

Commentary
It is now clear that changes in astrocyte channels, transporters, and metabolism play a critical role in seizure susceptibility and epilepsy. In particular, alterations in astrocyte potassium, water, glucose, and adenosine homeostasis and gap junctional coupling have all been associated with hyperexcitability (largely in temporal lobe epilepsy). Distinct astrocytic changes have been identified in other types of epilepsy, such as tuberous sclerosis, tumor-associated epilepsy, and posttraumatic epilepsy. Together, the emerging literature on astrocytes and epilepsy provides a powerful rationale for distinct new therapeutic targets that are astrocyte specific.
Epilepsy as an “astrocytopathy”
Astrocytes play an established role in removal of glutamate at synapses and the sequestration and redistribution of K+ and H2O during neural activity (1, 2) (Figure 1). It is becoming increasingly clear that changes in astrocyte channels, transporters, and metabolism play a direct role in seizure susceptibility and the development of epilepsy (2–8). Stimulation of astrocytes leads to prolonged neuronal depolarization and epileptiform discharges (6). Astrocytes release neuroactive molecules and modulate synaptic transmission through modifications in channels, gap junctions, receptors, and transporters (2, 3, 6, 9–14). Furthermore, striking changes in astrocyte form and function occur in epilepsy. Astrocytes adopt reactive morphology (4, 15), become uncoupled (16), and lose domain organization (17) in epileptic tissue. These and other changes, such as altered expression of various astrocytic enzymes, for example, adenosine kinase (16) and glutamine synthetase (18), astroglial proliferation, dysregulation of water and ion channel and glutamate transporter expression (2, 19, 20), alterations in secretion of neuroactive molecules, and increased activation of inflammatory pathways (2, 4, 8, 15, 21–24) may all contribute to hyperexcitability and epileptogenesis.
FIGURE 1.
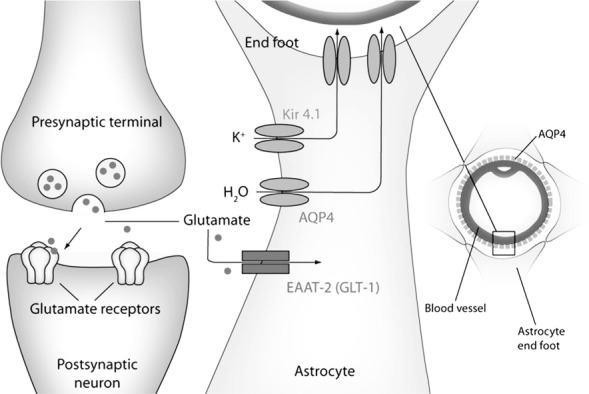
Astrocyte regulation of water, potassium, and glutamate homeostasis at the tripartite synapse. Colocalization of AQP4, K ir4.1, and GLT1 in distinct astrocyte membrane domains (perisynaptic, perivascular) provides the basis for a critical role of astrocytes in control of water, potassium, and glutamate homeostasis. Reproduced with permission from: Benarroch E. Aquaporin-4, homeostasis, and neurologic disease. Neurology 2007;69:2266–2268.
Alterations in astrocyte channels, transporters, and enzymes Kir channels
During neuronal hyperactivity, K+ released by active neurons is thought to be primarily taken up by astrocytes. Any impairment of astrocyte K+ uptake would be expected to be proconvulsant: in the hippocampus, millimolar and even submillimolar increases in extracellular K+ concentration powerfully enhance epileptiform activity. A primary mechanism for K+ reuptake is via glial inwardly rectifying K+ channels (Kir channels). However, downregulation of astroglial Kir channels occurs in the injured or diseased CNS. Kir currents are reduced following injury-induced reactive gliosis in vitro, entorhinal cortex lesion, freeze lesion-induced cortical dysplasia, and traumatic and ischemic brain injury. In addition, several studies have indicated downregulation of Kir currents in specimens from patients with temporal lobe epilepsy (TLE). Using ion-sensitive microelectrodes, Heinemann's group compared glial Ba2+-sensitive K+ uptake in the CA1 region of hippocampal slices obtained from patients with or without mesial temporal sclerosis (MTS) (24, 25). Ba2+, a blocker of Kir channels, augmented stimulus-evoked K+ elevation in nonsclerotic but not in sclerotic specimens, suggesting impairment in K+ buffering in sclerotic tissue. Direct evidence for downregulation of Kir currents in the sclerotic CA1 region of hippocampus came from a comparative patch-clamp study in which a reduction in astroglial Kir currents was observed in sclerotic compared with nonsclerotic hippocampi (23). These data indicate that dysfunction of astroglial Kir channels could underlie impaired K+ buffering and contribute to hyperexcitability in epileptic tissue (21). When and how this dysfunction develops during epileptogenesis is not yet clear.
Aquaporin-4 water channels
Alterations in astroglial water regulation could also powerfully affect excitability (2). Brain tissue excitability is exquisitely sensitive to osmolarity and the size of the extracellular space (ECS) (26). Decreasing ECS volume produces hyperexcitability and enhanced epileptiform activity; conversely, increasing ECS volume with hyperosmolar medium attenuates epileptiform activity. These experimental data parallel extensive clinical experience indicating that hypo-osmolar states, such as hypo-natremia lower seizure threshold, while hyperosmolar states elevate seizure threshold (27).
