

Abstract
The tautomerism of 1-phenyl-1,2-dihydro-3H-pyrazol-3-one was investigated. An X-ray crystal structure analysis exhibits dimers of 1-phenyl-1H-pyrazol-3-ol units. Comparison of NMR (nuclear magnetic resonance) spectra in liquid state (1H, 13C, 15N) with those of “fixed” derivatives, as well as with the corresponding solid state NMR spectra reveal this compound to exist predominantly as 1H-pyrazol-3-ol molecule pairs in nonpolar solvents like CDCl3 or C6D6, whereas in DMSO-d6 the corresponding monomers are at hand. Moreover, the NMR data of different related 1H-pyrazol-3-ol derivatives are presented.
Keywords: prototropic tautomerism; 1H-pyrazol-3-ol; 1,2-dihydro-3H-pyrazol-3-ones; NMR (1H; 13C; 15N); solid state NMR; X-ray structure analysis
1. Introduction
Pyrazolones are interesting chemical entities not only due to their importance as building blocks for the synthesis of various bio-active compounds [1,2,3,4,5,6], but also in respect to their capability to prototropic tautomerism and to the—more uncommon—phenomenon of desmotropy [7]. Whereas for 1-substituted 1H-pyrazol-5-ols and their tautomers (2-substituted 2,4-dihydro-3H-pyrazol-3-ones and 2-substituted 1,2-dihydro-3H-pyrazol-3-ones), a considerably large number of experimental and theoretical studies concerning their tautomerism has been published [8,9,10,11,12], there is much less known about their structural isomers with a 1-substituted 1H-pyrazol-3-ol motif. The latter compounds are in as much of interest as they can serve as starting materials for further functionalization [13], the construction of anellated systems [14], as well as for the synthesis of biologically active compounds [15,16,17]. Hence, this study is devoted to investigations with 1-substituted 1H-pyrazol-3-ols (tautomers to the corresponding 1,2-dihydro-3H-pyrazol-3-ones) and some derivatives carrying different substituents at pyrazole C-4.
2. Results and Discussion
In principle, for the title compounds, two tautomeric forms are possible, namely the OH-form and the NH-form (Figure 1). In Chemical Abstracts such compounds carrying an alkyl or a (hetero)aryl substituent at the pyrazole nitrogen atom are always listed as 3H-pyrazol-3-ones. Hence, as many authors prefer the latter denomination, in the course of this study we investigated which tautomer is really relevant in a solid state and, especially, in solution.
Figure 1.
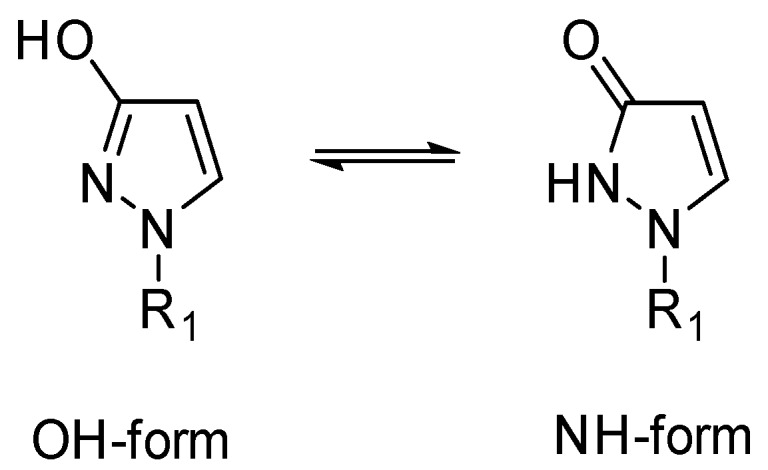
Possible tautomeric forms of 1-substituted 3-hydroxy-1H-pyrazoles (1-substituted 1,2-dihydro-3H-pyrazol-3-ones).
2.1. X-ray Analysis of 1-Phenyl-1,2-dihydro-3H-pyrazol-3-one (1-Phenyl-1H-pyrazol-3-ol) (1)
In the solid state an unambiguous determination of structure and, thus, a safe discrimination between OH and NH-form is possible. In view of this fact crystals of 1-phenyl-1,2-dihydro-3H-pyrazol-3-one (1-phenyl-1H-pyrazol-3-ol) (1)—obtained by crystallization from ethanol/water—were subjected to X-ray structure analysis. It turned out that this compound is present in the 1H-pyrazol-3-ol form constituting dimeric units connected by two identical intermolecular hydrogen bonds (Figure 2). In principle, the formation of similar dimeric structures would be also possible by combination of two identical NH-isomers establishing two intermolecular hydrogen bonds between C=O and the NH of the second molecule. However, the electron density map clearly shows the position of the hydrogen atom at the oxygen and thus excludes the latter alternative (Figure 2, further details can be found in the Experimental Section).
Figure 2.

Difference electron density map of 1. Determination of the H position at O1. The distance of O1 to the electron density position (green shaded) in the direct donor acceptor line is 0.8927 (10) Å. The according distance to N2′ is 1.8339 (10) Å. For further details please follow the CCDC Code.
2.2. Solid State NMR (SSNMR) of 1
The same material as used for the X-ray analysis was subjected to solid state NMR (CP/MAS). As the X-ray analysis revealed the presence of the 1H-pyrazol-3-ol form, the solid state NMR spectra also should exclusively origin from this species. Here, due to its simplicity the 15N-NMR spectrum is particularly valuable, showing the “pyridine-like” pyrazole N-atom (N-2) at 243.1 ppm, whereas the “pyrrole-like” N-atom (N-1) is resonating at 192.6 ppm referenced against external 15NH4Cl (39.3 ppm with respect to liquid NH3) (Figure 3). The distinct chemical shift difference of N-1 compared to N-2 (Δδ = 50.5 ppm) clearly reflects the fact that the two nitrogen atoms are of different types.
Figure 3.

15N CP/MAS NMR spectrum of 1.
In addition, in 15N-NQS (non-quaternary suppression) experiments with different pre-scan dephasing delays (20 µs, 100 µs, 200 µs) the intensities of both 15N-NMR resonances remained constant. This behaviour suggests that the two nitrogen atoms are of the same type regarding their protonation status what is only the case for the OH isomer.
2.3. NMR Spectra in Solution
Whereas in the solid state, an unambiguous determination of individual tautomeric forms is smoothly possible by X-ray structure analysis, the situation in solution is much more complex. Here, depending on a plethora of different influencing variables, tautomeric equilibria with the simultaneous presence of several tautomeric forms are possible, whereas time-averaged signals are obtained in case of fast exchange. Amongst the appropriate methods for investigating such tautomeric equilibria in solution NMR spectroscopic methods play a prominent role [8,9,18,19,20]. A frequently used concept is the comparison of the data obtained in solution with those of “fixed” derivatives (representing the individual “frozen” tautomeric forms) or with the data of the individual tautomeric forms obtained from solid state NMR experiments. Although this approach comes along with some difficulties (i.e. estimating the difference between the tautomer and the model compound) in many cases fairly good results can be obtained, particularly when one tautomeric form is strongly dominating. However, in cases when several forms are present to a significant extent the precise determination of the percentage composition by interpolation is difficult.
As compound 1 is present as 1H-pyrazol-3-ol in the solid state, a comparison of the crucial SSNMR chemical shifts with those in solution should provide valuable information. As outlined in Figure 4, the 13C and the 15N chemical shifts at the pyrazole nucleus show a high degree of accordance between the solid state and those in CDCl3 or in C6D6 solution which leads to the conclusion that the 3-hydroxy form is far dominating in these solvents.
Figure 4.

Crucial chemical shifts of 1 in solid state and in different solvents. 1H-NMR chemical shifts are represented in italics, 13C-NMR chemical shifts in plain text, 15N-NMR chemical shifts in bold.
In DMSO-d6 solution it is noticeable that the signal of pyrazole N-2 is clearly shifted downfield compared to the recordings in CDCl3 or C6D6. A possible explanation for this phenomenon is the fact that in the latter nonpolar solvents 1 is obviously present as a dimer of 1H-pyrazol-3-ols (like in the solid state) and, thus, the pyrazole N-2 atom is involved in an intramolecular hydrogen bond, whereas—in contrast—in the strong acceptor solvent DMSO-d6 these intermolecular hydrogen bonds are broken and now monomers are dominating. It is well-known that involvement of a nitrogen’s lone-pair in hydrogen bonds (or—to a larger extent—oxidation, alkylation, or complexation) leads to a marked upfield shift of the corresponding 15N resonance [21,22,23]. In 3-methoxy-1-phenyl-1H-pyrazole (2) (Figure 5) the above mentioned dimerization and, thus, participation of pyrazole N-2 into intermolecular hydrogen bonding is not possible, what is reflected by a larger chemical shift of the latter. Hence, δ(N-2) (261.7 ppm) is now comparable to the value in DMSO-d6 (262.1 ppm), whereas δ(N-1) in compound 2 (195.6 ppm) and in 1 in different solvents (191.7–194.5 ppm) is very similar. Hence, it can be concluded that 1 is also present as 1H-pyrazol-3-ol in DMSO-d6 solution, however, not in the dimeric form stabilized by intermolecular hydrogen bonds.
