materials were recovered almost quantitatively. A similar result was achieved using a catalytic amount of either DBU (entry 8) or acetic acid in dichloromethane, where they did not catalyse any reaction and even after 72 h the starting material was recovered.
The best results were observed by exploiting the approach previously reported by Dyachenko et al. [41,42], who employed acetic acid as solvent. With our delight, benzofuran-2-one 15 was obtained with a 45% yield keeping the temperature at 120 °C for 12 h (entry 10). Differently to Lewis acids, the employment of acetic acid does not suffer from the formation of an unreactive substrate/catalyst complex. Therefore, the mechanistic pathway should involve (see Scheme SM-1): (i) the initial activation of the alkylating agent 8 by the acidic solvent that promotes (ii) the Friedel–Crafts alkylation at the ortho position of substrate 1 to obtain the key intermediate A; (iii) a subsequent acid-assisted intramolecular transesterification to deliver the expected 3-hydroxylactone 15.
The subsequent optimization of the applied protocol particularly benefited from a more concentrated reaction mixture and decrease in the reaction time (entries 11 and 12). Finally, the desired lactone 15 was isolated in 80% yield after only 4 h at 120 °C and using 3 mL of acetic acid as solvent. In almost all cases, the desired product was successfully formed in quite good yield (up to 86%). Noteworthy, in phenol 3 containing a hydroxyl group in the meta position, the introduction of an alkyl group in the ortho position considerably enhanced the overall reactivity (86% yield, entry 14), with respect to the corresponding non-methylated phenol 2, which gave the desired lactone 16 with 72% yield (entry 13), as well as the phenol 5, which possesses two additional hydroxyl groups in both the meta positions and reacted even less efficiently furnishing the lactone 19 in 66% yield (entry 16). Moreover, the presence of a catecholic group (entries 15 and 17) did not interfere with the reaction path, as proven by compounds 18 and 20, that were obtained in comparable yields (respectively 78% and 76%) after 4 h.
Additionally, hydroxyl phenols 4–6 were tested using AcOH as solvent and diethylketomalonate 7 as alkylating agent at high temperature (120 °C), but unfortunately a complex reaction mixture was obtained and the corresponding products 12–14 were not detected, which confirmed the extreme lability of such compounds.
2.2. Antioxidant Capacity Evaluation
In the last decades, there has been an increasing interest in antioxidants, principally in those proposed to prevent the potential injurious effects of free radicals [43]. Therefore, it is very appealing to have convenient and quick methods for estimating the efficacy of a substance as antioxidant. For this purpose, numerous in vitro methods have been developed to evaluate different antioxidant effects [44,45]. Depending upon the reactions involved, these assays can be classified into assays based on hydrogen atom transfer (HAT) reactions, and assays based on electron transfer (ET). The former assays analyse competitive reaction kinetics, quantified through the kinetic curves, while the ET-based assays monitor one redox reaction with the oxidant as an indicator of the reaction endpoint. Both methods are able to measure the radical (or oxidant) scavenging capacity, instead of the preventive antioxidant capacity of a sample [46]. In light of such features, we evaluated the antioxidant capacity of our compounds by DPPH assay and Cyclic Voltammetry analyses.
2.2.1. DPPH Assay
The DPPH assaying of compounds 9–11 and 15–20 was performed using Trolox as reference compound. Actually, to achieve more reliable results and to obtain information on the antioxidant activity of these compounds, the assay was carried out both in alcoholic media (methanol, MeOH) as well as in an aprotic solvent (acetonitrile, ACN), i.e., solvents with different abilities in solvating molecules and forming hydrogen bonds. Specifically, to the degassed solutions (2 mL) of antioxidant, with a final concentration from 0 to 60 μM, were added 2 mL of 140 μM DPPH• stock solution (70 μM DPPH final concentration). The solution was stirred at room temperature in the absence of light, and, after 60 min, the absorbance was measured at 517 nm. The working DPPH• concentration was calculated from the absorbance response at 517 nm of the initial solution in the absence of antioxidant and using the calibration curves reported for methanol in Figure 1 and acetonitrile in Figure 2, respectively. The DPPH• scavenging percentage or inhibition [I%] was calculated from the measured absorbance using the following expression:
where A0 and A represent respectively the absorbance in the absence of antioxidant and the absorbance at a given antioxidant concentration. The plot of I(%) versus the molar ratio of reagents (molsantioxidant/molsDPPH•) presented a linear response (ŷ = ax ± b) until the plateau was reached, that is, when the inhibition arrived to its maximum [44,47]. The predicted relative IC50 (rIC50, the half inhibitory concentration in terms of molsantioxidant/molsDPPH•) was estimated replacing the ŷ-value by 50 in the regression line (see the Supplementary Material). Sometimes it is more useful to discuss in terms of antiradical power (ARP), that is the inverse of rIC50, therefore the higher the ARP value is, the more efficient the antioxidant is.
Figure 2.

Regression line of the tested compounds 9–11 and 15–20 in DPPH assay in methanol.
DPPH Assay in Methanol
Figure 2 shows the plot of inhibition percentage versus moles antioxidant/moles DPPH• for the synthesized compounds 9–11 and 15–20.
By comparing the recorded absorbances (Figure 2 and Table 3), remarkably compounds 9, 15, 18, and 20 achieve a ratio molar (molsantioxidant/molsDPPH•) between 0.18 and 0.31, which is quite close to the value obtained with Trolox (0.23), whereas the other synthesised benzofuran-2-ones (10, 11, 16, 17, and 19) exhibit a poor to moderate reactivity towards the DPPH•. From the regression lines, the rIC50 in terms of molsantioxidant/molsDPPH• was subsequently estimated as well as the corresponding coefficient of determination R2, which undoubtedly validated the robustness of our analyses (Table 3). Additionally, the antiradical power (ARP), the stoichiometry, and the number of DPPH• reduced were calculated, as reported in Table 3.
Table 3.
Antioxidant capacity of compounds 9–11 and 15–20 towards DPPH• in methanol a.
| Entry | Antioxidant | rIC50 (molsantiox.t/molsDPPH∙) | N° DPPH• Reduced b |
|---|---|---|---|
| 1 | Trolox | 0.23 | 2.16 |
| 2 | 9 | 0.31 | 1.63 |
| 3 | 10 | 3.62 | 0.14 |
| 4 | 11 | 2.03 | 0.25 |
| 5 | 15 | 0.22 | 2.28 |
| 6 | 16 | 3.52 | 0.14 |
| 7 | 17 | 1.07 | 0.47 |
| 8 | 18 | 0.24 | 2.07 |
| 9 | 19 | 0.62 | 0.80 |
| 10 | 20 | 0.18 | 2.72 |
a All the measures were performed in triplicate and the values were reported as mean ± SD; b The number of DPPH• molecules, reduced by one molecule of antioxidant, is the inverse of the stoichiometry.