The aquaporins (AQPs) are a family of membrane proteins that function as “water channels” in many cell types and tissues in which fluid transport is crucial (28). Aquaporin-4 (AQP4) is expressed ubiquitously by glial cells, especially at specialized membrane domains including astroglial end feet in contact with blood vessels and astrocyte membranes that ensheathe glutamatergic synapses. Mice deficient in AQP4 have markedly decreased accumulation of brain water (cerebral edema) following water intoxication and focal cerebral ischemia (29) and impaired clearance of brain water in models of vasogenic edema (30), suggesting a functional role for AQP4 in brain water transport. In addition, AQP4−/− mice have remarkably impaired K+ clearance and prolonged seizures in response to in vivo hippocampal stimulation (31). These data suggest that AQP4 downregulation may trigger hyperexcitability.
Alteration in the expression and subcellular localization of AQP4 has been described in sclerotic hippocampi obtained from patients with MTS. Using immunohistochemistry, RT-PCR, and gene chip analysis, Lee et al. (32) demonstrated an overall increase in AQP4 expression in sclerotic hippocampi. However, using quantitative immunogold electron microscopy, the same group found mislocalization of AQP4 in the human epileptic hippocampus, with reduction in perivascular membrane expression (33). The authors hypothesized that the loss of perivascular AQP4 perturbs water flux, impairs K+ buffering, and results in an increased propensity for seizures.
Subsequently, very similar AQP4 dysregulation has been confirmed in animal models of epilepsy. In particular, downregulation and/or mislocalization of AQP4 occur during the early epileptogenic phase in the rat pilocarpine (34, 35), rat kainic acid (36), and mouse kainic acid (19, 20) models of epilepsy. Based on these data, restoration of AQP4 homeostasis may represent a novel antiepileptogenic strategy (2).
Glutamate transporters and glutamine synthetase
Several CNS cell types express glutamate transporters, but astrocytes are primarily responsible for glutamate uptake. Studies using mice with deletion (37) or antisense oligonucleotide-mediated inhibition (38) of the astroglial transporter glutamate transporter 1(GLT1), also called excitatory amino acid transporter-2 (EAAT2), revealed that this subtype is responsible for the bulk of extracellular glutamate clearance in the CNS (39). Several studies have suggested an involvement of glutamate transporters in seizure development. GLT1 knockout in mice caused spontaneous seizures and hippocampal pathology resembling alterations in TLE patients with MTS (37). A more recent follow-up paper by the same group using region-specific GLT1 knockouts confirmed spontaneous seizures when GLT1 was deleted from forebrain astrocytes (40).
What is the evidence for alteration in astrocyte glutamate transporters in human epilepsy specimens and in animal models? Decreased GLT1 immunoreactivity has been reported in the sclerotic human hippocampus, although glutamate aspartate transporter (GLAST) immunoreactivity was reported as unchanged (41) or decreased (42). These findings support the hypothesis that reduced or dysfunctional glial glutamate transporters in the hippocampus may trigger spontaneous seizures in patients with MTS (43). A recent study in the mouse intrahippocampal kainic acid model found a significant initial increase in dorsal hippocampal GLT1 immunoreactivity and protein levels 1 day after status epilepticus followed by a marked downregulation at 4 and 7 days post status epilepticus, a time period during which spontaneous seizures arise in this model (19). Therefore, early GLT1 dysregulation may precede spontaneous seizures, and thus contribute to epileptogenesis.
Another potential mechanism for glutamate dysregulation is loss of the astrocyte enzyme glutamate synthetase in the sclerotic versus nonsclerotic hippocampus of TLE patients (44). After uptake of glutamate into astrocytes, this enzyme rapidly converts glutamate into glutamine. In sclerotic TLE hippocampus, downregulation of glutamate synthetase caused a slowing of the glutamate-glutamine cycling and accumulation of glutamate in astrocytes and in the extracellular space (44). This conclusion was compatible with findings in animal models of epilepsy and earlier data demonstrating slowed glutamate-glutamine cycling in sclerotic human epileptic hippocampus with magnetic resonance spectroscopy (45).
Gap junctions
Functional coupling analysis, obtained by patch-clamping astrocytes and filling the astrocyte syncytium with dyes to quantitatively measure astrocyte-astrocyte gap junction coupling, has led to recent seminal findings of astrocyte “uncoupling” in human and animal TLE (46). In this study, the gap junctional connectivity of astrocytes from 119 specimens from patients with mesial temporal lobe epilepsy (MTLE) with and without sclerosis was examined. In MTLE specimens with typical hippocampal sclerosis, there is a complete absence of typical “classical” astrocytes and astrocyte gap junctional coupling. In contrast, coupled astrocytes were abundant in nonsclerotic hippocampus. In the intracortical kainic acid (ICKA) model of TLE, mice exhibited decreased astrocytic coupling 4 to 5 days post injection and completely lacked coupling 3 and 6 months after status epilepticus in the sclerotic hippocampus (46). In the nonsclerotic hippocampus, however, coupling remained intact. Interestingly, decreased astrocyte coupling preceded apoptotic neuronal death and the onset of spontaneous seizures. Decreased gap junction coupling also impaired K+ clearance 4 hours post injection. The authors found that proinflammatory cytokines induced the uncoupling of hippocampal astrocytes in vivo (46), which agreed with similar in vitro findings that pro-inflammatory cytokines have an inhibitory effect on astrocytic gap junctional coupling (47). Their data suggest that inflammation may contribute to rapid uncoupling of astrocytes and the decoupling of astrocytes may be involved in epileptogenesis.