Figure 5.

1H-NMR (in italics), 13C-NMR, and 15N-NMR (in bold) chemical shifts of 1 and its “fixed” derivatives 2 and 3 (in CDCl3).
In addition, employing the concept of the fixed derivatives we compared the 1H-, 13C- and 15N-NMR chemical shifts, as well as characteristic spin coupling constants of 1 with its O-methyl (2) and N-methyl derivative (3), the “fixed” OH- and NH-forms, respectively.
From the data given in Figure 5 the above conclusions are confirmed, namely that compound 1, which in principle is capable of prototropic tautomerism, is predominantly existing as OH-isomer in CDCl3 solution. This is supported by the fact that the 1H-, 13C-, and 15N-NMR chemical shifts of 1 resemble closely to those of the fixed O-methyl congener 2. Especially the 15N-chemical shifts are valuable indicators, as the N-methyl derivative 3 exhibits two sp3-type nitrogen atoms with similar 15N-NMR chemical shifts, whereas in 1 and 2 the large chemical shift differences between the two nitrogen atoms in the corresponding molecule hint to different types of N-atoms (sp3 and sp2). For comparison only, with 1-phenylpyrazolidin-3-one, which formally is the dihydro derivative of the NH form of 1, we found 105.1 ppm for N-1 and 151.8 ppm for N-2 in DMSO-d6 solution. Moreover, 1 and 2 show equal sizes of the vicinal 3J(H4,H5) coupling constant at the pyrazole nucleus (2.6 Hz), whereas in 3 this coupling is considerably larger (3.6 Hz) (Figure 5). An additional difference consists in the 13C-NMR chemical shift of Ph C-2/6 which is akin in 1 (118.6 ppm) and 2 (117.8 ppm). In contrast, 3 shows a markedly larger chemical shift for these carbon atoms (123.0 ppm) which can be attributed to some distorsion of phenyl and pyrazole ring obviously induced by the sterical hindrance of the N-methyl group [24,25]. Additionally, the large differences in 13C-NMR chemical shifts of pyrazole C-5 (1: 129.1 ppm, 2: 127.7 ppm) in comparison to that of 3 (142.3 ppm) provide an extra confirmation.
Moreover, 1H-, 13C-, and 15N-NMR spectra of compound 1 were additionally taken from C6D6, DMSO-d6 and CD3OD solutions. As all the significant criteria discussed above were almost similar to those in CDCl3 solution, it is reasoned that, also in these solvents, the hydroxy form is far more predominant. The regarding data are presented in the Experimental Section.
In the following, congeners of 1 carrying a halogen atom or an acyl moiety at pyrazole C-4 were investigated (compounds 4–8). Again, all these species clearly exist as pyrazol-3-ols in CDCl3, as well as in DMSO-d6 solution based on the 13C- and 15N-NMR chemical shift considerations outlined above. Regarding the 4-bromo derivative 5 a “fixed” 3-methoxy derivative 9 has been described already by us, whose data resemble the “free” 1H-pyrazol-3-ol 5 [26]. The same is the case for the pair 7 and 10 (Figure 6). When switching from CDCl3 to DMSO-d6 solution, the 13C chemical shift of the carbonyl C-atom in 7 receives an upfield shift of 4.0 ppm (195.7 ppm → 191.7 ppm) which hints to the existence of an intramolecular hydrogen bond—but now—between carbonyl O-atom and OH proton in CDCl3 solution, which is broken in the strong acceptor solvent DMSO-d6 [27]. In principle, also 3-O-acyl derivatives of 1 (compounds 11–13) can be regarded as fixed 3-OH derivatives, although the 3-O-acyl rest seems to be less comparable to OH than an OCH3 group. However, despite larger differences regarding the 13C chemical shifts of the pyrazole C-atoms between 1 and 11–13 appear, the data of the phenyl ring closely resemble as well as the 3J(H4,H5) coupling constant at the pyrazole nucleus, which for 11–13 is the same as in 1 (2.5–2.6 Hz).
Figure 6.

Investigated 4-substituted 1-phenyl-1H-pyrazol-3-ols (4–8) and 3-methoxy congeners (9, 10), as well as 3-O-acyl derivatives of 1 (11–13).
The triple 14–16 provides another example of comparing a free 1H-pyrazol-3-ol (14) with the corresponding O-alkyl (15) and N-alkyl derivative (16), respectively. Again, the selected data depicted in Figure 7 clearly hint that 14 predominantly exists as OH-isomer and not as pyrazol-3-one.
Figure 7.

1H (in italics), 13C and 15N (in bold) NMR chemical shifts of 14 and its “fixed” derivatives 15 and 16 (in CDCl3).
In addition, we investigated 1H-pyrazol-3-ols carrying a methyl (17) and a benzyl substituent (18), respectively, at pyrazole N-1. From the relevant data of these compounds, depicted in Figure 8, the conclusion can be drawn, that also 17 and 18 exist in the 3-hydroxy form in CDCl3, DMSO-d6, and C6D6 solution. Again, as found with 1-phenyl-1H-pyrazol-3-ol 1 and compounds 17, 18, the markedly larger chemical shifts of pyrazole N-2 in DMSO-d6 compared to those in CDCl3 or C6D6 hint to the absence of dimers stabilized by intermolecular hydrogen bonds in this solvent, what is supported by a distinctly smaller 1H-NMR chemical shift of the OH-proton in DMSO-d6 (Figure 8).
Figure 8.

Crucial 1H (in italics), 13C, and 15N (in bold) NMR chemical shifts of 1-methyl-1H-pyrazol-3-ol (17) and 1-benzyl-1H-pyrazol-3-ol (18) in CDCl3, DMSO-d6, and C6D6 solution.
3. Experimental Section
3.1. General Information
Melting points were determined on a Büchi M-565 melting point apparatus (Büchi Labortechnik AG, Flawil, Switzerland) and are uncorrected. IR (infrared) spectra (KBr pellets) were recorded on a Bruker Tensor 27 spectrometer (Bruker Optik GmbH, Ettlingen, Germany) and are reported in wave numbers (cm−1). High-resolution ESI-TOF mass spectra were measured on a Bruker maXis spectrometer (Bruker Daltonik GmbH, Bremen, Germany). Elemental analyses were performed at the Microanalytical Laboratory, University of Vienna. 1H and 13C-NMR spectra were recorded on a Bruker Avance 500 spectrometer (500.13 MHz for 1H, 125.77 MHz for 13C) (Bruker BioSpin GmbH, Rheinstetten, Germany–valid for all mentioned Bruker NMR spectrometers), a Bruker Avance III 400 spectrometer (400.23 MHz for 1H, 100.64 MHz for 13C) or a Varian UnityPlus300 spectrometer (299.95 MHz for 1H, 75.43 MHz for 13C) (Varian, Palo Alto, CA, USA) at 293 K. The centre of the solvent signal was used as an internal standard which was related to TMS with δ 7.26 ppm (1H in CDCl3), δ 2.49 ppm (1H in DMSO-d6), δ 7.16 ppm (1H in C6D6), δ 3.31 ppm (1H in CD3OD), δ (δ 77.0 ppm (13C in CDCl3), δ 39.5 ppm (13C in DMSO-d6), δ 128.06 ppm (13C in C6D6) and δ 49.00 ppm (13C in CD3OD). The digital resolutions were 0.20 Hz/data point in the 1H and 0.33 Hz/data point in the 13C-NMR spectra. 15N-NMR spectra were obtained on Bruker Avance 500 (50.69 MHz) and Bruker Avance III 400 (40.56 MHz) spectrometers (both equipped with “direct” detection broadband z-gradient observe probes) or on a Bruker Avance III 700 (70.96 MHz) equipped with a 5 mm TCI 1H-13C/15N/D z-gradient cryoprobe, and were measured against external nitromethane (coaxial capillary) and recalculated to liquid ammonia. Solid-state NMR spectra (CP/MAS, MAS: 10 kHz) were recorded on a Bruker Avance III 500 instrument with a broadband MAS-probe for 3.2 mm rotors. CP contact times were 2 ms for (1H, 13C) and 3 ms for (1H, 15N). 1H RF of 100 kHz was used for spinal64 broadband decoupling. 1H, 13C-HETCOR spectra were recorded using FSLG homonuclear decoupling during t1-evolution and mixing times of 50 µs and 200 µs. 15N-NMR spectra were referenced to 15NH4Cl and recalculated to the liquid ammonia scale (δ 15NH4Cl 39.3 ppm). 13C spectra were referenced to the methylene carbon signals of adamantane and recalculated to the TMS scale (δ 13CH2 38.5 ppm). 1H chemical shifts were referenced to the NH3+ resonance in α-Glycine and recalculated to the TMS scale (δ 15NH3+ 8.5 ppm). Product yields were not optimised.