As depicted above, compounds 9, 15, 18, and 20 presented rIC50 values from 0.18 to 0.31 (entries 2, 5, 8, and 10 respectively), representing the best antioxidants of the analysed series in the employed conditions. Specifically, compounds with a single phenolic group (i.e., 9, 15, and 18) seemed to reduce approximately two molecules of DPPH•, whereas the benzolactone 20, decorated with two hydroxyl groups on the aromatic ring, reduced almost three molecules of DPPH•. These experimental results highlight the possible involvement of the lactone ring in the reduction mechanism, although no exhaustive mechanistic study was undertaken. Conversely, the remaining compounds (i.e., 10, 11, 16, 17, and 19), bearing a hydroxyl group in meta position (with respect the oxygen of the lactone ring), show quite high values of rIC50 (0.62–3.62) and consequently, a limited antioxidant capacity toward DPPH• (entries 3, 4, 6, 7, and 9 respectively). In order to further validate these outcomes, the DPPH assay was also carried out in an aprotic solvent (acetonitrile), and the results were consequently compared to those obtained in alcoholic media.
DPPH Assay in Acetonitrile
The assay was performed as described above, using acetonitrile as solvent (see experimental part for calibration curve of DPPH in ACN, SM). The absorbance measurements of each solution containing a known concentration of antioxidant and DPPH• were collected. Figure 3 depicted the plot of inhibition percentage versus molsantioxidant/molsDPPH• for the synthesized compounds 9–11 and 15–20.
Figure 3.

Regression line of the tested compounds 9–11 and 15–20 in DPPH assay in acetonitrile (ACN).
By the comparison of achieved results (Figure 3 and Table 4), only compound 20 shows a ratio molar (molsantioxidant/molsDPPH•) of 0.17, which suggests an even better antioxidant capacity than that of Trolox, whereas the other synthesised benzofuran-2-ones (9–11 and 15–19) exhibit a lower reactivity towards the DPPH•. Table 4 shows the corresponding regression lines with the values of R2 as well as the rIC50 and all the deducible parameters, i.e., the ARP, the stoichiometric value, and the number of DPPH• molecules that were reduced.
Table 4.
Antioxidant capacity of compounds 9–11 and 15–20 towards DPPH• in acetonitrile a.
| Entry | Antiox. | rIC50 (molsantiox./molsDPPH∙) | N° DPPH• Reduced b |
|---|---|---|---|
| 1 | Trolox | 0.22 | 2.24 |
| 2 | 9 | 4.26 | 0.12 |
| 3 | 10 | 4.12 | 0.12 |
| 4 | 11 | 3.92 | 0.13 |
| 5 | 15 | 1.69 | 0.30 |
| 6 | 16 | 4.47 | 0.11 |
| 7 | 17 | 3.25 | 0.15 |
| 8 | 18 | 0.54 | 0.93 |
| 9 | 19 | 2.23 | 0.22 |
| 10 | 20 | 0.17 | 3.02 |
a All the measures were performed in triplicate and the values were reported as mean ± SD; b The number of DPPH• molecules, reduced by one molecule of antioxidant, is the inverse of the stoichiometry.
The results depicted above are surprisingly different from the previous ones recorded in methanol. Indeed, almost all the examined compounds showed an inadequate antioxidant capacity toward DPPH• in acetonitrile, since compounds 9–17 and 19 exhibited rIC50 between 1.69 and 4.47, not reducing any molecules of DPPH•. Conversely, compound 18 exhibited a better antioxidant capacity, although not to the level of Trolox, with a rIC50 of 0.54 and a number of reduced molecules of DPPH• amounting approximately to one. Exceptionally, with our delight, the 3-hydroxy benzofuran-2-one derivative 20, bearing two hydroxyl groups in ortho and meta positions, presented a rIC50 of 0.17, which reduced three molecules of DPPH•, as observed before in methanol.
These experimental results highlight the different behavior of compounds in the DPPH assay with respect to the employed solvent. In this regard, the rate constants for hydrogen atom abstraction from compounds 9, 15, 18, and 20 by the DPPH• were determined both in methanol and acetonitrile. Such a kind of analyses should help us to verify if the obtained results could have been invalidated by solvent interference.
2.2.2. Measurement of Rate Constants for the Reaction of Compounds 9, 15, 18, and 20 with DPPH•
Several studies on the kinetic solvent effects (KSEs) of DPPH•/phenol reactions established that the large KSEs observed for H-atom abstractions from phenols are mainly a consequence of hydrogen bonding to the solvent, when it is a hydrogen bond acceptor (HBA) [48,49,50]. Precisely, Ingold et al. [51,52,53] endorsed that the bimolecular rate constant enhancement (ksPhOH/DPPH•, Scheme 1, reaction a) is due to the partial ionization of the phenol in those solvents that supports an ionization process (Scheme 1, reaction b), especially alcohols, and a very fast electron transfer from the phenoxide anion to the DPPH• (Scheme 1, reaction c), leading to profound kinetic consequences. Additionally, enhanced rate constants were a general feature of PhOH/DPPH• reaction also for phenols with low pKa values, in a non-hydroxylic, polar solvent, such as n-butyl ether, acetonitrile, THF, and DMSO.
Scheme 1.

Reaction involved in the kinetic solvent effects.
In order to better comprehend the possible kinetic solvent effects in the performed DPPH assays of compounds 9, 15, 18, and 20, the rate constant measurement for the reactions of these compounds with DPPH• was carried out. Practically, the decay of DPPH• absorbance in the presence of a known concentration of phenols was followed at 517 nm and analysed as a pseudo-first-order process to yield the kex/s−1. Because of this, the antioxidants were always used in large excess (final concentration 1–6 mM) over DPPH• (final concentration 85 µM). Afterwards, the second-order rate constants (ksPhOH/DPPH•) of the synthesised compounds were evaluated by plotting the pseudo-first-order constant (kex) versus antioxidant concentration: this plot is linear and its slope gives the bimolecular constants (see Supporting Material Table SM-5 and Table SM-6). Table 5 shows the ks of analysed compounds, as well as the corresponding rIC50 are reported again for a more explicit comparison.
Table 5.