How general a mechanism is astrocytic uncoupling for other forms of epilepsy? In a follow-up paper, uncoupling of astrocytes was also observed in a febrile seizure model (48). This body of work suggests that restoration of gap junctional coupling in astrocytes, perhaps via modulation of the toll-like receptor 4 (TLR4) pathway, represents a novel therapeutic strategy.
Adenosine
Adenosine levels are elevated during seizure activity. Adenosine exerts a powerful inhibitory effect on excitatory synaptic transmission primarily through its interaction with presynaptic A1 adenosine receptors (A1Rs) to suppress neurotransmitter release. Once released from neurons and astrocytes, ATP is rapidly converted into adenosine monophosphate (AMP) and then into adenosine by extracellular nucleotidases. The reuptake of adenosine occurs through equilibrative nucleoside transporters, and phosphorylation by the astrocyte-specific enzyme adenosine kinase (ADK) breaks down adenosine and clears excess adenosine from the extracellular space. Therefore, alterations in ADK are especially relevant to the generation of seizures. Increased ADK expression has been linked to seizure activity in both human tissue and experimental models of epilepsy (16, 49–51).
Collectively, the above findings support the adenosine kinase hypothesis of epileptogenesis (49, 50), including the dysregulation of ADK and its contribution to the epileptogenic cascade. Adenosine, adenosine receptor agonists, and ADK inhibitors have well established anticonvulsant efficacy (52–55). Intracranial injection of adenosine prevents seizures in rats (56). In addition, the use of transgenic mice revealed that reduced forebrain ADK protects against epileptogenesis (57). Other studies involving adenosine augmentation therapies (AAT) include a silk protein–based release system for adenosine (58) and the local release of adenosine from grafted cells (59), both of which resulted in seizure suppression. Focal adenosine delivery, such as slow-release polymers, cellular implants, gene therapy, or pump systems, has been suggested as a new pharmacologic tool to treat refractory epilepsy with minimal side effects (60).
Particularly exciting is the recent finding that even a transient adenosine augmentation may have longer-lasting epigenetic effects that are antiepileptogenic (61, 62). Probably the most effective treatment would be a brain-permeant peripherally administered small molecule inhibitor of ADK. This would hopefully obviate systemic side effects seen with direct adenosine delivery. If effective in triggering long-lasting antiepileptogenesis, such a drug would ideally need to be given only during an isolated therapeutic window just after an epileptogenic stimulus.
Examples of astrocyte dysfunction in specific epilepsy syndromes
Tuberous sclerosis
Tuberous sclerosis (TS) is a multisystem genetic disorder resulting from autosomal dominant mutations of either the TSC1 or TSC2 genes. The TSC1 gene encodes the protein hamartin and TSC2 encodes tuberin, which are thought to be regulators of cell signaling and growth. Epilepsy occurs in 80 to 90 percent of cases of TS, frequently involves multiple seizure types, and is often medically refractory. Cortical tubers represent the pathologic substrate of TS, and microscopically consist of a specific type of dysplastic lesion with astrocytosis and abnormal giant cells. While this suggests that astrocytes are involved in the pathologic lesion, in itself this is not evidence for a causative role of astrocytes in TS epileptogenesis. However, evidence using astrocyte-specific TSC1 conditional knockout mice has provided insight into a potential role of astrocytes in the etiology of TS. These mice, which have conditional inactivation of the TSC1 gene in glial fibrillary acidic protient (GFAP) -expressing cells (TSC1FAPCKO mice), develop severe spontaneous seizures by 2 months of age and die prematurely (63). Intriguingly, the time point of onset of spontaneous seizures in these mice is concordant with increased astroglial proliferation. Furthermore, two functions of astrocytes—glutamate and K+ reuptake—are impaired in these mice. These mice display reduced expression of the astrocyte glutamate transporters GLT1 and GLAST (64). In addition, recent evidence indicates that astrocytes from Tsc1GFAPCKO mice exhibit reduced Kir channel activity, and hippocampal slices from these mice demonstrated increased sensitivity to K+-induced epileptiform activity (65). A more recent inducible Tsc1 knockout mouse in which Tsc1 gene inactivation in GFAP-expressing cells was done at 2 weeks of age was sufficient to cause astrogliosis and mild epilepsy (but the phenotype was less severe than prenatal Tsc1 gene activation) (66). Together, these studies demonstrate that in this model, changes in glial properties may be a direct cause of epileptogenesis.
Tumor-associated epilepsy
Tumor-associated epilepsy is an important clinical problem, seen in approximately one-third of tumors. Surgical removal of tumors usually results in seizure control, but many tumors cannot safely be resected, and tumor-associated seizures are often resistant to anticonvulsant therapy. Classic epilepsy-associated brain tumors include astrocytoma, oligodendroglioma, ganglioglioma, dysembryoplastic neuroepithelial tumor, and pleomorphic xanthoastrocytoma (67). Microdialysis studies of gliomas have revealed reduced glutamate in the tumor compared with peritumoral tissue (68). A “glutamate hypothesis” of tumor-associated epilepsy has been advanced, which suggests that tumors excite surrounding tissue by glutamate overstimulation. Two lines of evidence are relevant to this hypothesis. First, the glutamate receptor subunit GluR2 has been found to be under-edited at the glutamine/arginine (Q/R) site in gliomas, which would increase a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor Ca+2 permeability and potentially result in increased glutamate release by glioma cells (69). Second, Sontheimer's group found that glioma cells release larger than normal amounts of glutamate in vitro (70). The release of glutamate was accompanied by a marked deficit in Na+-dependent glutamate uptake, reduced expression of astrocytic glutamate transporters, and upregulation of cystine-glutamate exchange (71). Hence, glioma cell glutamate release at the margins of the tumor may initiate seizures in peritumoral neurons. Another distinct potential mechanism underlying tumor-associated epilepsy is altered K+ homeostasis. In support of this hypothesis, both reduced Kir currents (72) and mislocalization of Kir4.1 channels (73) have been found in malignant astrocytes. Recent studies have shown that a hypothesized glutamate release pathway, cysteine-glutamate transporter (SXC), is active in a subset of gliomas (74). SLC7A11/xCT, the catalytic subunit of SXC, demonstrated elevated expression in approximately 50% of patient tumors. Compared with tumors lacking this transporter, SLC7A11-positive tumors were associated with faster growth, peritumoral glutamate excitotoxicity, seizures, and decreased survival. In a translational pilot study, use of the FDA-approved SXC inhibitor sulfasalazine in nine patients with biopsy-proven SXC expression led to inhibition of glutamate release from the tumor in vivo as assessed by magnetic resonance spectroscopy (74). This exciting study demonstrates that phenotyping tumors for glial-associated transport molecules will lead to selective pharmacologic targeting to prevent or ameliorate tumor-associated epilepsy, and addresses the pathologic mechanism of glutamate release from tumor cells rather than standard antiepileptic drug approaches of globally suppressing synaptic transmission.