3.2. Data of Investigated Compounds
1-Phenyl-1H-pyrazol-3-ol (1). Compound 1 was prepared by oxidation of 1-phenylpyrazolidin-3-one with FeCl3 according to Reference [28] and recrystallized from EtOH–H2O. 1H-NMR (CDCl3): δ 12.16 (br s, 1H, OH), 7.67 (d, 3J = 2.6 Hz, 1H, pyrazole H-5), 7.52 (m, 2H, Ph H-2,6), 7.45 (m, 2H, Ph H-3,5), 7.25 (m, 1H, Ph H-4), 5.92 (d, 3J = 2.6 Hz, 1H, pyrazole H-4). 13C-NMR (CDCl3): δ 164.0 (2J(C3,H4) = 2.2 Hz, 3J(C3,H5) = 10.4 Hz, pyrazole C-3), 139.4 (Ph C-1), 129.6 (Ph C-3,5), 129.1 (1J(C5,H5) = 187.0 Hz, 2J(C5,H4) = 8.4 Hz, pyrazole C-5), 125.9 (Ph C-4), 118.6 (Ph C-2,6), 94.2 (1J(C4,H4) = 180.2 Hz, 2J(C4,H5) = 7.7 Hz, pyrazole C-4). 15N-NMR (CDCl3): δ 192.1 (pyrazole N-1), 245.9 (pyrazole N-2). 1H-NMR (DMSO-d6): δ 10.28 (s, 1H, OH), 8.18 (d, 3J = 2.6 Hz, 1H, pyrazole H-5), 7.67 (m, 2H, Ph H-2,6), 7.40 (m, 2H, Ph H-3,5), 7.15 (m, 1H, Ph H-4), 5.82 (d, 3J = 2.6 Hz, 1H, pyrazole H-4). 13C-NMR (DMSO-d6): δ 162.8 (2J(C3,H4) = 2.5 Hz, 3J(C3,H5) = 10.3 Hz, pyrazole C-3), 139.9 (Ph C-1), 129.4 (Ph C-3,5), 128.4 (1J(C5,H5) = 188.2 Hz, 2J(C5,H4) = 8.9 Hz, pyrazole C-5), 124.7 (Ph C-4), 116.8 (Ph C-2,6), 94.4 (1J(C4,H4) = 178.0 Hz, 2J(C4,H5) = 8.0 Hz, pyrazole C-4). 15N-NMR (DMSO-d6): δ 194.4 (pyrazole N-1), 262.1 (pyrazole N-2). 1H-NMR (C6D6): δ 12.69 (br s, 1H, OH), 7.33 (m, 2H, Ph H-2,6), 7.06 (m, 2H, Ph H-3,5), 6.97 (d, 3J = 2.6 Hz, 1H, pyrazole H-5), 6.86 (m, 1H, Ph H-4), 5.76 (d, 3J = 2.6 Hz, 1H, pyrazole H-4). 13C-NMR (C6D6): δ 165.1 (2J(C3,H4) = 2.2 Hz, 3J(C3,H5) = 10.4 Hz, pyrazole C-3), 139.9 (Ph C-1), 129.7 (Ph C-3,5), 129.3 (1J(C5,H5) = 187.1 Hz, 2J(C5,H4) = 8.5 Hz, pyrazole C-5), 125.8 (Ph C-4), 118.8 (Ph C-2,6), 94.6 (1J(C4,H4) = 179.8 Hz, 2J(C4,H5) = 7.7 Hz, pyrazole C-4). 15N-NMR (C6D6): δ 191.7 (pyrazole N-1), 246.1 (pyrazole N-2). 1H-NMR (CD3OD): δ 7.89 (3J = 2.6 Hz, 1H, pyrazole H-5), 7.57 (m, 2H, Ph H-2,6), 7.38 (m, 2H, Ph H-3,5), 7.17 (m, 1H, Ph H-4), 5.82 (d, 3J = 2.6 Hz, 1H, pyrazole H-4). 13C-NMR (CD3OD): δ 164.5 (2J(C3,H4) = 2.6 Hz, 3J(C3,H5) = 10.2 Hz, pyrazole C-3), 141.3 (Ph C-1), 130.4 (Ph C-3,5), 129.7 (1J(C5,H5) = 187.6 Hz, 2J(C5,H4) = 8.6 Hz, pyrazole C-5), 126.3 (Ph C-4), 118.8 (Ph C-2,6), 94.8 (1J(C4,H4) = 178.6 Hz, 2J(C4,H5) = 7.9 Hz, pyrazole C-4). 15N-NMR (CD3OD): δ 193.8 (pyrazole N-1), 256.3 (pyrazole N-2). 13C-SSNMR: δ 164.9 (pyrazole C-3), 140.1 (Ph C-1), 132.9, 131.6 (pyrazole C-5), 131.0, 127.1, 118.8, 115.5, 95.9 (pyrazole C-4). 1H-SSNMR: δ 11.2 (pyrazole OH), 7.1, 6.9, 6.9, 6.7, 5.8, 5.4 (pyrazole H-5), 5.1 (pyrazole H-4). 13C/1H-HETCOR-SSNMR: δ 132.9/6.9, 131.6/5.4, 131.0/7.1, 127.1/6.7, 118.8/6.9, 115.5/5.8, 95.9/5.1 (pyrazole C-4). 15N-SSNMR: δ 243.1 (pyrazole N-2), 192.6 (pyrazole N-1).
3-Methoxy-1-phenyl-1H-pyrazole (2) [29]. 1H-NMR (CDCl3): δ 7.72 (d, 3J = 2.6 Hz, 1H, pyrazole H-5), 7.61 (m, 2H, Ph H-2,6), 7.40 (m, 2H, Ph H-3,5), 7.19 (m, 1H, Ph H-4), 5.89 (d, 3J = 2.6 Hz, 1H, pyrazole H-4), 3.98 (s, 3H, OCH3). 13C-NMR (CDCl3): δ 165.0 (2J(C3,H4) = 2.0 Hz, 3J(C3,H5) = 10.2 Hz, 3J(C3,OMe) = 4.0 Hz, pyrazole C-3), 140.2 (Ph C-1), 129.3 (Ph C-3,5), 127.7 (1J(C5,H5) = 186.2 Hz, 2J(C5,H4) = 8.3 Hz, pyrazole C-5), 125.2 (Ph C-4), 117.8 (Ph C-2,6), 93.4 (1J(C4,H4) = 179.2 Hz, 2J(C4,H5) = 8.1 Hz, pyrazole C-4), 56.3 (1J = 145.3 Hz, OCH3). 15N-NMR (CDCl3): δ 195.6 (pyrazole N-1), 261.7 (pyrazole N-2).
2-Methyl-1-phenyl-1,2-dihydro-3H-pyrazol-3-one (3) [30]. 1H-NMR (CDCl3): δ 7.44 (m, 2H, Ph H-3,5), 7.39 (d, 3J = 3.6 Hz, 1H, pyrazole H-5), 7.33 (m, 1H, Ph H-4), 7.18 (m, 2H, Ph H-2,6), 5.59 (d, 3J = 3.6 Hz, 1H, pyrazole H-4), 3.24 (s, 3H, NCH3). 13C-NMR (CDCl3): δ 168.2 (pyrazole C-3), 142.3 (1J(C5,H5) = 188.5 Hz, 2J(C5,H4) = 7.7 Hz, pyrazole C-5), 137.8 (Ph C-1), 129.9 (Ph C-3,5), 127.8 (Ph C-4), 123.0 (Ph C-2,6), 98.1 (1J(C4,H4) = 182.6 Hz, 2J(C4,H5) = 6.3 Hz, pyrazole C-4), 30.3 (1J = 140.7 Hz, NCH3). 15N-NMR (CDCl3): δ 159.1 (pyrazole N-1), 162.5 (pyrazole N-2).