Bimolecular rate constants (M−1·s−1) for H-atom abstraction from compounds 9, 15, 18, and 20 by DPPH• a.
| Entry | Antiox. | ks (M−1·s−1) b | rIC50 (molsantioxidant/molsDPPH•) | ks (M−1·s−1) b | rIC50 (molsantioxidant/molsDPPH•) |
|---|---|---|---|---|---|
| MeOH | MeOH | ACN | ACN | ||
| 1 | 9 | 3.26 | 0.31 | 8.54 | 4.26 |
| 2 | 15 | 3.77 | 0.22 | 1.26 | 1.69 |
| 3 | 18 | 1.40 | 0.24 | 0.23 | 0.54 |
| 4 | 20 | 0.77 | 0.18 | 2.22 | 0.17 |
a All the measurements were performed in triplicate and processed using a Sigma Plot software, 12.0, Systat software, Inc., San Jose, CA, USA); b The second-order rate constants were the slopes of the plots of kes vs. compound concentration (See Supplementary Material).
As shown above, compound 9 (entry 1) gave a high value of ks in acetonitrile, although the corresponding rIC50 revealed a moderate antioxidant capacity. Moreover, even though the kinetic constant is lower in methanol than in acetonitrile, the significant value of ks suggests a partial ionization of the phenol and slight interference by the employed solvent. Instead, compounds 15 and 18 (entries 2 and 3) display values of ks quite in agreement with the corresponding rIC50: in methanol, a partial ionization of the phenols altered the results obtained with DPPH assay in methanol (rIC50 0.22 and 0.24, respectively), while in acetonitrile, the value of rIC50 was reliable. Finally, the ks values of compound 20 (entry 4), which presented a similar antioxidant capacity towards DPPH• in both solvents, were mildly in disagreement, in fact in acetonitrile the ks was higher than in methanol. Consequently, the obtained result in the DPPH assay, considering the reaction kinetics and possible solvent effects, were reassessed and benzofuran-2-one 20 presented a real quite good antioxidant capacity toward DPPH•.
2.2.3. Cyclic Voltammetry
Cyclic voltammetry (CV) is an electrochemical technique frequently used (often along with DPPH assay) for the determination of the antioxidant activity of target compounds [54,55]. It consists of a potential scanning from a starting value to a final one, and then returning to the initial potential, while registering the current flowing between two electrodes. The potential at which an increase of current is registered corresponds to the oxidation (or reduction) of a species in solution. Thus, the oxidation potential measured by cyclic voltammetry can be taken as a measure of the ease of the oxidation process. Specifically, low oxidation potentials are associated with a greater facility of a given molecule for electro-donation and thus to act as antioxidant [54,56,57,58].
Moreover, CV can be carried out in aqueous medium as well as in organic solvents, e.g., in the presence of a supporting electrolytes to ensure the electrical conductivity of the solution [58].
The redox chemistry of the synthesized compounds 9–11 and 15–20 was evaluated using CV, recording the voltammograms in aqueous medium (H2O) as well as in an organic solvent (acetonitrile) and comparing the first oxidation peak potential to that of Trolox as a reference compound. Table 6 summarizes the observed results.
Table 6.
First oxidation peaks (Epox) from CV in aqueous medium and acetonitrile relative to compounds 9–11 and 15–20 (Figure 3 and Figure 4).
| Entry | Compounds | Epox (V) 1 (H2O) | Epox (V) 1 (ACN) |
|---|---|---|---|
| 1 | Trolox | 0.52 | 1.08 |
| 2 | 9 | 0.72 | 1.62 |
| 3 | 10 | 1.13 | 1.44 |
| 4 | 11 | 1.11 | 1.65 |
| 5 | 15 | 0.62 | 1.72 |
| 6 | 16 | 1.01 | 1.92 |
| 7 | 17 | 1.05 | 1.77 |
| 8 | 18 | 0.73 | 0.92 |
| 9 | 19 | 1.03 | 1.88 |
| 10 | 20 | 0.85 | 1.81 |
1 All the peak potentials are referred to SCE.
Cyclic Voltammetry in Aqueous Medium of Compounds 9–11 and 15–20
The CV in aqueous medium was performed in a cell containing a three-electrode system: (i) the reference electrode (Saturated Calomel Electrode, SCE); (ii) the working electrode (Glassy Carbon, GC); and (iii) a counter electrode (platinum wire). Each antioxidant (2 × 10−3 M final concentration), dissolved in EtOH, was added to 40 mL of a solution of water, containing 0.5 M NaCl, as supporting electrolyte, and then the voltammogram was recorded.
As Figure 4 and Table 6 highlight, all the considered compounds showed a similar voltammetric behavior, i.e., an irreversible anodic oxidation whose Epox (oxidation peak potential) is between +0.62 and +1.13 V (vs SCE). All Epox of the considered products are of higher potential than that of the reference compound, Trolox (Epox 0.52 V vs. SCE), which corresponds to a minor easing of the oxidation process. Nevertheless, among these, products 9, 15, and 18 exhibited the lowest values of potential, which, being under 1V (Epox 0.72, 0.62, and 0.73 respectively) and quite close to the Trolox one, is indicative of antioxidant capacity. These results suggested that in these electrochemical measurements, the fundamental structural features necessary to increase the antioxidant capacity are the phenolic groups in positions 5 and 7 (in compounds 9, 15, and 18, respectively). Moreover, the presence of a CF3 group seems essential for the ease in the oxidation process, lowering Epox by 60–120 mV (compare compound 9 vs. 15, 10 vs. 16 and 11 vs. 17 in Table 6).
Figure 4.

Voltammetric curves of compounds 9–11 and 15–20 (c = 2 × 10−3 M) in aqueous medium (H2O-0.5 M NaCl). Starting potential: −0.5 V; reversal potential: +1.5 V (GC working electrode, ν = 0.2 V·s−1, SCE reference electrode, 25 °C, N2).
Cyclic Voltammetry in Acetonitrile of Compounds 9–11 and 15–20
The CV in acetonitrile was performed in a cell containing a three-electrode system involving: (i) the reference electrode (modified SCE electrode, −0.029 V vs. SCE electrode− i.e., containing an organic junction); (ii) the working electrode (Glassy Carbon, GC); and (iii) a counter electrode (platinum wire). Each antioxidant (2 × 10−3 M final concentration), dissolved in ACN, was added to 5 mL of a solution of ACN, containing 0.1 M TEABF4 (tetraethylammonium tetrafluoroborate), as supporting electrolyte. The corresponding results are depicted in Figure 5 and Table 6.
Figure 5.

Voltammetric curves of compounds 9–11 and 15–20 in acetonitrile (c = 2 × 10−3 M) in acetonitrile (MeCN-0.1 M Et4NBF4). Starting potential: −0.5 V; reversal potential: +1.5 V (GC working electrode, ν = 0.2 V·s−1, SCE reference electrode, 25 °C, N2).