Posttraumatic epilepsy
Posttraumatic epilepsy (PTE) refers to a recurrent seizure disorder whose cause is traumatic brain injury (TBI). PTE develops in a variable proportion of TBI survivors depending on the severity of the injury and the time after injury (75, 76). Anticonvulsant prophylaxis is ineffective at preventing the occurrence of late seizures (77–79). Various animal models of PTE have demonstrated characteristic structural and functional changes in the hippocampus, such as death of dentate hilar neurons and mossy fiber sprouting (80–82). Recently, studies have also implicated altered astrocyte function in PTE models. Recordings from glial cells in hippocampal slices 2 days after fluid-percussion injury demonstrated reduction in transient outward and inward K+ currents, and antidromic stimulation of CA3 led to abnormal extracellular K+ accumulation in posttraumatic slices compared with controls (83). This was accompanied by the appearance of electrical after-discharges in CA3. Thus, this study suggests impaired K+ homeostasis in posttraumatic hippocampal glia. Another study demonstrated reduction in expression of the astrocyte glutamate transporter GLT1 in a PTE model induced by intracortical ferrous chloride injection, suggesting impaired glutamate transport (84). Further studies of the role of glial cells appear warranted now that reliable PTE animal models have been developed (85). In particular, long-term changes in astrocyte channels and transporters after TBI that may correlate with PTE should be investigated.
Because TBI is associated with breakdown of the blood–brain barrier (BBB) at the time of the initial event, studies of BBB disruption-induced epileptogenesis are also relevant to mechanisms of PTE. Indeed, transient opening of the BBB is sufficient for focal epileptogenesis (86). Extravasated albumin can be taken up by astrocytes, which activates the transforming growth factor-β (TGF-β) pathway leading to focal epileptogenesis. Weissberg et al. have discovered a mechanism by which albumin induces excitatory synaptogenesis through astrocyte TGF-β/ALK5 signaling. This mechanism provides an astrocytic basis for BBB disruption-induced epileptogenesis and suggests antiepileptogenic therapeutic approaches (TGF-β inhibition). Indeed, losartan, a TGF-β inhibitor and FDA-approved antihypertensive medication, was found to exert antiepileptogenic effects in these BBB disruption models (88, 89). It will be of interest in the future to test similar strategies in PTE models for antiepileptogenic efficacy.
Conclusions
An understanding of the manifold structural and functional changes in astrocytes that occur during epileptogenesis is gradually emerging. It is clear that based on the critical role of astrocytes in K+, glutamate, and water homeostasis at the synapse (Figure 1), alterations of these and closely related metabolic mechanisms can lead to multiple astrocytic sources of hyperexcitability (see Figure 2 for a recent unifying hypothesis related to astrocyte swelling mechanisms) (90).
FIGURE 2.