4-Chloro-1-phenyl-1H-pyrazol-3-ol (4) [29]. 1H-NMR (CDCl3): δ 11.30 (br s, 1H, OH), 7.72 (s, 1H, pyrazole H-5), 7.47 (m, 4H, Ph H-2,3,5,6), 7.30 (m, 1H, Ph H-4). 13C NMR (CDCl3): δ 159.2 (3J(C3,H5) = 8.5 Hz, pyrazole C-3), 139.0 (Ph C-1), 129.8 (Ph C-3,5), 126.8 (1J(C5,H5) = 192.3 Hz, pyrazole C-5), 126.6 (Ph C-4), 118.7 (Ph C-2,6), 98.2 (2J(C4,H5) = 4.3 Hz, pyrazole C-4). 15N-NMR (CDCl3): δ 187.6 (pyrazole N-1), 246.5 (pyrazole N-2). 1H-NMR (DMSO-d6): δ 11.02 (s, 1H, OH), 8.52 (s, 1H, pyrazole H-5), 7.66 (m, 2H, Ph H-2,6), 7.43 (m, 2H, Ph H-3,5), 7.20 (m, 1H, Ph H-4). 13C-NMR (DMSO-d6): δ 158.1 (3J(C3,H5) = 8.5 Hz, pyrazole C-3), 139.3 (Ph C-1), 129.4 (Ph C-3,5), 126.4 (1J(C5,H5) = 194.4 Hz, pyrazole C-5), 125.2 (Ph C-4), 116.7 (Ph C-2,6), 97.2 (2J(C4,H5) = 4.6 Hz, pyrazole C-4). 15N-NMR (DMSO-d6): δ 190.0 (pyrazole N-1), 262.0 (pyrazole N-2).
4-Bromo-1-phenyl-1H-pyrazol-3-ol (5) [29]. 1H-NMR (CDCl3): δ 11.33 (br s, 1H, OH), 7.73 (s, 1H, pyrazole H-5), 7.48 (m, 4H, Ph H-2,3,5,6), 7.30 (m, 1H, Ph H-4). 13C-NMR (CDCl3): δ 160.6 (pyrazole C-3, 3J(C3,H5) = 8.7 Hz), 139.1 (Ph C-1), 129.8 (Ph C-3,5), 129.1 (1J(C5,H5) = 192.5 Hz, pyrazole C-5), 126.7 (Ph C-4), 118.8 (Ph C-2,6), 82.2 (2J(C4,H5) = 4.6 Hz, pyrazole C-4). 15N-NMR (CDCl3): δ 191.7 (pyrazole N-1), 247.9 (pyrazole N-2). 1H-NMR (DMSO-d6): δ 10.99 (s, 1H, OH), 8.51 (s, 1H, pyrazole H-5), 7.67 (m, 2H, Ph H-2,6), 7.43 (m, 2H, Ph H-3,5), 7.21 (m, 1H, Ph H-4). 13C-NMR (DMSO-d6): δ 159.4 (3J(C3,H5) = 8.9 Hz, pyrazole C-3), 139.3 (Ph C-1), 129.4 (Ph C-3,5), 128.5 (1J(C5,H5) = 194.7 Hz, pyrazole C-5), 125.3 (Ph C-4), 116.8 (Ph C-2,6), 82.1 (2J(C4,H5) = 5.2 Hz, pyrazole C-4). 15N-NMR (DMSO-d6): δ 193.7 (pyrazole N-1), 262.5 (pyrazole N-2).
4-Iodo-1-phenyl-1H-pyrazol-3-ol (6) [29]. 1H-NMR (CDCl3): δ 11.40 (br s, 1H, OH), 7.72 (s, 1H, pyrazole H-5), 7.48 (m, 4H, Ph H-2,3,5,6), 7.31 (m, 1H, Ph H-4). 13C-NMR (CDCl3): δ 163.6 (pyrazole C-3, 3J(C3,H5) = 9.1 Hz), 139.1 (Ph C-1), 133.3 (1J(C5,H5) = 192.3 Hz, pyrazole C-5), 129.8 (Ph C-3,5), 126.7 (Ph C-4), 118.8 (Ph C-2,6), 46.6 (2J(C4,H5) = 5.0 Hz, pyrazole C-4). 15N-NMR (CDCl3): δ 197.2 (pyrazole N-1), 248.1 (pyrazole N-2). 1H-NMR (DMSO-d6): δ 10.89 (s, 1H, OH), 8.40 (s, 1H, pyrazole H-5), 7.67 (m, 2H, Ph H-2,6), 7.41 (m, 2H, Ph H-3,5), 7.18 (m, 1H, Ph H-4). 13C-NMR (DMSO-d6): δ 162.7 (3J(C3,H5) = 9.4 Hz, pyrazole C-3), 139.3 (Ph C-1), 132.5 (1J(C5,H5) = 193.8 Hz, pyrazole C-5), 129.4 (Ph C-3,5), 125.2 (Ph C-4), 116.8 (Ph C-2,6), 49.0 (2J(C4,H5) = 6.3 Hz, pyrazole C-4). 15N-NMR (DMSO-d6): δ 199.3 (pyrazole N-1), 263.0 (pyrazole N-2).
1-(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)ethan-1-one (7) [29,31]. 1H-NMR (CDCl3): δ 9.40 (s, 1H, OH), 8.13 (s, 1H, pyrazole H-5), 7.67 (m, 2H, Ph H-2,6), 7.46 (m, 2H, Ph H-3,5), 7.33 (m, 1H, Ph H-4), 2.48 (s, 3H, COCH3). 13C-NMR (CDCl3): δ 195.7 (C=O), 164.0 (3J(C3,H5) = 8.7 Hz, pyrazole C-3,), 139.0 (Ph C-1), 129.6 (Ph C-3,5), 128.1 (1J(C5,H5) = 188.0 Hz, pyrazole C-5), 127.4 (Ph C-4), 119.1 (Ph C-2,6), 108.4 (2J(C4,H5) = 7.8 Hz, 3J(C4,CH3) = 1.6 Hz, pyrazole C-4), 27.0 (1J = 127.9 Hz, CH3). 15N-NMR (CDCl3): δ 200.4 (pyrazole N-1), 264.2 (pyrazole N-2). 1H-NMR (DMSO-d6): δ 11.10 (s, 1H, OH), 8.84 (s, 1H, pyrazole H-5), 7.80 (m, 2H, Ph H-2,6), 7.48 (m, 2H, Ph H-3,5), 7.29 (m, 1H, Ph H-4), 2.38 (s, 3H, COCH3). 13C-NMR (DMSO-d6): δ 191.7 (C=O), 161.4 (3J(C3,H5) = 9.0 Hz, pyrazole C-3), 138.8 (Ph C-1), 131.2 (1J(C5,H5) = 191.8 Hz, pyrazole C-5), 129.4 (Ph C-3,5), 126.4 (Ph C-4), 118.0 (Ph C-2,6), 110.8 (2J(C4,H5) = 6.8 Hz, 3J(C4,CH3) = 1.4 Hz, pyrazole C-4), 28.5 (1J = 127.4 Hz, CH3). 15N-NMR (DMSO-d6): δ 198.5 (pyrazole N-1), 263.1 (pyrazole N-2).
(3-Hydroxy-1-phenyl-1H-pyrazol-4-yl)(phenyl)methanone (8) [32]. To a stirred suspension of anhydrous aluminum chloride (16.0 g, 0.12 mol) in 20 mL of carbon disulfide, maintained at room temperature with a water bath, a slurry of 1-phenyl-1H-pyrazol-3-yl benzoate (12) (2.64 g, 10 mmol) in 70 mL of carbon disulfide was added. After the addition was complete, the reaction mixture was refluxed for 8 h. After the solvent was removed under reduced pressure the residual paste was cooled in an ice bath and a solution of 13.3 mL of 6N hydrochloric acid in 33 mL of ice water was added slowly under stirring to decompose the aluminum chloride salts, then the mixture was allowed to stand overnight. The solid was filtered off, washed with water, dried and recrystallized from EtOH to afford 818 mg (31 %) of 8, m.p. 138–140 °C (EtOH). 1H-NMR (CDCl3): δ 9.91 (s, 1H, OH), 8.17 (s, 1H, pyrazole H-5), 7.89 (m, 2H, CPh H-2,6), 7.69 (m, 2H, NPh H-2,6), 7.63 (m, 1H, CPh H-4), 7.54 (m, 2H, CPh H-3,5), 7.45 (m, 2H, NPh H-3,5), 7.32 (m, 1H, NPh H-4). 13C-NMR (CDCl3): δ 191.7 (C=O), 165.6 (3J(C3,H5) = 8.8 Hz, pyrazole C-3), 139.0 (NPh C-1), 138.0 (CPh C-1), 132.8 (CPh C-4), 129.5 (NPh C-3,5), 129.1 ((1J(C5,H5) = 189.6 Hz, pyrazole C-5), 128.9 (CPh C-3,5), 128.2 (CPh C-2,6), 127.4 (NPh C-4), 119.2 (NPh C-2,6), 106.9 (2J(C4,H5) = 8.0 Hz, pyrazole C-4). 15N-NMR (CDCl3): δ 202.1 (pyrazole N-1), 264.0 (pyrazole N-2). IR (KBr): 1627 (C=O) cm−1. MS m/z (%): 265 ([M + H]+, 100). Anal. Calcd. for C16H12N2O2: C, 72.72; H, 4.58; N, 10.60. Found: C, 73.01; H, 4.65; N, 10.27.