As displayed in Table 6, the electrochemical results obtained from CV in acetonitrile were relatively in accord with those observed in aqueous medium. Specifically, compounds 9 and 10 showed lower potential (Epox 1.62 and 1.44, respectively), although they were higher than the value of Trolox (Epox 1.08 V). Remarkably, compound 18 exhibited the first potential peak at 0.92 V, lower than Trolox. This result showed that an essential structural feature which makes the 3-hydroxy-3Hbenzofuranone scaffold a possible antioxidant compound is the presence of phenolic group in ortho position and a CF3 group.
3. Materials and Methods
Solvents and common reagents were purchased from a commercial source and used without further purification. All reactions were monitored by thin-layer chromatography (TLC) carried out on Merck F-254 silica glass plates and visualized with UV light or by 5% phosphomolibdic acid/ethanol test. Flash chromatography was performed on Sigma-Aldrich silica gel (60, particle size: 0.040–0.063 mm). 1H-NMR and 13C-NMR were recorded in CDCl3 (99.8% in deuterium) using a Varian Gemini 300 spectrometer (300 MHz, Varian Inc., Palo Alto, CA, USA). All chemical shifts are expressed in parts per million (δ scale) and are referenced to the residual protons of the NMR solvent (CDCl3, δ 7.24 ppm). Coupling constant (J) was expressed in Hz. Infrared spectra (FT-IR) were obtained using a Bruker Vector 22 spectrometer (Bruker, Billerica, MA, USA); data are presented as the frequency of absorption (cm−1). High-resolution mass spectrometry (HRMS) spectra were recorded with Micromass Q-TOF micro mass spectrometer (Waters Corporations, Milford, MA, USA) and Micromass LCT (ESI, Waters Corporations, Milford, MA, USA) with Lock-Spray-Injector (Injection Loop-Modus in a HPLC system, Waters, Alliance 2695, Waters Corporations, Milford, MA, USA). UV-Vis measurements were performed with a Shimadzu-UV-2401PC spectrophotometer. Cyclic Voltammetry measurements were acquired on a AMEL552 electrochemical workstation. The standard three-electrode arrangement was employed. In all cases, a Pt wire auxiliary electrode was used, the working electrode was a 3 mm diameter glassy carbon, and the solution was degassed with N2. Melting points were determined on a Mel-Temp apparatus.
3.1. General Procedure for the Lewis-Acid-Catalysed Friedel–Crafts/Lactonization Reaction
The alkylating agent (2.2 mmol) was added in one portion to a stirred solution of the appropriate phenol (2.0 mmol) in anhydrous CHCl3 (9 mL), and then TiCl4 (1 M in anhydrous CH2Cl2; 0.4 mL, 10 mol-%) was added. The system was kept under an argon atmosphere. The clear reddish solution was stirred at the reported temperature until the substrate had been completely consumed (TLC monitoring). Afterwards, the reaction mixture was poured into cold water (18 mL), and the aqueous phase was extracted several times with EtOAc (4 × 20 mL). The combined organic layers were washed with brine, dried with anhydrous Na2SO4, and concentrated under vacuum. The residue was purified by flash chromatography on silica gel to give the corresponding products as described below.
3.2. Characterization Data for Benzofuran 9–11
Ethyl 3,5-Dihydroxy-2-oxo-2,3-Dihydrobenzofuran-3-Carboxylate, 9. Following the general procedure, the single product 9 was obtained as a white solid in 70% yield after purification by flash chromatography on silica gel (nHexane/EtOAc = 7/3). m.p. 139–142 °C. IR (CHCl3): = 3468–3412, 3018, 2979, 2914, 1759, 1725, 1608 cm−1. 1H-NMR (CDCl3, 300 MHz, 25 °C): δ (ppm) = 8.59 (bs, 1H, OHphen), 7.08 (d, J = 8.6 Hz, 1H, CHarom), 6.98–6.88 (m, 2H, CHarom), 6.50 (bs, 1H, OH), 4.30–4.15 (m, 2H, CH2CH3), 1.16 (t, J = 7.1 Hz, 3H, CH2CH3). 13C-NMR (CDCl3, 75 MHz, 25 °C): δ (ppm) 173.2, 168.5, 155.4, 147.7, 128.2, 118.6, 112.5, 111.7, 77.9, 63.3, 14.0. HRMS: exact mass calculated for (C11H10NaO6) requires m/z 261.0370, found m/z 261.0371.
Ethyl 3,6-Dihydroxy-2-oxo-2,3-Dihydrobenzofuran-3-Carboxylate, 10. Following the general procedure, the single product 10 was obtained as a white solid in 62% yield after purification by flash chromatography on silica gel (nHexane/EtOAc = 4/6). m.p. 142–144 °C. IR (CHCl3): = 3468–3420, 3010, 2972, 2921, 1760, 1727, 1615 cm−1. 1H-NMR (CDCl3, 300 MHz, 25 °C): δ (ppm) = 9.17 (bs, 1H, OHphen), 7.25 (d, J = 7.9 Hz, 1H, CHarom), 6.72 (m, 2H, CHarom), 6.37 (bs, 1H, OH), 4.29–4.08 (m, 2H, CH2CH3), 1.15 (t, J = 7.1 Hz, 3H, CH2CH3). 13C-NMR (CDCl3, 75 MHz, 25 °C): δ (ppm) 173.2, 168.7, 161.1, 155.9, 126.0, 118.0, 112.5, 99.5, 77.2, 63.0, 14.0. HRMS: exact mass calculated for (C11H10NaO6) requires m/z 261.0370, found m/z 261.0372.
Ethyl 3,6-Dihydroxy-7-Methyl-2-oxo-2,3-Dihydrobenzofuran-3-Carboxylate, 11. Following the general procedure, the single product 11 was obtained as a white solid in 81% yield after purification by flash chromatography on silica gel (nHexane/EtOAc = 4/6). m.p. 126–128 °C. IR (CHCl3): = 3460–3420, 3262, 3005, 2970, 2919, 1801, 1730, 1629 cm−1. 1H-NMR (CDCl3, 300 MHz, 25 °C): δ (ppm) = 8.32 (bs, 1H, OHphen), 7.02 (d, J = 7,9 Hz, 1H, CHarom), 6.61 (m, 2H, CHarom), 5.98 (bs, 1H, OH), 4.41–3.99 (m, 2H, CH2CH3), 2.15 (s, 3H, CCH3), 1.15 (t, J = 7.1 Hz, 3H, CH2CH3). 13C-NMR (CDCl3, 75 MHz, 25 °C): δ (ppm) 174.3, 169.7, 159.6, 154.9, 122.7, 117.8, 111.6, 109, 78.3, 63.5, 14.1, 9.6. HRMS: exact mass calculated for (C12H12NaO6) requires m/z 275.0526, found m/z 275.052.