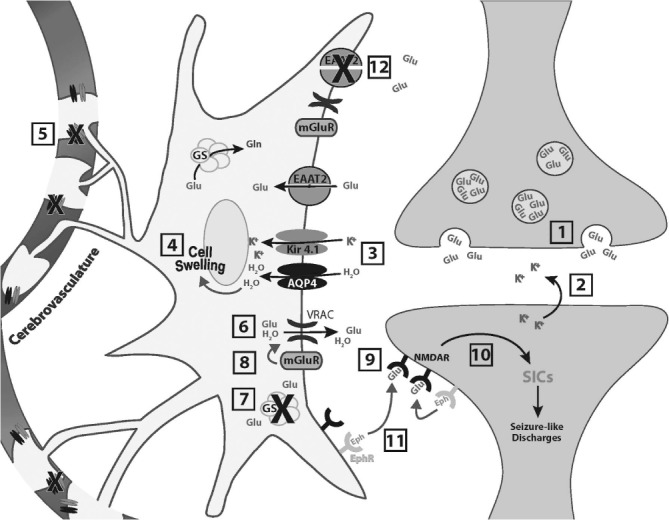
Model for role of astrocyte swelling in neuronal excitability. At an excitatory synapse the neurotransmitter glutamate (Glu) is released from vesicles within the presynaptic terminal [1]. Glu binds to receptors on the postsynaptic membrane and opens ion channels (not shown) allowing the movement of ions across the membrane, including the release of potassium ions [2]. Elevated levels of K+ promote uptake by perisynaptic astrocytes along with water (H2O) predominantly by Kir4.1 potassium channels and aquaporin-4 (AQP4) channels, respectively [3]. This leads to cell swelling and a reduction in extracellular space [4]. Loss or redistribution of AQP4 and Kir4.1 away from astrocytic end feet [5] would exacerbate astrocyte swelling because water flux into astrocytes at synapses would be increased while efflux via end feet into the cerebrovasculature would be decreased. Astrocyte swelling opens volume-regulated anion channels (VRACs), which release glutamate into the extracellular space [6]. In addition, reduced expression of glutamine synthetase (GS) in the epileptic condition [7] elevates the cytoplasmic concentration of glutamate in astrocytes, providing more glutamate to be released. Upregulation of metabotropic glutamate receptors (mGluRs) in reactive astrocytes could enhance AQP4-dependent swelling and swelling-evoked release of glutamate through VRACs [8]. Astrocytically released glutamate can then bind to extrasynaptic N-methyl-D-aspartate receptors (NMDARs) [9], generating slow inward currents (SICs) and potentially interictal and ictal (seizure-like) discharges [10]. Facilitated by the close proximity of adjacent cellular membranes during cell swelling, Eph receptor (EphR)–ephrin (Eph) ligand interactions may further enhance the stimulation of NMDA receptors [11]. EphRs, ephrins, and certain NMDA receptor subunits are upregulated after neural injury on different cell types, including reactive astrocytes and neurons. Reduced excitatory amino transporter 2 (EAAT2) expression in epileptic tissue may lead to delayed clearance of glutamate from the extrasynaptic space [12]. All of the above mechanisms may contribute to astrocyte control of neuronal excitability. Reproduced with permission from: Hubbard JA, Hsu MS, Fiacco TA, Binder DK. Glial cell changes in epilepsy: overview of the clinical problem and therapeutic opportunities. Neurochem Int 2013;63:638–651.
While animal models and human tissue studies have concordantly suggested astrocytic involvement in epilepsy, both levels of investigation have certain limitations. Animal studies may not accurately represent the disease progression as seen in humans; and human tissue obtained from resected specimens does not allow determination of whether observed cellular and molecular changes are a cause or a consequence of epilepsy. Future studies should focus on characterizing astrocyte alterations that occur prior to spontaneous seizure onset (i.e., during early epileptogenesis) in distinct models of epilepsy, as this could lead to a greater understanding of disease pathogenesis. The term “reactive gliosis” is too descriptive and should be replaced by careful morphologic, biochemical, and electrophysiologic studies of identified glial cell subtypes in human tissue and animal models, paying particular attention to astrocyte heterogeneity (91, 92). In addition to changes in preexisting glial cell populations, newly generated glial cells with distinct properties may migrate into the hippocampus and contribute to enhanced seizure susceptibility (93, 94). The available data likely represent only the “tip of the iceberg” in terms of the functional role of astroglial cells in epilepsy (for more information please see Ref. 95). Further study of astrocyte alterations in epilepsy should continue to unearth new molecular targets and open new avenues for the development of creative and restorative antiepileptic therapies.
References
- 1. Ransom B, Behar T, Nedergaard M.. New roles for astrocytes (stars at last). Trends Neurosci 2003; 26: 520– 522. [DOI] [PubMed] [Google Scholar]
- 2. Binder DK, Nagelhus EA, Ottersen OP.. Aquaporin-4 and epilepsy. Glia 2012; 60: 1203– 1214. [DOI] [PubMed] [Google Scholar]
- 3. Binder DK, Steinhäuser C.. Functional changes in astroglial cells in epilepsy. Glia 2006; 54: 358– 368. [DOI] [PubMed] [Google Scholar]
- 4. Clasadonte J, Haydon PG.. Astrocytes and epilepsy. : Jasper's Basic Mechanisms of the Epilepsies. ( Noebles JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, ) Bethesda: National Center for Biotechnology Information, 2012: 19. [PubMed] [Google Scholar]
- 5. Friedman A, Kaufer D, Heinemann U.. Blood-brain barrier breakdown-inducing astrocytic transformation: novel targets for the prevention of epilepsy. Epilepsy Res 2009; 85: 142– 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tian G, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Zielke R, Kang J, Nedergaard M.. An astrocytic basis of epilepsy. Nat Med 2005; 11: 973– 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seifert G, Carmignoto G, Steinhäuser C.. Astrocyte dysfunction in epilepsy. Brain Res Rev 2010; 63: 212– 221. [DOI] [PubMed] [Google Scholar]
- 8. Seifert G, Schilling K, Steinhäuser C.. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci 2006; 7: 194– 206. [DOI] [PubMed] [Google Scholar]