4-Bromo-3-methoxy-1-phenyl-1H-pyrazole (9). The synthesis and the 1H and 13C-NMR spectra of 9 are described in lit. [26]. 15N-NMR (CDCl3): δ 194.7 (pyrazole N-1), 262.6 (pyrazole N-2).
1-(3-Methoxy-1-phenyl-1H-pyrazol-4-yl)ethan-1-one (10) [29]. 1H-NMR (CDCl3): δ 8.25 (s, 1H, pyrazole H-5), 7.63 (m, 2H, Ph H-2,6), 7.44 (m, 2H, Ph H-3,5), 7.28 (m, 1H, Ph H-4), 4.08 (s, 3H, OCH3), 2.46 (s, 3H, COCH3). 13C-NMR (CDCl3): δ 192.3 (C=O), 162.6 (3J(C3,H5) = 8.8 Hz, 3J(C3,OCH3) = 3.8 Hz, pyrazole C-3), 139.1 (Ph C-1), 130.4 (1J(C5,H5) = 189.4 Hz, pyrazole C-5), 129.5 (Ph C-3,5), 126.8 (Ph C-4), 118.5 (Ph C-2,6), 111.6 (2J(C4,H5) = 6.6 Hz, 3J(C4,CH3) = 1.4 Hz, pyrazole C-4), 56.6 (1J = 146.5 Hz, OCH3), 29.1 (1J = 127.8 Hz, CH3). 15N-NMR (CDCl3): δ 200.0 (pyrazole N-1), 262.9 (pyrazole N-2).
1-Phenyl-1H-pyrazol-3-yl acetate (11) [29]. 1H-NMR (CDCl3): δ 7.83 (d, 3J = 2.5 Hz, 1H, pyrazole H-5), 7.62 (m, 2H, Ph H-2,6), 7.42 (m, 2H, Ph H-3,5), 7.26 (m, 1H, Ph H-4), 6.36 (d, 3J = 2.5 Hz, 1H, pyrazole H-4), 2.32 (s, 3H, COCH3). 13C-NMR (CDCl3): δ 167.9 (2J(CO;CH3) = 7.0 Hz, C=O), 156.4 (2J(C3,H4) = 1.2 Hz, 3J(C3,H5) = 10.9 Hz, pyrazole C-3), 139.6 (Ph C-1), 129.4 (Ph C-3,5), 127.7 (1J(C5,H5) = 188.5 Hz, 2J(C5,H4) = 8.5 Hz, pyrazole C-5), 126.4 (Ph C-4), 118.6 (Ph C-2,6), 98.8 (1J(C4,H4) = 189.9 Hz, 2J(C4,H5) = 8.1 Hz, pyrazole C-4), 20.9 (1J = 130.3 Hz, CH3). 15N-NMR (CDCl3): δ 202.9 (pyrazole N-1), 263.8 (pyrazole N-2).
1-Phenyl-1H-pyrazol-3-yl benzoate (12). A solution of 1 (480 mg, 3 mmol), benzoyl chloride (436 mg, 3.1 mmol) and pyridine (0.1 mL) in toluene (4 mL) was heated at 100 °C for 30 minutes. Then the reaction mixture was poured into water (10 mL), the precipitate was filtered off, washed with water and recrystallized from 50% aqueous ethanol to afford 490 mg (62%) of 11 as colourless crystals, m.p. 59–60 °C. IR (KBr): 1745 (C=O) cm−1. 1H-NMR (CDCl3): δ 8.26 (m, 2H, CPh H-2,6), 7.90 (d, 3J = 2.6 Hz, 1H, pyrazole H-5), 7.67 (m, 2H, NPh H-2,6), 7.64 (m, 1H, CPh H-4), 7.51 (m, 2H, CPh H-3,5), 7.45 (m, 2H, NPh H-3,5), 7.28 (m, 1H, NPh H-4), 6.52 (d, 3J = 2.6 Hz, 1H, pyrazole H-4). 13C-NMR (CDCl3): δ 163.8 (C=O), 156.7 (2J(C3,H4) = 1.6 Hz, 3J(C3,H5) = 11.1 Hz, pyrazole C-3), 139.7 (NPh C-1), 133.8 (CPh C-4), 130.4 (CPh C-2,6), 129.4 (NPh C-3,5), 128.8 (CPh C-1), 128.6 (CPh C-3,5), 127.8 (1J(C5,H5) = 188.5 Hz, 2J(C5,H4) = 8.6 Hz, pyrazole C-5), 126.5 (NPh C-4), 118.7 (NPh C-2,6), 99.1 (1J(C4,H4) = 184.1 Hz, 2J(C4,H5) = 8.0 Hz, pyrazole C-4). 15N-NMR (CDCl3): δ 203.2 (pyrazole N-1), 277.7 (pyrazole N-2). MS m/z (%): 265 ([M + H]+, 100). Anal. Calcd. for C16H12N2O2: C, 72.72; H, 4.58; N, 10.60. Found: C, 72.93; H, 4.45; N, 10.34.
1-Phenyl-1H-pyrazol-3-yl thiophene-2-carboxylate (13). A solution of 1 (3.20 g, 20 mmol) and 2-thiophenecarbonyl chloride (2.93 g, 20 mmol) in dry toluene (25 mL) was refluxed for 3.5 h. The reaction mixture was poured into water (40 mL), the phases were separated and the aqueous phase was extracted with toluene (2 × 15 mL). The combined organic phases were dried (Na2SO4) and, after filtration, evaporated under reduced pressure. The residue was recrystallized from EtOH-H2O to afford 3.54 g (65%) of 13 as almost colorless crystals, m.p. 62–63 °C. IR (KBr): 1736 (C=O) cm−1. 1H-NMR (CDCl3): δ 8.02 (dd, 3J(H3,H4) = 3.8 Hz, 4J(H3,H5) = 1.1 Hz, 1H, Th H-3), 7.88 (d, 3J = 2.6 Hz, 1H, pyrazole H-5), 7.67 (dd, 3J(H5,H4) = 4.9 Hz, 4J(H5,H3) = 1.3 Hz, 1H, Th H-5), 7.65 (m, 2H, Ph H-2,6), 7.43 (m, 2H, Ph H-3,5), 7.27 (m, 1H, Ph H-4), 7.16 (dd, 3J(H4,H3) = 3.8 Hz, 3J(H4,H5) = 4.9 Hz, 1H, Th H-4), 6.49 (d, 3J = 2.6 Hz, pyrazole H-4). 13C-NMR (CDCl3): δ 159.1 (C=O), 156.2 (2J(C3,H4) = 1.5 Hz, 3J(C3,H5) = 11.1 Hz, pyrazole C-3), 139.6 (NPh C-1), 135.1 (1J = 171.3 Hz, 2J = 5.6 Hz, 3J = 9.2 Hz, Th C-3), 134.1 (1J = 185.7 Hz, 2J = 7.3 Hz, 3J = 11.2 Hz, Th C-5), 131.9 (2J = 5.8 Hz, 3J(C2,H4) = 9.7 Hz, 3J(C3,H5) = 5.8 Hz, Th C-2), 129.3 (Ph C-3,5), 128.0 (1J = 170.6 Hz, 2J(C4,H3) = 5.0 Hz, 2J(C4,H5) = 4.0 Hz, Th C-4), 127.7 (1J(C5,H5) = 188.6 Hz, 2J(C5,H4) = 8.5 Hz, pyrazole C-5), 126.4 (Ph C-4), 118.6 (Ph C-2,6), 99.0 (1J(C4,H4) = 184.4 Hz, 2J(C4,H5) = 8.1 Hz, pyrazole C-4). 15N-NMR (CDCl3): δ 203.3 (pyrazole N-1), 277.8 (pyrazole N-2). MS m/z (%): 271 ([M + H]+, 100). Anal. Calcd. for C14H10N2O2S: C, 62.21; H, 3.73; N, 10.36. Found: C, 62.51; H, 4.01; N, 9.97.
4-Bromo-1-(4-bromophenyl)-1H-pyrazol-3-ol (14). The synthesis and spectral data of 14 are given in [33].
Preparation of compounds 15 and 16. 4-Bromo-1-(4-bromophenyl)-1H-pyrazol-3-ol 14 (1.0 g, 3.15 mmol) was dissolved in DMF (20 mL) and potassium hydroxide (204 mg, 3.65 mmol) was added to the solution. The mixture was stirred for 15 min, then allyl bromide (442 mg, 3.65 mmol) was added, and stirring was continued for 30 min. The reaction mixture was poured into water (40 mL) and extracted with ether (3 × 30 mL). The combined organic extracts were dried over anhydrous sodium sulphate, the solvent was then evaporated under reduced pressure, and the residue subjected to column chromatography (silica gel, eluent: hexane–ethyl acetate 3:1) to yield 824 mg (73%) of compound 15 (Rf 0.68) and 58 mg (5%) of compound 16 (Rf 0.37) as oily substances.