3.3. General Procedure for the Domino Friedel-Crafts/Lactonization Reaction Performed in AcOH
The alkylating agent (5.5 mmol) was added in one portion to a stirred solution of the appropriate phenol (5.0 mmol) in acetic acid (3 mL). The system was kept under an argon atmosphere. The clear reddish solution was stirred at reflux temperature until the substrate had been completely consumed (TLC and HPLC monitoring). Afterwards, the reaction mixture was concentrated under vacuum and the residue was purified by flash chromatography on silica gel to give the products as described below.
3.4. Characterization Data for Benzofuran-2(3H)-One 15–20
3,5-Dihydroxy-3-(trifluoromethyl)benzofuran-2(3H)-One, 15. Following the general procedure, the single product 15 was obtained as a white solid in 35% yield after purification by flash chromatography on silica gel (nHexane/Acetone = 8/2). m.p. 136–138 °C. IR (CHCl3): = 3460–3190, 3005, 2970, 2919, 1801, 1730, 1629, 1498 cm−1. 1H-NMR (CDCl3, 300 MHz, 25 °C): δ (ppm) = 8.72 (bs, 1H, OHphen), 7.24–6.95 (m, 4H, CHarom + OH). 13C-NMR (CDCl3, 75 MHz, 25 °C): δ (ppm) 168.7, 153.8, 145.7, 121.8, 121.6 (q, 1JCF = 283.3 Hz), 118.1, 111.4, 111.0, 74.0 (q, 2JCF = 32.5 Hz). HRMS: exact mass calculated for (C9H5NaF3O4) requires m/z 257.0032, found m/z 257.0033.
3,6-Dihydroxy-3-(trifluoromethyl)benzofuran-2(3H)-One, 16. Following the general procedure, the single product 16 was obtained as a white solid in 28% yield after purification by flash chromatography on silica gel (nHexane/Et2O = 1/1). m.p. 138–140 °C. IR (CHCl3): = 3490–3230, 3010, 2970, 2923, 1806, 1735, 1637 cm−1. 1H-NMR (CDCl3, 300 MHz, 25 °C): δ (ppm) = 9.40 (bs, 1H, OHphen), 7.45 (d, J = 9 Hz, 2H, CHarom), 7.02 (bs, 1H, OH), 6.80 (d, J = 9 Hz, 1H, CHarom), 6.74 (s, 1H, CHarom). 13C-NMR (CDCl3, 75 MHz, 25 °C): δ (ppm) 170.5, 162.1, 155.9, 127.7, 123.6 (q, 1JCF = 283.9 Hz), 113.2, 113.0, 99.5, 75.2 (q, 2JCF = 32.8 Hz). HRMS: exact mass calculated for (C9H5NaF3O4) requires m/z 257.0032, found m/z 257.0031.
3,6-Dihydroxy-7-methyl-3-(trifluoromethyl)benzofuran-2(3H)-One, 17. Following the general procedure, the single product 17 was obtained as a white solid in 61% yield after purification by flash chromatography on silica gel (nHexane/Et2O=1/1). m.p. 129–131 °C. IR (CHCl3): = 3470–3220, 3000, 2975, 2913, 1826, 1724, 1630 cm−1. 1H-NMR (CDCl3, 300 MHz, 25 °C): δ (ppm) = 7.32 (bs, 1H, OHphen), 7.08 (d, J = 8.4 Hz, 1H, CHarom), 6.58 (d, J = 8.3 Hz, 1H, CHarom), 4.78 (bs, 1H, OH), 2.22 (s, 3H, CCH3). 13C-NMR (CDCl3, 75 MHz, 25 °C): δ (ppm) 170.5, 159.3, 153.4, 123.2, 122.8 (q, 1JCF = 283.8 Hz), 112.1, 111.6, 108.6, 75.2 (q, 2JCF = 32.8 Hz), 8.12. HRMS: exact mass calculated for (C10H7NaF3O4) requires m/z 271.0189, found m/z 271.0190.
3,7-Dihydroxy-3-(trifluoromethyl)benzofuran-2(3H)-One, 18. Following the general procedure, the single product 18 was obtained as a white solid in 78% yield after purification by flash chromatography on silica gel (nHexane/Et2O = 1/1). m.p. 142–144 °C. IR (CHCl3): = 3498–3256, 3012, 2988, 2909, 1831, 1744, 1629 cm−1. 1H-NMR (CDCl3, 300 MHz, 25 °C): δ (ppm) = 9.20 (bs, 1H, OHfen), 7.23–7.07 (m, 4H, CHarom + OH).13C-NMR (CDCl3, 75 MHz, 25 °C): δ (ppm) 169.2, 141.6, 125.9, 123.6, 122.8 (q, 1JCF = 284.1 Hz), 120.3, 119.8, 116.4, 75.0 (q, 2JCF = 32.6 Hz). HRMS: exact mass calculated for (C9H5NaF3O4) requires m/z 257.0032, found m/z 257.0032.
3,4,6-Trihydroxy-3-(trifluoromethyl)benzofuran-2(3H)-One, 19. Following the general procedure, the single product 19 was obtained as a white solid in 66% yield after purification by flash chromatography on silica gel (nHexane/Acetone=7/3). m.p. 174–176 °C. IR (CHCl3): = 3500–3280, 3023, 3000, 2959, 1821, 1754, 1607 cm−1. 1H-NMR (CDCl3, 300 MHz, 25 °C): δ (ppm) = 9.30 (bs, 1H, OHfen), 9.22 (bs, 1H, OHphen), 6.71 (bs, 1H, OHfen), 6.28 (s, 1H, CHarom), 6.23 (s, 1H, CHarom).13C-NMR (CDCl3, 75 MHz, 25 °C): δ (ppm) 167.3, 162.0, 159.4, 158.0, 123.2 (q, 1JCF = 285.7 Hz), 122.7, 98.4, 95.6, 75.4 (q, 2JCF = 33.6 Hz). HRMS: exact mass calculated for (C9H5NaF3O5) requires m/z 272.9981, found m/z 272.9983.
3,6,7-Trihydroxy-3-(trifluoromethyl)benzofuran-2(3H)-One, 20. Following the general procedure, the single product 20 was obtained as a white solid in 76% yield after purification by flash chromatography on silica gel (nHexane/Et2O = 3/7). m.p. 148–150 °C. IR (CHCl3): = 3517–3239, 3045, 3018, 2960, 1819, 1734, 1615 cm−1. 1H-NMR (CDCl3, 300 MHz, 25 °C): δ (ppm) = 9.03 (bs, 1H, OHphen), 8.55 (bs, 1H, OHphen), 6.95 (d, J = 8.4 Hz, 1H, CHarom), 6.81 (d, J = 8.4 Hz, 1H, CHarom), 6.65 (bs, 1H, OH).13C-NMR (CDCl3, 75 MHz, 25 °C): δ (ppm) 169.7, 149.6, 142.0, 130.0, 122.9 (q, 1JCF = 284.0 Hz), 116.6, 113.8, 111.9, 75.0 (q, 2JCF = 35.1 Hz). HRMS: exact mass calculated for (C9H5NaF3O5) requires m/z 272.9981, found m/z 272.9980.