- 9. Beenhakker MP, Huguenard JR.. Astrocytes as gatekeepers of GABAB receptor function. J Neurosci 2010; 30: 15262– 15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang F, Smith NA, Xu Q, Fujita T, Baba A, Matsuda T, Takano T, Bekar L, Nedergaard M.. Astrocytes modulate neural network activity by Ca2+-dependent uptake of extracellular K+. Sci Signal 2012; 5: ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santello M, Bezzi P, Volterra A.. TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 2011; 69: 988– 1001. [DOI] [PubMed] [Google Scholar]
- 12. Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C.. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008; 322: 1551– 1555. [DOI] [PubMed] [Google Scholar]
- 13. Volterra A, Steinhäuser C.. Glial modulation of synaptic transmission in the hippocampus. Glia 2004; 47: 249– 257. [DOI] [PubMed] [Google Scholar]
- 14. Halassa MM, Fellin T, Haydon PG.. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med 2007; 13: 54– 63. [DOI] [PubMed] [Google Scholar]
- 15. Heinemann U, Jauch GR, Schulze JK, Kivi A, Eilers A, Kovacs R, Lehmann TN.. Alterations of glial cell functions in temporal lobe epilepsy. Epilepsia 2000; 41 Suppl 6: S285– S189. [DOI] [PubMed] [Google Scholar]
- 16. Aronica E, Zurolo E, Iyer A, de Groot M, Anink J, Carbonell C, van Vliet EA, Baayen JC, Boison D, Gorter JA.. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 2011; 52: 1645– 1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, Nedergaard M.. Loss of astrocytic domain organization in the epileptic brain. J Neurosci 2008; 28: 3264– 3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coulter DA, Eid T.. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 2012; 60: 1215– 1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hubbard JA, Szu JI, Yonan JM, Binder DK.. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp Neurol 2016; 283 Pt A: 85– 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee DJ, Hsu MS, Seldin MM, Arellano JL, Binder DK.. Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp Neurol 2012; 235: 246– 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Steinhäuser C, Seifert G.. Glial membrane channels and receptors in epilepsy: imact for generation and spread of seizure activity. Eur J Pharmacol 2002; 447: 227– 237. [DOI] [PubMed] [Google Scholar]
- 22. de Lanerolle NC, Lee T.. New facets of the neuropathology and molecular profile of human temporal lobe epilepsy. Epilepsy Behav 2005; 7: 190– 203. [DOI] [PubMed] [Google Scholar]
- 23. Hinterkeuser S, Schröder W, Hager G, Seifert G, Blümcke I, Elger CE, Schramm J, Steinhäuser C.. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci 2000; 12: 2087– 2096. [DOI] [PubMed] [Google Scholar]
- 24. Kivi A, Lehmann TN, Kovács R, Eilers A, Jauch R, Meencke HJ, von Deimling A, Heinemann U, Gabriel S.. Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci 2000; 12: 2039– 2048. [DOI] [PubMed] [Google Scholar]
- 25. Heinemann U, Gabriel S, Jauch R, Schulze K, Kivi A, Eilers A, Kovacs R, Lehmann TN.. Alterations of glial cell function in temporal lobe epilepsy. Epilepsia 2000;( 41 Suppl 6: S185– S189. [DOI] [PubMed] [Google Scholar]
- 26. Schwartzkroin PA, Baraban SC, Hochman DW.. Osmolarity, ionic flux, and changes in brain excitability. Epilepsy Res 1998; 32: 275– 285. [DOI] [PubMed] [Google Scholar]
- 27. Andrew RD, Fagan M, Ballyk BA, Rosen AS.. Seizure susceptibility and the osmotic state. Brain Res 1989; 498: 175– 180. [DOI] [PubMed] [Google Scholar]
- 28. Verkman AS. More than just water channels: unexpected cellular roles of aquaporins. J Cell Sci 2005; 118 Pt 15: 3225– 3232. [DOI] [PubMed] [Google Scholar]
- 29. Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS.. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 2000; 6: 159– 163. [DOI] [PubMed] [Google Scholar]
- 30. Papadopoulos MC, Manley GT, Krishna S, Verkman AS.. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J 2004; 18 11: 1291– 1293. [DOI] [PubMed] [Google Scholar]
- 31. Binder DK, Yao X, Sick TJ, Verkman AS, Manley GT.. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 2006; 53: 631– 636. [DOI] [PubMed] [Google Scholar]
- 32. Lee TS, Eid T, Mane S, Kim JH, Spencer DD, Ottersen OP, de Lanerolle NC.. Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol (Berl) 2004; 108: 493– 502. [DOI] [PubMed] [Google Scholar]
- 33. Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjornsen LP, Spencer DD, Agre P, Ottersen OP, de Lanerolle NC.. Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci U S A 2005; 102: 1193– 1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim JE, Ryu HJ, Yeo SI, Seo CH, Lee BC, Choi IG, Kim DS, Kang TC.. Differential expressions of aquaporin subtypes in astroglia in the hippocampus of chronic epileptic rats. Neuroscience 2009; 163: 781– 789. [DOI] [PubMed] [Google Scholar]
- 35. Kim JE, Yeo SI, Ryu HJ, Kim MJ, Kim DS, Jo SM, Kang TC.. Astroglial loss and edema formation in the rat piriform cortex and hippocampus following pilocarpine-induced status epilepticus. J Comp Neurol 2010; 518: 4612– 4628. [DOI] [PubMed] [Google Scholar]
- 36. Alvestad S, Hammer J, Hoddevik EH, Skare O, Sonnewald U, Amiry-Moghaddam M, Ottersen OP.. Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res 2013; 105: 30– 41. [DOI] [PubMed] [Google Scholar]
- 37. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K.. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997; 276: 1699– 1702. [DOI] [PubMed] [Google Scholar]
- 38. Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF.. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996; 16: 675– 686. [DOI] [PubMed] [Google Scholar]
- 39. Danbolt NC. Glutamate uptake. Prog Neurobiol 2001; 65: 1– 105. [DOI] [PubMed] [Google Scholar]