4-Bromo-1-(4-bromophenyl)-3-(prop-2-en-1-yloxy)-1H-pyrazole (15). 1H-NMR (CDCl3): δ 7.74 (s, 1H, pyrazole H-5), 7.51 (m, 2H, Ph H-2,6), 7.42 (m, 2H, Ph H-3,5), 6.12 (m, 1H, CH=CH2), 5.46 (dd, 2J = 1.4 Hz, 3J = 17.2 Hz, 1H, CH=CH2(trans)), 5.31 (2J = 1.4 Hz, 3J = 10.5 Hz, 1H, CH=CH2(cis)), 4.83 (m, OCH2). 13C-NMR (CDCl3): δ 160.5 (3J(C3,H5) = 5.1 Hz, 3J(C3,OCH2) = 2.8 Hz, pyrazole C-3), 138.6 (Ph C-1), 132.6 (CH=CH2), 132.4 (Ph C-3,5), 127.5 (1J = 191.9 Hz, pyrazole C-5), 118.9 (Ph C-2,6), 118.6 (Ph C-4), 118.4 (CH=CH2), 83.1 (2J(C4,H5) = 5.1 Hz, pyrazole C-4), 70.2 (OCH2). 15N-NMR (CDCl3): δ 192.2 (pyrazole N-1), 262.6 (pyrazole N-2). MS m/z (%): 361/359/357 ([M + H]+, 49/100/51). Anal. Calcd. for C12H10Br2N2O: C, 40.26; H, 2.82; N, 7.82. Found: C, 40.40; H, 2.90; N 7.69.
4-Bromo-1-(4-bromophenyl)-2-(prop-2-en-1-yl)-1,2-dihydro-3H-pyrazol-3-one (16). 1H-NMR (CDCl3): δ 7.58 (m, 2H, Ph H-3,5), 7.52 (s, 1H, pyrazole H-5), 7.08 (m, 2H, Ph H-2,6), 5.63 (m, 1H, CH=CH2), 5.09 (m, 1H, CH=CH2(cis)), 4.91 (m, 1H, CH=CH2(trans)), 4.34 (m, NCH2). 13C-NMR (CDCl3): δ 164.2 (pyrazole C-3), 142.5 (1J = 193.9 Hz, pyrazole C-5), 136.7 (Ph C-1), 133.2 (Ph C-3,5), 130.9 (CH=CH2), 125.2 (Ph C-2,6), 122.2 (Ph C-4), 119.0 (CH=CH2), 90.1 (2J(C4,H5) = 3.0 Hz, pyrazole C-4), 46.3 (NCH2). 15N-NMR (CDCl3): δ 150.8 (pyrazole N-1), 170.8 (pyrazole N-2). IR (KBr): 1659 (C=O) cm−1. MS m/z (%): 361/359/357 ([M + H]+, 49/100/51). Anal. Calcd. for C12H10Br2N2O: C, 40.26; H, 2.82; N, 7.82. Found: C, 40.51; H, 3.15; N, 8.13.
1-Methyl-1H-pyrazol-3-ol (17) [34]. 1H-NMR (CDCl3): δ 12.00 (s, 1H, OH), 7.07 (d, 3J = 2.4 Hz, 1H, pyrazole H-5), 5.56 (d, 3J = 2.4 Hz, 1H, pyrazole H-4), 3.69 (s, 3H, NCH3). 13C-NMR (CDCl3): δ 162.6 (pyrazole C-3), 131.8 (pyrazole C-5), 90.6 (pyrazole C-4), 38.3 (NCH3). 15N-NMR (CDCl3): δ 172.1 (pyrazole N-1), 252.0 (pyrazole N-2). 1H-NMR (DMSO-d6): δ 9.53 (s, 1H, OH), 7.30 (d, 3J = 2.2 Hz, 1H, pyrazole H-5), 5.39 (d, 3J = 2.2 Hz, 1H, pyrazole H-4), 3.58 (s, 3H, NCH3). 13C-NMR (DMSO-d6): δ 160.9 (pyrazole C-3), 131.3 (pyrazole C-5), 89.8 (pyrazole C-4), 38.2 (NCH3). 15N-NMR (DMSO-d6): δ 177.2 (pyrazole N-1), 271.8 (pyrazole N-2). 1H-NMR (C6D6): δ 12.56 (s, 1H, OH), 6.28 (d, 3J = 2.4 Hz, 1H, pyrazole H-5), 5.62 (d, 3J = 2.4 Hz, 1H, pyrazole H-4), 2.90 (s, 3H, NCH3). 13C-NMR (C6D6): δ 163.7 (pyrazole C-3), 131.5 (pyrazole C-5), 90.8 (pyrazole C-4), 37.4 (NCH3). 15N-NMR (C6D6): δ 171.8 (pyrazole N-1), 252.7 (pyrazole N-2).
1-Benzyl-1H-pyrazol-3-ol (18) [34]. 1H-NMR (CDCl3): δ 11.30 (s, 1H, OH), 7.33 (m, 3H, Ph H-3,4,5), 7.25 (m, 2H, Ph H-2,6), 7.11 (d, 3J = 2.5 Hz, 1H, pyrazole H-5), 5.62 (d, 3J = 2.5 Hz, 1H, pyrazole H-4), 5.07 (s, 2H, NCH2). 13C-NMR (CDCl3): δ 162.6 (2J(C3,H4) = 2.2 Hz, 3J(C3,H5) = 10.1 Hz, pyrazole C-3), 136.0 (Ph C-1), 130.9 (1J(C5,H5) = 185.8 Hz, 2J(C5,H4) = 8.2 Hz, 3J(C5,NCH2) = 3.2 Hz, pyrazole C-5), 128.8 (Ph C-3,5), 128.1 (Ph C-4), 127.8 (Ph C-2,6), 91.3 (1J(C4,H4) = 178.9 Hz, 2J(C4,H5) = 8.0 Hz, pyrazole C-4), 55.4 (1J = 139.3 Hz, NCH2). 15N-NMR (CDCl3): δ 183.5 (pyrazole N-1), 251.2 (pyrazole N-2). 1H-NMR (DMSO-d6): δ 9.63 (s, 1H, OH), 7.50 (d, 3J = 2.3 Hz, 1H, pyrazole H-5), 7.32 (m, 2H, Ph H-3,5), 7.27 (m, 1H, Ph H-4), 7.19 (m, 2H, Ph H-2,6), 5.47 (d, 3J = 2.3 Hz, 1H, pyrazole H-4), 5.05 (s, 2H, NCH2). 13C-NMR (DMSO-d6): δ 161.3 (2J(C3,H4) = 2.5 Hz, 3J(C3,H5) = 10.1 Hz, pyrazole C-3), 138.0 (Ph C-1), 131.2 (1J(C5,H5) = 185.8 Hz, 2J(C5,H4) = 8.5 Hz, 3J(C5,NCH2) = 3.1 Hz, pyrazole C-5), 128.3 (Ph C-3,5), 127.4 (Ph C-2,6), 127.3 (Ph C-4), 90.3 (1J(C4,H4) = 176.2 Hz, 2J(C4,H5) = 8.6 Hz, pyrazole C-4), 54,5 (1J = 139.2 Hz, NCH2). 15N-NMR (DMSO-d6): δ 187.9 (pyrazole N-1), 269.9 (pyrazole N-2). 1H-NMR (C6D6): δ 12.40 (s, 1H, OH), 6.93–7.05 (m, 5H, Ph H), 6.45 (d, 3J = 2.4 Hz, 1H, pyrazole H-5), 5.63 (d, 3J = 2.4 Hz, 1H, pyrazole H-4), 4.51 (s, 2H, NCH2). 13C-NMR (C6D6): δ 164.2 (2J(C3,H4) = 2.2 Hz, 3J(C3,H5) = 10.1 Hz, pyrazole C-3), 137.0 (Ph C-1), 131.4 (1J(C5,H5) = 185.0 Hz, 2J(C5,H4) = 8.2 Hz, 3J(C5,NCH2) = 3.1 Hz, pyrazole C-5), 129.2 (Ph C-3,5), 128.4 (Ph C-4), 128.4 (Ph C-2,6), 92.0 (1J(C4,H4) = 178.2 Hz, 2J(C4,H5) = 8.1 Hz, pyrazole C-4), 55.6 (1J = 139.2 Hz, NCH2). 15N-NMR (C6D6): δ 183.3 (pyrazole N-1), 251.9 (pyrazole N-2).