4. Conclusions
In summary, we have synthesized a series of 3,3-disubstituted-3H-benzofuran-2-one derivatives (9–11 and 15–20) by improving our previous findings concerning the domino Friedel–Crafts/lactonization reaction. With respect the chosen electrophile as well as the various polyphenols (1–6) used as nucleophilic counterpart, two different protocols were followed: (i) the use of TiCl4 as catalyst in CHCl3 at 60 °C; and (ii) the employment of AcOH as solvent at 120 °C without the addition of any further catalyst. Afterwards, the antioxidant capacity of the synthesized compounds (9–11 and 15–20) was evaluated using DPPH assay and Cyclic Voltammetry, performing the experiments in both MeOH and acetonitrile. Specifically, the benzofuran-2-ones 9, 15, 18, and 20 presented the best values of rIC50. Among them, compound 20 exhibited a remarkable rIC50 of 0.18 (MeOH) and 0.17 (ACN), and therefore possessed the greatest antioxidant activity by reducing three molecules of DPPH• in both solvents, a result that is validated despite the possible interference of the solvent which was evaluated and excluded almost completely by measuring the rate constants for the reaction with DPPH•. Additionally, the electrochemical measurements (CV), recorded in in aqueous medium as well as in acetonitrile, established compounds 18 and 20 as the best reducing agents among the tested molecules. The observed results undeniably suggest that the 3-hydroxy-benzofuran-2-one scaffold should be involved in the antioxidant mechanism: the presence of a hydroxyl group at C-7 position as well as a strong electron withdrawing group at C-3 position are the essential structural features. Detailed additional in vitro tests to further endorse the notable and promising antioxidant activity of the studied compounds are ongoing in our laboratories and will be reported in due course.
Acknowledgments
The authors gratefully acknowledge Dipartimento di Scienze and Sezione di Nanoscienze e Nanotecnologie (Università di Roma Tre, Roma, Italy) for the financial support.
Supplementary Materials
The following are available online at http://www.mdpi.com/1420-3049/23/4/710/s1. Experimental procedures, characterization data of synthesized compounds, explanation of the reaction mechanism, and the complete set of antioxidant assay are reported in the Supplementary Material.
Author Contributions
Tecla Gasperi and Martina Miceli conceived and designed the experiments; Martina Miceli and Elia Roma performed the experiments; Martina Miceli, Paolo Rosa, M. Antonietta Loreto, Daniela Tofani, and Marta Feroci analyzed the data; Tecla Gasperi and Marta Feroci contributed reagents/materials/analysis tools; Tecla Gasperi, Martina Miceli, Marta Feroci, and Paolo Rosa wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 9–11 and 15–20 are available from the authors.
References
- 1.Halliwell B. Antioxidants in human health and disease. Ann. Rev. Nutr. 1996;16:33–50. doi: 10.1146/annurev.nu.16.070196.000341. [DOI] [PubMed] [Google Scholar]
- 2.Lobo V., Patil A., Phatak A., Chandra N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010;4:118–126. doi: 10.4103/0973-7847.70902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lushchak V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014;224:164–175. doi: 10.1016/j.cbi.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Valko M., Rhodes C.J., Moncol J., Izakovic M., Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Valko M., Leibfritz D., Moncol J., Cronin M.T.D., Mazur M., Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Rahman K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging. 2007;2:219–236. [PMC free article] [PubMed] [Google Scholar]
- 7.Guerra-Araiza C., Alvarez-Mejia A.L., Sanchez-Torres S., Farfan-Garcia E., Mondragon-Lozano R., Pinto-Almazan R., Salgado-Ceballos H. Effect of natural exogenous antioxidants on aging and on neurodegenerative diseases. Free Radic. Res. 2013;47:451–462. doi: 10.3109/10715762.2013.795649. [DOI] [PubMed] [Google Scholar]
- 8.Halliwell B., Gutteridge J.M.C. Free Radicals in Biology and Medicine. 2nd ed. Clarendon Press; Oxford, UK: 1989. [Google Scholar]
- 9.Reuter S., Gupta S.C., Chaturvedi M.M., Aggarwal B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phaniendra A., Jestadi D.B., Periyasamy L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015;30:11–26. doi: 10.1007/s12291-014-0446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta R.K., Patel A.K., Shah N., Choudhary A.K., Jha U.K., Yadav U.C., Gupta P.K., Pakuwal U. Oxidative stress and antioxidants in disease and cancer: A review. Asian Pac. J. Cancer Prev. 2014;15:4405–4409. doi: 10.7314/APJCP.2014.15.11.4405. [DOI] [PubMed] [Google Scholar]
- 12.Xu D.P., Li Y., Meng X., Zhou T., Zhou Y., Zheng J., Zhang J.J., Li H.B. Natural antioxidants in foods and medicinal plants: Extraction, assessment and resources. Int. J. Mol. Sci. 2017;18:96. doi: 10.3390/ijms18010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sen C.K., Khanna S., Roy S. Tocotrienols: Vitamin e beyond tocopherols. Life Sci. 2006;78:2088–2098. doi: 10.1016/j.lfs.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aggarwal B., Nesaretnam K. Vitamin E tocotrienols: Life beyond tocopherols. Genes Nutr. 2012;7:1. doi: 10.1007/s12263-011-0234-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dutta N.K., Mazumdar K., Mishra B.K., Dastidar S.G., Park J.-H. Isolation and identification of a flavone (quercetin) from Butea frondosa bark. Pharm. Chem. J. 2007;41:37–39. doi: 10.1007/s11094-007-0059-5. [DOI] [Google Scholar]