- 40. Sugimoto J, Tanaka M, Sugiyama K, Ito Y, Aizawa H, Soma M, Shimizu T, Mitani A, Tanaka K.. Region-specific deletions of the glutamate transporter GLT1 differentially affect seizure activity and neurodegeneration in mice. Glia 2018; 66: 777– 788. [DOI] [PubMed] [Google Scholar]
- 41. Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP, Chimelli L, Born DE, Sakamoto AC, Assirati JA, Fried I, Peacock WJ, Ojemann GA, Adelson PD.. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 1999; 52: 453– 472. [DOI] [PubMed] [Google Scholar]
- 42. Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, van Veelen CW, van Rijen PC, van Nieuwenhuizen O, Gispen WH, de Graan PN.. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 2002; 125 Pt 1: 32– 43. [DOI] [PubMed] [Google Scholar]
- 43. During MJ, Spencer DD.. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993; 341: 1607– 1610. [DOI] [PubMed] [Google Scholar]
- 44. Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, de Lanerolle NC.. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 2004; 363: 28– 37. [DOI] [PubMed] [Google Scholar]
- 45. Petroff OA, Errante LD, Rothman DL, Kim JH, Spencer DD.. Glutamate-glutamine cycling in the epileptic human hippocampus. Epilepsia 2002; 43: 703– 710. [DOI] [PubMed] [Google Scholar]
- 46. Bedner P, Dupper A, Huttmann K, Muller J, Herde MK, Dublin P, Deshpande T, Schramm J, Haussler U, Haas CA, Henneberger C, Theis M, Steinhäuser C.. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 2015; 138 Pt 5: 1208– 1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meme W, Calvo CF, Froger N, Ezan P, Amigou E, Koulakoff A, Giaume C.. Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: potentiation by beta-amyloid. FASEB J 2006; 20: 494– 496. [DOI] [PubMed] [Google Scholar]
- 48. Khan D, Dupper A, Deshpande T, Graan PN, Steinhauser C Bedner P.. Experimental febrile seizures impair interastrocytic gap junction coupling in juvenile mice. J Neurosci Res 2016; 94: 804– 813. [DOI] [PubMed] [Google Scholar]
- 49. Boison D, Chen J, Fredholm BB.. Adenosine signalling and function in glial cells. Cell Death Diff 2010; 17: 1071– 1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boison D. Adenosine dysfunction in epilepsy. Glia 2012; 60: 1234– 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gouder N, Scheurer L, Fritschy J, Boison D.. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci 2004; 24: 692– 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jacobson KA, Gao ZG.. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 2006; 5: 247– 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spedding M, William M.. Developments in purine and pyridimidine receptor-based therapeutics. Drug Dev Res 1996; 39: 436– 441. [Google Scholar]
- 54. Williams M. Development in P2 receptor targeted therapeutics. Prog Brain Res 1999; 120: 93– 106. [DOI] [PubMed] [Google Scholar]
- 55. Williams M, Jarvis MF.. Purinergic and pyrimidinergic receptors as potential drug targets. Biochem Pharmacol 2000; 59: 1173– 1185. [DOI] [PubMed] [Google Scholar]
- 56. Anschel DJ, Ortega EL, Kraus AC, Fisher RS.. Focally injected adenosine prevents seizures in the rat. Exp Neurol 2004; 190: 544– 547. [DOI] [PubMed] [Google Scholar]
- 57. Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D.. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest 2008; 118: 571– 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilz A, Pritchard EM, Li T, Lan J, Kaplan DL, Boison D.. Silk polymer based adenosine release: therapeutic potential for epilepsy. Biomaterials 2008; 29: 3609– 3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huber A, Padrun V, Déglon N, Aebischer P, Möhler H, Boison D.. Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. PNAS 2001; 98: 7611– 7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boison D, Stewart K.. Therapeutic epilepsy research: from pharmacological rationale to focal adenosine augmentation. Biochem Pharmacol 2009; 78: 1428– 1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boison D. Adenosinergic signaling in epilepsy. Neuropharmacology 2016; 104: 131– 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Williams-Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM, Kaplan DL, Boison D.. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest 2013; 123 8: 3552– 3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada K, Gutmann DH.. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol 2002; 52: 285– 296. [DOI] [PubMed] [Google Scholar]
- 64. Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, Mennerick S, Yamada KA, Gutmann DH.. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol 2003; 54: 251– 256. [DOI] [PubMed] [Google Scholar]
- 65. Jansen LA, Uhlmann EJ, Crino PB, Gutmann DH, Wong M.. Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia 2005; 46: 1871– 1880. [DOI] [PubMed] [Google Scholar]
- 66. Zou J, Zhang B, Gutmann DH, Wong M.. Postnatal reduction of tuberous sclerosis complex 1 expression in astrocytes and neurons causes seizures in an age-dependent manner. Epilepsia 2017; 58: 2053– 2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Luyken C, Blumcke I, Fimmers R, Urbach H, Elger CE, Wiestler OD, Schramm J.. The spectrum of long-term epilepsy-associated tumors: long-term seizure and tumor outcome and neurosurgical aspects. Epilepsia 2003; 44: 822– 830. [DOI] [PubMed] [Google Scholar]
- 68. Bianchi L, De Micheli E, Bricolo A, Ballini C, Fattori M, Venturi C, Pedata F, Tipton KF, Della Corte L.. Extracellular levels of amino acids and choline in human high grade gliomas: an intraoperative microdialysis study. Neurochem Res 2004; 29: 325– 334. [DOI] [PubMed] [Google Scholar]