3.3. X-ray Crystal Structure Analysis
The X-ray intensity data was measured on a Bruker X8 APEXII equipped with multilayer monochromators, with a Mo K/a INCOATEC micro focus sealed tube, and a Kryoflex II cooling device. The structure was solved by direct methods and refined by full-matrix least-squares techniques. Non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen located at O1 was refined without any restraints or constraints. All other hydrogen atoms were inserted at calculated positions and refined with a riding model. The following software was used: Frame integration, Bruker SAINT software package [35] using a narrow-frame algorithm, Absorption correction, SADABS [36], structure solution, SHELXL-2013 [37], refinement, SHELXL-2013 [37], OLEX2 [38], SHELXLE [39], molecular diagrams, OLEX2 [38]. Experimental data and CCDC-Code [40] can be found in Table 1. Crystal data, data collection parameters, and structure refinement details are given in Table 2 and Table 3. Molecular structures in “Ortep View” are displayed in Figure 2 and Figure 9. Bond length details are given in Table 4.
Table 1.
Experimental parameter and CCDC-Code.
| Sample | Machine | Source | Temp. | Detector Distance | Time/Frame | #Frames | Frame Width | CCDC |
|---|---|---|---|---|---|---|---|---|
| Bruker | [K] | [mm] | [s] | [°] | ||||
| 1 | X8 | Mo | 100 | 35 | 25 | 3739 | 0.5 | 1586020 |
Table 2.
Sample and crystal data of 1.
| Chemical Formula | C9H8N2O | Crystal System | Orthorhombic | |
|---|---|---|---|---|
| Formula weight (g/mol) | 160.17 | Space group | Pbca | |
| Temperature (K) | 100 | Z | 8 | |
| Measurement method | Φ and ω scans | Volume (Å3) | 1509.4(3) | |
| Radiation (Wavelength (Å)) | MoKα (λ = 0.71073) | Unit cell dimensions (Å) and (°) | 13.6517(13) | 90 |
| Crystal size (mm3) | 0.22 × 0.12 × 0.04 | 6.3663(6) | 90 | |
| Crystal habit | clear colourless plate | 17.3673(17) | 90 | |
| Density (calculated) (g/cm3) | 1.41 | Absorption coefficient/(mm−1) | 0.096 | |
| Abs. correction Tmin | 0.703 | Abs. correction Tmax | 0.746 | |
| Abs. correction type | multi-scan | F(000) (e−) | 672 | |
Table 3.
Data collection and structure refinement of 1.
| Index ranges | −19 ≤ h ≤ 19, −9 ≤ k ≤ 8, −24 ≤ l ≤ 24 | Theta range for data collection (°) | 4.69 to 60.5 | |
| Reflections number | 62937 | Data/restraints/parameters | 2243/0/113 | |
| Refinement method | Least squares | Final R indices | all data | R1 = 0.0473, wR2 = 0.1264 |
| Function minimized | Σ w(Fo2 − Fc2)2 | I > 2σ(I) | R1 = 0.0407, wR2 = 0.1202 | |
| Goodness-of-fit on F2 | 1.085 | Weighting scheme | w = 1/(σ2(Fo2) + (0.0657P)2 + 0.6517P) | |
| Largest diff. peak and hole (e Å−3) | 0.39/−0.21 | where P = (Fo2 + 2Fc2)/3 | ||
Figure 9.

Asymmetric unit of 1, drawn with 50% displacement ellipsoids.
Table 4.
Proof of the bond length for the position of H at O1 [43].
| Bond Lengths in Crystalline Organic Compounds | Compound 1 | ||||||
|---|---|---|---|---|---|---|---|
| d | m | σ | ql | qu | |||
| in pyrazole: (N1–N2) | 1.366 | 1.366 | 0.019 | 1.350 | 1.375 | N1 N2 single bond | 1.376 |
| in pyrazole: (N2=C3) | 1.329 | 1.331 | 0.014 | 1.315 | 1.339 | N1 C7 double bond | 1.329 |
| in pyrazole: (N1–C5) | 1.357 | 1.359 | 0.012 | 1.347 | 1.365 | N1 C9 single bond | 1.354 |
| in enols: C=C–OH | 1.333 | 1.331 | 0.017 | 1.324 | 1.342 | C7 O1 single bond | 1.339 |
| in phenols: Caromatic-OH | 1.362 | 1.364 | 0.015 | 1.353 | 1.373 | ||
| in lactams: (C=O) | 1.240 | 1.241 | 0.003 | 1.237 | 1.243 | ||
| in benzoquinones: (C=O) | 1.222 | 1.220 | 0.013 | 1.211 | 1.231 | ||
d is the unweighted mean in Å of all the values for that bond length found in the sample; m is the median in Å of all values; σ is the standard deviation in the sample; ql is the lower quartile for the sample; qu is the upper quartile for the sample.
The difference electron density map (Figure 2) gives very detailed information about the position of the searched hydrogen atom. The electron density distance of the latter to N2′—as displayed in Figure 2—excludes the possibility of a single bond to the nitrogen for the concerning hydrogen atom. Additionally, its distance to O1 for the electron density proves the position of the hydrogen is located at the oxygen. The free refinement of the hydrogen position without using any restraints or constraints clears all doubts about the non-tautomeric geometry of the molecule in the solid state. Furthermore, at least two very close molecules [41,42] were already measured and interpreted in the same way. In these samples also two identical hydrogen bonds build up molecule pairs because of symmetry reasons.
Pyrazole is well known in crystallography and its different bonds are well characterized by the Handbook of Chemistry and Physics [43]. Table 4 compares the results from compound 1 with corresponding bonds in pyrazole. In detail the double bond N2=C7 in 1 is with 1.329 Å identical to the unweighted mean of the table value for pyrazoles N2=C3 from the Handbook of Chemistry and Physics. The single bond C7-O1 with 1.339 Å is also in strong correlation to expected values like in enols, 1.333 Å, given. Double bond values like in lactams, 1.240 Å, and benzoquinones, 1.222 Å are in contrast to the measured 1.339 Å too small. Finally we proved the position of the searched hydrogen at O1 and we can exclude the NH form in the solid crystalline state.
Acknowledgments
The solid state NMR experiments were performed at the Upper Austrian-South Bohemian Research Infrastructure Center “RERI-uasb” in Linz, co-financed by the European Union in the context of the project EFRE RU2-EU-124/100-2010 (ETC Austria-Czech Republic 2007–2013, project M00146).
Author Contributions
E.A., S.K., A.M., and V.M. accomplished the syntheses; M.B. recorded the solid state NMR spectra; A.R. performed the X-ray structure analysis; A.B. and W.H. conducted the liquid state NMR studies; and W.H. and A.Š. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 1, 4 and 5 are available from the authors.