- 16.Valentova K., Kanova K., Di Meo F., Pelantova H., Chambers C.S., Rydlova L., Petraskova L., Krenkova A., Cvacka J., Trouillas P., et al. Chemoenzymatic preparation and biophysical properties of sulfated quercetin metabolites. Int. J. Mol. Sci. 2017;18:2231. doi: 10.3390/ijms18112231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trujillo M., Mateos R., Collantes de Teran L., Espartero J.L., Cert R., Jover M., Alcudia F., Bautista J., Cert A., Parrado J. Lipophilic hydroxytyrosyl esters. Antioxidant activity in lipid matrices and biological systems. J. Agric. Food Chem. 2006;54:3779–3785. doi: 10.1021/jf060520z. [DOI] [PubMed] [Google Scholar]
- 18.Kubo I., Xiao P., Fujita K. Antifungal activity of octyl gallate: Structural criteria and mode of action. Bioorg. Med. Chem. Lett. 2001;11:347–350. doi: 10.1016/S0960-894X(00)00656-9. [DOI] [PubMed] [Google Scholar]
- 19.Kubo I., Masuoka N., Xiao P., Haraguchi H. Antioxidant activity of dodecyl gallate. J. Agric. Food Chem. 2002;50:3533–3539. doi: 10.1021/jf011250h. [DOI] [PubMed] [Google Scholar]
- 20.Miranda A.R., Albrecht C., Cortez M.V., Soria E.A. Pharmacology and toxicology of polyphenols with potential as neurotropic agents in non-communicable diseases. Curr. Drug Targets. 2018;19:97–110. doi: 10.2174/1389450117666161220152336. [DOI] [PubMed] [Google Scholar]
- 21.Justino G.C., Correia C.F., Mira L., Dos Santos R.M.B., Simoes J.A.M., Silva A.M., Santos C., Gigante B. Antioxidant activity of a catechol derived from abietic acid. J. Agric. Food Chem. 2006;54:342–348. doi: 10.1021/jf052062k. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Z.W., Li W.C., Hu Y., Wang B., Ren G., Feng L.H. Synthesis of the intermediate for fumimycin: A natural peptide deformylase inhibitor. Res. Chem. Int. 2013;39:3049–3054. doi: 10.1007/s11164-012-0817-1. [DOI] [Google Scholar]
- 23.Gross P.J., Furche F., Nieger M., Braese S. Asymmetric total synthesis of (+)-fumimycin via 1,2-addition to ketimines. Chem. Commun. 2010;46:9215–9217. doi: 10.1039/c0cc03399e. [DOI] [PubMed] [Google Scholar]
- 24.Nicolaou K.C., Snyder S.A., Huang X.H., Simonsen K.B., Koumbis A.E., Bigot A. Studies toward diazonamide A: Initial synthetic forays directed toward the originally proposed structure. J. Am. Chem. Soc. 2004;126:10162–10173. doi: 10.1021/ja040090y. [DOI] [PubMed] [Google Scholar]
- 25.Bassarello C., Bifulco G., Montoro P., Skhirtladze A., Kemertelidze E., Pizza C., Piacente S. Gloriosaols A and B, two novel phenolics from Yucca gloriosa: Structural characterization and configurational assignment by a combined NMR-quantum mechanical strategy. Tetrahedron. 2007;63:148–154. doi: 10.1016/j.tet.2006.10.034. [DOI] [Google Scholar]
- 26.Fedorova T.E., Ivanova S.Z., Fedorov S.V., Babkin V.A. Larisinol, a new spirobiflavonoid from Larix gmelinii bark. Chem. Nat. Compd. 2007;43:208–209. doi: 10.1007/s10600-007-0080-7. [DOI] [Google Scholar]
- 27.Kontogianni V.G., Tomic G., Nikolic I., Nerantzaki A.A., Sayyad N., Stosic-Grujicic S., Stojanovic I., Gerothanassis I.P., Tzakos A.G. Phytochemical profile of Rosmarinus officinalis and Salvia officinalis extracts and correlation to their antioxidant and anti-proliferative activity. Food Chem. 2013;136:120–129. doi: 10.1016/j.foodchem.2012.07.091. [DOI] [PubMed] [Google Scholar]
- 28.Andolfi A., Cimmino A., Vurro M., Berestetskiy A., Troise C., Zonno M.C., Motta A., Evidente A. Agropyrenol and agropyrenal, phytotoxins from Ascochyta agropyrina var. nana, a fungal pathogen of Elitrigia repens. Phytochemistry. 2012;79:102–108. doi: 10.1016/j.phytochem.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 29.Chien Y.-C., Lin C.-H., Chiang M.Y., Chang H.-S., Liao C.-H., Chen I.-S., Peng C.-F., Tsai I.-L. Secondary metabolites from the root of Ehretia longiflora and their biological activities. Phytochemistry. 2012;80:50–57. doi: 10.1016/j.phytochem.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Gasperi T., Punzi P., Migliorini A., Tofani D. An organocatalytic approach to the synthesis of six-membered heterocycles. Curr. Org. Chem. 2011;15:2098–2146. doi: 10.2174/138527211796150714. [DOI] [Google Scholar]
- 31.Piacente S., Montoro P., Oleszek W., Pizza C. Yucca schidigera bark: Phenolic constituents and antioxidant activity. J. Nat. Prod. 2004;67:882–885. doi: 10.1021/np030369c. [DOI] [PubMed] [Google Scholar]
- 32.Marzocco S., Piacente S., Pizza C., Oleszek W., Stochmal A., Pinto A., Sorrentino R., Autore G. Inhibition of inducible nitric oxide synthase expression by yuccaol C from Yucca schidigera roezl. Life Sci. 2004;75:1491–1501. doi: 10.1016/j.lfs.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 33.Olas B., Wachowicz B., Stochmal A., Oleszek W. Inhibition of blood platelet adhesion and secretion by different phenolics from Yucca schidigera roezl. Bark. Nutrition. 2005;21:199–206. doi: 10.1016/j.nut.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 34.Balestrieri C., Felice F., Piacente S., Pizza C., Montoro P., Oleszek W., Visciano V., Balestrieri M.L. Relative effects of phenolic constituents from Yucca schidigera roezl. Bark on kaposi’s sarcoma cell proliferation, migration, and PAF synthesis. Biochem. Pharmacol. 2006;71:1479–1487. doi: 10.1016/j.bcp.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 35.Wenzig E.M., Oleszek W., Stochmal A., Kunert O., Bauer R. Influence of phenolic constituents from Yucca schidigera bark on arachidonate metabolism in vitro. J. Agric. Food Chem. 2008;56:8885–8890. doi: 10.1021/jf801289m. [DOI] [PubMed] [Google Scholar]
- 36.De Figueiredo R.M., Mazziotta A., de Sant’Ana D.P., Palumbo C., Gasperi T. Active methylene compounds in asymmetric organocatalytic synthesis of natural products and pharmaceutical scaffolds. Curr. Org. Chem. 2012;16:2231–2289. doi: 10.2174/138527212803520290. [DOI] [Google Scholar]