- 69. Maas S, Patt S, Schrey M, Rich A.. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci U S A 2001; 98: 14687– 14692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ye ZC, Sontheimer H.. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res 1999; 59: 4383– 4391. [PubMed] [Google Scholar]
- 71. Ye ZC, Rothstein JD, Sontheimer H.. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J Neurosci 1999; 19: 10767– 10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bordey A, Sontheimer H.. Electrophysiological properties of human astrocytic tumor cells in situ: enigma of spiking glial cells. J Neurophysiol 1998; 79: 2782– 2793. [DOI] [PubMed] [Google Scholar]
- 73. Olsen ML, Sontheimer H.. Mislocalization of Kir channels in malignant glia. Glia 2004; 46: 63– 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Robert SM, Buckingham SC, Campbell SL, Robel S, Holt KT, Ogunrinu-Babarinde T, Warren PP, White DM, Reid MA, Eschbacher JM, Berens ME, Lahti AC, Nabors LB, Sontheimer H.. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med 2015; 7: 289ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Annegers JF, Hauser WA, Coan SP, Rocca WA.. A population-based study of seizures after traumatic brain injuries. N Engl J Med 1998; 338: 20– 24. [DOI] [PubMed] [Google Scholar]
- 76. Caveness WF, Meirowsky AM, Rish BL, Mohr JP, Kistler JP, Dillon JD, Weiss GH.. The nature of posttraumatic epilepsy. J Neurosurg 1979; 50s: 545– 553. [DOI] [PubMed] [Google Scholar]
- 77. Temkin NR, Dikmen SS, Wilensky AJ, Keihm J, Chabal S, Winn HR.. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med 1990; 323: 497– 502. [DOI] [PubMed] [Google Scholar]
- 78. Temkin NR, Dikmen SS, Anderson GD, Wilensky AJ, Holmes MD, Cohen W, Newell DW, Nelson P, Awan A, Winn HR.. Valproate therapy for prevention of posttraumatic seizures: a randomized trial. J Neurosurg 1999; 91: 593– 600. [DOI] [PubMed] [Google Scholar]
- 79. D'Ambrosio R, Perucca E.. Epilepsy after head injury. Curr Opin Neurol 2004; 17: 731– 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK.. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci 1992; 12: 4846– 4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Golarai G, Greenwood AC, Feeney DM, Connor JA.. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci 2001; 21: 8523– 8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Santhakumar V, Ratzliff AD, Jeng J, Toth Z, Soltesz I.. Long-term hyper-excitability in the hippocampus after experimental head trauma. Ann Neurol 2001; 50: 708– 717. [DOI] [PubMed] [Google Scholar]
- 83. D'Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D.. Impaired K+ homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J Neurosci 1999; 19: 8152– 8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Samuelsson C, Kumlien E, Flink R, Lindholm D, Ronne-Engstrom E.. Decreased cortical levels of astrocytic glutamate transport protein GLT-1 in a rat model of posttraumatic epilepsy. Neurosci Lett 2000; 289: 185– 188. [DOI] [PubMed] [Google Scholar]
- 85. Hunt RF, Scheff SW, Smith BN.. Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol 2009; 215: 243– 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Seiffert E, Dreier JP, Ivens S, Bechmann I, Tomkins O, Heinemann U, Friedman A.. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci 2004; 24: 7829– 7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Weissberg I, Wood L, Kamintsky L, Vazquez O, Milikovsky DZ, Alexander A, Oppenheim H, Ardizzone C, Becker A, Frigerio F, Vezzani A, Buckwalter MS, Huguenard JR, Friedman A, Kaufer D.. Albumin induces excitatory synaptogenesis through astrocytic TGF-beta/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol Dis 2015; 78: 115– 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bar-Klein G, Cacheaux LP, Kamintsky L, Prager O, Weissberg I, Schoknecht K, Cheng P, Kim SY, Wood L, Heinemann U, Kaufer D, Friedman A.. Losartan prevents acquired epilepsy via TGF-beta signaling suppression. Ann Neurol 2014; 75: 864– 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Friedman A, Bar-Klein G, Serlin Y, Parmet Y, Heinemann U, Kaufer D.. Should losartan be administered following brain injury? Expert Rev Neurother 2014; 14: 1365– 1375. [DOI] [PubMed] [Google Scholar]
- 90. Murphy TR, Binder DK, Fiacco TA.. Turning down the volume: Astrocyte volume change in the generation and termination of epileptic seizures. Neurobiol Dis 2017; 104: 24– 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Degen J, Dublin P, Zhang J, Dobrowolski R, Jokwitz M, Karram K, Trotter J, Jabs R, Willecke K, Steinhauser C, Theis M.. Dual reporter approaches for identification of Cre efficacy and astrocyte heterogeneity. FASEB J 2012; 26: 4576– 4583. [DOI] [PubMed] [Google Scholar]
- 92. Hoft S, Griemsmann S, Seifert G, Steinhäuser C.. Heterogeneity in expression of functional ionotropic glutamate and GABA receptors in astrocytes across brain regions: insights from the thalamus. Philos Trans R Soc Lond B Biol Sci 2014; 369: 20130602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hüttmann K, Sadgrove M, Wallraff A, Hinterkeuser S, Kirchhoff F, Steinhäuser C, Gray WP.. Seizures preferentially stimulate proliferation of radial glia-like astrocytes in the adult dentate gyrus: functional and immunocytochemical analysis. Eur J Neurosci 2003; 18: 2769– 2778. [DOI] [PubMed] [Google Scholar]
- 94. Parent JM, von dem Bussche N, Lowenstein DH.. Prolonged seizures recruit caudal subventricular zone glial progenitors into the injured hippocampus. Hippocampus 2006; 16: 321– 328. [DOI] [PubMed] [Google Scholar]
- 95. Hubbard JA, Binder DK.. Astrocytes and Epilepsy. New York: Academic Press, 2016. [Google Scholar]