References
- 1.Elguero J., Goya P., Jagerovic N., Silva A.M.S. Pyrazoles as drugs: Facts and fantasies. In: Attanasi O.A., Spinelli D., editors. Targets in Heterocyclic Systems. Volume 6. Royal Society of Chemistry; Cambridge, UK: 2002. pp. 52–98. [Google Scholar]
- 2.Perez-Fernandez R., Goya P., Elguero J. A review of recent progress (2002–2012) on the biological activities of pyrazoles. ARKIVOC. 2014;ii:233–293. [Google Scholar]
- 3.Schmidt A., Dreger A. Recent advances in the chemistry of pyrazoles. Properties, biological activities, and syntheses. Curr. Org. Synth. 2011;15:1423–1463. doi: 10.2174/138527211795378263. [DOI] [Google Scholar]
- 4.Fustero S., Sánchez-Roselló M., Barrio P., Simón-Fuentes A. From 2000 to Mid-2010: A fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011;111:6984–7034. doi: 10.1021/cr2000459. [DOI] [PubMed] [Google Scholar]
- 5.Ansari A., Ali A., Asif M., Shamsuzzaman Biologically active pyrazole derivatives. New J. Chem. 2017;41:16–41. doi: 10.1039/C6NJ03181A. [DOI] [Google Scholar]
- 6.Lamberth C. Pyrazole Chemistry in Crop Protection. Heterocycles. 2007;71:1467–1502. doi: 10.3987/REV-07-613. [DOI] [Google Scholar]
- 7.Holzer W., Claramunt R.M., Lopez C., Alkorta I., Elguero J. A study in desmotropy. Solid State Nucl. Magn. Reson. 2008;34:68–76. doi: 10.1016/j.ssnmr.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Elguero J., Marzin C., Katritzky A.R., Linda P. The Tautomerism of Heterocycles. Academic Press; New York, NY, USA: 1976. [Google Scholar]
- 9.Minkin V.I., Garnovskii A.D., Elguero J., Katritzky A.R., Denisko O.V. The tautomerism of heterocycles: Five-membered rings with two or more heteroatoms. Adv. Heterocycl. Chem. 2000;76:157–323. [Google Scholar]
- 10.Holzer W., Mereiter K., Plagens B. 4-Acyl-5-methyl-2-phenylpyrazolones: NMR and X-ray structure investigations. Heterocycles. 1999;50:799–818. doi: 10.3987/COM-98-S(H)74. [DOI] [Google Scholar]
- 11.Holzer W., Hallak L. Synthesis and NMR spectroscopic investigations with 3-amino-, 3-hydroxy-, and 3-methoxy-4-acyl-1-phenyl-2-pyrazolin-5-ones. Heterocycles. 2004;63:1311–1334. doi: 10.3987/COM-04-10046. [DOI] [Google Scholar]
- 12.Holzer W., Kautsch C., Laggner C., Claramunt R.M., Pérez-Torralba M., Alkorta I., Elguero J. On the Tautomerism of Pyrazolones: The Geminal 2J[Pyrazole C-4,H-3(5)] Spin Coupling Constant as a Diagnostic Tool. Tetrahedron. 2004;60:6791–6805. doi: 10.1016/j.tet.2004.06.039. [DOI] [Google Scholar]
- 13.Guillou S., Janin Y.L. 5-Iodo-3-Ethoxypyrazoles: An Entry Point to New Chemical Entities. Chem. Eur. J. 2010;16:4669–4677. doi: 10.1002/chem.200903442. [DOI] [PubMed] [Google Scholar]
- 14.Arbačiauskienė E., Laukaitytė V., Holzer W., Šačkus A. Metal-free intramolecular alkyne-azide cycloaddition leading to the novel pyrazolo[4,3-f][1,2,3]triazolo[5,1-c][1,4]oxazepine ring system. Eur. J. Org. Chem. 2015:5663–5670. [Google Scholar]
- 15.Li Y., Liu R., Yan Z., Zhang X., Zhu H. Synthesis, Crystal Structure and Fungicidal Activities of New Type Oxazolidinone-Based Strobilurin Analogues. Bull. Korean Chem. Soc. 2010;31:3341–3347. doi: 10.5012/bkcs.2010.31.11.3341. [DOI] [Google Scholar]
- 16.Li Y., Liu Y.-Y., Chen N.-Q., Lue K.-Z., Xiong X.-H., Li J. One-Pot Regioselective Synthesis of Novel Oximino Ester-Containing 1-Aryl-4-chloro-3-oxypyrazoles as Potential Fungicides. Helv. Chim. Acta. 2014;97:1269–1282. doi: 10.1002/hlca.201300407. [DOI] [Google Scholar]
- 17.Liu Y., Li Y., Chen N., Lv K., Zhou C., Xiong X., Li F. Synthesis and Fungicidal Activity of Novel Chloro-Containing 1-Aryl-3-oxypyrazoles with an Oximino Ester or Oximino Amide Moiety. Molecules. 2014;19:8140–8150. doi: 10.3390/molecules19068140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elguero J., Katritzky A.R., Denisko O.V. Advances in Heterocyclic Chemistry. Volume 76. Academic Press; Cambridge, MA, USA: 2000. Prototropic Tautomerism of Heterocycles: Heteroaromatic Tautomerism—General Overview and Methodology; pp. 1–84. [Google Scholar]
- 19.Sanz D., Claramunt R.M., Alkorta I., Elguero J. The use of chemical shifts vs. coupling constants for studying tautomerism: A combined experimental and theoretical approach. Struct. Chem. 2007;18:703–708. doi: 10.1007/s11224-007-9208-4. [DOI] [Google Scholar]
- 20.Katritzky A.R., Karelson M., Harris P.A. Prototropic tautomerism of heteroaromatic compounds. Heterocycles. 1991;32:329–369. doi: 10.3987/REV-90-421. [DOI] [Google Scholar]
- 21.Von Philipsborn W., Müller R. 15N-NMR Spectroscopy-New Methods and Application. Angew. Chem. Int. Ed. Engl. 1986;25:383–413. doi: 10.1002/anie.198603833. [DOI] [Google Scholar]
- 22.Gil V.M.S., von Philipsborn W. Effect of electron lone-pairs on nuclear spin-spin coupling constants. Magn. Reson. Chem. 1989;27:409–430. doi: 10.1002/mrc.1260270502. [DOI] [Google Scholar]
- 23.Berger S., Braun S., Kalinowski H.-O. NMR-Spektroskopie von Nichtmetallen: 15N-NMR-Spektroskopie. Volume 2. Thieme; Stuttgart, Germany: New York, NY, USA: 1992. pp. 42–43. [Google Scholar]
- 24.Begtrup M. Hydrogen-1 and carbon-13-NMR spectra of phenyl-substituted azole derivatives. II. Conformational study. Acta Chem. Scand. B. 1974;28:61–77. doi: 10.3891/acta.chem.scand.28b-0061. [DOI] [Google Scholar]
- 25.Carrillo J.R., Cossio F.P., Diaz-Ortiz A., Gomez-Escalonilla M.J., de la Hoz A., Lecea B., Moreno A., Prieto P. A complete model for the prediction of 1H- and 13C-NMR chemical shifts and torsional angles in phenyl-substituted pyrazoles. Tetrahedron. 2001;57:4179–4187. doi: 10.1016/S0040-4020(01)00291-5. [DOI] [Google Scholar]
- 26.Kleizienė N., Arbačiauskienė E., Holzer W., Šačkus A. 4-Bromo-3-methoxy-1-phenyl-1H-pyrazole. Molbank. 2009:M639. doi: 10.3390/M639. [DOI] [Google Scholar]
- 27.Kalinowski H.-O., Berger S., Braun S. 13C-NMR Spektroskopie. Thieme; Stuttgart, Germany: New York, NY, USA: 1984. p. 194. [Google Scholar]
- 28.Arbačiauskienė E., Vilkauskaite G., Eller G.A., Holzer W., Sackus A. Pd-Catalyzed Cross-Coupling Reactions of Halogenated 1-Phenylpyrazol-3-ols and Related Triflates. Tetrahedron. 2009;65:7817–7824. doi: 10.1016/j.tet.2009.07.017. [DOI] [Google Scholar]
- 29.O’Brien D.F., Gates J.W., Jr. Some Reactions of 3-Hydroxy-1-phenylpyrazole. J. Org. Chem. 1966;31:1538–1542. doi: 10.1021/jo01343a054. [DOI] [Google Scholar]
- 30.Begtrup M. Reactions between azolium salts and nucleophilic reagents. IX. Cine-substitution of pyrazolium salts. Acta Chem. Scand. 1973;27:2051–2074. doi: 10.3891/acta.chem.scand.27-2051. [DOI] [Google Scholar]
- 31.Arbačiauskienė E., Martynaitis V., Krikštolaitytė S., Holzer W., Šačkus A. Synthesis of 3-substituted 1-phenyl-1H-pyrazole-4-carbaldehydes and the corresponding ethanones by Pd-catalysed cross-coupling reactions. ARKIVOC. 2011;xi:1–21. [Google Scholar]
- 32.Mikhaleva M.A., Mamaev V.P. Interaction of 3-hydroxy- and 4-hydroxypyrazoles with benzylidenebisurea. Izv. Sib. Otd. Akad. Nauk SSSR Seriya Khimicheskikh Nauk. 1969;6:93–98. [Google Scholar]
- 33.Nedzelskyte E., Martynaitis V., Šačkus A., Eller G.A., Holzer W. Synthesis of Mono- and Dibromo-Derivatives of 1-Phenylpyrazol-3-ol. Molbank. 2007:M551. doi: 10.3390/M551. [DOI] [Google Scholar]
- 34.Sucrow W., Mentzel C., Slopianka M. Enehydrazines. 9. 1-Alkyl-3-hydroxypyrazoles from hydrazones or hydrazines. Chem. Ber. 1974;107:1318–1328. doi: 10.1002/cber.19741070428. [DOI] [Google Scholar]
- 35.Bruker SAINT, V7.68A. Copyright © 2005–2018 Bruker AXS; Madison, WI, USA: 2012. [Google Scholar]
- 36.Sheldrick G.M. SHELXS. University of Göttingen; Göttingen, Germany: 1996. [Google Scholar]
- 37.Sheldrick G.M. A short history of SHELX. Acta Cryst. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 38.Dolomanov O.V., Bourhis L.J., Gildea R.J., Howard J.A.K., Puschmann H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009;42:339–341. doi: 10.1107/S0021889808042726. [DOI] [Google Scholar]
- 39.Huebschle C.B., Sheldrick G.M., Dittrich B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011;44:1281–1284. doi: 10.1107/S0021889811043202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.CCDC 1586020 Contains the Supplementary Crystallographic Data for This Paper. [(accessed on 16 November 2017)]; Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html.
- 41.Bechtel F., Gaultier J., Hauw C. 1-Phenyl-5-methyl-3-pyrazolone, C10H10N2O. Cryst. Struct. Commun. 1973;2:473–476. [Google Scholar]
- 42.Ren X.-Y., Wang J.-G., Li Y.-Y. 1-(4-Chlorophenyl)-1H-pyrazol-3-ol. Acta Cryst. 2010;E66:o186. doi: 10.1107/S1600536809053641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Handbook of Chemistry and Physics, 96th Edition 2015/16. CRC Press; Boca Raton, FL, USA: 2015. Bond Lengths in Crystalline Organic Compounds. [Google Scholar]