- 37.Gross P.J., Braese S. The total synthesis of (±)-fumimycin. Chem. Eur. J. 2010;16:12660–12667. doi: 10.1002/chem.201001036. [DOI] [PubMed] [Google Scholar]
- 38.Kwon Y.-J., Sohn M.-J., Zheng C.-J., Kim W.-G. Fumimycin: A peptide deformylase inhibitor with an unusual skeleton produced by Aspergillus fumisynnematus. Org. Lett. 2007;9:2449–2451. doi: 10.1021/ol0703231. [DOI] [PubMed] [Google Scholar]
- 39.Vetica F., Pelosi A., Gambacorta A., Loreto M.A., Miceli M., Gasperi T. Catalytic Friedel-Crafts/lactonization domino reaction: Facile access to 3-hydroxybenzofuran-2-one scaffold. Eur. J. Org. Chem. 2014;2014:1899–1906. doi: 10.1002/ejoc.201301686. [DOI] [Google Scholar]
- 40.Vetica F., de Figueiredo R.M., Cupioli E., Gambacorta A., Loreto M.A., Miceli M., Gasperi T. First asymmetric organocatalyzed domino Friedel-Crafts/lactonization reaction in the enantioselective synthesis of the GABAB receptor modulator (S)-BHFF. Tetrahedron Lett. 2016;57:750–753. doi: 10.1016/j.tetlet.2016.01.015. [DOI] [Google Scholar]
- 41.Dyachenko V.I., Kolomiets A.F., Fokin A.V. Preparation of substituted 3-hydroxy-3-trifluoromethyl-2(3H)benzofuranones. Bull. Acad. Sci. USSR Div. Chem. Sci. 1987;36:2332–2337. doi: 10.1007/BF00957310. [DOI] [Google Scholar]
- 42.Dyachenko V.I., Galakhov M.V., Kolomiets A.F., Fokin A.V. 2-hydroxy-2-trifluoromethyl-2,3H-1,4-benzoxazinone-3. Bull. Acad. Sci. USSR Div. Chem. Sci. 1988;37:1056. doi: 10.1007/BF00957102. [DOI] [Google Scholar]
- 43.Prior R.L., Cao G.H. In vivo total antioxidant capacity: Comparison of different analytical methods. Free Radic. Biol. Med. 1999;27:1173–1181. doi: 10.1016/S0891-5849(99)00203-8. [DOI] [PubMed] [Google Scholar]
- 44.Sanchez-Moreno C. Review: Methods used to evaluate the free radical scavenging activity in foods and biological systems. Food Sci. Technol. Int. 2002;8:121–137. doi: 10.1177/1082013202008003770. [DOI] [Google Scholar]
- 45.Aruoma O.I. Methodological considerations for characterizing potential antioxidant actions of bioactive components in plant foods. Mutation Res.-Fundam. Mol. Mech. Mutagen. 2003;523:9–20. doi: 10.1016/S0027-5107(02)00317-2. [DOI] [PubMed] [Google Scholar]
- 46.Huang D.J., Ou B.X., Prior R.L. The chemistry behind antioxidant capacity assays. J. Agric. Food Chem. 2005;53:1841–1856. doi: 10.1021/jf030723c. [DOI] [PubMed] [Google Scholar]
- 47.Brand-Williams W., Cuvelier M.E., Berset C. Use of a free-radical method to evaluate antioxidant activity. Food Sci. Technol.-Lebensm.-Wiss. Technol. 1995;28:25–30. doi: 10.1016/S0023-6438(95)80008-5. [DOI] [Google Scholar]
- 48.Foti M.C. Antioxidant properties of phenols. J. Pharm. Pharmacol. 2007;59:1673–1685. doi: 10.1211/jpp.59.12.0010. [DOI] [PubMed] [Google Scholar]
- 49.Foti M.C., Daquino C., Geraci C. Electron-transfer reaction of cinnamic acids and their methyl esters with the DPPH center dot radical in alcoholic solutions. J. Org. Chem. 2004;69:2309–2314. doi: 10.1021/jo035758q. [DOI] [PubMed] [Google Scholar]
- 50.Foti M.C., Daquino C., Mackie I.D., DiLabio G.A., Ingold K.U. Reaction of phenols with the 2,2-diphenyl-1-picrylhydrazyl radical. Kinetics and DFT calculations applied to determine Aro-H bond dissociation enthalpies and reaction mechanism. J. Org. Chem. 2008;73:9270–9282. doi: 10.1021/jo8016555. [DOI] [PubMed] [Google Scholar]
- 51.Valgimigli L., Banks J.T., Ingold K.U., Lusztyk J. Kinetic solvent effects on hydroxylic hydrogen-atom abstractions are independent of the nature of the abstracting radical—2 extreme tests using vitamin-E and phenol. J. Am. Chem. Soc. 1995;117:9966–9971. doi: 10.1021/ja00145a005. [DOI] [Google Scholar]
- 52.Litwinienko G., Ingold K.U. Abnormal solvent effects on hydrogen atom abstractions. 1. The reactions of phenols with 2,2-diphenyl-1-picrylhydrazyl (DPPH•) in alcohols. J. Org. Chem. 2003;68:3433–3438. doi: 10.1021/jo026917t. [DOI] [PubMed] [Google Scholar]
- 53.Litwinienko G., Ingold K.U. Solvent effects on the rates and mechanisms of reaction of phenols with free radicals. Acc. Chem. Res. 2007;40:222–230. doi: 10.1021/ar0682029. [DOI] [PubMed] [Google Scholar]
- 54.Chevion S., Roberts M.A., Chevion M. The use of cyclic voltammetry for the evaluation of antioxidant capacity. Free Radic. Biol. Med. 2000;28:860–870. doi: 10.1016/S0891-5849(00)00178-7. [DOI] [PubMed] [Google Scholar]
- 55.Chevion S., Chevion M. Antioxidant status and human health—Use of cyclic voltammetry for the evaluation of the antioxidant capacity of plasma and of edible plants. In: Chiueh C.C., editor. Reactive Oxygen Species: From Radiation to Molecular Biology: A Festschrift in Honor of Daniel L. Gilbert. Volume 899. New York Academy of Sciences; New York, NY, USA: 2000. pp. 308–325. [DOI] [PubMed] [Google Scholar]
- 56.Arteaga J.F., Ruiz-Montoya M., Palma A., Alonso-Garrido G., Pintado S., Rodriguez-Mellado J.M. Comparison of the simple cyclic voltammetry (CV) and DDPH assays for the determination of antioxidant capacity of active principles. Molecules. 2012;17:5126–5138. doi: 10.3390/molecules17055126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Papanikos A., Eklund J., Jackson W.R., Kenche V.B., Campi E.M., Robertson A.D., Jarrott B., Beart P.M., Munro F.E., Callaway J.K. Cyclic voltammetry as an indicator of antioxidant activity. Aust. J. Chem. 2002;55:205–212. doi: 10.1071/CH01193. [DOI] [Google Scholar]
- 58.Chevion S., Berry E.M., Kitrossky N., Kohen R. Evaluation of plasma low molecular weight antioxidant capacity by cyclic voltammetry. Free Radic. Biol. Med. 1997;22:411–421. doi: 10.1016/S0891-5849(96)00337-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.